
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of a novel resorcin[4]arene–glucose conjugate and its catalysis of the CuAAC reaction for the synthesis of 1,4-disubstituted 1,2,3-triazoles in water†
Ali A. Husain and
Kirpal S. Bisht *
*
Department of Chemistry, University of South Florida, 4202 East Fowler Avenue, CHE 205, Tampa, FL 33620, USA. E-mail: kbisht@usf.edu
First published on 29th March 2019
Abstract
The Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) in aqueous media using resorcin[4]arene glycoconjugate (RG) is reported. The eight β-D-glucopyranoside moieties constructed on the resorcin[4]arene upper rim provide a pseudo-saccharide cavity that offers a suitable host environment for water-insoluble hydrophobic azido and/or alkyne substrates in water. The utility of RG was established as an efficient inverse phase transfer catalyst for the CuAAC in water as a green approach for the synthesis of 1,4-disubstituted 1,2,3-triazole species. The catalytic utility of RG (1 mol%) was demonstrated in a multicomponent one-pot CuAAC for various azido/alkyne substrates. The RG acts as a molecular host and a micro-reactor resulting in the 1,4-disubstituted 1,2,3-triazoles in excellent yield.
Introduction
The classical Huisgen1 cycloaddition reaction for the synthesis of 1,2,3-triazole involves thermal 1,3-dipolar cycloaddition of organic azides with alkynes, though in low yield and mixed regioselectivity. Sharpless'2 and Meldal's3 research groups later independently developed the improved procedure involving the copper(I)-catalyzed Huisgen 1,3-dipolar cycloaddition reaction, which is the widely studied ‘click’ reaction (Scheme 1). The impact of the copper catalyzed azide–alkyne click reaction in various branches of science is increasing exponentially as evidenced from numerous recent reviews available in the literature since 2010.4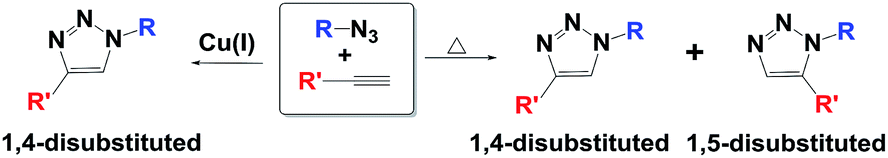 | ||
| Scheme 1 Azide–alkyne cycloaddition (AAC) reaction under different reaction conditions. | ||
The three most common facile protocols for CuAAC include (i) use of copper(I) salts (mostly in organic solvents), (ii) the reduction of a copper(II to I), and (iii) oxidation of Cu(0 to I). Of the three protocols described above, the method employing in situ reduction of copper(II) salts is known to be more practical and can be carried out in aqueous conditions. From review of literature, it is easy to conclude that water is an appropriate choice as a solvent for the CuSO4/sodium ascorbate catalyzed click protocol, which results in the formation of the triazole in high yields and with excellent regioselectivity. However, despite the efficiency of the CuAAC reaction, there are limitations to using the procedure especially when the substrates are not water-soluble. The protocol in essence requires deoxygenated conditions in the presence of mixed aprotic organic solvents such as THF, CH3CN, CH2Cl2, toluene, etc. and due to the oxidative tendency of the copper(I), a higher catalyst concentration throughout the reaction is needed. To stabilize the catalyst, several phosphine-based complexes and amine-based (bound with different heterocyclic donors) ligands have been used for rate acceleration.5 Additionally, a number of heterogeneous Cu catalysts,6 including amberlyst resin-supported,7 polymer-supported,8 and zeolite-supported9 have been explored to catalyze the triazole formation. To speed up the azide–alkyne reaction, use of surfactants and phase transfer catalyst,10 the microwave11 and ultrasound irradiations12 have also been reported.13
Recently, our research group reported resorcin[4]arene cavitand glycoconjugates (RCGs)14 as inverse phase transfer catalysts with abilities to catalyze organic reactions in aqueous media. We also reported on the RCGs ability to catalyze the formation of 1,4-disubstituted 1,2,3-triazoles in water without the addition of any co-organic solvents.14 It is noteworthy to mention that we were the first to establish the concept of the spatial directionality of β-D-glycopyranoside units on the resorcin[4]arene rigid structure “cavitand”. The RCGs possesses a unique molecular host system “pseudo-saccharide bucket” which can encapsulate organic substrates and catalyze chemical reactions in water.14
In this manuscript, we describe the synthesis of a resorcin[4]arene glycoconjugate (RG) (Fig. 1) and its application as a micro-reactor for the synthesis of 1,4-disubstituted 1,2,3-triazole species in aqueous media via the CuAAC reaction. RG structure consists of eight β-D-glucopyranoside moieties constructed on the phenolic parts on the resorcin[4]arene upper rim via multiple 1,4-disubstituted 1,2,3-triazole linkages. The eight arm resorcin[4]arene glycoconjugate offers an enlarged flexible pseudo-saccharide cavity that can act as a molecular vessel for water-insoluble azido and/or alkyne substrates in aqueous environment.
