
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrophilic activation of nitroalkanes in efficient synthesis of 1,3,4-oxadiazoles†
Alexander V. Aksenov *a,
Vladislav Khamraeva,
Nicolai A. Aksenova,
Nikita K. Kirilova,
Dmitriy A. Domenyukb,
Vladimir A. Zelenskyb and
Michael Rubin*ac
*a,
Vladislav Khamraeva,
Nicolai A. Aksenova,
Nikita K. Kirilova,
Dmitriy A. Domenyukb,
Vladimir A. Zelenskyb and
Michael Rubin*ac
aDepartment of Chemistry, North Caucasus Federal University, 1a Pushkin St., Stavropol 355009, Russian Federation. E-mail: alexaks05@rambler.ru
bDepartment of General Practice Dentistry and Child Dentistry, Stavropol State Medical University, 310 Mira Street, Stavropol 355017, Russian Federation
cDepartment of Chemistry, University of Kansas, 1567 Irving Hill Road, Lawrence, KS 66045-7582, USA. E-mail: mrubin@ku.edu; Tel: +1-785-864-5071
First published on 26th February 2019
Abstract
A novel methodology for general and chemoselective preparation of non-symmetric 1,3,4-oxadiazoles is developed. This unusual reaction proceeds via polyphosphoric acid-assisted activation of nitroalkanes towards nucleophilic attack with acylhydrazides.
Introduction
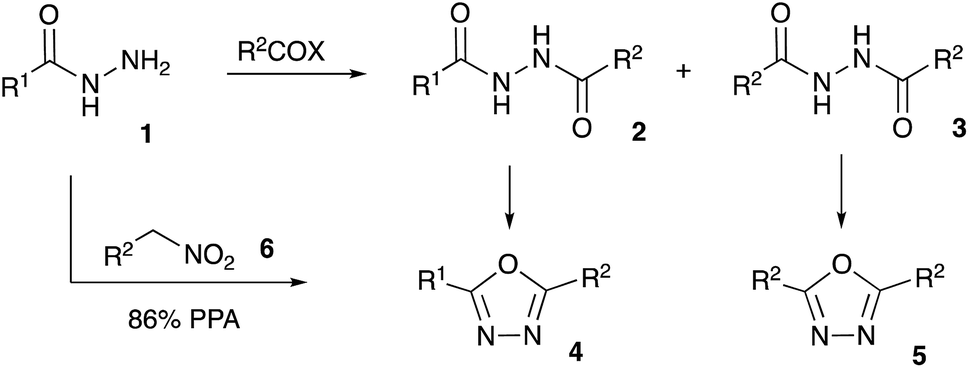
Bioisosteric to carboxylate and carboxamide functionalities, 1,3,4-oxadiazoles are often considered among other privileged heterocyclic scaffolds for drug discovery.1 Molecules possessing these key structural fragments have demonstrated a wide variety of important biological properties, including antibacterial, antimycobacterial, antifungal, insecticidal, herbicidal, anti-inflammatory, analgesic, anticonvulsant and anticancer activities.2 Several medicinal agents featuring this heterocyclic ring have been marketed, including anti-HIV drug Raltegravir, antihypertensive Nesapidil, and anti-cancer agent Zibotentan (Fig. 1). This heterocyclic building block was also used for the construction of chiral catalysts3 and metal-selective chemo-sensors.4 A symmetrically substituted version of this system (5) can be easily obtained by cyclo-condensation of hydrazine hydrate with a variety of carbonyl compounds.5 Assembly of 1,3,4-oxazoles 4 with two different substituents is much more challenging, as it involves the selective reaction of hydrazine with two different carbonyl compounds, or their synthetic equivalents, to obtain non-symmetric N,N′-diacylhydrazide precursor 2 (Scheme 1). Such processes, especially when carried out in one-pot fashion, can be significantly complicated by side reactions involving trans-acylation of N,N′-diacylhydrazide intermediates with excess of the second carbonyl compound and leading to the formation of a mixture of non-symmetric (4) and symmetric (5) products. A variety of methods were suggested in an attempt to address this issue, most of which are dealing with the moderation of the reactivity of the acylating reagent.6 In addition, different schemes involving cyclo-condensations of N′-alkylidene acylhydrazides,7 or re-cyclizations of tetrazoles,8 were also suggested. Herein we wish to report a new preparative method for selective assembly of non-symmetrically substituted 1,3,4-oxadiazoles employing nitroalkanes electrophilicity activated in the presence of polyphosphoric acid that serves as an acylating agent equivalent.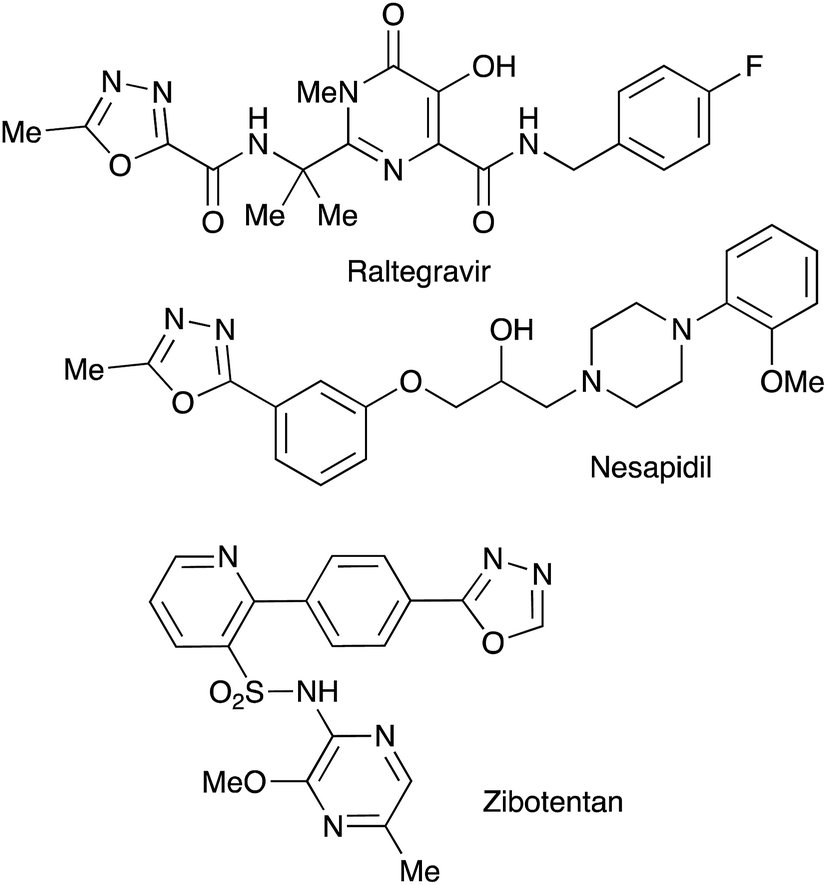 | ||
