
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlled grafting of dialkylphosphonate-based ionic liquids on γ-alumina: design of hybrid materials with high potential for CO2 separation applications†
M. A. Pizzoccaro-Zilamy‡
a,
S. Muñoz Piña ab,
B. Rebiereb,
C. Danielc,
D. Farrussengc,
M. Drobeka,
G. Sillyb,
A. Julbea and
G. Guerrero*b
ab,
B. Rebiereb,
C. Danielc,
D. Farrussengc,
M. Drobeka,
G. Sillyb,
A. Julbea and
G. Guerrero*b
aInstitut Européen des Membranes, UMR5635, CNRS-UM-ENSCM, Université de Montpellier (CC047), Place Eugène Bataillon, 34095 Montpellier Cedex 5, France
bInstitut Charles Gerhardt, UMR5253, CNRS-UM-ENSCM, Université de Montpellier, Place Eugène Bataillon, 34095 Montpellier Cedex 5, France. E-mail: gilles.guerrero@umontpellier.fr; Tel: +33-467-144-223
cIRCELYON, UMR5256, CNRS-Université Lyon 1, 2 Avenue Albert Einstein, 69626 Villeurbanne Cedex, France
First published on 25th June 2019
Abstract
In this work we provide a detailed study on grafting reactions of various dialkylphosphonate-based ILs. Special attention has been devoted to a comprehensive investigation on how the nature of the anion and the organic spacer composition (hydrophilic or hydrophobic groups) could impact the grafting densities and bonding modes of phosphonate-based ILs anchored to γ-alumina (γ-Al2O3) powders. For the first time, the bonding of phosphonate-based ILs with only surface hexacoordinated aluminum nuclei was established using both solid-state 31P–27Al D-HMQC and 31P NMR experiments. It has been demonstrated that the grafting of dialkylphosphonate-based ILs is competing with a hydrolysis and/or precipitation process which could be attractively hindered by changing the anion nature: bis(trifluoromethane)sulfonylimide anion instead of bromide. In additon, independently of the chosen spacer, similar reaction conditions led to equivalent grafting densities with different bonding mode configurations. The CO2 physisorption analysis on both pure ILs and grafted ILs on alumina powders confirmed that the initial sorption properties of ILs do not change upon grafting, thus confirming the attractive potential of as-grafted ILs for the preparation of hybrid materials in a form of selective adsorbers or membranes for CO2 separation applications.
Introduction
Organic–inorganic hybrid materials based on metal oxide supports functionalized with ionic liquids (ILs) are emerging as an important class of components for the adsorptive separation of acidic gases (e.g., CO2, SO2) from dilute gas streams.1–7 Compared to the amine-oxide hybrid materials,8 the ILs systems are known to interact strongly and reversibly with CO2.1–3 The remarkable properties of ILs such as limited vaporization coupled with good chemical and thermal stability have led to the realization of Supported Ionic Liquid (SIL) materials applied as adsorbents or membranes for CO2 separation applications.2–7,9Such membranes are composed of two parts: a high surface area inorganic (ceramic) support and the IL2,3 linked together either by weak (i.e. van der Waals forces or hydrogen bonds), or strong (i.e. covalent or coordination bonds) interactions, respectively referenced as Class I and Class II materials (see Fig. 1). Class I materials are typically prepared by impregnation techniques like vacuum/pressure-assisted infiltration or immersion of the support in the IL solution, followed by solvent evaporation. In this way, ILs are only physisorbed or mechanically trapped in the pores of the support, preserving their initial bulk physico-chemical properties. Despite of their promising performance,9–14 these systems exhibit inherent limitations such as non-uniformity or limited stability. In order to design hybrid materials with higher chemical and thermo-mechanical stability, covalent linking (grafting) of ILs on ceramic supports appears as an attractive alternative strategy (Class II systems). However, controlling the chemical grafting of ILs on the surface of porous materials is not obvious when comparing to a simple impregnation. In Class II systems, tailoring the chemical nature of the support, as well as its microstructure, and governing both the fixation of the ILs and their distribution on the surface are challenging and lead to Grafted Ionic Liquid (GIL) materials with novel properties.5 In addition, the Class II systems limit the ILs leaching and improve the long-term stability of ILs based materials.5
