
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComprehensive quali-quantitative profiling of neutral and sialylated O-glycome by mass spectrometry based on oligosaccharide metabolic engineering and isotopic labeling†
Lijing Nana,
Jiao Liab,
Wanjun Jinab,
Ming Weiab,
Mengjun Tangab,
Chengjian Wangab,
Guiping Gongab,
Linjuan Huangab,
Ying Zhang*a and
Zhongfu Wang *ab
*ab
aKey Laboratory of Resource Biology and Biotechnology in Western China, Ministry of Education and Provincial Key Laboratory of Biotechnology, College of Life Sciences, Northwest University, Xi'an 710069, P. R. China. E-mail: wangzhf@nwu.edu.cn
bCollege of Food Science and Technology, Northwest University, Xi'an 710069, P. R. China
First published on 20th May 2019
Abstract
Mass spectrometry (MS) analysis combined with stable isotopic labeling is of great importance for quantitatively profiling abnormal sialylated O-glycans associated with disease development, but technically hindered by the poor releasing efficiency of O-glycans from glycoprotein or the labile nature of sialic acid residues at glycans. Herein, we developed an isotopic precursor based metabolic amplification and labeling (IPMAL) technique for relative quantitative profiling of the repertoire O-glycans between normal and tumor cells by ESI-MS. Two groups of cells were incubated with peracetylated benzyl-α-N-acetylgalactosamine (Ac3GalNAc-α-Bnd0) or a heavy labeled peracetylated benzyl-α-N-acetylgalactosamine (Ac3GalNAc-α-Bnd5) precursor respectively to amplify the repertoire of O-glycans as Bnd0/d5-O-glycans which could achieve the quantitative O-glycome analysis by ESI-MS after derivatization. The established method demonstrates desirable feasibility, accuracy (relative error (RE) ≤ 4.20%), reproducibility (coefficient of variation (CV) ≤ 7.61%, n = 3) and good quantitation linearity (R2 > 0.99, n = 3) for five Bn-O-glycans with 2 orders of magnitude. Finally, the method has been successfully applied to quantitative analysis of the repertoire O-glycome changes between normal human liver cell line L02 and human hepatoma cell line SMMC-7721. Moreover, the α-2,3/2,6 sialic acid isomers of Bn-O-glycans from these two cells have been further quantitatively distinguished when involved a sialic acid specific derivatization procedure.
Introduction
Mucin-type O-glycans at glycoproteins are covalently attached to Ser/Thr residues of the core protein through the GalNAc residue and play important roles in a variety of biological processes such as inter-cellular and intra-cellular interactions, communication, disease development and signal transductions.1–3 Terminal α-2,3 and/or α-2,6 sialic acids (N-acetylneuraminic acid or N-glycolylneuraminic acid) at O-glycans have shown significant biological importance in receptor recognition processes such as inflammation, bacterial and virus infections.4–7 Moreover, aberrant O-glycans at glycoprotein with or without sialic acids in both structures and their expression levels are associated with various human diseases such as tumors or cancer.8–11 Therefore, the quali-quantitative determination as well as sialic acid linkage specific identification of O-glycans expressed in normal and tumor cells will be beneficial for functional elucidation and diagnostic or prognostic purposes during disease development procedures as required in pathology or glycomics.Mass spectrometry with high sensitivity and resolution, has played an important role in the structural analysis of glycoconjugates and quantitative analysis of glycans changes between two samples using “light” and “heavy” stable isotopic labels.12–14 One common method employing NaOH solution to release and instantaneously reduce the aldehyde ends at O-glycans with “light” and “heavy” isotopic reagents, NaBH4 and NaBD4, respectively, has been widely used for MS quantification profiling of O-glycome changes between two samples.15 Besides, the O-glycans derived from two samples could be permethylated with 12CH3I/13CH3I or CD3I/CH3I isotopic labeling strategy to achieve the quantitation determination of overall O-glycome by MS.16,17 Wang et al.18 have developed an one-pot O-glycans releasing and d0/d5-PMP isotope labeling assay for qualitative and quantitative profiling of O-glycans from glycoproteins by MS. Recently, Kudelka had reported an isotopic labeling with cellular O-glycome reporter/amplification (ICORA) for comparative O-glycomics of cultured cells by MS.19 Despite all progress in quantification determination of O-glycans in glycomics studies, further investigation is needed to be conducted to improve/resolve these corresponding defects, such as misunderstanding of natural peaks of glycans with the isotope-labeled one due to the small molecular weight difference (1 Da), lack of universal hydrolytic enzyme used for effectively releasing of all O-glycans from glycoproteins, large amounts of glycoproteins used which might be not practicable for biological sample analysis and difficulty in quali-quantitative identification of α-2,3/2,6 sialic acid terminal ends of O-glycan isomers by MS.20–22
Although mass spectrometry has become an important tool to determine glycan structures, poor ionization of native glycans or sialic acids signal loss at glycans were easily happen when without a derivatization procedure.23,24 Moreover, MS and/or MS/MS glycomics profiling might face challenge in quali-quantitative determination of α-2,3/2,6-linked sialic acids at the terminal ends of glycan isomers.25 Due to the pathological importance of the O-glycan terminal sialic acids,26 it is of great significance to establish a high-throughput and effective method for MS-based quantification of overall neutral and sialylated O-glycans with linkage specificity between normal and tumor cells.