 | ||
| Fig. 1 Resorcin[4]arene glycoconjugate (RG & RCGs14). | ||
Results and discussion
Synthesis of eight arm resorcin[4]arene glycoconjugate (RG)
For the synthesis of the novel RG, resorcin[4]arene 1 (ref. 14 and 15) was first synthesized upon the acid-catalyzed cyclocondensation reaction of methyl resorcinol with heptanal. Compound 1 was then treated with propargyl bromide in the presence of potassium carbonate in refluxing acetone to achieve the octa-propargyl resorcin[4]arene intermediate 2 (Scheme 2).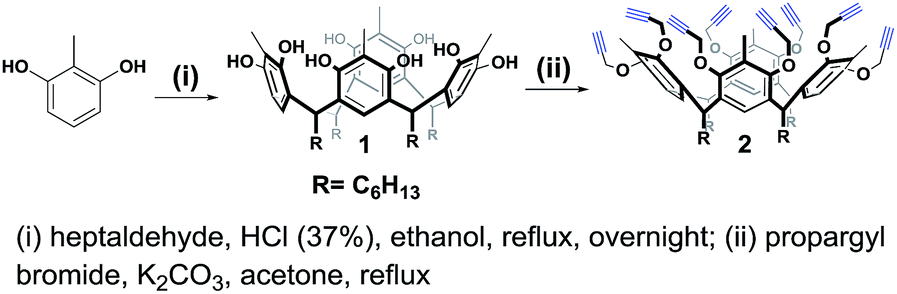 | ||
| Scheme 2 Synthesis of octa-alkyne resorcin[4]arene 2. | ||
Resorcin[4]arene 2 was characterized conclusively from its NMR and spectral data. In its 1H-NMR spectrum, the benzylic protons (–CH3, Ha) were found as a singlet at 2.25 ppm. The alkyne protons (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH, Hf) were observed as a triplet at 2.50 ppm (J = 2.4 Hz) and the propargyl methylene protons (–OCH2C, Hc,c′) showed as two set of double doublets with J = 15.3 and 2.4 Hz at 4.20 and 4.36 ppm. Its 13C-NMR spectrum had resonances for the alkyne carbons Ce and Cf at 74.8 and 79.5 ppm, respectively, and the propargyl methylene carbons (–OCH2C, Cc) were at 60.3 ppm (Fig. 2). The molecular formula (C80H96O8) of compound 2 was confirmed from its m/z measurement using ESI-Q-TOF HRMS: observed 1207.7034 (M + Na)+, calculated 1207.7003 (M + Na)+.
CH, Hf) were observed as a triplet at 2.50 ppm (J = 2.4 Hz) and the propargyl methylene protons (–OCH2C, Hc,c′) showed as two set of double doublets with J = 15.3 and 2.4 Hz at 4.20 and 4.36 ppm. Its 13C-NMR spectrum had resonances for the alkyne carbons Ce and Cf at 74.8 and 79.5 ppm, respectively, and the propargyl methylene carbons (–OCH2C, Cc) were at 60.3 ppm (Fig. 2). The molecular formula (C80H96O8) of compound 2 was confirmed from its m/z measurement using ESI-Q-TOF HRMS: observed 1207.7034 (M + Na)+, calculated 1207.7003 (M + Na)+.