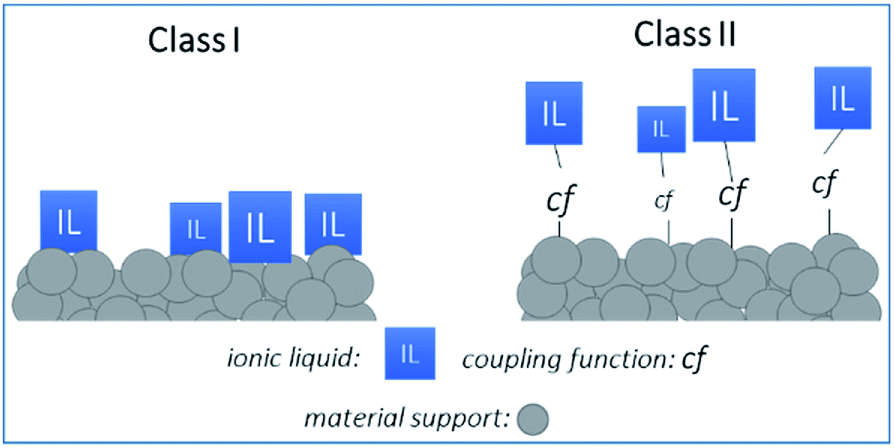 | ||
| Fig. 1 The two classes of supported ionic liquid systems: Class I (physisorbed IL species – weak interactions with the support) and Class II (grafted IL species – strong interactions with the support). | ||
As a result, each newly-developed Class II/SIL system has a critical impact on both environmental and economic aspects. Several Class II/SIL materials are developed for catalytic applications and recently, special attention has been paid to the preparation of grafted ionic liquid membranes (GILMs) for potential gas separation applications.3,4 Indeed, due to the exceptional solubility of acidic gases in ILs, the preparation of supported ionic liquid materials with ILs covalently bounded to selected ceramic membrane supports is of great interest.3,4,10,11
In our previous studies we demonstrated that γ-alumina can be grafted with two imidazolium bromide-based ionic liquids bearing phosphonyl groups on the cationic part (either dialkyl or bis(trimethylsilyl)ester phosphonate) under specific reaction conditions.15 The grafting process was found to be time-dependent and the possible formation of bulk aluminum phosphonate phases was avoided. In the conclusion of this study, the applied reaction conditions with the dialkylphosphonate-based IL ([ImPE][Br]) were found to be the most promising route for grafting the surface of γ-alumina supports.15
In this work, the following four ILs were studied (Fig. 2): [ImPE][Br] (1-methyl-3-(3-(diethylphosphinyl)propyl)-imidazolium bromide), [ImPE][Tf2N] (1-methyl-3-(3-(diethyl-phosphinyl)propyl)imidazolium bis(trifluoromethanesulfonimide)), [ImC12PE][Tf2N] (1-methyl-3-(3-(diethylphosphinyl)dodecyl)imidazolium bis(trifluoromethanesulfonimide)) and [ImPEGPE][Tf2N] (1-methyl-3-(3-(diethylphosphinyl)2-(2-(2-(2-ethoxy)ethoxy)ethoxy)ethyl)-imidazolium bis(trifluoro methanesulfonimide)). These ILs are expected to be promising coupling agents for CO2 transport applications. As shown in Fig. 2 the ILs are composed of the same cationic moiety (i.e. N-methylimidazolium salt) and differ in their counter anion (Br− or Tf2N−) and/or organic spacer parts (oligoethylene glycol, short (propyl) or long (dodecyl) alkyl chain).
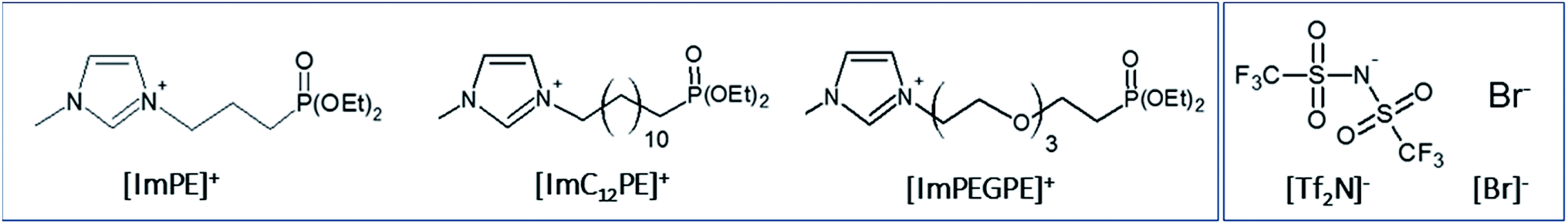 | ||
| Fig. 2 Structure of cationic (left) and anionic (right) parts of the dialkylphosphonate-based ILs used in this study. | ||
The principal objective of this study is to provide an extended insight into an efficient control of the grafting reaction for various dialkylphosphonate-based ILs. A special attention was devoted to the choice of ILs composition (cation, anion and organic spacer) in order to observe any possible influence on the grafting duration and bonding modes. The as-prepared ILs were thoroughly characterized to identify the key parameters for the design of hybrid membranes for CO2 separation applications. The effectiveness of IL grafting on the γ-alumina surface, the influence of the IL composition on the surface properties and microstructure were studied by using a set of characterization techniques including Energy Dispersive X-ray spectroscopy (EDX), N2 physisorption measurements, Fourier-transform infrared spectroscopy (FTIR). Solid-state multi-Nuclear Magnetic Resonance (NMR) spectroscopy (1H, 31P, 27Al and 19F nuclei). In particular, 31P–27Al D-HMQC experiments were applied for the first time to this type of systems. The influence of IL composition on the CO2 sorption isotherms was also investigated on grafted γ-alumina powders and compared to a blank γ-alumina.