In our previous work, a sialic acid specific derivatization combined with Girard's reagent T (GT) on-target labeling has been exploited for MS-based linkage specific determination of sialic acids at kinds of glycans.27 Importantly, it has been reported recently the dimethylamidation is compatible with traditional permethylation to achieve the linkage-specific identification of sialic acids at N-glycan ends by MS.28 These research findings inspire us to utilize the strengths of chemical derivatization and oligosaccharide metabolic engineering and isotopic labeling strategy, to develop an isotopic precursor based metabolic amplification and labeling (IPMAL) assay for quali-quantitatively profiling of sialic acids linkage specificity of O-glycans between normal and tumor cells by MS.29
Within our IPMAL strategy, normal and tumor cells were respectively incubated with Ac3GalNAc-α-Bnd0 and Ac3GalNAc-α-Bnd5 (5 Da mass difference) substrates, which could enter cells and be used by enzymes in Golgi to amplify the repertoire of O-glycome to be Bn-O-glycans.19,29 Comprehensive quali-quantitative profiling of Bn-O-glycans from two cells after 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixing could be achieved by MS, as illustrated in Scheme 1. We have evaluated the method in terms of feasibility, reproducibility, accuracy and linear properties. The established assay has been applied to quali-quantitatively profile and determine the O-glycans between normal human liver cell line L02 and human hepatoma cell line SMMC-7721 after permethylation.30,31 Furthermore, the whole O-glycan isomers with/without α-2,3/2,6 sialic acid ends have been quali-quantitatively distinguished when a sialic acid specific derivatization procedure was involved.28
:1 mixing could be achieved by MS, as illustrated in Scheme 1. We have evaluated the method in terms of feasibility, reproducibility, accuracy and linear properties. The established assay has been applied to quali-quantitatively profile and determine the O-glycans between normal human liver cell line L02 and human hepatoma cell line SMMC-7721 after permethylation.30,31 Furthermore, the whole O-glycan isomers with/without α-2,3/2,6 sialic acid ends have been quali-quantitatively distinguished when a sialic acid specific derivatization procedure was involved.28
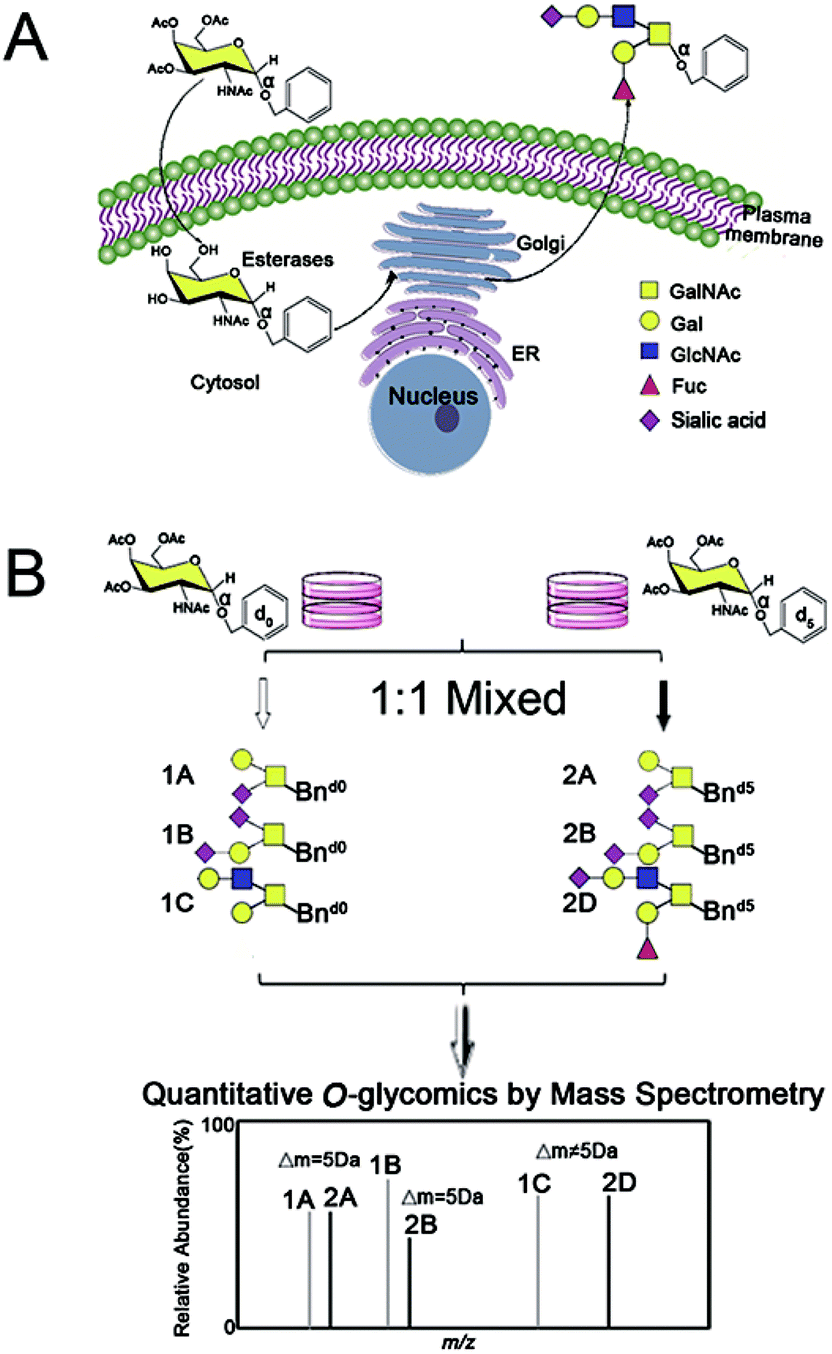 | ||
| Scheme 1 Isotopic precursor based metabolic amplification and labeling (IPMAL) strategy for quantitatively profiling the repertoire of O-glycans between normal and tumor cells. Structure formulas: yellow square, N-acetylgalactosamine (GalNAc); blue square, N-acetylglucosamine (GalNAc); yellow circle, galactose (Gal); red triangle, fucose (Fuc); purple diamond, sialic acid (N-acetylneuraminic acid, Neu5Ac; N-glycolylneuraminic acid, Neu5Gc). | ||
Materials and methods
Chemicals and materials
N-Acetyl-D-galactosamine, 1,2-dichloroethane, copper trifluoromethanesulfonate (Cu(OTf)2), benzyl alcohol, 4-nitrobenzyl alcohol, dimethylamine hydrochloride, 1-ethyl-3-(3-(dimethylamino)propyl)-carbodiimide (EDC) hydrochloride and 1-hydroxybenzotriazole (HOBt) monohydrate were purchased from Aladdin Inc. (Shanghai, China). Dimethyl sulfoxide, benzyl-2,3,4,5,6-d5 alcohol and 4-ethynylbenzyl alcohol were purchased from Sigma-Aldrich (St. Louis, MO, USA). High-glucose Dulbecco's modified Eagle's medium (DMEM) was purchased from HyClone (Logan, UT, USA). Amicon Ultra 4 mL centrifugal filters (10 kDa) were purchased from Millipore (Bedford, MA, USA). 3′-Sialyllactose (3′-SL, α2,3-linked sialic acid) and 6′-sialyllactose (6′-SL, α2,6-linked sialic acid) were purchased from Carbosynth (Compton, U.K.). Sep-Pak C18 (3 cc) solid phase extraction (SPE) cartridges were obtained from Water Corporation (Milford, MA, USA). Human hepatoma cells SMMC-7721 and human normal liver cells L02 cell lines were purchased from Shanghai cell bank, Chinese Academy of Sciences. 1H and 13C NMR spectra were recorded with Bruker AC-600 spectrometers using CDCl3 as solvent. Mass spectra were measured under a Thermo Fisher Scientific LTQ Orbitrap XL spectrometer. The chemical reaction was monitored by thin layer chromatography (silica gel, 25 μm layer thickness) and detected by a UV lamp (254 nm) combined with sulfuric acid-lichen phenol staining. All of these reagents were of analytical grade.Synthesis of O-glycan precursors Ac3GalNAc-α-Bnd0 and Ac3GalNAc-α-Bnd5
Synthesis of O-glycan precursors were carried out according to a reported procedure with minor modification.32–34 To a solution of glycosamine pentaacetates (200 mg, 0.514 mmol) in dry 1,2-dichloroethane (10 mL), anhydrous Cu(OTf)2 (0.15 equiv.) and d0-benzyl alcohol (4 equiv.) or heavy labeled benzyl alcohol (benzyl-2,3,4,5,6-d5 alcohol, 4 equiv.) were added. The reaction mixture was stirred vigorously under an N2 atmosphere reflux at 130 °C for 6 h. When the reaction was completed, the saturated NaHCO3 aqueous solution was added to the reaction mixture to stop the reaction. The organic layer was totally layered after stirring. The collected organic layer was concentrated under the reduced pressure after washing with water twice. The concentrated organic residues were concentrated and loaded onto silica gel column separation using petroleum ether–EtOAc (1:1 or other ratios based on TLC separation result) as mobile phase to acquire the desired product Ac3GalNAc-α-Bnd0 (Rf = 0.75, 161.77 mg, 72%) or Ac3GalNAc-α-Bnd5 (Rf = 0.75, 161.35 mg, 71%), which were further confirmed by MS and NMR data, as presented in Fig. 1B and S1A–D (ESI† file).
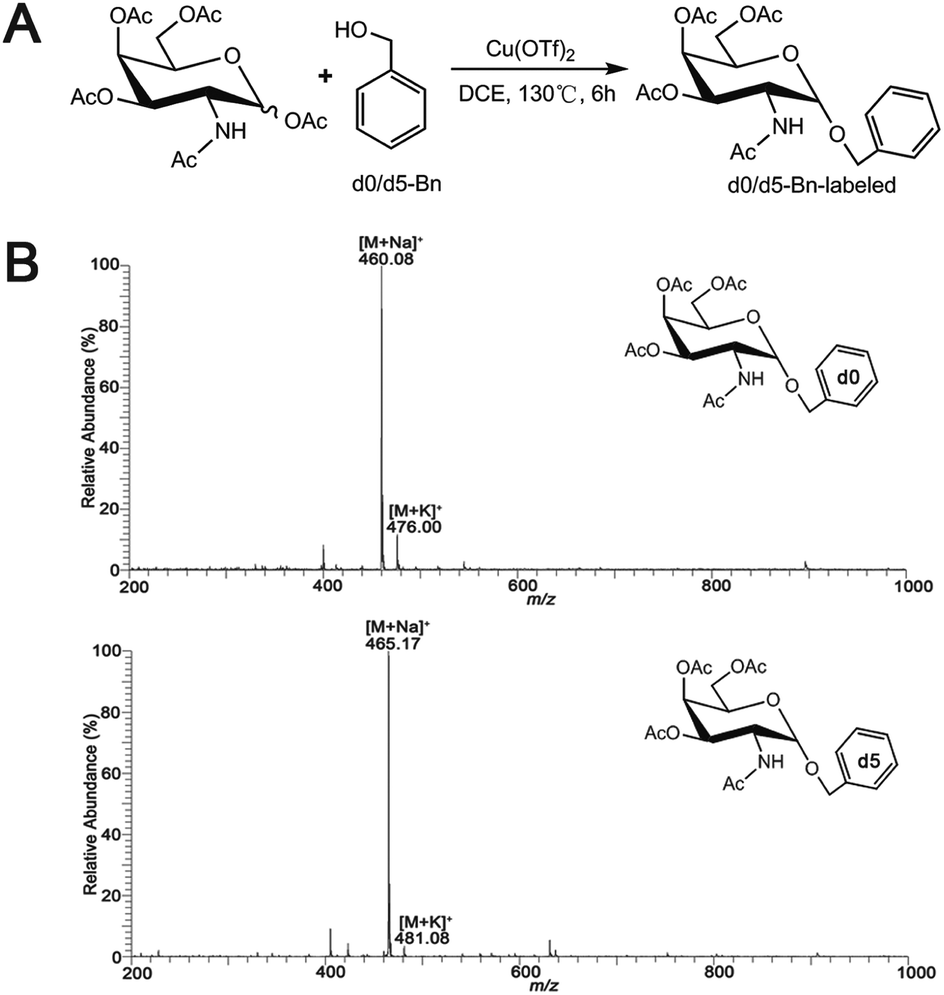 | ||
| Fig. 1 (A) Synthetic scheme for Ac3GalNAc-α-Bn precursor and (B) ESI-MS spectrum of the synthesized Ac3GalNAc-α-Bnd0 and Ac3GalNAc-α-Bnd5. | ||
Ac3GalNAc-α-Bnd0, purity 98.6% (HPLC); 1H NMR (600 MHz, CDCl3), δ 7.37 (d, J = 7.5 Hz, 2H), 7.31 (d, J = 6.8 Hz, 1H), 7.26 (s, 1H), 5.58 (d, J = 9.7 Hz, 1H), 5.38 (d, J = 2.5 Hz, 1H), 5.17 (dd, J = 11.3, 3.2 Hz, 1H), 4.97 (d, J = 3.6 Hz, 1H), 4.72 (d, J = 11.7 Hz, 1H), 4.61–4.57 (m, 1H), 4.49 (d, J = 11.7 Hz, 1H), 4.21 (s, 1H), 4.09 (dd, J = 18.6, 6.6 Hz, 2H), 2.16 (s, 2H), 2.05 (s, 2H), 1.98 (s, 2H), 1.90 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 173.59, 173.09, 173.02, 172.63, 139.20, 131.35, 131.04, 130.89, 99.63, 79.90, 79.68, 79.47, 72.77, 71.06, 70.00, 69.58, 64.56, 50.34, 25.95, 23.43, 23.40. ESI-MS: calcd for C21H27NO9, [M + Na]+ m/z 460.35, found m/z 460.08.