 | ||
| Fig. 2 Partial 1H-, 13C- and DEPT NMR spectra (250 and 62.5 MHz, CDCl3) of 2. | ||
2-Azidoethyl β-D-glucopyranoside tetraacetate (3) was prepared following the typical procedures found in literature (Scheme 3) and unambiguously characterized.14,16
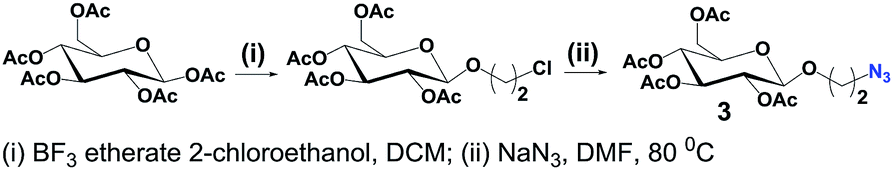 | ||
| Scheme 3 Synthesis of 3. | ||
Octa-propargyl resorcin[4]arene 2 and azido glucopyranoside 3 were then coupled together via the CuAAC reaction. The reaction was carried out in refluxing chloroform in presence of CuI (10 mol%) and DIPEA (6.0 equiv.) to yield the octaacetoxy-RG (4) in excellent yield. Global deacetylation using NaOMe solution (0.1 M) resulted in RG in gram quantity (Scheme 4).
 | ||
| Scheme 4 Synthesis of RG. | ||
The structures of the RG and its octaacetoxy precursor 4 were established from the respective 1H- and 13C-NMR data. Namely, the absence of the alkyne protons (Hf), the emergence of the triazole protons (1H, at 7.69 ppm) and the shift of the propargyl protons (Hc, from 4.11 ppm to 4.50 ppm) confirmed the structure of 4 (Fig. 3). In the 1H-NMR spectra of RG (Fig. 3b), the disappearance of the acetate protons (–OCOCH3, OAc) from the region of 1.70–2.10 ppm was confirmatory of its structure. The molecular weight of the products {for 4 m/z (ESI-Q-TOF): observed 2283.8955 (M + 2Na)2+, calculated 2283.8984 (M + 2Na)2+; for RG m/z (ESI-Q-TOF): observed 1611.7374 (M + 2Na)2+, calculated 1611.7294 (M + 2Na)2+} confirmed the respective molecular formula.
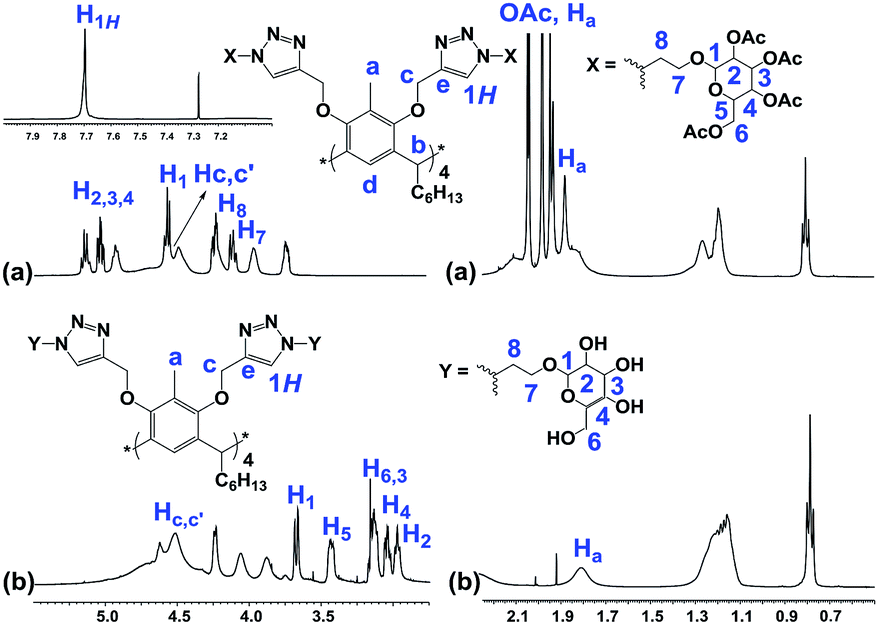 | ||
| Fig. 3 Partial 1H-NMR spectra of (a) 4 (500 MHz, CDCl3) and (b) RG (500 MHz, DMSO-d6). | ||
Optimizing CuAAC in water catalyzed by RG
The CuAAC reactions was investigated to examine the catalytic activity in aqueous environment. For optimization, the CuAAC reaction of benzyl azide (1.0 mmol) with phenyl acetylene (1.05 equiv.) with/without the addition of a catalyst (1 mol%) was carried out (Fig. 4). The reactions were performed in the presence of copper sulfate (1 mol%) and sodium L-ascorbate (3 mol%) in 10 mL of distilled deionized water at 80 °C (Fig. 4). The chemical structures and synthetic procedure for RCG catalysts evaluated in Fig. 4 have been previously reported by our research group.14 | ||
| Fig. 4 CuAAC of benzyl azide and phenyl acetylene at 80 °C. RCG catalyst structures in Fig. 1. | ||
The coupling reaction was much slower in absence of added RCG catalyst and in presence of compound 14 (Fig. 1),14 which lacks the spatial directionality of the RCGs. While the RCGs catalyzed the CuAAC reaction between benzyl azide and phenyl acetylene, there were significant differences in substrate conversion to suggest a dependence of catalytic activity on chemical structure (Fig. 4). Remarkably, the CuAAC reaction was almost completed with more than 93% conversion only in 10 minutes when RG (1 mol%) was added but only 26% conversion was observed in its absence (no catalyst). Obviously, the fast CuAAC in the presence of RG indicates that it provides a unique molecular environment that is capable of catalyzing the CuAAC reaction efficiently.