Experimental
Starting materials
Triethyl phosphite (98%) and 1-methylimidazole (≥99%) were purchased from Sigma-Aldrich (Saint-Quentin-Fallavier, France) and were used as received. 1,3-Dibromopropane (98%) was provided by Fisher Chemical (Illkirch, France). Tetraethylene glycol (99%), triphenylphosphine (99%), and N-bromosuccinimide (99%) were purchased from Alfa Aesar. The diethyl(3-bromododecyl)phosphonate (99%) was provided by Sikemia. Lithium bis(trifluoromethane)sulfonimide (99%) was obtained from Solvionic. Anhydrous ethanol, methanol, pentane, tetrahydrofuran (THF) and methylene chloride (CH2Cl2) were provided by Sigma-Aldrich. These solvents were used as received, except for THF which was dried on a silica–alumina drying column (PureSolv-Innovative Technology).Boehmite (Pural SB type) with high crystallinity and surface area (249 m2 g−1) was supplied by CTI S.A. (Salindres, France). It was used to prepare a batch of γ-Al2O3 powder (SBET ∼ 200 m2 g−1) using a sol–gel process based on colloid chemistry in aqueous media followed by a final thermal treatment at 600 °C/3 h in air, according to a protocol described in our previous work.15 Before use, the γ-alumina powder was pretreated at 400 °C under N2 and flasks containing equal amounts of powder (400 mg) were prepared and stored in a glovebox under argon.15 This final step ensured an absence of water on the alumina surface yielding comparable samples for the grafting reactions. Details on the γ-Al2O3 powder synthesis are provided in the ESI.†
Organic synthesis
The syntheses of [ImPE][Br], [ImPE][Tf2N], [ImC12PE][Tf2N] and [ImPEGPE][Tf2N] are detailed in ESI.† Briefly, [ImPE][Br] was obtained as a yellow oil in high yield by nucleophilic substitution of 1-methylimidazole with diethyl(3-bromopropyl)phosphonate in dry tetrahydrofuran (THF) (δ(31P) = 29.80 ppm (CDCl3)). The syntheses of both [ImC12PE][Br] and [ImPEGPE][Br] were adapted from the conditions described by Rout et al.16 Anion exchange reactions between LiTf2N and the different bromide phosphonate-based ILs were carried out in aqueous solution and led to the following ILs: [ImPE][Tf2N] (δ(31P) = 30.39 ppm (DMSO)), [ImPEGPE][Tf2N] (δ(31P) = 28.66 ppm (DMSO)) and [ImC12PE][Tf2N] (δ(31P) = 32.07 ppm (DMSO)).Preparation of the IL/γ-Al2O3 grafted powders
The reaction conditions applied for the graftings of [ImPE][Br], [ImPE][Tf2N], [ImC12PE][Tf2N] and [ImPEGPE][Tf2N] ILs on the surface and inside the pores of γ-Al2O3 powder are reported in Table 1. Typical experiments are described below. Phosphonate-based IL solutions were prepared by dissolving a 12-fold excess (7.2 mmol) of the pure [ImPE][Br] or [ImPE][Tf2N] and a 6-fold excess (3.6 mmol) of pure [ImPEGPE][Tf2N] or [ImC12PE][Tf2N] in the selected solvent. Then, 400 mg of the γ-alumina powder (stored under argon) were dispersed within 10 mL of the prepared solution directly in a Teflon® autoclave. The autoclave was sealed and the suspension was heated at 130 °C for the selected reaction time. After cooling down to room temperature, the suspension was centrifuged at 8500 rpm for 5 minutes. The supernatant was removed and the remaining suspension was re-dispersed in 5 mL of a (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) ethanol–water solution and stirred at room temperature for 5 minutes to remove any physisorbed species from its surface. The supernatant was removed after centrifugation (8500 rpm, 5 minutes) and the washing step with the (1:1) ethanol–water solution was repeated twice. An additional washing treatment had to be carried out using a Soxhlet extractor for the [Tf2N]− grafted samples. The grafted powders were placed in a cellulose thimble and closed with cotton wool. The treatment was conducted with 80 mL of absolute ethanol at 115 °C for 24 h performing about 26 washing cycles. All the as-washed samples were finally dried at 70 °C for ∼16 hours.