Ac3GalNAc-α-Bnd5, purity 97.8% (HPLC); 1H NMR (600 MHz, CDCl3), δ 7.26 (s, 6H), 5.56 (d, J = 9.6 Hz, 1H), 5.37 (t, J = 9.8 Hz, 1H), 5.18 (dd, J = 11.3, 3.1 Hz, 1H), 4.98 (d, J = 3.5 Hz, 1H), 4.72 (d, J = 11.7 Hz, 1H), 4.64–4.53 (m, 1H), 4.50 (d, J = 11.7 Hz, 1H), 4.22 (t, J = 6.4 Hz, 2H), 4.10 (m, 2H), 2.16 (s, 3H), 2.06 (d, J = 2.2 Hz, 3H), 1.95 (s, 2H), 1.90 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 173.56, 173.04, 172.97, 172.59, 102.31, 99.65, 79.87, 79.66, 79.45, 77.12, 72.72, 71.09, 70.02, 69.60, 64.55, 63.03, 50.39, 25.92, 23.69, 23.40, 23.33. ESI-MS: calcd for C21H32NO9 [M + Na]+ m/z 465.08, found m/z 465.17.
Cell culture
Cell culture was carried out according to the reported procedure.35 SMMC-7721 and L02 cells were cultured with 10 mL DMEM medium containing 10% (by volume) fetal bovine serum and 100 U mL−1 penicillin–streptomycin mix. Cells were kept in an incubator at 37 °C, 5% CO2 and 95% (volume fraction) air-saturated humidity. When the cell density was increased to 80% (cells 2 × 107), the medium with 10% fetal bovine serum was replaced with medium containing 8% (volume fraction) fetal bovine serum. Then Ac3GalNAc-α-Bn (100 μM) was added to medium and kept for further three days.Acquisition and purification of Bn-O-glycans
Acquisition and purification of Bn-O-glycans were carried out according to reported procedure with minor modification.29 After three days of culture, the culture medium was collected and filtered through a 10 kDa ultrafiltration membrane (2500g, 30 min). The filtered solution was purified through a C18 cartridge (200 mg/4 mL) according to the following procedure. The column was equilibrated with 4 mL of acetonitrile, 8 mL of 50% (v/v) acetonitrile and 10 mL of 0.1% (v/v) trifluoracetic acid (TFA). Then the filtered media was loaded onto the column and washed with 2 mL of 0.1% (v/v) trifluoracetic acid (TFA) four times. Bn-O-glycans were then eluted with 3 mL of 50% (v/v) acetonitrile containing 0.1% TFA. The purified sample was collected and lyophilized to dry.General permethylation of Bn-O-glycans
General permethylation of O-glycan samples was carried out according to the reported procedures with minor modification.30,31 The dried sample was dissolved in 300 μL DMSO–NaOH suspension and 75 μL CH3I mixture. After shaking 30 min in dark, 500 μL water was added into sample to quench the reaction. Then 500 μL dichloromethane was added. The supernatant was then partitioned between water and dichloromethane. The organic layer was washed with 500 μL water twice, dried and re-dissolved in 200 μL 50% methanol for further MS analysis.The whole sialic acid specific derivatization procedure for Bn-O-glycans
The whole derivatization procedure has two steps, including linkage specific derivatization of sialic acid and general permethylation. Linkage specific derivatization of sialic acid was conducted according to the previous references with minor modification.28,36 25 μL of 500 mM 1-hydroxybenzotriazole (HOBt, dissolved in DMSO), 25 μL of 250 mM 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC, dissolved in DMSO) and 25 μL of 250 mM dimethylamine dissolved in DMSO, were added to the dried glycan sample (5 pmol to 10 nmol) and incubated at 60 °C for 1 h. The excess dimethyamine was evaporated in a Speed Vac at 60 °C for 20 min. Then the sample was treated with 200 μL of NaOH–DMSO slurry and 100 μL of methyl iodide for 30 min with agitation at room temperature. The reaction was terminated by adding 800 μL of water. Subsequently, 200 μL of dichloromethane was added. The mixture was stirred and centrifuged to facilitate partitioning. The top aqueous layer was then removed, while the dichloromethane layer was washed with 1 mL water three times additionally. Then the organic layer was dried and submitted to the general permethylation procedure as described above.Feasibility evaluation of the assay
Two groups of human hepatoma cells SMMC-7721 cells were parallelly cultured with 100 μM Ac3GalNAc-α-Bnd0 and Ac3GalNAc-α-Bnd5, respectively, for 3 days. Both cell media was collected and mixed at a ratio of 1:1. The mixed culture medium was then submitted to purification, permethylation and MS-based quantification analysis.
Evaluation the linearity of the assay
Two groups of SMMC-7721 cells were in parallel cultured with 100 μM Ac3GalNAc-α-Bnd0 and Ac3GalNAc-α-Bnd5, respectively, for 3 days. The media from two conditions was mixed in a gradient ratios (10:1, 7.5:1, 5:1, 2.5:1, 1:1, 1:2.5, 1:5, 1:7.5 and 1:10, n = 3). Linearity evaluation of the assay was plotted using relative intensities ratios of major O-glycan pair peaks observed in MS spectra as the y-axis versus the adding linear gradient ratios (10:1, 7.5:1, 5:1, 2.5:1, 1:1, 1:2.5, 1:5, 1:7.5 and 1:10) of O-glycans mixture as the x-axis. The calibration graphs for the assay were calculated by the equation: y = mx + c.