Scoping the CuAAC in water using RG
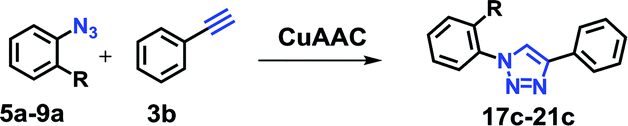
To scope the CuAAC reactions catalyzed by RG in aqueous media, we have investigated coupling of substituted benzyl azides 1a–4a with aromatic and aliphatic alkynes 1b–4b (Table 1).
As recorded in Table 1, the RG catalyzed CuAAC reactions of a variety of substituted benzyl azides in water and all reactions led to the desired 1,4-disubstituted 1,2,3-triazole products (2c–16c) in high yields (>90% isolated). Not surprisingly, it was determined that the reaction took longer to reach completion as the hydrophobicity of the substrate pair increased, as evidenced by their clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values (Table 1). For example, the coupling between the 4-bromo benzyl azide (4a, clogP = 4.13) and 1-hexyne (4b, clogP = 2.51) took nearly 65 minutes to completion while the reactions between propargyl alcohol (1b, clogP = −0.34) and 3-methoxy benzyl azide (1a, clogP = 3.18) was completed in 15 minutes.
P values (Table 1). For example, the coupling between the 4-bromo benzyl azide (4a, clogP = 4.13) and 1-hexyne (4b, clogP = 2.51) took nearly 65 minutes to completion while the reactions between propargyl alcohol (1b, clogP = −0.34) and 3-methoxy benzyl azide (1a, clogP = 3.18) was completed in 15 minutes.
To further evaluate the effectiveness of the RG as a catalyst in CuAAC of hydrophobic substrates, we carried out the coupling of phenyl acetylene (3b) with alkylated ortho-azido phenols (5a–9a) of increasing steric bulk and clogP values from 2.55–4.95. A comparative study of the CuAAC reaction in absence and presence of the RG is shown in Table 2.
|
|||||
|---|---|---|---|---|---|
| Entry | Azide (clogP value) |
Product | Time (min) | Catalyst (% yield)b | |
| None | RG | ||||
| a Reaction condition: azidophenol derivative (1 mmol) and phenyl acetylene 1b (1.05 mmol), Cu(II) (1 mol%), Na-L-ascorbate (3 mol%), cat. (1 mol%), water (10 mL), 80 °C.b Isolated yield. | |||||
| 1 | 5a R = OH (2.55) | 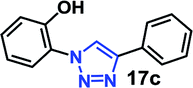 |
30 | 64 | 96 |
| 2 | 6a R = OMe (3.18) | 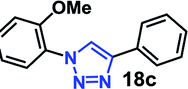 |
30 | 22 | 97 |
| 3 | 7a R = OEt (3.71) |  |
30 | 16 | 98 |
| 4 | 8a R = OBu (4.77) | 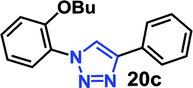 |
30 | 12 | 98 |
| 5 | 9a R = OBn (4.95) | 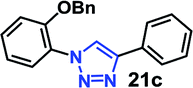 |
30 | <10 | 99 |
As expected, the coupling of o-azido phenols (5a, clogP = 2.44) with phenyl acetylene 1b catalyzed by RG resulted in triazole 17c in 96% yield and even without added catalyst the yield was 64% due to its higher hydrophilicity (entry 1). However, replacing the hydroxyl functionality with alkoxy groups, i.e. –OMe, –OEt, –OBu, –OPh of progressively higher bulk, hydrophobicity, and clogP value led to much slower reactions in absence of the RG. The reactions catalyzed by the RG resulted in near quantitative conversion in about 30 minutes irrespective of the bulk or hydrophobicity of the azide substrates, attesting to its effectiveness as a catalyst in the CuAAC reactions in water even for the much bulkier and hydrophobic substrates.