:1) ethanol–water solution and stirred at room temperature for 5 minutes to remove any physisorbed species from its surface. The supernatant was removed after centrifugation (8500 rpm, 5 minutes) and the washing step with the (1:1) ethanol–water solution was repeated twice. An additional washing treatment had to be carried out using a Soxhlet extractor for the [Tf2N]− grafted samples. The grafted powders were placed in a cellulose thimble and closed with cotton wool. The treatment was conducted with 80 mL of absolute ethanol at 115 °C for 24 h performing about 26 washing cycles. All the as-washed samples were finally dried at 70 °C for ∼16 hours.
| Sample | Cation | Anion | Grafting duration (h) | n-fold excess | Solvent (v:v) |
wt% Pa | Pb nm−2 | P/Br or P/S atomic ratio (expected)a |
|---|---|---|---|---|---|---|---|---|
| a From EDX analysis.b Average number of coupling molecules per nm2. | ||||||||
| ImPE-Br(1) | [ImPE]+ | [Br]− | 40 | 12 | H2O | 1.73 ± 0.08 | 2.2 | P/Br = 0.91 (1.0) |
| ImPE-Br(2) | [ImPE]+ | [Br]− | 45 | 12 | H2O | 2.41 ± 0.25 | 3.0 | P/Br = 1.05 (1.0) |
| ImPE-Br(3) | [ImPE]+ | [Br]− | 92 | 12 | H2O | 4.17 ± 1.15 | 5.2 | P/Br = 1.15 (1.0) |
| ImPE-Tf2N(1) | [ImPE]+ | [Tf2N]− | 40 | 12 | H2O:EtOH (1:1) |
1.34 ± 0.08 | 2.2 | P/S = 0.54 (0.5) |
| ImPE-Tf2N(2) | [ImPE]+ | [Tf2N]− | 48 | 12 | H2O:EtOH (1:1) |
1.29 ± 0.08 | 2.1 | P/S = 0.72 (0.5) |
| ImPE-Tf2N(3) | [ImPE]+ | [Tf2N]− | 92 | 12 | H2O:EtOH (1:1) |
0.94 ± 0.10 | 1.5 | P/S = 0.58 (0.5) |
| ImC12PE-Tf2N | [ImC12PE]+ | [Tf2N]− | 40 | 6 | H2O:EtOH (1:1) |
1.43 ± 0.39 | 2.6 | P/S = 0.75 (0.5) |
| ImPEGPE-Tf2N | [ImPEGPE]+ | [Tf2N]− | 40 | 6 | H2O:EtOH (1:1) |
1.47 ± 0.06 | 2.6 | P/S = 0.51 (0.5) |
Powder X-ray diffraction
Powder X-ray diffraction patterns were recorded using a PANanalytical X'Pert PRO diffractometer at the wavelength of Cu Kα (λ = 1.5405 Å) (X-ray power: 40 kV, 20 mA) in Bragg–Brentano scanning mode. The program scanned angles (2θ) from 5 to 55° with a 0.017° step and a step time of 40 s.Specific surface area measurements
The specific surface areas (SBET) and BET constants (CBET) of the samples were obtained from N2 adsorption experiments at 77 K by using Tristar instrument (Micromeritics) for the grafted powders and ASAP 2020 (Micromeritics) for the pristine γ-alumina powder. Prior to measurements, samples were degassed under vacuum overnight at 100 °C for the grafted powders and 300 °C for the pristine γ-alumina powder.CO2 gas sorption experiments
CO2 sorption measurements were conducted with the help of a BELSORP Max I system (Microtrac Bel) on both blank pristine γ-Al2O3 powder and selected grafted samples which are considered as CO2-phile (i.e. ImPE-Tf2N(1), ImC12PE-Tf2N and ImPEGPE-Tf2N). The blank pristine γ-Al2O3 powder has been exposed to the same reaction conditions as during the hydrothermal grafting treatment but without any phosphonate-based IL (reference sample). Before measurements, powders were outgassed during 17 h under primary vacuum and 3 h under secondary vacuum at 100 °C.Energy dispersive X-ray spectroscopy (EDX)
The weight percentages of phosphorus, sulfur and bromine in the samples were determined by EDX using a Zeiss SEM EVO HD15 at 10 kV with the Oxford instruments software. Samples were prepared as pellets for the analysis and deposited on double-sided carbon tape.FTIR spectroscopy
FTIR spectra were obtained with a PerkinElmer Spectrum 2 spectrophotometer and were recorded in the 1400–800 cm−1 range using 4 scans at a nominal resolution of 4 cm−1 in ATR mode (with γ-alumina as a background spectrum).Solid-state NMR experiments
Solid-state NMR spectra were acquired on a Varian VNMRS 600 spectrometer (1H: 599.95 MHz, 31P: 242.93 MHz, 27Al: 156.37 MHz, 19F: 564.511 MHz). A 3.2 mm Varian T3 HXY magic angle spinning (MAS) probe was used for 1H, 27Al One-pulse and 31P CP experiments, and a 3.2 mm Varian T3 HX magic angle spinning (MAS) probe for the 19F One-pulse experiments. All NMR experiments were performed under temperature regulation in order to ensure that the temperature inside the rotor is 20 °C.27Al MAS NMR experiments were acquired at a spinning frequency of 20 kHz. The single pulse experiments were performed with a ∼15° solid pulse of 1 μs and 1H decoupling during acquisition. A recycle delay of 5 s was used (corresponding in both cases to full relaxation of 27Al nuclei). 27Al chemical shifts were referenced to external Al(NO3)3 at 0 ppm.