Statistical analysis
Results of all experiments performed for the relative intensity ratios of peak pairs in MS-based relative quantification in triplicates were indicated by mean ± SD. Data was analyzed by one-way analysis of variance (ANOVA) using SPSS 16.0. Statistical differences were determined using a two-tailed Student's t-test, with p < 0.05 indicating statistical significance.ESI-MS and MS/MS conditions
The ESI-MS and MS/MS data was acquired with an LTQ XL ion-trap mass spectrometer equipped with an electrospray ion (ESI) source (Thermo Scientific, USA). The samples were injected into electrospray ion source via a 2 μL Rheodyne loop in a stream of 50% methanol at a flow rate of 100 μL min−1. The spray voltage was set at 4000 V, with a sheath gas (nitrogen gas) flow rate of 20 arb, an auxiliary gas (nitrogen gas) flow rate of 5.0 arb, a capillary voltage of 37 V, a tube lens voltage of 250 V, and a capillary temperature of 300 °C, positive ion mode scan, scan range is m/z 200–2000. MS/MS analysis was carried out using helium (He) as the collision gas, a normalized collision energy degree of 35–45% and an isotope width of m/z 2.00. The ESI-MS and MS/MS data were acquired with LTQ Tune software Xcalibur (Thermo). Other parameters were acquiescent.Results and discussions
Kudelka recently have developed an isotopic labeling with cellular O-glycome reporter/amplification (ICORA) strategy, which employed two O-glycan precursors, Ac3GalNAc-BnH7 and Ac3GalNAc-BnD7.19 The assay provides a useful tool for exploring the dynamic changes in O-glycan between two cells. However, the incapability of determination α2,3/2,6-sialylated O-glycan isomers with linkage specificity by MS and/or MS/MS might be limit its application at some extent. Moreover, dimethylamidation has been used to distinguish α-2,3/2,6-linked sialic acids at N-glycans, which is compatible with common permethylation.28 Herein, an innovative strategy for the first MS-based quali-quantitative profiling and linkage-specific identification of O-glycans with or without sialic acids ends through isotopic precursor based metabolic amplification and labeling (IPMAL) assay could be achieved, when involved the sialic acid specific derivatization.28Principle of the assay
A schematic procedure for quantitative profiling of the repertoire of O-glycome through isotopic precursor based metabolic amplification and labeling (IPMAL) assay, is depicted in Scheme 1. We designed a novel strategy for rapid synthesis of the normal and/or heavy labeled precursors, as depicted in Fig. 1A. One step O-glycosylation of glycosamine pentaacetates with a normal or heavy labeled benzyl alcohol could afford the final products Ac3GalNAc-α-Bnd0 or Ac3GalNAc-α-Bnd5 in good yields (>70%), as confirmed by NMR (Fig. S1A–D, 1H and 13C NMR, ESI† file) and ESI-MS (Fig. 1B). Both neutral and sialylated O-glycans could be amplified through oligosaccharide metabolic engineering in the presence of precursors Ac3GalNAc-α-Bnd0 or Ac3GalNAc-α-Bnd5 (5 Da mass difference) within two cells. Then quali-quantitative profiling of O-glycans as well as sialic acid linkage specific characterization of O-glycans could be achieved by MS and MS/MS when involved a permethylation29 or the sialic acid specific derivatization procedure,28 as illustrated in Fig. S2 (ESI† file).Feasibility of the assay
Two groups of SMMC-7721 cells in parallel were incubated with equal molar amounts of Ac3GalNAc-α-Bnd0 and Ac3GalNAc-α-Bnd5 O-glycan precursors (5 Da mass difference) respectively, to amplify the whole O-glycans. Two groups of the amplified O-glycans were mixed in 1:1 ratio and submitted to permethylation. After direct permethylation, ESI-MS analysis of Bnd0/d5-O-glycans was illustrated in Fig. 2A. Ten pairs of the Bnd0/d5-O-glycans peaks with signals at m/z 955.25/960.33, 1200.33/1205.33, 1316.33/1321.33, 1404.42/1409.42, 1561.33/1566.42, 1578.17/1583.25, 1649.25/1654.42, 1677.42/1682.33, 1765.42/1770.50 and 1939.33/1944.50 ([M + Na]+ adducts) were observed in MS profile. Structural characterizations of one pair of O-glycan peaks with signals at m/z 1316.33/1321.33 and other nine pairs of O-glycan peaks were achieved by ESI-MS/MS fragmentation in Fig. 2B and S3A–I,† correspondingly. According to the MS data (n = 3), the R values (the intensity ratio of the Bnd0-O-glycan to Bnd5-O-glycan) of each set of peaks were 1.02, 1.05, 0.99, 1.03, 0.92, 1.02, 1.07, 1.07, 0.93 and 0.98, which afforded an averaged R value of 1.01 that was very close to the theoretical ratio of 1.0, demonstrating the good feasibility and reliability of the established method for relative quantitative analysis of O-glycans in two cells.
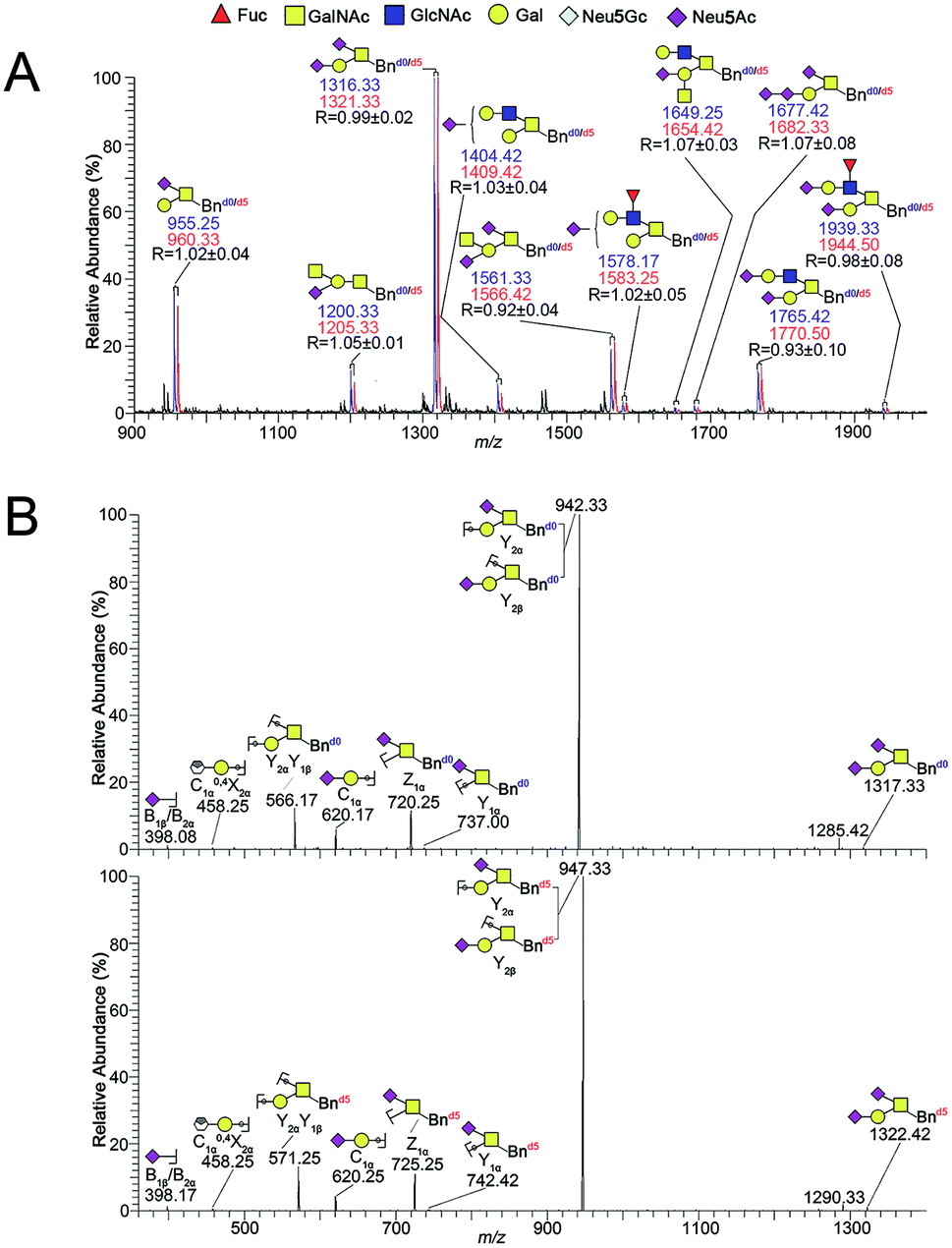 | ||
| Fig. 2 (A) ESI-MS spectra profile of the 1:1 mixture of Bnd0-O-glycans (blue) and Bnd5-O-glycans (red) from SMMC-7721 cells and (B) comparative sequencing of Bnd0/d5-O-glycans from SMMC-7721 by ESI-MS/MS. Each peak was fragmented by CID, respectively. Structure formulas: yellow square, N-acetylgalactosamine (GalNAc); blue square, N-acetylglucosamine (GalNAc); yellow circle, galactose (Gal); red triangle, fucose (Fuc); purple diamond, sialic acid (Neu5Ac); gray diamond, sialic acid (Neu5Gc). | ||
Linearity and accuracy verification of the assay
Linearity of five major pairs of Bnd0/d5-O-glycans peaks with signals at m/z 955.25/960.33, 1200.33/1205.33, 1316.33/1321.33, 1561.33/1566.42 and 1765.42/1770.50 ([M + Na]+ adducts) were plotted according to their relative intensities ratios in the positive mode ESI-MS spectra versus the adding gradient ratios of O-glycans mixture (10:1, 7.5:1, 5:1, 2.5:1, 1:1, 1:2.5, 1:5, 1:7.5 and 1:10, n = 3), as shown in Fig. 3. The experiment revealed a desirable linear dependency between the observed relative intensities ratios of five major peaks of O-glycans in two groups of SMMC-7721 cells by MS and concentration ratios of the two samples, with the correlation coefficient being ≥0.99 in all cases (n = 3, Table 1). The assay clearly presented a desirable linearity with a 100 folds linear dynamic range for sample measurements.