| Fig. 1 1,3,4-Oxazoles in drug discovery and medicinal chemistry. | ||
 | ||
| Scheme 1 | ||
Results and discussion
For several years our group had a great interest in the development of novel acid-mediated cascade transformations of nitroalkenes and nitroalkanes targeting material science and medicinal chemistry applications. It was demonstrated that nitroalkanes 6 dissolved in polyphosphoric acid (PPA) transform into phosphorylated nitronate 7 showing strong electrophilic properties. This unusual species can be used to design one-pot multi-step transformations involving various carbon-based nucleophiles.9 The utilization of nucleophilic amines was also demonstrated.10 Mechanistically, the latter process is related to the classical Nef reaction,11 employing aniline species 8 instead of water. This reaction provides imidinium ion 9, that can be further employed as a convenient building block for the synthesis of heterocyclic compounds: oxazoles 10, imidazoles 11, and diazines 12–13 (Scheme 2).10 We wondered about the possibility of employing acylhydrazides 1 as nucleophilic components en route to the 1,3,4-oxazoles scaffolds. Indeed, such a nucleophilic attack to nitronate species 7 would afford (2-acylhydrazineyl)alkaniminium species 14,12 that should be well suited for an intramolecular 5-exo-trig cyclization involving the carbonyl group of the hydrazide function and providing 4,5-dihydro-1,3,4-oxadiazol-3-ium ion 15. The latter after deprotonation is expected to form 2,3-dihydro-1,3,4-oxadiazole 16, which after the elimination of O-phosphorylated hydroxylamine would provide desired 1,3,4-oxadizole 4 (Scheme 2).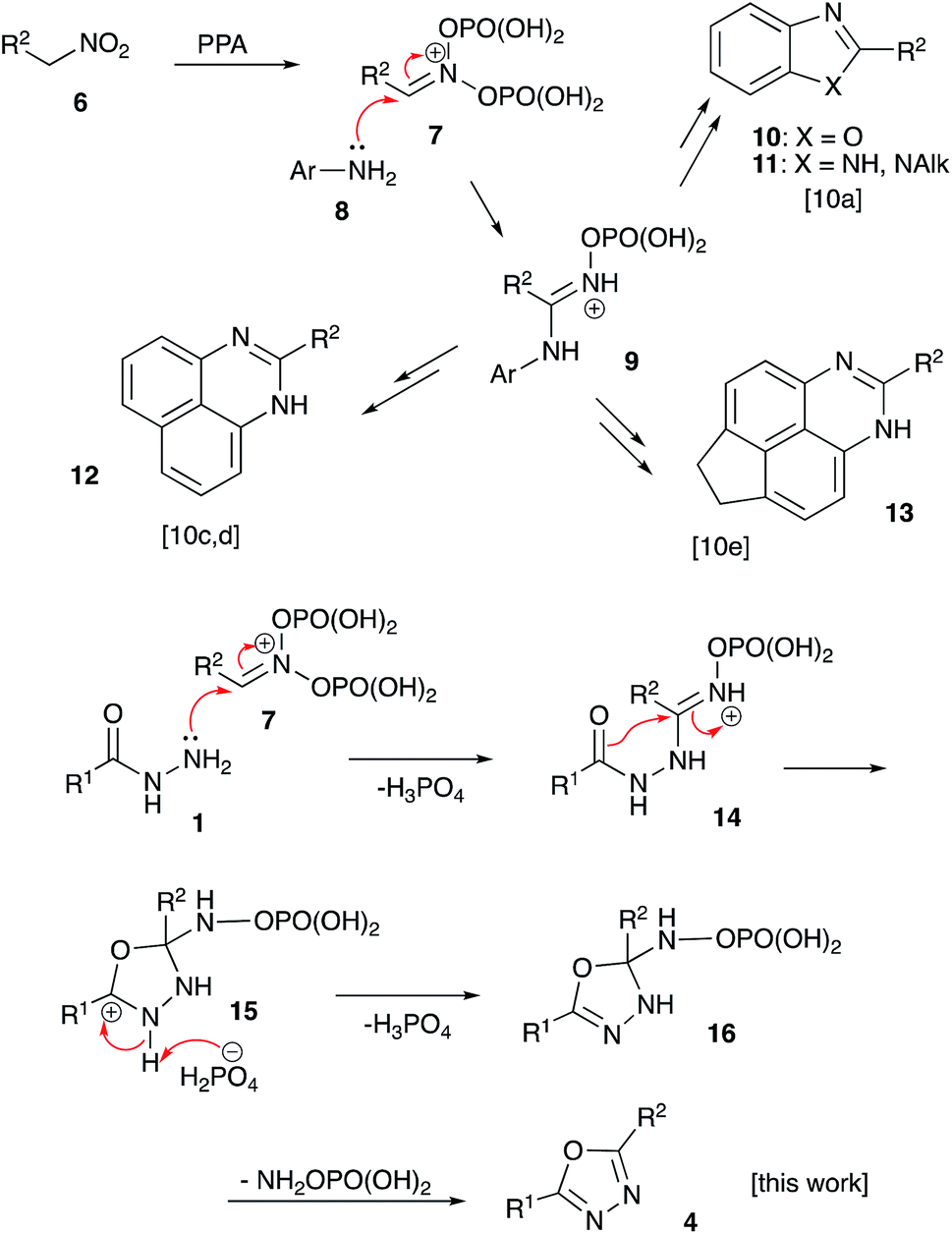 | ||
| Scheme 2 | ||
To test this idea, a mixture benzohydrazide (1a, R1 = Ph) and nitroethane (6a, R2 = Me, 2.00 equiv.) was stirred in polyphosphoric acid (86% P2O5) at 70 °C (Table 1, entry 1). However, even after 90 min no reaction was observed. At 90 °C the reaction proceeded slowly and after 30 min the conversion reached 14%, however the main product of the reaction was 2,5-diphenyl-1,3,4-oxadiazole (5a, R2 = Ph). At 110 °C (boiling point of nitroethane) the process proceeded to completion affording mixtures of 10% of non-symmetric product 4aa and 55% symmetric oxadiazole 5a (entry 3). The rest of the starting material polymerized forming highly polar resins. Employment of nitroethane (6a) in excess (3.00 equiv.) did not notably improve the reaction outcome (entries 4 and 5). Evidently, the symmetric product (5a) was formed via acid-mediated trans-acylation of 1a to produce intermediate symmetric N,N′-diacylhydrazide 3a (R2 = Ph). In order to suppress this undesired process, we decided to add starting hydrazide 1a in small portions over an extended period of time. This resulted in only marginal improvement, however, in the reactions carried out in the presence of stoichiometric amount of nitroethane (entry 6). Gradual increase of the concentration of nitroethane (entries 7–10) resulted in both the conversion and chemoselectivity of the process to achieve exclusive formation of product 5a at 99% yield (entry 10). We also tested the possibility of employing Eaton's reagent (P2O5 in MeSO3H) as a mixture that is often used as an alternative to PPA in synthesis, but in this medium hydrazide 6a underwent complete hydrolysis to afford benzoic acid as the only detectible product of the reaction (entries 11 and 12).