31P CP-MAS NMR experiments were recorded at a spinning frequency of 20 kHz. The number of acquisitions was 64 and the recycle delays were 7 s. A 90° pulse width of 4 μs with 1.5 ms CP contact time was employed. An acquisition time of 20.48 ms was used and the 1H channel was decoupled on this period. 31P chemical shifts were referenced to external hydroxyapatite at 2.80 ppm (used as a secondary reference).
31P–27Al correlation experiments were performed using a D-HMQC sequence (Dipolar Hetero-nuclear Multiple-Quantum Coherences). A spin echo selective to the central transition was first applied on the 27Al side (using 27Al π/2 and π pulses of 8 and 16 μs respectively, these pulse times being optimized directly on the sample). 1H π/2 pulse of 2.5 μs was applied on either side of the 27Al π pulse. In the case of the 31P–27Al correlation experiments, 31P π/2 pulse of 3 μs was applied on either side of the 27Al π pulse. The dipolar recoupling scheme (SR421) was rotor-synchronized and the recoupling time, τ, is integer multiples, p, of the rotor period (τ = pTR). The recycle delay was set to 0.25 s (for the 1H–27Al D-HMQC) or 1 s (for the 31P–27Al D-HMQC), and the total number of scans acquired ranged from 1536 to 3072, depending on the experiences and samples. All 2D experiments were recorded under rotor-synchronised conditions along the indirect F1 dimension.
CO2 solubility measurements
The CO2 solubility in the pure ILs [ImPE][Tf2N], [ImPEGPE][Tf2N] and [ImC12PE][Tf2N] were measured at 30 °C using the isochoric saturation method. The description of the system is provided in the ESI.† Prior to measurements, the ILs were degassed for 10 h (vacuum pressure of 5 × 10−5 mbar) and the resulting mass of each degassed IL was determined. The pressure decrease (resulting from CO2 absorption into the IL) was monitored over time. Depending on the type and quantity of the IL, the equilibrium was reached after 1 to 3 h. The CO2 solubility data measured for [emim][Tf2N] (used as a standard) were in good agreement with those published in the literature.Results and discussion
Optimization of the grafting reaction conditions
The balance between the grafting of organic moieties on inorganic surfaces and the formation of bulk aluminum phosphonate phases is closely related to the nature of the starting materials (γ-alumina), of the reagents (ILs) and of the grafting reaction conditions. As reported in our previous work,15 the grafting density increases with the concentration of the dialkylphosphonate IL solution when applying forcing reaction conditions. This term refers to an application of both large excess of coupling agent (necessary to achieve a full support surface coverage), and an increased reaction temperature superior to the boiling point of the solvent used (130 °C). It has been demonstrated that IL solutions containing a 6 or 12-fold excess (respectively 3.6 or 7.2 mmol) of the IL in either ethanol–water co-solvent (for [ImPE][Tf2N], [ImC12PE][Tf2N] and [ImPEGPE][Tf2N]) or in aqueous medium (for [ImPE][Br]) led to a significant enhancement of the grafted species quantity. The reaction parameters (time, solvent, concentration) used for each sample are detailed in Table 1. At the end of the grafting process, the samples were centrifuged and washed with an ethanol–water solution to remove unreacted and physisorbed species. A second washing treatment using the Soxhlet method (dry ethanol, 110 °C, 24 h) was employed for the samples grafted with Tf2N− anion. All samples were then dried at 70 °C before analysis.The reaction of organophosphorus derivatives on γ-Al2O3 surface is supposed to involve: (i) the coordination of the oxygen atom from the phosphoryl group (P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) to Lewis acid sites, and (ii) the condensation reactions of P–OH or P-OX functions (X = –CH3, –CH2CH3 or –Si(CH3)3) with Al–OH surface groups.17 Up to three P–O–Al bonds for each phosphonate unit can be formed during the grafting reaction resulting in three possible bonding modes (monodentate, bidentate or tridentate).
O) to Lewis acid sites, and (ii) the condensation reactions of P–OH or P-OX functions (X = –CH3, –CH2CH3 or –Si(CH3)3) with Al–OH surface groups.17 Up to three P–O–Al bonds for each phosphonate unit can be formed during the grafting reaction resulting in three possible bonding modes (monodentate, bidentate or tridentate).
Furthermore, these bonds can be either bridging (each oxygen atom binds to a different metal atom) or chelating (two or three oxygen atoms bind to the same metal atom).18 Hydrogen bonds can also exist between residual P–OH or PO groups and hydroxyl surface groups of the γ-Al2O3. All these options yield a large variety of possible bonding modes (few of them are represented in Fig. 3).