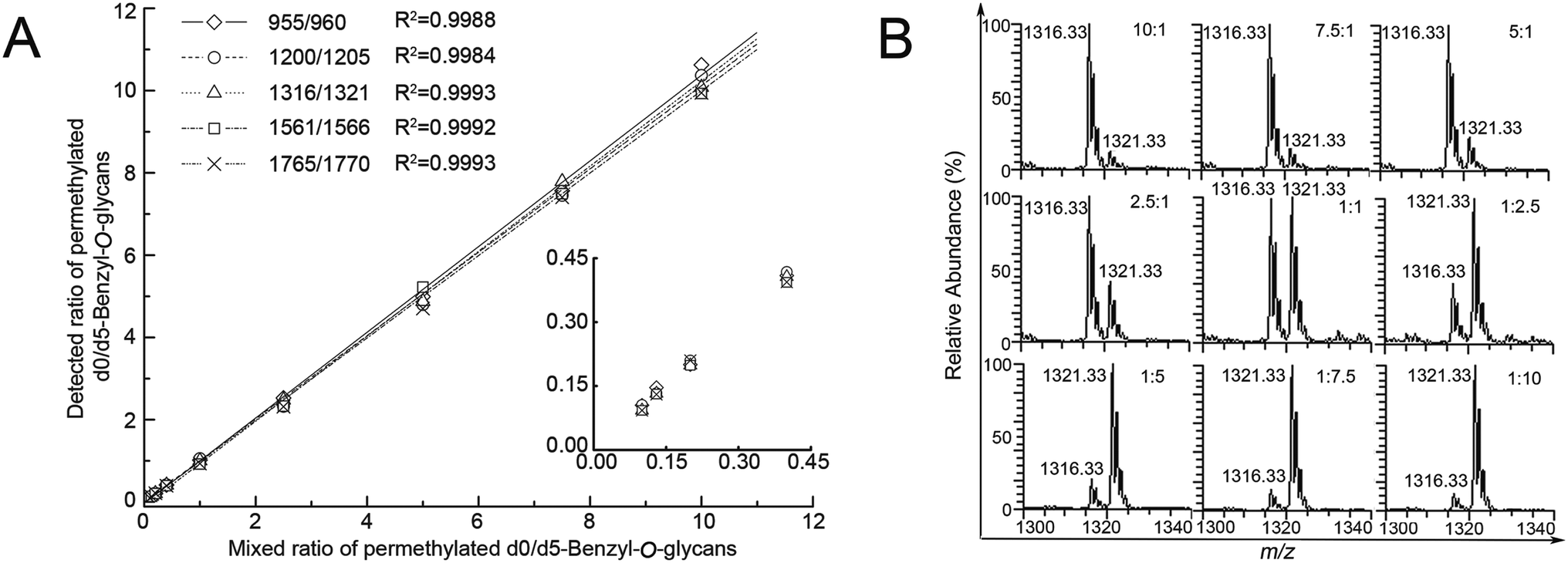 | ||
| Fig. 3 (A) The linearity graph of the observed ESI-MS peak abundance ratios of Bnd0-O-glycans to Bnd5-O-glycans versus the added ratios of Bnd0-O-glycans to Bnd5-O-glycans and (B) ESI-MS peak abundance ratios of one random pair peaks (m/z 1316.33/1321.33) of Bnd0-O-glycans to Bnd5-O-glycans versus the added ratios of Bnd0-O-glycans to Bnd5-O-glycans peaks. | ||
| Glycan MS signals (m/z) | Regression equation | Correlation coefficient (R2) | Mixed molar ratios of d0/d5-Bn-labeled glycans | Measured molar ratios of d0/d5-Bn-labeled glycans (mean ± SD, n = 3) | Mean reproducibility (%CV, n = 3) | Mean relative error (%) |
|---|---|---|---|---|---|---|
| 955 | Y = 1.0415x − 0.0399 | 0.9988 | 1:10 |
0.105 ± 0.009 | 9.04 | 4.89 |
| 1:7.5 |
0.147 ± 0.008 | 5.36 | 9.87 | |||
| 1:5 |
0.200 ± 0.009 | 4.36 | 0.75 | |||
| 1:2.5 |
0.409 ± 0.022 | 5.28 | 2.31 | |||
| 1:1 |
1.018 ± 0.043 | 4.18 (4.72) | 1.78 (3.10) | |||
| 2.5:1 |
2.526 ± 0.092 | 3.66 | 1.04 | |||
| 5:1 |
4.992 ± 0.227 | 4.56 | 0.17 | |||
| 7.5:1 |
7.566 ± 0.327 | 4.32 | 0.88 | |||
| 10:1 |
10.622 ± 0.178 | 1.68 | 6.22 | |||
| 1200 | Y = 1.017x − 0.047 | 0.9984 | 1:10 |
1.106 ± 0.004 | 3.66 | 5.86 |
| 1:7.5 |
0.134 ± 0.016 | 11.93 | 0.18 | |||
| 1:5 |
0.209 ± 0.004 | 2.14 | 4.85 | |||
| 1:2.5 |
0.418 ± 0.019 | 4.66 | 4.43 | |||
| 1:1 |
1.052 ± 0.008 | 0.72 (4.07) | 5.20 (3.99) | |||
| 2.5:1 |
2.323 ± 0.039 | 1.69 | 7.07 | |||
| 5:1 |
4.798 ± 0.011 | 0.23 | 4.04 | |||
| 7.5:1 |
7.455 ± 0.357 | 4.79 | 0.60 | |||
| 10:1 |
10.367 ± 0.709 | 6.84 | 3.67 | |||
| 1316 | Y = 1.015x − 0.0201 | 0.9993 | 1:10 |
0.093 ± 0.006 | 6.58 | 7.04 |
| 1:7.5 |
0.133 ± 0.011 | 8.26 | 0.56 | |||
| 1:5 |
0.199 ± 0.010 | 5.09 | 0.71 | |||
| 1:2.5 |
0.406 ± 0.019 | 4.65 | 1.55 | |||
| 1:1 |
0.999 ± 0.013 | 1.35 (4.74) | 0.04 (2.00) | |||
| 2.5:1 |
2.477 ± 0.075 | 3.02 | 0.91 | |||
| 5:1 |
4.874 ± 0.140 | 2.87 | 2.52 | |||
| 7.5:1 |
7.789 ± 0.519 | 6.67 | 3.85 | |||
| 10:1 |
10.080 ± 0.417 | 4.13 | 0.80 | |||
| 1561 | Y = 1.0018x − 0.0182 | 0.9992 | 1:10 |
0.093 ± 0.009 | 9.37 | 7.28 |
| 1:7.5 |
0.131 ± 0.006 | 4.89 | 1.70 | |||
| 1:5 |
0.199 ± 0.022 | 10.99 | 0.69 | |||
| 1:2.5 |
0.393 ± 0.024 | 6.19 | 1.68 | |||
| 1:1 |
0.921 ± 0.045 | 4.88 (6.76) | 7.86 (3.48) | |||
| 2.5:1 |
2.331 ± 0.134 | 5.75 | 6.76 | |||
| 5:1 |
5.225 ± 0.034 | 0.64 | 4.50 | |||
| 7.5:1 |
7.492 ± 0.584 | 7.79 | 0.11 | |||
| 10:1 |
9.929 ± 1.029 | 10.37 | 0.71 | |||
| 1765 | Y = 0.9892x − 0.0454 | 0.9993 | 1:10 |
0.095 ± 0.019 | 9.25 | 7.14 |
| 1:7.5 |
0.130 ± 0.013 | 9.67 | 2.27 | |||
| 1:5 |
0.210 ± 0.011 | 5.34 | 5.09 | |||
| 1:2.5 |
0.394 ± 0.028 | 7.17 | 1.45 | |||
| 1:1 |
0.932 ± 0.104 | 11.17 (7.61) | 6.76 (4.20) | |||
| 2.5:1 |
2.316 ± 0.084 | 3.63 | 7.37 | |||
| 5:1 |
4.702 ± 0.495 | 10.53 | 5.95 | |||
| 7.5:1 |
7.409 ± 0.311 | 4.19 | 1.22 | |||
| 10:1 |
9.944 ± 0.752 | 7.57 | 0.56 |
Clearly, the observed MS intensity ratios were very identical to the theoretical molar ratios as well as the samples amount ratios, as listed in Table 1. Moreover, these average ratio data illustrated the averaged CVs (coefficients of variation) values (4.72, 4.07, 4.74, 6.76 and 7.61%, n = 3) lower than 8%, demonstrating the desirable reproducibility and precision of the established method, after repeating the experiment in triplicate.