| 6a (equiv.) | Medium | T, °C (time) | 4aa![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5aa :5aa |
|
|---|---|---|---|---|
| a NMR yields of compounds 4a and 5a are shown.b Benzohydrazide was added in a single portion.c Benzohydrazide was slowly added by small portions.d Complete hydrolysis of benzohydrazide was observed, affording benzoic acid as sole product. | ||||
| 1 | 1.00 | 86% PPA | 70 (1.5 h)b | 0:0 |
| 2 | 1.00 | 90 (0.5 h)b | 0:14 |
|
| 3 | 1.00 | 110 (0.5 h)b | 10:55 |
|
| 4 | 3.00 | 90 (0.5 h)b | 0:17 |
|
| 5 | 3.00 | 110 (1 h)b | 31:61 |
|
| 6 | 1.00 | 110 (1 h)c | 12:38 |
|
| 7 | 2.00 | 110 (1 h)c | 63:25 |
|
| 8 | 3.00 | 110 (1 h)c | 64:15 |
|
| 9 | 5.00 | 110 (1.5 h)c | 94:6 |
|
| 10 | 7.00 | 110 (1.5 h)c | 99:0 |
|
| 11 | 3.00 | Eaton's reagent 1:7 |
110 (1 h) | 0:0d |
| 12 | 3.00 | Eaton's reagent 1:10 |
110 (1 h) | 0:0d |
With the optimized reaction conditions in hand we carried out the reaction between 1a and 6a in preparative scale (1.00 mmol). We were pleased to find, that oxadiazole 4aa was obtained in this reaction as sole product and isolated in high yield (Scheme 3, entry 1). Hydrazides 1b–g, derived from other aryl- and hetarylcarboxylic acids, also reacted with nitroethane (6a) smoothly, affording the corresponding heterocyclic products (entries 2–7). Remarkably, non-protected phenols 1e and 1f also reacted uneventfully, which showcases the dual role of polyphosphoric acid as a versatile reaction medium with acidic properties and as a reagent for the reversible installation of temporary protecting phosphatyl groups.
 | ||
| Scheme 3 | ||
Thorough analysis of literature revealed that synthetic access to monosubstituted oxadiazoles is especially challenging, since preparation of non-symmetric bis-hydrazides 2 required for this cyclo-condensation is severely complicated by relatively high strength formic acid and elevated electrophilicity of formohydrazide derivatives.13 This problem can be typically circumvented by usage of orthoformates as acylating agents.14 We felt, however, that in our featured methodology, there should not be a dramatic difference between reactivity of nitroethane (6a) and nitromethane (6b). This would allow for the development of a very general synthetic protocol to access 1,3,4-oxadiazoles with different substitution patterns. To test this possibility, we carried out reactions with a series of benzohydrazides (1a–g) in the presence of excess nitromethane (6b) at 100 °C (boiling point of nitromethane). Gratifyingly, these reactions proceeded smoothly affording the corresponding 2-aryl-1,3,4-oxadiazoles 4ab–4gb in high yields (Scheme 3, entries 8–14). Along the same lines, we tested the possibility of employing (2-nitroethyl)benzenes (6c–d) as electrophilic components in the featured transformation. These reactions were more sluggish, probably due to excessive steric hindrance in the phosphorylated nitronate intermediates 7. It was found, however, that the cyclo-condensation could be pushed to complete conversion at 140 °C to afford the corresponding 5-benzyloxadiazoles 4ac, 4ad, and 4cd albeit in somewhat lower yields (entries 15–17).