Di-CuAAC reactions in water using RG
Simultaneous multiple CuAAC reactions have found interest in synthesis of the polymers and dendrimers and the need for efficient catalyst that can catalyze reactions in water has never been greater.17 We have therefore investigated RG for catalyzing the di-CuAAC reactions in water of di-propargyl benzene derivatives 5b–7b with substituted benzyl azides 1a–4a (Table 3). Interestingly, the di-CuAAC reaction were completed within 45 min resulting in the desired bis-1,2,3-triazole products 22c–33c in gram quantities.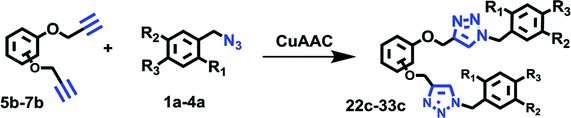

Multicomponent one-pot CuAAC reactions in water
Organic azides are not always considered safe for handling because of their toxic and shock sensitive nature and there have been alternative methods employed for their in situ synthesis. The azides can be prepared from their corresponding halides upon the addition of sodium azide. Hence, a multicomponent one-pot CuAAC reaction between in situ generated azide from its corresponding precursor and alkyne is highly desirable.18We have investigated the multicomponent one-pot CuAAC reactions of phenyl acetylene 3b with the aryl bromides and sodium azide catalyzed by the RG. Complete conversion were accomplished within 25–55 minutes even for a bulkier aryl system (naphthyl) to achieve the 1,2,3-triazoles in excellent isolated yield (90–96%) (Table 4, entries 1–5). In addition, reaction with α-bromo acetophenones (entries 6 and 7), α-bromoesters (entry 8) and ally bromide (entry 9) led to quantitative conversion within 25–30 min to the desired triazoles 35c–37c in 91–95% isolated yield. However, the multi component CuAAC failed when saturated aliphatic bromides were used as substrates, i.e., butyl, heptyl and dodecyl bromides (entries 10–12). We suspect the π–π interactions play an important role in the encapsulation, in water, of the substrates molecule in the hydrophobic cavity of the RG and the lack of these interactions in the aliphatic alkyl bromide does not allow their encapsulation in the RG cavity and hence no observed reaction.
|
|||
|---|---|---|---|
| Entry | Product | Time (min) | Yieldb (%) |
| a Reaction condition: phenyl acetylene (1 mmol), bromide derivative (1.05 mmol) and sodium azide (1.1 mmol), RG (1 mol%), water (10 mL), 80 °C.b Isolated yields, N/A = not isolated. | |||
| 1 |  |
25 | 92 |
| 2 |  |
30 | 94 |
| 3 |  |
30 | 96 |
| 4 |  |
50 | 90 |
| 5 |  |
55 | 91 |
| 6 | 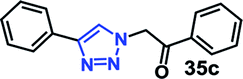 |
30 | 92 |
| 7 | 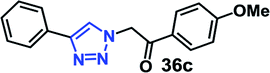 |
30 | 95 |
| 8 | 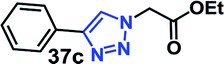 |
25 | 93 |
| 9 |  |
25 | 91 |
| 10 | 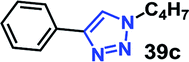 |
200 | <10 |
| 11 |  |
200 | N/A |
| 12 | 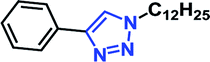 |
200 | N/A |
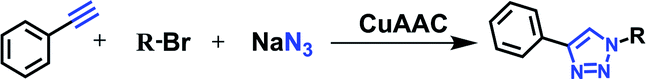
Inclusion complex of RG with benzyl azide (2a) and phenyl acetylene (3b) in D2O
We set out to probe the encapsulation of the substrates by the RG. Specifically, we have studied the encapsulation of benzyl azide and phenyl acetylene by RG using NMR. The 1H-NMR spectra of the guest (2a & 3b) were recorded in the presence of the host (RG) in a 1:1 molar ratio for 2 mM concentrated solution in D2O at 25 °C (Fig. 5). In the 1H-NMR, the aromatic protons (H2, H3, H4) in 2a were shifted up field from 7.35 ppm to 6.90 ppm upon addition of the RG (Fig. 5a). Similarly, the aromatic protons (H6, H7, H8) in 3b were shifted upfield from 7.35 and 7.45 ppm to 6.98 and 7.15 ppm, respectively upon addition of RG (Fig. 5b). The shielding of the guest 1H NMR resonances upon addition of RG indicated their encapsulation in the RG cavity. The encapsulation of the substrates in the RG cavity may explain the catalytic activity of the RG. In addition, the observation that the CuAAC reactions of the aromatic and π bond containing azides were accelerated in the presence of RG (Table 4), the encapsulation of the guest most likely involves π–π host–guest interactions.