The accuracy of the method was evaluated by the measured ratios of d0/d5-Bn-labeled O-glycans versus the theoretical ratios of d0/d5-Bn-labeled O-glycans to give some relative errors (REs). The mean RE (3.10, 3.99, 2.00, 3.48 and 4.20%, n = 3) was obtained, as listed in Table 1. Five pairs of the Bn-O-glycans peaks were also presented CV value below 8% and RE value below 5%, respectively (Table 1, n = 3), which are indicative of rather good precision and acceptable accuracy of the established method.
Quali-quantitative profiling of Bn-O-glycans between L02 and SMMC-7721 cells
The established IPMAL method has been applied to quali-quantitatively profiling of the repertoire O-glycans from normal human liver cell line L02 and human hepatoma cell line SMMC-7721. In parallel, L02 and SMMC-7721 cells were cultured with Ac3GalNAc-α-Bnd0 or Ac3GalNAc-α-Bnd5 precursor, respectively, and presented the MS-based quantitative results as illustrated in Fig. 4. As shown in Fig. 4, 9 and 10 Bn-O-glycans were observed in L02 and SMMC-7721 cells, respectively. The characterized structural details of these Bn-O-glycans were further identified by MS/MS, in consistent with the previous ref. 29, 37 and 38 One random MS/MS data of the two peaks with signals at m/z 1043.33 and 1346.33 ([M + Na]+ adducts, from L02 cells) was illustrated in Fig. S4.†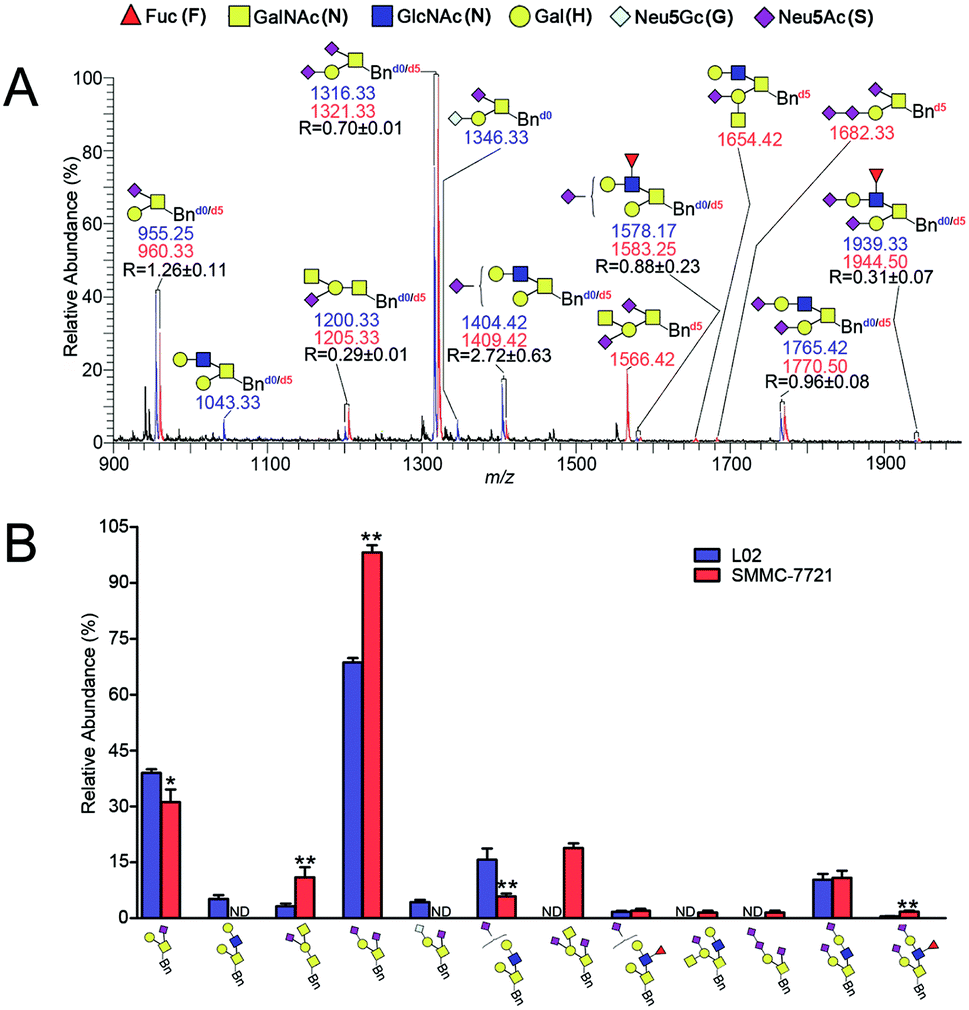 | ||
| Fig. 4 (A) Quali-quantitative profiling of Bn-O-glycans between L02 and SMMC-7721 cells by ESI-MS and (B) bar charts derived from the relative intensities of the MS signals in (A). The intensity ratio of the Bnd0-O-glycans to its corresponding Bnd5-O-glycans is indicated as R (n = 3, averaged). Note: *p < 0.05, **p < 0.01. Structure formulas: yellow square, N-acetylgalactosamine (GalNAc, N); blue square, N-acetylglucosamine (GlcNAc, N); yellow circle, galactose (Gal, H); red triangle, fucose (Fuc, F); purple diamond, sialic acid (Neu5Ac, S); white gray diamond, sialic acid (Neu5Gc, G). | ||
ESI-MS combined with MS/MS data has demonstrated that L02 cells amplified 9 Bn-O-glycans, one neutral Bn-O-glycans ([M + Na]+ ion peak with signal at m/z 1043.33) and eight sialylated Bn-O-glycans ([M + Na]+ adducts with signals at m/z 955.25, 1200.33, 1316.33, 1346.33, 1404.42, 1578.17, 1765.42 and 1939.33), as shown in Fig. 4. Whereas, SMMC-7721 expressed 10 Bn-O-glycans ([M + Na]+ adducts with signals at m/z 960.33, 1205.33, 1321.33, 1409.42, 1566.42, 1583.25, 1654.42, 1682.33, 1770.50 and 1944.50), which were either high sialylated or fucosylated ones as later confirmed by MS/MS. In Fig. 4, seven pairs of O-glycan peaks with a 5 Da difference including m/z 955.25/960.33, 1200.33/1205.33, 1316.33/1321.33, 1404.42/1409.42, 1578.17/1583.25, 1765.42/1770.50 and 1939.33/1944.50, were observed in MS profile. From relative intensities signals in MS, the relative quantitation of these O-glycans in L02 and 7721 cells were indicated to be R = 1.26, 0.29, 0.70, 2.72, 0.88, 0.96 and 0.31. Two single peaks with signals at m/z 1043.33 and 1346.33 were detected in L02 only, whereas three single O-glycan peaks with signals at m/z 1566.42, 1654.42 and 1682.33 with high sialylation level, were observed in SMMC-7721 cells only. Furthermore, based on relative abundance values of each pair of O-glycans peaks in MS, relative quantification of O-glycans between normal and tumor cells was achieved, as illustrated in Fig. 4B.
As shown in Fig. 4, O-glycans in SMMC-7721 cells with sugar compositions of N2H1S1, N1H1S2, N2H2F1S2 (molecular ion peaks [M + Na]+ with signals at m/z 1205.33, 1321.33 and 1944.50) were significantly higher than those of L02 cells. Moreover, N2H2F1S1 and N2H2S2 ([M + Na]+ ion peaks with signals at m/z 1583.25 and 1770.50) was slightly higher than those of L02 cells. Fucosylated core 2 O-glycans were up-regulated in SMMC-7721 cells. None of neutral O-glycan was observed in SMMC-7721 cells. Data indicated that SMMC-7721 express more O-glycans and higher levels of sialylated and/or fucosylated O-glycans over that of normal cell line L02, in line with the facts that abnormal sialylated and fucosylated O-glycans in biological processes were closely related to the occurrence, development and deterioration of liver tumor.35
Quali-quantitative profiling and distinction of both neutral and sialylated Bn-O-glycans between L02 and SMMC-7721 cells
Due to the biological importance of the glycan terminal sialylation associated with infection or disease development, it is important to establish novel and effective method for linkage-specific distinction of sialic acids. Despite sialylated Bn-O-glycans can be successfully determined by ESI-MS, it is very challenging to assign sialic acids specific linkages by ESI-MS and/or MS/MS, as presented in Fig. 2B, S3 and S4 (ESI† file). Fortunately, a linkage-specific assignation of O-glycans terminal sialic acid could be achieved by combining our IPMAL assay with sialic acid derivatization (a mild dimethylamidation and permethylation), which could selectively convert α2,6-linked sialic acids to dimethylamidation and α2,3-linked sialic acid to lactones, as shown in Fig. S2.†To ensure the feasibility of the assay, standard sialylated trisaccharides, α2,3-Neu5Ac-Lac (3′-SL) and α2,6-Neu5Ac-Lac (6′-SL), were submitted to the sialic acids specific derivatization and profiled by ESI-MS, as shown in Fig. S5 (ESI† file). According to the structural changes of glycans with sialic acid ends after the specific derivatization reactions, a mass difference (13 Da) between α2,3- and α2,6-sialic acid can be detected, facilitating the isomeric assignment of α2,3- and/or α2,6-sialylated glycans. As we expected, distinct [M + Na]+ionized peaks at m/z 838 and 851 (13 Da) were assigned to be 3′-SL and 6′-SL, respectively, demonstrating the feasibility of the assay for linkage specific characterization of glycan isomers with α2,3- and α2,6-sialic acid ends.