Another major challenge in chemistry of 1,3,4-oxadiazoles is associated with incorporation of ester substituents at C-2 of this heterocyclic scaffold.15 To address this issue we attempted the reaction of various benzohydrazides (1a,b,e) with ethyl 2-nitroacetate. We were pleased to discover that this reaction also proceeded smoothly under standard reaction conditions (temperature was raised to 130 °C to ensure complete conversion) affording the corresponding 1,3,4-oxadiazole-2-carboxylates as the sole isolable product (Scheme 3, entries 18–20).
The formation and structural identity of compounds 5a, 4ga, and 4ca was unambiguously confirmed by a single crystal X-ray crystallography (Fig. 2).
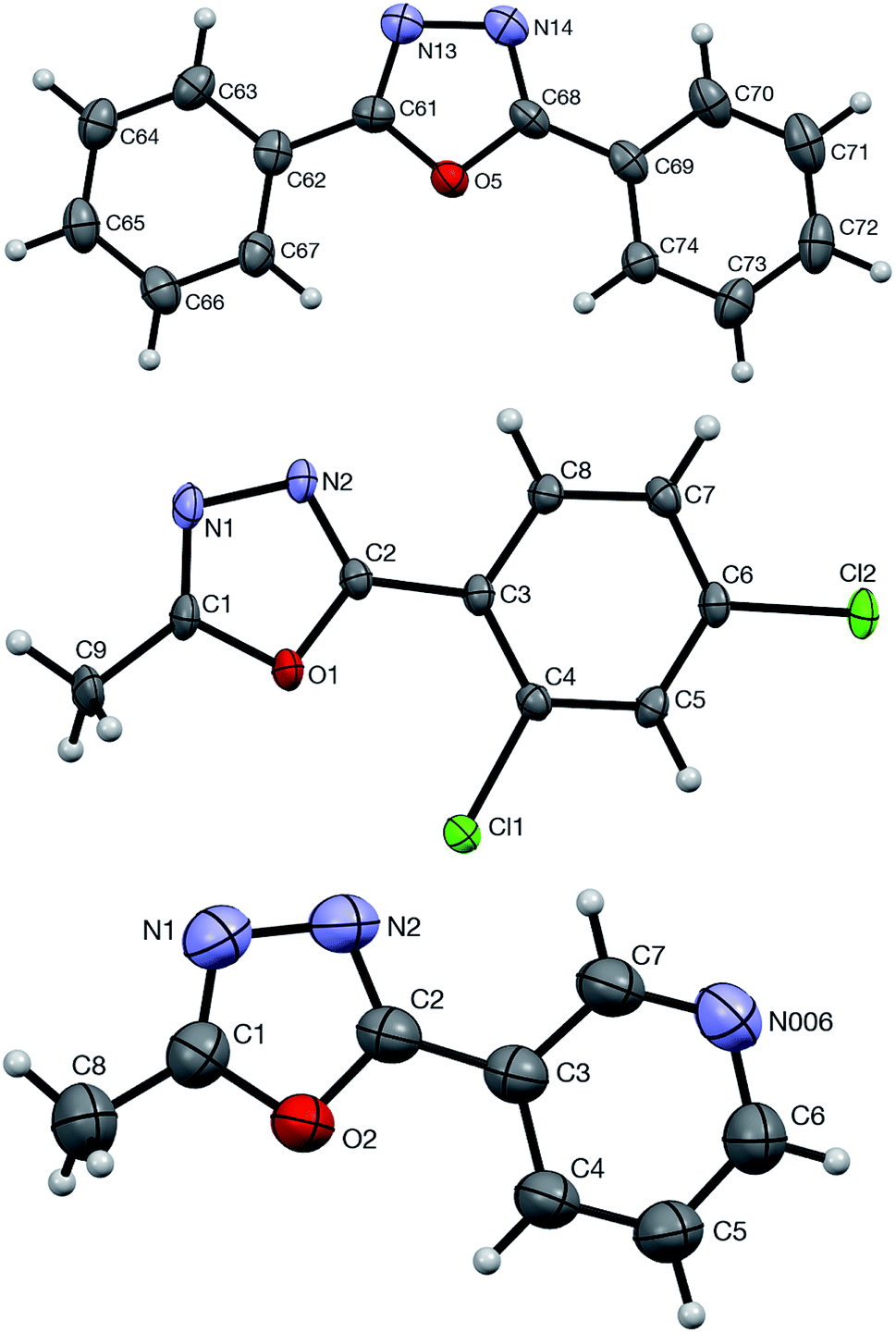 | ||
| Fig. 2 ORTEP drawings of 5a (top, CCDC #1875720), 4ga (middle, CCDC #1875721), and 4ca (bottom, CCDC #1875723) showing atom numbering schemes and 50% probability ellipsoids. | ||
Conclusion
In conclusion, the novel one-pot multistep reaction sequence involving cyclo-condensation of acylhydrazides with electrophilicity-activated nitroalkanes was investigated. Careful optimization of the reaction conditions resulted in excellent chemo-selectivity of this newly developed synthetic protocol towards monosubstituted and non-symmetrically disubstituted 1,3,4-oxadiazoles. High yields of the resulting heterocyclic products and great functional group compatibility of this synthetic methodology was demonstrated.Experimental part
General information
1H and 13C NMR spectra were recorded on a Bruker Avance-III spectrometer (400 or 100 MHz, respectively) equipped with a BBO probe in CDCl3 or DMSO-d6, using TMS as an internal standard. High-resolution mass spectra were registered with a Bruker Maxis spectrometer (electrospray ionization, in MeCN solution, using HCO2Na–HCO2H for calibration). Melting points were measured with a Stuart smp30 apparatus. All reactions were performed in oven-dried 5 mL round-bottomed flasks open to the atmosphere, employing overhead stirring. The reaction progress and purity of isolated compounds were controlled by TLC on Silufol UV-254 plates, with EtOAc as eluent. PPA was obtained by dissolving of P2O5 in 85% orthophosphoric acid according to the published protocol.16 All reagents and solvents were purchased from commercial vendors and used as received. Physical and spectral properties of compound 5a were identical to those reported in literature.17:1 to obtain the titled compound as a colorless solid, mp 63–65 °C (EtOAc), Rf 0.39 (EtOAc/petroleum ether, 1:1). Yield 146 mg (0.91 mmol, 91%). 1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 7.8 Hz, 2H), 7.55–7.46 (m, 3H), 2.61 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 165.0, 163.8, 131.7, 129.1, 126.8, 124.0, 11.3; IR (KBr, film, cm−1): 3060, 2943, 1582, 1556, 1487, 1450, 1252, 1095, 1073, 1021, 959; HRMS (ESI TOF) calculated for C9H8N2NaO (M + Na)+ 183.0529, found 183.0531 (1.3 ppm).:3). Yield 199 mg (83%). 