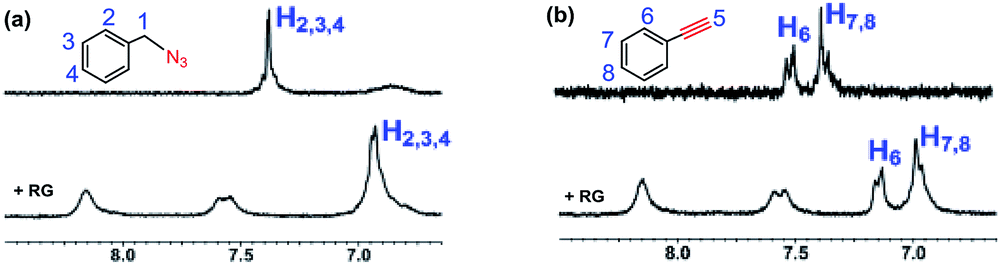 | ||
| Fig. 5 Partial 1H-NMR spectra (250 MHz, D2O, 2 mM) at 25 °C of (a) benzyl azide and benzyl azide + RG (1:1); (b) phenylacetylene and phenylacetylene + RG (1:1). | ||
Proposed mechanism for the CuAAC using RG
Based upon the reaction catalysed and the encapsulation observed, the proposed mechanism for the CuAAC reaction catalyzed by RG may proceed via step shown in Fig. 6. Starting with inclusion of the alkyne and azide substrates in the pseudo-β-D-glucopyranoside cavity of RG. The closed proximity of the alkyne and azide in the presence of Cu(I) catalyst accelerate the cycloaddition process resulting in the copper–triazole complex followed by protonation and dissociation of the desired 1,4-di substituted 1,2,3-triazole (Fig. 6).19 The binding of the Cu(I) to the multiple triazoles in the RG may also facilitate the coupling reaction inside the RG cavity. | ||
| Fig. 6 Proposed mechanism for the CuAAC in aqueous media catalyzed by RG. | ||
Conclusion
In conclusion, a new resorcinarene sugar conjugate (RG) is reported. It was determined that RG is an efficient catalyst for the CuAAC reaction in water at only 1 mol%. Based upon the reaction catalysis and the encapsulation observed, RG catalytic mechanism, we believe, begins with inclusion of the alkyne and azide substrates resulting in the copper–triazole complex followed by protonation and dissociation to the desired 1,4-di substituted 1,2,3-triazole. RG was found to catalyze the coupling of the alkyne/azide pair and also of the multicomponent alkyl bromide/sodium azide/alkyne to the 1,4-disubstituted 1,2,3-triazole products in excellent yield within short period of time in water.Experimental section
General
1H- and 13C-NMR spectra were recorded on a Bruker DRX-250, a Inova-400 and a DD-500 spectrometers. Sample concentrations were about 10% (w/v) in CDCl3 or DMSO-d6 and the J values are given in Hz. The mass spectral analyses were performed on an Aligent Technologies 6540 UHD Accurate-Mass Q-TOF LC/MS. The clogP values were calculated using ChemDraw Professional, Version 15.1.0.144.
Materials and reagents
All reagents were used with no further purification unless otherwise specified. 2-Methyl resorcinol (98%) was purchased from Acros Organics Chemical Company. Octa-hydroxy resorcin[4]arene (1) and 2-azidoethyl β-D-glucopyranoside tetraacetate (3) were synthesized following synthetic procedures reported previously.14–16Typical procedure for screening and scoping the CuAAC in water catalyzed by RG
Aryl azide (1 mmol) and aryl/alkyl alkyne (1.05 equiv.) were added to a solution of copper(II) sulfate pentahydrates, Na-ascorbate and RG (1:3:1 mol%) in 10 mL de-ionized distilled water. The reaction mixture was then heated to 80 °C for 15–65 min. After completion, the solid reaction product was filtered off, dried, and weighed to calculate the isolated yield. When the product was not solid (10c and 11c), the reaction mixture was extracted with DCM (2 × 5 mL). The combined organic layer was collected and dried over MgSO4. DCM was then removed using a rotary evaporator and 1H-NMR was taken in CDCl3 or DMSO-d6.