We further applied our IPMAL method combined with sialic acids specific derivatization to achieve quali-quantitative profiling and distinction of both neutral and sialylated Bn-O-glycans between L02 and SMMC-7721 cells. After combining the IPAML assay with sialic acid derivatization, not only the sialic acids residues at O-glycans in L02 and SMMC-7721 were further stabilized, but also the linkage specificity as well as the relative quantitation could be well characterized, as shown in Fig. 5.
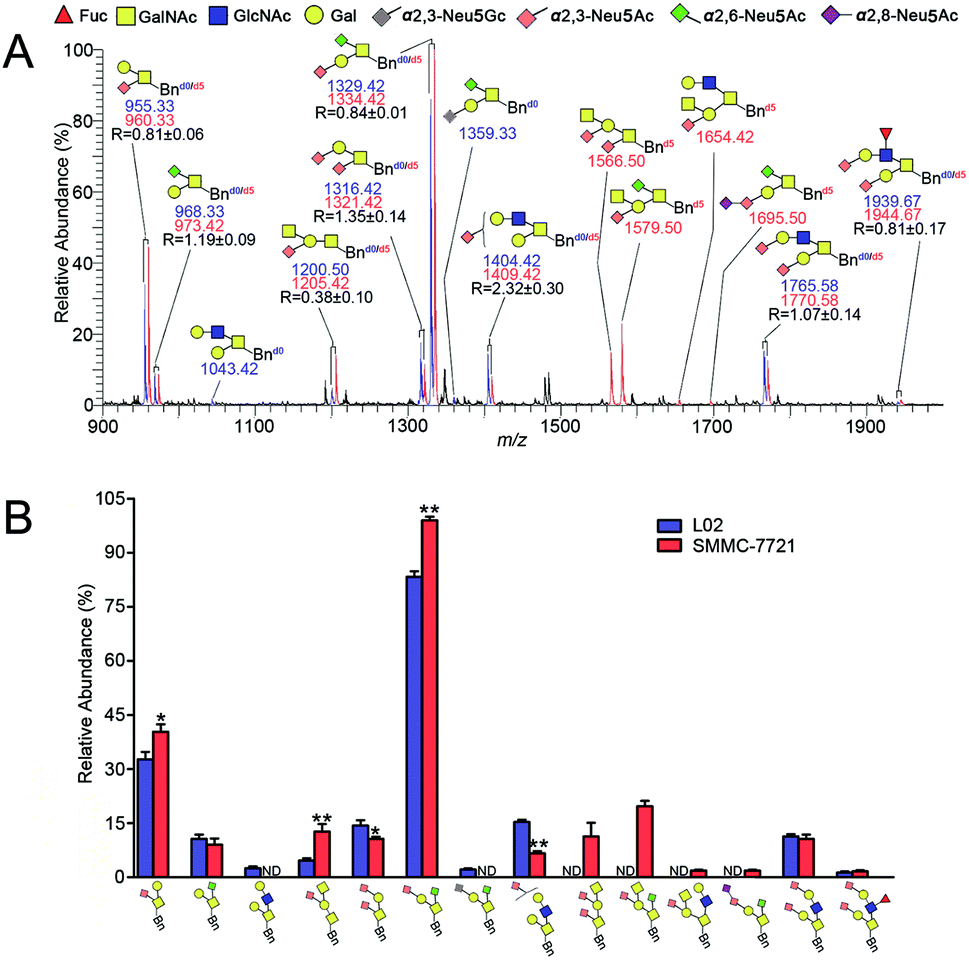 | ||
| Fig. 5 (A) Quali-quantitative profiling and distinction of both neutral and sialylated Bn-O-glycans between L02 and SMMC-7721 cells and (B) bar charts derived from the relative intensities of the MS signals in (A). The intensity ratio of the Bnd0-O-glycans to its corresponding Bnd5-O-glycans is indicated as R (n = 3, averaged). Notes: *p < 0.05; **p < 0.01. Structure formulas: yellow square, N-acetylgalactosamine (GalNAc); blue square, N-acetylglucosamine (GlcNAc); yellow circle, galactose (Gal); red triangle, fucose (Fuc); gray diamond, α2,3-Neu5Gc; pink diamond, α2,3-Neu5Ac; green diamond, α2,6-Neu5Ac; purple diamond, α2,8-Neu5Ac. | ||
Compared with the relative intensity of Bn-O-glycans in L02 and SMMC-7721 (Fig. 4A) in MS, MS signals at m/z 955.33/960.33, 1043.42, 1200.50/1205.42, 1316.42/1321.42, 1404.42/1409.42, 1566.50, 1654.42, 1765.58/1770.58 and 1939.67/1944.67 ([M + Na]+ ion peaks) did not change, indicating the presence of neutral or sialylated Bn-O-glycans with α2,3-linked sialic acid (Neu5Ac), as shown in Fig. 5A. Whereas, Bn-O-glycans with α2,6-linked sialic acid (Neu5Ac) terminals yielded a 13 Da mass increase. For example, peaks at m/z 955.25/960.33, 1316.33/1321.33, 1566.42 and 1682.33, in Fig. 4A were shifted to m/z 968.33/973.42, 1329.42/1334.42, 1579.50 and 1695.50 in Fig. 5A, suggesting the presence of α2,6-sialylated O-glycans. MS signal at m/z 1359.33 was assigned to be O-glycan with a α2,3-linked sialic acid (Neu5Gc) and a α2,6-linked sialic acid (Neu5Ac). Structural details of all other peaks, including neutral, di- and tri-sialylated O-glycans have been determined by MS/MS, providing the proposed information of O-glycans terminal sialic acids linkage specificity, as shown in Fig. 5A. The quali-quantitative profiling and distinction of both neutral and sialylated Bn-O-glycans with linkage-specificity was indicated in Fig. 5B. As shown in Fig. 5B, 8 O-glycans were up-expressed in SMMC-7721 cells, while 4 O-glycans were down-expressed in L02 cells. Moreover, most of the up-expressed O-glycans are highly sialylated. The expression level of specific O-glycans such as the disialyl core 1 Neu5Ac-α-2,3-Gal-β-1,3-(Neu5Ac-α-2,6)-GalNAc-α-O-Thr in SMMC-7721 cells was higher over that of L02 cells, in line with the previous works.8,35 Therefore, the good compatibility of our IPMAL assay with sialic acid derivatization (a mild dimethylamidation and permethylation), shows great potential in quali-quantitative determination of the O-glycans with or without sialic acids between normal and disease.
Conclusions
An innovative and effective approach termed as IPMAL for quali-quantitative profiling of neutral and sialylated Bn-O-glycans by ESI-MS in positive-ion mode has been developed. The “light” and “heavy” O-glycan precursors could be chemically synthesized by one-step O-glycosylation, which shows methodological advantage over previous work and will make easy generalization and wide application of the established assay. Through metabolic oligosaccharide engineering and isotopic labeling, the MS-based quali-quantitative profiling of Bn-O-glycans derived from two cells could be achieved simultaneously through the mass differences (5 Da) in pairs of peaks in MS. The quantitation feasibility, reproducibility, reliability and linearity of this strategy were fully investigated, yielding 100 fold linear dynamic range, good reproducibility (CV, 4.07–7.61%) and accuracy (RE, 2–4.2%). The quali-quantitative determination of Bn-O-glycans from L02 and SMMC-7721, were achieved successfully, which is beneficial for the mechanism elucidation of O-glycosylation as required in clinical and pathological research.When the IPAML approach was combined with sialic acid specific derivatization, quali-quantitative determination of neutral and sialylated O-glycans terminal sialic acids with α2,3- and/α2,6-linkages based on MS profile could be accomplished the first time. The proposed assay has been successfully applied to quali-quantitative profiling and sialic acid linkage specific discrimination of the whole O-glycans derived from L02 and SMMC-7721 cell. As the total sialic acids expression level as well as their linkage specificity could be used to predict the risk of various diseases such as tumor, the proposed assay which facilitates quali-quantitatively profiling the whole O-glycans especially sialylated ones, shows great potential for clinical biomarker discovery.