1H NMR (400 MHz, CDCl3) δ 7.89 (d, J = 7.7 Hz, 1H), 7.74 (d, J = 8.0 Hz, 1H), 7.44 (t, J = 7.6 Hz, 1H), 7.38 (t, J = 7.7 Hz, 1H), 2.64 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 164.3, 164.0, 134.6, 132.5, 131.7, 127.7, 125.5, 121.7, 11.3; IR (KBr, film, cm−1) 3071, 2932, 1586, 1435, 1347, 1241, 1098, 1021, 959; HRMS (ESI TOF) calculated for C9H7BrN2NaO (M + Na)+ 260.9634, found 260.9630 (1.7 ppm).:1). Yield 159 mg (0.99 mmol, 99%). 1H NMR (400 MHz, CDCl3) δ 9.25 (s, 1H), 8.77 (d, J = 4.7 Hz, 1H), 8.38 (d, J = 8.0 Hz, 1H), 7.50 (dd, J = 7.9, 5.0 Hz, 1H), 2.65 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 164.5, 162.7, 151.8, 147.2, 134.7, 124.2, 120.9, 11.9; IR (KBr, film, cm−1) 3082, 3049, 2932, 1593, 1575, 1549, 1468, 1432, 1351, 1259, 1087, 1010, 988, 959; HRMS (ESI TOF) calculated for C8H7N3NaO (M + Na)+ 184.0481, found 183.0482 (0.2 ppm).:1). Yield 191 mg (0.93 mmol, 93%). 1H NMR (400 MHz, CDCl3) δ 8.37 (d, J = 8.7 Hz, 2H), 8.22 (d, J = 8.6 Hz, 2H), 2.67 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 164.9, 163.4, 149.6, 129.6, 127.8, 124.5, 11.3; IR (KBr, film, cm−1) 3108, 3071, 3016, 2932, 1578, 1553, 1520, 1351, 1333, 1311, 1292, 1234, 1087, 1032, 867; HRMS (ESI TOF) calculated for C9H7N3NaO3 (M + Na)+ 228.0380, found 228.0378 (0.8 ppm).:3). Yield 143 mg (0.87 mmol, 87%). 1H NMR (400 MHz, CDCl3) δ 10.12 (br, s, 1H), 7.73 (dd, J = 7.9, 1.5 Hz, 1H), 7.47–7.38 (m, 1H), 7.12 (d, J = 8.4 Hz, 1H), 7.04–6.94 (m, 1H), 2.65 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 164.7, 162.6, 157.6, 133.6, 126.5, 120.0, 117.7, 108.3, 11.2; IR (KBr, film, cm−1) 3189, 3071, 2976, 2943, 1629, 1593, 1549, 1490, 1439, 1410, 1355, 1300, 1241, 1080, 1040, 963; HRMS (ESI TOF) calculated for C9H8N2NaO2 (M + Na)+ 199.0478, found 199.0476, (1.0 ppm).:1). Yield 146 mg (0.83 mmol, 83%). 1H NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H), 7.79 (d, J = 8.7 Hz, 2H), 6.93 (d, J = 8.7 Hz, 2H), 2.53 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 164.1, 163.1, 160.6, 128.3, 116.2, 114.4, 10.6. IR (KBr, film, cm−1) 3156, 3020, 2947, 1611, 1589, 1494, 1377, 1289, 1234, 1164, 1120, 1073, 933, 853; HRMS (ESI TOF) calculated for C9H8N2NaO2 (M + Na)+ 199.0478, found 199.0482 (1.9 ppm).:1). Yield 199 mg (0.87 mmol, 87%). 1H NMR (400 MHz, CDCl3) δ 7.92 (d, J = 8.5 Hz, 1H), 7.56 (d, J = 1.9 Hz, 1H), 7.39 (dd, J = 8.5, 1.9 Hz, 1H), 2.64 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 164.4, 162.7, 138.1, 133.9, 131.9, 131.3, 127.7, 121.9, 11.3; IR (KBr, film, cm−1) 3104, 3086, 3009, 2936, 1589, 1564, 1454, 1417, 1270, 1241, 1106, 1018, 963, 879; HRMS (ESI TOF) calculated for C9H6Cl2N2NaO (M + Na)+ 250.9749, found 250.9747 (0.8 ppm).:3). Yield 119 mg (0.81 mmol, 81%). 1H NMR (400 MHz, CDCl3) δ 8.48 (s, 1H), 8.11–8.05 (m, 2H), 7.57–7.49 (m, 3H); 13C NMR (101 MHz, CDCl3) δ 164.9, 152.8, 132.2, 129.3, 127.2, 123.6; IR (KBr, film, cm−1) 3064, 1578, 1556, 1487, 1450, 1355, 1248, 1091, 1076, 1025, 959, 930; HRMS (ESI TOF) calculated for C8H6N2NaO (M + Na)+ 169.0372, found 169.0373 (0.3 ppm).:3). Yield 162 mg (0.72 mmol, 72%). 1H NMR (400 MHz, CDCl3) δ 8.57 (s, 1H), 7.94 (d, J = 7.7 Hz, 1H), 7.77 (d, J = 7.9 Hz, 1H), 7.48 (t, J = 7.5 Hz, 1H), 7.41 (t, J = 7.7 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 163.9, 153.2, 134.7, 132.9, 131.9, 127.8, 125.1, 121.9; IR (KBr, film, cm−1) 3119, 3002, 1597, 1575, 1516, 1457, 1432, 1234, 1109, 1084, 1025, 952; HRMS (ESI TOF) calculated for C8H5BrN2NaO (M + Na)+ 246.9477, found 246.9479 (0.6 ppm).:1). Yield 116 mg (0.79 mmol, 79%). 