Typical procedure for three component one-pot CuAAC catalyzed by RG
Phenyl acetylene 1b (1 mmol) and aryl/alkyl bromides (1.05 equiv.) with sodium azide (1.1 equiv) were added to a solution of copper(II) sulfate pentahydrates, Na-ascorbate and RG (1:3:1 mol%) in 10 mL de-ionized distilled water. The reaction mixture was then heated to 80 °C for 20–55 min. After completion, the solid reaction product was filtered off, dried, and weighed to calculate the isolated yield. When the conversion was low (entries 10–12, Table 4), the reaction mixture was extracted with DCM (2 × 5 mL). The combined organic layer was collected and dried over MgSO4. The solvent was then removed using a rotary evaporator and 1H-NMR was taken in CDCl3.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Department of Chemistry and the Department of Sponsored research at the University of South Florida. The authors acknowledge the support of the Department of Chemistry NMR and Peptide core facilities directors, Dr Edwin Rivera and Dr Mohanraja Kumar, in acquisition of the NMR and HRMS spectral data.Notes and references
- R. Huisgen, G. Szeimies and L. Mobius, Chem. Ber., 1967, 100, 2494–2507 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Folkin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- C. W. TornØe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef.
- Selected reviews: C. J. Pickens, S. N. Johnson, M. M. Pressnall, M. A. Leon and C. J. Berkland, Bioconjugate Chem., 2018, 29, 686–701 CrossRef CAS PubMed; A. D. Pehere, X. Zhang and A. D. Abell, Aust. J. Chem., 2017, 70, 138–151 CrossRef; V. K. Tiwari, B. B. Mishra, K. B. Mishra, N. Mishra, A. S. Singh and X. Chen, Chem. Rev., 2016, 116, 3086–3240 CrossRef PubMed; C. Wang, D. Ikhlef, S. Kahlal, J.-Y. Saillard and D. Astruc, Coord. Chem. Rev., 2016, 316, 1 CrossRef; X. Wang, B. Huang, X. Liu and P. Zhan, Drug Discovery Today, 2016, 21, 118 CrossRef PubMed; M. S. Singh, S. Chowdhury and S. Koley, Tetrahedron, 2016, 72, 5257–5283 CrossRef; M. Arseneault, C. Wafer and J. F. Morin, Molecules, 2015, 20, 9263–9294 CrossRef PubMed; P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905–4979 CrossRef PubMed; M. V. Walter and M. Malkoch, Chem. Soc. Rev., 2012, 41, 4593 RSC; L. Liang and D. Astruc, Coord. Chem. Rev., 2011, 255, 2933 CrossRef; G. Franc and A. K. Kakkar, Chem. Soc. Rev., 2010, 39, 1536 RSC; K. D. Hanni and D. A. Leigh, Chem. Soc. Rev., 2010, 39, 1240 RSC; A. H. El-Sagheer and T. Brown, Chem. Soc. Rev., 2010, 39, 1388–1405 RSC; J. M. Holub and K. Kirshenbaum, Chem. Soc. Rev., 2010, 39, 1325–1337 RSC.
- N. Touj, A. Chakchouk-Mtibaa, L. Mansour, A. H. Harrath, J. H. Al-Tamimi, I. Ozdemir, L. Mellouli, S. Yasar and N. Hamdi, J. Organomet. Chem., 2017, 853, 49 CrossRef CAS; A. Keivanloo, M. Bakherad and M. Lotfi, Tetrahedron, 2017, 73, 5872–5882 CrossRef; A. A. Ali, M. Chetia, P. J. Saikiab and D. Sarma, RSC Adv., 2014, 4, 64388 RSC; H. A. Michaels and L. Zhu, Chem.–Asian J., 2011, 6, 2825 CrossRef PubMed; S. I. Presolski, V. Hong, S. H. Cho and M. Finn, J. Am. Chem. Soc., 2010, 132, 14570 CrossRef PubMed; G. C. Kuang, H. A. Michaels, J. T. Simmons, R. J. Clark and L. Zhu, J. Org. Chem., 2010, 75, 6540 CrossRef PubMed; P. S. Donnelly, S. D. Zanatta, S. C. Zammit, J. M. White and S. J. Williams, Chem. Commun., 2008, 2459 RSC; V. O. Rodionov, S. I. Presolski, S. Gardinier, Y. H. Lim and M. G. Finn, J. Am. Chem. Soc., 2007, 129, 12696–12704 CrossRef PubMed; V. O. Rodionov, S. I. Presolski, D. D. Diaz, V. V. Fokin and M. G. Finn, J. Am. Chem. Soc., 2007, 129, 12705 CrossRef PubMed; Q. Wang, T. R. Chan, R. Hilgraf, V. V. Fokin, K. B. Sharpless and M. G. Finn, J. Am. Chem. Soc., 2003, 125, 3192–3193 CrossRef PubMed.