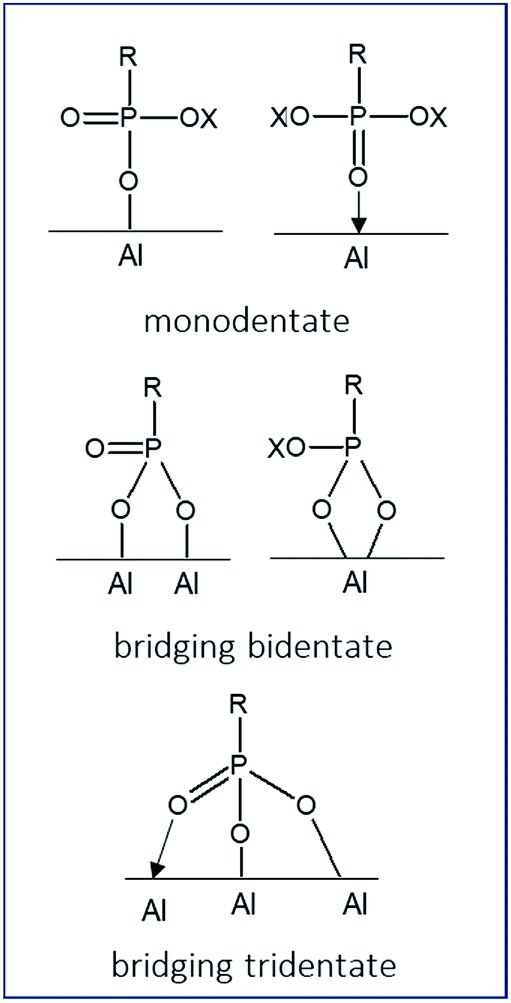 | ||
| Fig. 3 Schematic representations of some possible bonding modes of phosphonate-based molecules on hydroxylated alumina surface. | ||
The FTIR spectra in the range 1400–800 cm−1 of the pure phosphonate-based ILs are presented in Fig. 4. The attribution of the vibration bands for [ImPE][Br] was previously detailed on the basis of the theoretical FTIR spectrum simulated from DFT calculations.15 The dominating bands are related to the phosphonate moieties and correspond to PO stretching vibration, C–H deformation vibration, asymmetric and symmetric P–O–C stretching vibrations, located respectively at 1230 cm−1, 1014 cm−1, 1042 and 958 cm−1.15 In comparison, the dominating bands in the spectra of [ImPE][Tf2N], [ImC12PE][Tf2N] and [ImPEGPE][Tf2N] (Fig. 4) are related to the Tf2N− anion: asymmetric and symmetric –SO2 stretching vibrations at respectively 1327–1346, 1195 and 1132 cm−1, –CF3 stretching vibration at 1224 cm−1 and –SNS stretching vibration at 1061 cm−1.19 The comparison of the experimental and DFT calculated spectra of [ImPE][Tf2N] allowed to link the vibration modes to the associated wavenumbers (Fig. S12†). It was then possible to identify the vibration bands related to the cation such as the PO stretching vibration at 1236 cm−1, the asymmetric and symmetric P–O–C stretching vibrations at 970 and 1050 cm−1,20,21 and the C–H and –CH2 stretching vibrations at 1017 cm−1 and 1195 cm−1, respectively.19 By using these attributions, the PO stretching vibrations for [ImPEGPE][Tf2N] and [ImC12PE][Tf2N] were associated respectively to the bands at 1250 cm−1 and 1236 cm−1. In the case of [ImPEGPE][Tf2N] and [ImC12PE][Tf2N], the asymmetric and symmetric P–O–C stretching vibrations were located at the same position as the corresponding bands observed for [ImPE][Tf2N].