1H NMR (400 MHz, CDCl3) δ 9.31 (s, 1H), 8.84–8.76 (m, 1H), 8.55 (s, 1H), 8.39 (d, J = 8.0 Hz, 1H), 7.49 (dd, J = 8.0, 4.9 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 162.9, 153.2, 152.9, 148.2, 134.6, 124.0, 120.2, 77.2; IR (KBr, film, cm−1) 3332, 3071, 2998, 1608, 1586, 1556, 1512, 1468, 1432, 1267, 1131, 1102, 1073, 1021, 952, 897; HRMS (ESI TOF) calculated for C7H5N3NaO (M + Na)+ 170.0325, found 170.0330 (3.1 ppm).:2). Yield 165 mg (0.86 mmol, 86%). 1H NMR (400 MHz, CDCl3) δ 8.58 (s, 1H), 8.40 (d, J = 8.9 Hz, 2H), 8.30 (d, J = 8.9 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 163.3, 153.6, 149.9, 129.1, 128.3, 124.6; IR (KBr, film, cm−1) 3163, 3108, 3075, 3009, 1611, 1560, 1520, 1333, 1311, 1296, 1113, 1065, 948, 853; HRMS (ESI TOF) calculated for C8H5N3NaO3 (M + Na)+ 214.0223, found 214.0225 (1.1 ppm).:3). Yield 131 mg (0.81 mmol, 81%); 1H NMR (400 MHz, CDCl3) δ 10.06 (s, 1H), 8.47 (s, 1H), 7.79 (d, J = 7.8 Hz, 1H), 7.47 (t, J = 7.7 Hz, 1H), 7.14 (d, J = 8.4 Hz, 1H), 7.02 (t, J = 7.5 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 164.5, 157.8, 151.5, 134.2, 126.9, 120.2, 117.8, 107.9; IR (KBr, film, cm−1) 3222, 3148, 3068, 1769, 1728, 1626, 1586, 1549, 1490, 1406, 1303, 1256, 1157, 1095, 1058, 952; HRMS (ESI TOF) calculated for C8H6N2NaO2 (M + Na)+ 185.0321, found 185.0320 (1.0 ppm).:1). Yield 130 mg (0.81 mmol, 81%). 1H NMR (400 MHz, CDCl3) δ 10.33 (s, 1H), 9.23 (s, 1H), 7.85 (d, J = 8.6 Hz, 2H), 6.95 (d, J = 8.6 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 163.8, 160.9, 153.8, 128.7, 116.2, 114.0; IR (KBr, film, cm−1) 3147, 3027, 1615, 1597, 1498, 1384, 1285, 1245, 1179, 1128, 1065, 966, 941, 853; HRMS (ESI TOF) calculated for C8H6N2NaO2 (M + Na)+: 185.0321, found 185.0326 (2.3 ppm).:2). Yield 192 mg (0.89 mmol, 89%). 1H NMR (400 MHz, CDCl3) δ 8.57 (s, 1H), 7.96 (d, J = 8.5 Hz, 1H), 7.58 (d, J = 1.9 Hz, 1H), 7.41 (dd, J = 8.5, 1.8 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 162.7, 153.2, 138.6, 134.2, 132.1, 131.4, 127.8, 121.4; IR (KBr, film, cm−1) 3314, 3204, 3093, 3027, 1659, 1633, 1593, 1512, 1457, 1377, 1307, 1252, 1106, 1058, 959, 893, 864; HRMS (ESI TOF) calculated for C8H4Cl2N2NaO (M + Na)+ 236.9593, found 236.9588 (2.1 ppm).:3). Yield 139 mg (0.59 mmol, 59%). 1H NMR (400 MHz, CDCl3) δ 8.04–7.97 (m, 2H), 7.52–7.45 (m, 3H), 7.36 (d, J = 4.3 Hz, 3H), 7.31 (dd, J = 8.6, 4.2 Hz, 1H), 4.29 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 165.4, 165.3, 134.0, 131.8, 129.12, 129.08, 129.0, 127.7, 127.0, 124.0, 32.1; IR (KBr, film, cm−1) 3075, 2928, 2858, 1567, 1553, 1490, 1454, 1424, 1256, 1095, 1065, 1010, 959; HRMS (ESI TOF) calculated for C15H12N2NaO (M + Na)+: 259.0842, found 259.0842 (0.0 ppm).:3). Yield 162 mg (0.60 mmol, 60%). 1H NMR (400 MHz, CDCl3) δ 8.00 (dd, J = 8.0, 1.4 Hz, 2H), 7.53–7.45 (m, 3H), 7.32 (q, J = 8.6 Hz, 4H), 4.25 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 165.4, 164.9, 133.7, 132.4, 131.9, 130.3, 129.3, 129.2, 127.0, 123.8, 31.4; IR (KBr, film, cm−1) 3068, 2928, 2855, 1553, 1490, 1450, 1380, 1194, 1087, 1010, 961; HRMS (ESI TOF) calculated for C15H11ClN2NaO (M + Na)+ 293.0452, found 293.0460 (2.7 ppm).:3). Yield 146 mg (0.54 mmol, 54%). 