In addition, the method with the merits of bypassing the releasing and relabeling of the glycan, affording the Mucin-type O-glycan itself with chromophore group, which not only improves the detection sensitivity of mass spectrometry, but also enables the further HPLC and LC-MS separation analysis. The proposed method can also be applied to comprehensive screening meaningful O-glycans from clinical samples such as tissue.
The method is effective and highly reproducible. With all the merits described above, we believe this method will make significant contribution to glycomics.
The excellent applicability of the suggested procedure was validated via quantitative comparison of the O-glycans of perch and salmon eggs by ESI-MS and RP-HPLC-UV-ESI-MS/MS.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by National Natural Science Foundation (No. 31300678, 31370804, 21375103 and 31670808), the Scientific Research Program Fund for Shaanxi Province Key Laboratory (14JS101), Shaanxi Basic Research Program of Natural Science (2016JQ3018), Outstanding Youth Backbone of Northwest University and Graduate Innovation Fund (YZZ17163 and YZZ15067).Notes and references
- L. A. Tabak, Semin. Cell Dev. Biol., 2010, 21, 616–621 CrossRef CAS PubMed.
- D. T. Tran and K. G. Ten Hagen, J. Biol. Chem., 2013, 288, 6921–6929 CrossRef CAS PubMed.
- M. R. Kudelka, T. Ju, J. Heimburg-Molinaro and R. D. Cummings, Adv. Cancer Res., 2015, 126, 53–135 CrossRef CAS PubMed.
- S. Yang, E. Jankowska, M. Kosikova, H. Xie and J. Cipollo, Anal. Chem., 2017, 89, 9508–9517 CrossRef CAS PubMed.
- T. Nishikaze, H. Tsumoto, S. Sekiya, S. Iwamoto, Y. Miura and K. Tanaka, Anal. Chem., 2017, 89, 2353–2360 CrossRef CAS PubMed.
- D. H. Dube and C. R. Bertozzi, Nat. Rev. Drug Discovery, 2005, 4, 477–488 CrossRef CAS PubMed.
- A. L. Lewis and W. G. Lewis, Cell Microbiol., 2012, 14, 1174–1182 CrossRef CAS PubMed.
- E. V. Chandrasekaran, J. Xue, J. Xia, R. D. Locke, S. A. Patil, S. Neelamegham and K. L. Matta, J. Proteome Res., 2012, 11, 2609–2618 CrossRef CAS PubMed.
- C. B. Madsen, K. Lavrsen, C. Steentoft, M. B. Vester-Christensen, H. Clausen, H. H. Wandall and A. E. Pedersen, PLoS One, 2013, 8, e72413 CrossRef CAS PubMed.
- S. Chugh, V. S. Gnanapragassam, M. Jain, S. Rachagani, M. P. Ponnusamy and S. K. Batra, Biochim. Biophys. Acta Rev. Canc., 2015, 1856, 211–225 CrossRef CAS PubMed.
- T. Ju, Y. Wang, R. P. Aryal, S. D. Lehoux, X. Ding, M. R. Kudelka, C. Cutler, J. Zeng, J. Wang, X. Sun, J. Heimburg-Molinaro, D. F. Smith and R. D. Cummings, Proteomics: Clin. Appl., 2013, 7, 618–631 CAS.
- K. J. Kim, Y. W. Kim, Y. G. Kim, H. M. Park, J. M. Jin, Y. Hwan Kim, Y. H. Yang, J. Kyu Lee, J. Chung, S. G. Lee and A. Saghatelian, Biotechnol. Prog., 2015, 31, 840–848 CrossRef CAS PubMed.
- L. R. Ruhaak, G. Xu, Q. Li, E. Goonatilleke and C. B. Lebrilla, Chem. Rev., 2018, 118, 7886–7930 CrossRef CAS PubMed.
- M. J. Bowman and J. Zaia, Anal. Chem., 2007, 79, 5777–5784 CrossRef CAS PubMed.
- Y. Xie, J. Liu, J. Zhang, J. L. Hedrick and C. B. Lebrilla, Anal. Chem., 2004, 76, 5186–5197 CrossRef CAS PubMed.
- G. Alvarez-Manilla, N. L. Warren, T. Abney, J. Atwood III, P. Azadi, W. S. York, M. Pierce and R. Orlando, Glycobiology, 2007, 17, 677–687 CrossRef CAS PubMed.
- P. Kang, Y. Mechref, Z. Kyselova, J. A. Goetz and M. V. Novotny, Anal. Chem., 2007, 79, 6064–6073 CrossRef CAS PubMed.
- C. Wang, P. Zhang, W. Jin, L. Li, S. Qiang, Y. Zhang, L. Huang and Z. Wang, J. Proteomics, 2017, 150, 18–30 CrossRef CAS PubMed.
- M. R. Kudelka, A. V. Nairn, M. Y. Sardar, X. Sun, E. L. Chaikof, T. Ju, K. W. Moremen and R. D. Cummings, Glycobiology, 2018, 28, 214–222 CrossRef CAS PubMed.
- P. H. Jensen, D. Kolarich and N. H. Packer, FEBS J., 2010, 277, 81–94 CrossRef CAS PubMed.
- X. You, H. Qin and M. Ye, J. Sep. Sci., 2018, 41, 248–261 CrossRef CAS PubMed.
- J. Furukawa, N. Fujitani and Y. Shinohara, Biomol. Ther., 2013, 3, 198–225 CAS.
- S. Sekiya, Y. Wada and K. Tanaka, Anal. Chem., 2005, 77, 4962–4968 CrossRef CAS PubMed.
- M. Toyoda, H. Ito, Y. K. Matsuno, H. Narimatsu and A. Kameyama, Anal. Chem., 2008, 80, 5211–5218 CrossRef CAS PubMed.
- H. Nie, Y. Li and X. L. Sun, J. Proteomics, 2012, 75, 3098–3112 CrossRef CAS PubMed.
- E. Casal, R. Lebron-Aguilar, F. J. Moreno, N. Corzo and J. E. Quintanilla-Lopez, Rapid Commun. Mass Spectrom., 2010, 24, 885–893 CrossRef CAS PubMed.
- Y. Zhang, B. Wang, W. Jin, Y. Wen, L. Nan, M. Yang, R. Liu, Y. Zhu, C. Wang, L. Huang, X. Song and Z. Wang, Anal. Chim. Acta, 2019, 1048, 105–114 CrossRef CAS PubMed.
- K. Jiang, H. Zhu, L. Li, Y. Guo, E. Gashash, C. Ma, X. Sun, J. Li, L. Zhang and P. G. Wang, Anal. Chim. Acta, 2017, 981, 53–61 CrossRef CAS PubMed.
- M. R. Kudelka, A. Antonopoulos, Y. Wang, D. M. Duong, X. Song, N. T. Seyfried, A. Dell, S. M. Haslam, R. D. Cummings and T. Ju, Nat. Methods, 2016, 13, 81–86 CrossRef CAS PubMed.
- P. Kang, Y. Mechref, I. Klouckova and M. V. Novotny, Rapid Commun. Mass Spectrom., 2005, 19, 3421–3428 CrossRef CAS PubMed.
- I. Ciucanu and C. E. Costello, J. Am. Chem. Soc., 2003, 125, 16213–16219 CrossRef CAS PubMed.
- D. Frem, D. Urban, S. Norsikian and J.-M. Beau, Eur. J. Org. Chem., 2017, 2017, 5094–5101 CrossRef CAS.
- R. Sommer, D. Hauck and A. Titz, ChemistrySelect, 2017, 2, 4187–4192 CrossRef CAS.
- J.-M. Beau, F.-D. Boyer, S. Norsikian, D. Urban, B. Vauzeilles and A. Xolin, Eur. J. Org. Chem., 2018, 2018, 5795–5814 CrossRef CAS.
- Y. Zhang, W. Zhao, Y. Zhao and Q. He, Glycoconjugate J., 2015, 32, 39–47 CrossRef CAS PubMed.
- H. Li, W. Gao, X. Feng, B. F. Liu and X. Liu, Anal. Chim. Acta, 2016, 924, 77–85 CrossRef CAS PubMed.
- P. Babu, S. J. North, J. Jang-Lee, S. Chalabi, K. Mackerness, S. R. Stowell, R. D. Cummings, S. Rankin, A. Dell and S. M. Haslam, Glycoconjugate J., 2009, 26, 975–986 CrossRef CAS PubMed.
- J. P. Zanetta, V. V. Gouyer, E. Maes, A. Pons, B. Hemon, A. Zweibaum, P. Delannoy and G. Huet, Glycobiology, 2000, 20, 565–575 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra01114e |
| This journal is © The Royal Society of Chemistry 2019 |