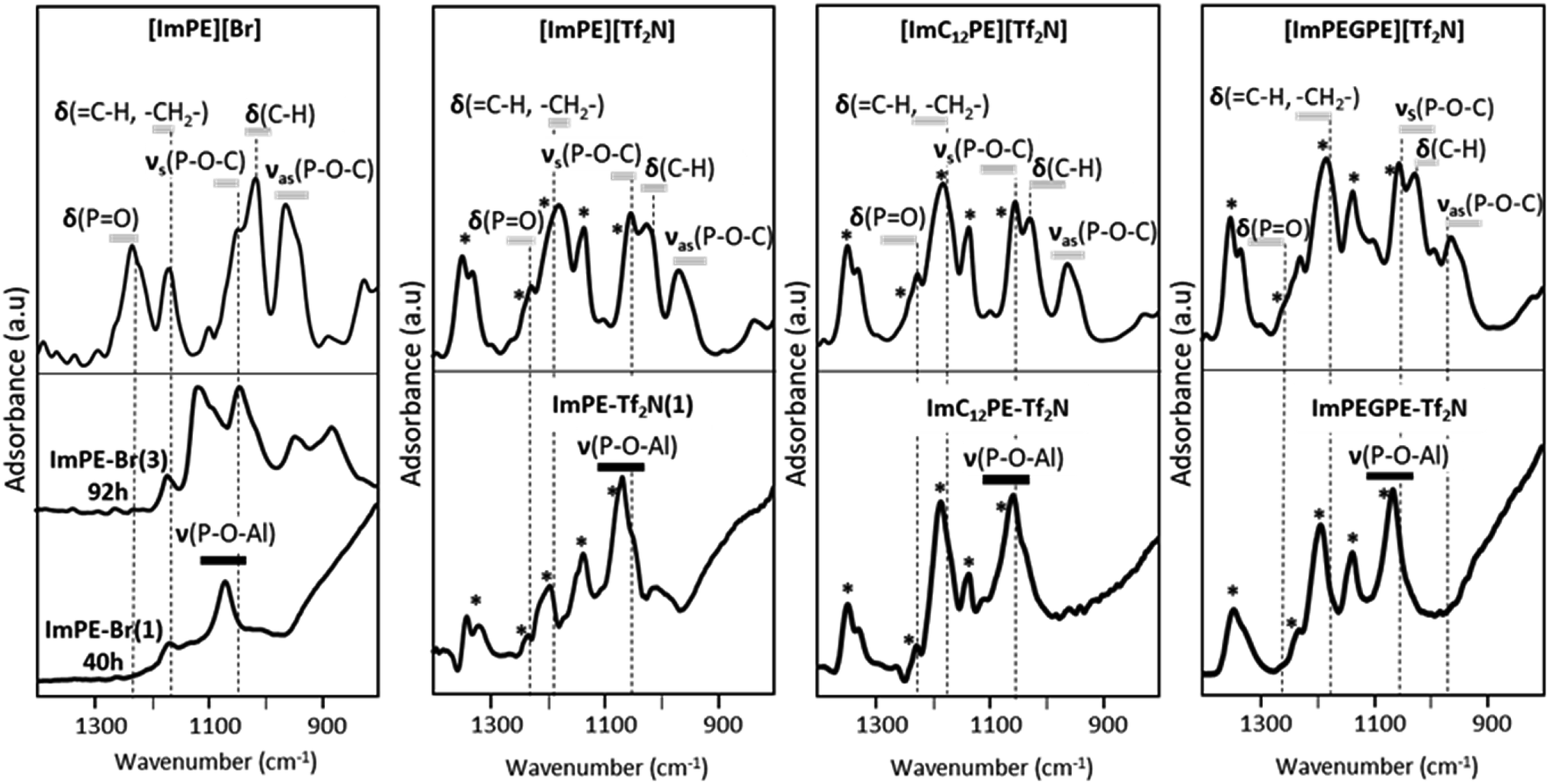 | ||
| Fig. 4 Experimental FTIR spectra of pure [ImPE][Br], [ImPE][Tf2N], [ImC12PE][Tf2N] and [ImPEGPE][Tf2N] ILs and corresponding grafted samples under forcing conditions after 40 h reaction, and also after 92 h reaction for [ImPE][Br]. * vibration due to Tf2N− anion. | ||
Concerning the grafted samples, the FTIR spectra of γ-alumina powders grafted with [ImPE][Br] during 40 h (ImPE-Br(1), Fig. 4) are similar to spectra of ImPE-Br observed in our previous study (130 °C, 17 h with a solution concentration in 12-fold excess)15 indicating that an increase of the grafting solution concentration and moderate grafting reaction time give rise to similar phosphonate bonding modes (i.e. with dominating bidentate and tridentate bonding modes). However, when the grafting reaction time increases from 40 h to 92 h (ImPE-Br(3), Fig. 4), the absorption bands become broader suggesting the presence of different P–O–Al bonds featuring multiple vibration bands and so on, an evolution of the phosphonate bonding modes. The FTIR spectrum of the ImPE-Tf2N(1) sample obtained after 40 h reaction time presents absorption bands associated with the Tf2N− anions at 1346, 1327, 1224, 1195, 1132 and 1061 cm−1. This result suggests that the integrity of the anion has been maintained as expected from EDX measurements (Table 1, e.g., ratio P/S = 0.54 (the theoretical value was 0.5)). 19F solid-state NMR confirmed this observation as well. The spectrum of ImPE-Tf2N(1) showed a broad signal centered at −80.6 ppm, corresponding to the –CF3 functions in the anion (Fig. S3†). Regarding the FTIR spectrum of the ImPE-Tf2N(1) sample, the stretching vibration of the phosphoryl group (PO) initially at ∼1236 cm−1 is no longer present (Fig. 4) and a strong absorption band at ∼1058 cm−1 relative to the formation of P–O–Al bands appeared. Furthermore, the presence of weak absorption bands at ∼1050 and 960 cm−1 (region of P–O–C stretching bands) can also be noticed, suggesting the existence of residual P–OEt groups.