1H NMR (400 MHz, CDCl3) δ 9.22 (s, 1H), 8.77 (d, J = 4.3 Hz, 1H), 8.35 (d, J = 7.9 Hz, 1H), 7.49 (dd, J = 7.8, 4.9 Hz, 1H), 7.33 (q, J = 8.5 Hz, 4H), 4.28 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 165.7, 163.1, 151.8, 147.2, 134.9, 134.0, 132.0, 130.4, 129.4, 124.2, 120.7, 31.4; IR (KBr, film, cm−1) 3090, 3064, 2958, 2925, 2855, 1567, 1490, 1413, 1256, 1186, 1084, 1007, 963, 853; HRMS calculated for C14H10ClN3NaO (M + Na)+ 294.0405, found 293.0395, (3.2 ppm).:3). Yield 136 mg (0.64 mmol, 64%). 1H NMR (400 MHz, CDCl3) δ 8.20–8.14 (m, 2H), 7.63–7.58 (m, 1H), 7.57–7.52 (m, 2H), 4.56 (q, J = 7.1 Hz, 2H), 1.48 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 166.6, 156.6, 154.6, 133.0, 129.4, 127.8, 122.9, 63.7, 14.3; IR (KBr, film, cm−1) 3078, 2994, 2941, 1748, 1625, 1546, 1478, 1452, 1377, 1242, 1190, 1163, 1070, 1017, 841, 792, 713, 691; HRMS (ESI TOF) calculated for C11H10N2NaO3 (M + Na)+ 241.0584, found 241.0577 (2.8 ppm).:3). Yield 205 mg (0.69 mmol, 69%). 1H NMR (400 MHz, CDCl3) δ 8.00 (dd, J = 7.6, 1.8 Hz, 1H), 7.79 (dd, J = 7.8, 1.3 Hz, 1H), 7.49 (td, J = 7.5, 1.4 Hz, 1H), 7.44 (td, J = 7.7, 1.9 Hz, 1H), 4.56 (q, J = 7.1 Hz, 2H), 1.48 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 165.5, 157.0, 154.5, 134.9, 133.5, 132.2, 127.9, 124.4, 122.2, 63.8, 14.2; IR (KBr, film, cm−1) 2985, 1748, 1598, 1535, 1441, 1377, 1298, 1253, 1186, 1167, 1100, 1028, 841, 770, 732; HRMS (ESI TOF) calculated for C11H9BrN2NaO3 (M + Na)+ 318.9689, found 318.9692 (1.0 ppm).:3). Yield 164 mg (0.70 mmol, 70%). 1H NMR (400 MHz, CDCl3) δ 9.92 (s, 1H), 7.87 (dd, J = 7.9, 1.6 Hz, 1H), 7.53–7.46 (m, 1H), 7.14 (d, J = 8.0 Hz, 1H), 7.06–7.00 (m, 1H), 4.56 (q, J = 7.1 Hz, 2H), 1.48 (t, J = 7.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 166.2, 158.4, 155.1, 154.2, 135.1, 127.4, 120.4, 118.0, 107.2, 63.9, 14.2; IR (KBr, film, cm−1) 3245, 2986, 1748, 1625, 1542, 1493, 1377, 1313, 1242, 1186, 1163, 1058, 1021, 852, 751, 710, 676; HRMS (ESI TOF) calculated for C11H10N2NaO4 (M + Na)+ 257.0533, found 257.0532 (0.5 ppm).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financed by the Russian Foundation for Basic Research (grant #18-33-20021 mol_a_ved) grants from the Ministry of Education and Science of the Russian Federation #4.1196.2017/4.6 and #4.4589.2017/6.7.Notes and references
- S. Sharma, P. K. Sharma, N. Kumar and R. Dudhe, Pharma. Chemica., 2010, 2, 253–263 CAS.
- H. Khalilullah, M. J. Ahsan, M. Hedaitullah, S. Khan and B. Ahmed, Mini-Rev. Med. Chem., 2012, 12, 789 CrossRef CAS PubMed.
- Y. Zhou, W. H. Wang, W. Dou, X.-L. Tang and W.-S. Liu, Chirality, 2008, 20, 110–114 CrossRef CAS PubMed.
- (a) X. L. Tang, X. H. Peng, W. Dou, J. Mao, J. R. Zheng, W. W. Qin, W.-S. Liu, J. Chang and X.-J. Yao, Org. Lett., 2008, 10, 3653–3656 CrossRef CAS PubMed; (b) J. Zhang, L. Zhou, H. A. Al-Attar, K. Shao, L. Wang, D. Zhu, Z. Su, M. R. Bryce and A. P. Monkman, Adv. Funct. Mater., 2013, 23, 4667–4677 CAS.