- N. Sun, Z. Yu, H. Yi, X. Zhu, L. Jin, B. Hu, Z. Shen and X. Hu, New J. Chem., 2018, 42, 1612 RSC; S. Chassaing, V. Beneteau and P. Pale, Catal. Sci. Technol., 2016, 6, 923–957 RSC; J. Albadi and M. Keshavarz, Synth. Commun., 2013, 43, 2019–2030 CrossRef CAS.
- C. Girard, E. Onen, M. Aufort, S. Beauviere, E. Samson and J. Herscovici, Org. Lett., 2006, 8, 1689–1693 CrossRef CAS PubMed.
- U. Sirion, Y. J. Bae, B. S. Lee and D. Y. Chi, Synlett, 2008, 15, 2326–2330 Search PubMed.
- C. Spiteri and J. E. Moses, Angew. Chem., Int. Ed., 2010, 49, 31–33 CrossRef CAS PubMed; S. Chassaing, M. Kumarraja, A. S. S. Sido, P. Pale and J. Sommer, Org. Lett., 2007, 9, 883–886 CrossRef PubMed.
- E. Tasca, G. La Sorella, L. Sperni, G. Strukul and A. Scarso, Green Chem., 2015, 17, 1414 RSC; J.-A. Shin, Y.-G. Lim and K.-H. Lee, J. Org. Chem., 2012, 77, 4117 CrossRef CAS PubMed; J.-A. Shin, S.-J. Oh, H.-Y. Lee and Y.-G. Lim, Catal. Sci. Technol., 2017, 7, 2450 RSC.
- A. D. Moorhouse and J. E. Moses, Synlett, 2008, 14, 2089–2092 Search PubMed; P. Appukkuttan, W. Dehaen, V. V. Fokin and E. Van der Eycken, Org. Lett., 2004, 6, 4223–4225 CrossRef CAS PubMed.
- G. Cravotto, V. V. Fokin, D. Garella, A. Binello, L. Boffa and A. Barge, J. Comb. Chem., 2010, 12, 13–15 CrossRef CAS PubMed; P. Cintas, K. Martina, B. Robaldo, D. Garella, L. Boffa and G. Cravotto, Collect. Czech. Chem. Commun., 2007, 72, 1014–1024 CrossRef.
- A. Barge, S. Tagliapietra, A. Binello and G. Cravotto, Curr. Org. Chem., 2011, 15, 189–203 CrossRef CAS.
- A. A. Husain, A. Maknenko and K. S. Bisht, Chem.–Eur. J., 2016, 22, 6223 CrossRef CAS PubMed.
- S. N. Parulekar, K. Muppalla, F. R. Fronczek and K. S. Bisht, Chem. Commun., 2007, 4901 RSC; S. N. Parulekar, K. Muppalla, A. A. Husain and K. S. Bisht, RSC Adv., 2015, 5, 25477 RSC; E. E. Dueno and K. S. Bisht, Tetrahedron, 2004, 60, 10859 CrossRef CAS; E. E. Dueno and K. S. Bisht, Chem. Commun., 2004, 954 RSC; R. Wu, T. F. Al-Azemi and K. S. Bisht, Chem. Commun., 2009, 1822 RSC; R. Wu, T. F. Al-Azemi and K. S. Bisht, RSC Adv., 2014, 4, 16864 RSC.
- R. Singh and A. Varma, Green Chem., 2012, 14, 348–356 RSC; P. Chong and P. Petillo, Org. Lett., 2000, 2, 1093–1096 CrossRef CAS PubMed.
- J. Garcia-Alvarez, J. Gimeno and A. J. L. Pombeiro, Adv. Organomet. Chem. Catal., 2014, 199–206 CAS.
- S. Hassana and T. J. Muller, Adv. Synth. Catal., 2015, 357, 617–666 CrossRef.
- B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457–460 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra for new compounds are included. See DOI: 10.1039/c9ra00972h |
| This journal is © The Royal Society of Chemistry 2019 |