- See, for example: V. K. Tandon and R. B. Chhor, Synth. Commun., 2001, 31, 1727–1732 CrossRef CAS.
- See, for example: (a) B. J. Brown, I. R. Clemens and J. K. Neesom, Synlett, 2000, 131–133 CAS; (b) P. S. N. Reddy and P. P. Reddy, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1988, 27B, 763–765 CAS.
- (a) K. Du, X. Cao, P. Zhang and H. Zheng, Bioorg. Med. Chem. Lett., 2014, 24, 5318–5320 CrossRef CAS PubMed; (b) C. Dobrota, C. C. Paraschivescu, I. Dumitru, M. Matache, I. Baciu and L. L. Ruta, Tetrahedron Lett., 2009, 50, 1886–1888 CrossRef CAS.
- (a) C. Trecant, A. Dlubala, P. George, P. Pichat, I. Ripoche and Y. Troin, Eur. J. Med. Chem., 2011, 46, 4035–4041 CrossRef CAS PubMed; (b) B. Verheyde and W. Dehaen, J. Org. Chem., 2001, 66, 4062–4064 CrossRef CAS PubMed; (c) X. Jiang, R. A. Register, K. A. Killeen, M. E. Thompson, F. Pschenitzka and J. C. Sturm, Chem. Mater., 2000, 12, 2542–2549 CrossRef CAS.
- (a) N. A. Aksenov, A. V. Aksenov, O. N. Nadein, D. A. Aksenov, A. N. Smirnov and M. Rubin, RSC Adv., 2015, 5, 71620–71626 RSC; (b) A. V. Aksenov, N. A. Aksenov, O. N. Nadein and I. V. Aksenova, Synlett, 2010, 2628–2630 CrossRef CAS.
- (a) N. A. Aksenov, A. V. Aksenov, O. N. Nadein, D. A. Aksenov, A. N. Smirnov and M. Rubin, RSC Adv., 2015, 5, 71620–71626 RSC; (b) A. V. Aksenov, A. N. Smirnov, N. A. Aksenov, A. S. Bijieva, I. V. Aksenova and M. Rubin, Org. Biomol. Chem., 2015, 13, 4289–4295 RSC; (c) A. V. Aksenov, N. A. Aksenov, D. S. Ovcharov, D. A. Aksenov, G. Griaznov, L. G. Voskressensky and M. Rubin, RSC Adv., 2016, 6, 82425–82431 RSC; (d) A. V. Aksenov, D. S. Ovcharov, N. A. Aksenov, D. A. Aksenov, O. N. Nadein and M. Rubin, RSC Adv., 2017, 7, 29927–29932 RSC; (e) A. V. Aksenov, N. A. Aksenov, S. V. Scherbakov, A. N. Smirnov, I. V. Aksenova, V. I. Goncharov and M. A. Rubin, Russ. J. Org. Chem., 2017, 53, 1081–1084 CrossRef CAS; (f) A. I. Konovalov, et al., Russ. J. Org. Chem., 2018, 54, 157–371 CrossRef CAS.
- For reviews, see: (a) R. Ballini and M. Petrini, Adv. Synth. Catal., 2015, 357, 2371–2402 CrossRef CAS; (b) R. Ballini and M. Petrini, Tetrahedron, 2004, 60, 1017–1047 CrossRef CAS.
- Formation of species 14 was unambiguously confirmed by high resolution ESI MS analysis of the reaction mixture. See ESI (page S55†) for details.
- (a) M.-F. Pouliot, L. Angers, J.-D. Hamel and J.-F. Paquin, Org. Biomol. Chem., 2012, 10, 988–993 RSC; (b) Q. Gao, S. Liu, X. Wu, J. Zhang and A. Wu, Org. Lett., 2015, 17, 2960–2963 CrossRef CAS PubMed.
- (a) N. Salvanna, G. C. Reddy, B. R. Rao and B. Das, RSC Adv., 2013, 3, 20538–20544 RSC; (b) K. K. Gnanasekaran, B. Nammalwar, M. Murie and R. A. Bunce, Tetrahedron Lett., 2014, 55, 6776–6778 CrossRef CAS; (c) T. Kawano, K. Hirano, T. Satoh and M. Miura, J. Am. Chem. Soc., 2010, 132, 6900–6901 CrossRef CAS PubMed; (d) L.-H. Zou, J. Reball, J. Mottweiler and C. Bolm, Chem. Commun., 2012, 48, 11307–11309 RSC.
- (a) J. Dost, M. Heschel and J. Stein, J. Prakt. Chem., 1985, 327, 109–116 CrossRef CAS; (b) G. I. Elliott, J. R. Fuchs, B. S. J. Blagg, H. Ishikawa, H. Tao, Z.-Q. Yuan and D. L. Boger, J. Am. Chem. Soc., 2006, 128, 10589–10595 CrossRef CAS PubMed; (c) J. Garfunkle, C. Ezzili, T. J. Rayl, D. G. Hochstatter, I. Hwang and D. L. Boger, J. Med. Chem., 2008, 51, 4392–4403 CrossRef CAS PubMed.
- (a) F. Uhlig, Angew. Chem., 1954, 66, 435–436 CrossRef CAS; (b) A. L. Huhti and P. A. Gartaganis, Can. J. Chem., 1956, 34, 790–797 CrossRef.
- Y.-D. Park, J.-J. Kim, H.-A. Chung, D.-H. Kweon, S.-D. Cho, S.-G. Lee and Y.-J. Yoon, Synthesis, 2003, 560–564 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1875720, 1875721 and 1875723. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra00976k |
| This journal is © The Royal Society of Chemistry 2019 |