The FTIR spectra of the [ImPEGPE][Tf2N] and [ImC12PE][Tf2N] grafted samples were also dominated by absorption bands related to the Tf2N− anions. As for ImPE-Tf2N(1) sample, the presence and conservation of the organic part was confirmed by EDX analysis and 19F solid-state NMR.
Concerning the coupling function, one can notice the decrease of the intensity of the phosphoryl (PO) stretching bands for both samples, with an increase of the P–O–Al stretching vibrations. This highlights the reactivity of the coupling function with the alumina surface, leading to grafted organic moieties.
EDX analysis of the grafted γ-Al2O3 samples were useful to quantify both the wt% P on the grafted samples and to estimate the grafting densities (P nm−2) on the γ-Al2O3 surface (Table 1). First, the results reveal the presence of phosphorus indicating the presence of IL coupling agents in all samples. By comparison with the best grafted sample prepared in our previous work (i.e. wt% P = 1.42 ± 0.03, reaction time = 17 h, 12-fold excess IL),15 higher phosphorus contents were measured for the new ImPE-Br samples series. As an example, the sample also prepared in 12-fold excess of [ImPE][Br] (40 h reaction time) exhibits a P content of 1.73 ± 0.08 wt%. The quantity of grafted species increases continuously with the reaction time. After 45 h, the P content reaches the value of 2.41 ± 0.25 wt%, corresponding to ∼75% of a full surface coverage (i.e., 3.2 wt% P correspond to a full surface coverage (monolayer), assuming an area of 25 Å2 per phosphonate molecule). After 92 h, the wt% P reached 4.17 ± 0.65 and so, exceeded the value corresponding to a full surface coverage meaning that ∼130% of the full monolayer is reached (e.g., 5.2 P nm−2). This high phosphorus content observed for ImPE-Br(3) suggests the formation of bulk aluminum phosphonate phases by a dissolution–precipitation mechanism.22
Concerning the ImPE-Tf2N sample series, the P concentration in the grafted samples never exceeds the value corresponding to full surface coverage (i.e., 2.5 wt% P for ImPE-Tf2N). For ImPE-Tf2N samples, the grafting density values (P nm−2) vary in the range 1.5–2.2, showing that a maximum of ∼55% of the full monolayer was achieved after a 40 h reaction time without any formation of bulk aluminum phosphonate phases, even after a 92 h treatment. This result shows that both the nature of the counter anion and the solvent (H2O:EtOH mixture instead of H2O) play key roles in governing the reaction pathways. The same conclusions were made for the ImPEGPE-Tf2N and ImC12PE-Tf2N samples (i.e., 2.3 and 2.2 wt% P). In both cases, the P nm−2 values were 2.6, representing respectively the formation of 62.5 and 65% of the full monolayer. In comparison with ImPE-Tf2N samples, the nature of the organic spacer slightly increases the grafting density and no formation of bulk aluminum phosphonate phases was also evidenced.
XRD patterns of ImPE-Br series, ImPE-Tf2N series, ImPEGPE-Tf2N and ImC12PE-Tf2N grafted samples are presented in Fig. 5. The grafting of organic coupling agents should not affect the γ-Al2O3 diffraction patterns. As expected, all the Tf2N− based phosphonate-ILs samples (Fig. 5b) and the ImPE-Br(1) sample (40 h reaction time) (Fig. 5a) reveal that the initial broad diffraction peaks characteristic for the γ-Al2O3 structure was maintained after the grafting reaction. However, additional diffraction peaks corresponding to the formation of a boehmite phase were detected, revealing a partial hydrolysis of the support (surface or bulk) during the grafting treatment. Both ImPE-Br(2) and ImPE-Br(3) samples exhibit additional low-angle diffraction peaks at 2θ ≈ 5° and 8°, respectively, attributed to the formation of bulk aluminum phosphonate phases in agreement, for ImPE-Br(3) sample, with the high wt% P value determined by EDX analysis. Unexpectedly, the XRD pattern of ImPE-Br(2) sample presents also low-angle diffraction peaks even with a wt% P not exceeding the full surface coverage. The aluminum phosphonate phases have characteristic lamellar structures, with interlamellar spacing d001 (related to inter-sheets distance) that can be respectively estimated at 10.3 (ImPE-Br(2)) and at 17.0 Å ImPE-Br(3). As an example, the interlamellar spacing for aluminium phenylphosphonate phases is classically in the range 14–15 Å, with phenyl groups in adjacent positions.23 However, this structural organization could vary depending on the reaction conditions; the interlamellar spacing can increase for less stacked arrangements.24 These XRD results suggest that for the grafted samples composed of the [ImPE]+ cation, a dissolution–precipitation mechanism occurs resulting from a hydrolysis of the γ-alumina support forming: (i) boehmite for [ImPE][Tf2N] and [ImPE][Br] and (ii) bulk aluminum phosphonate phases for [ImPE][Br].
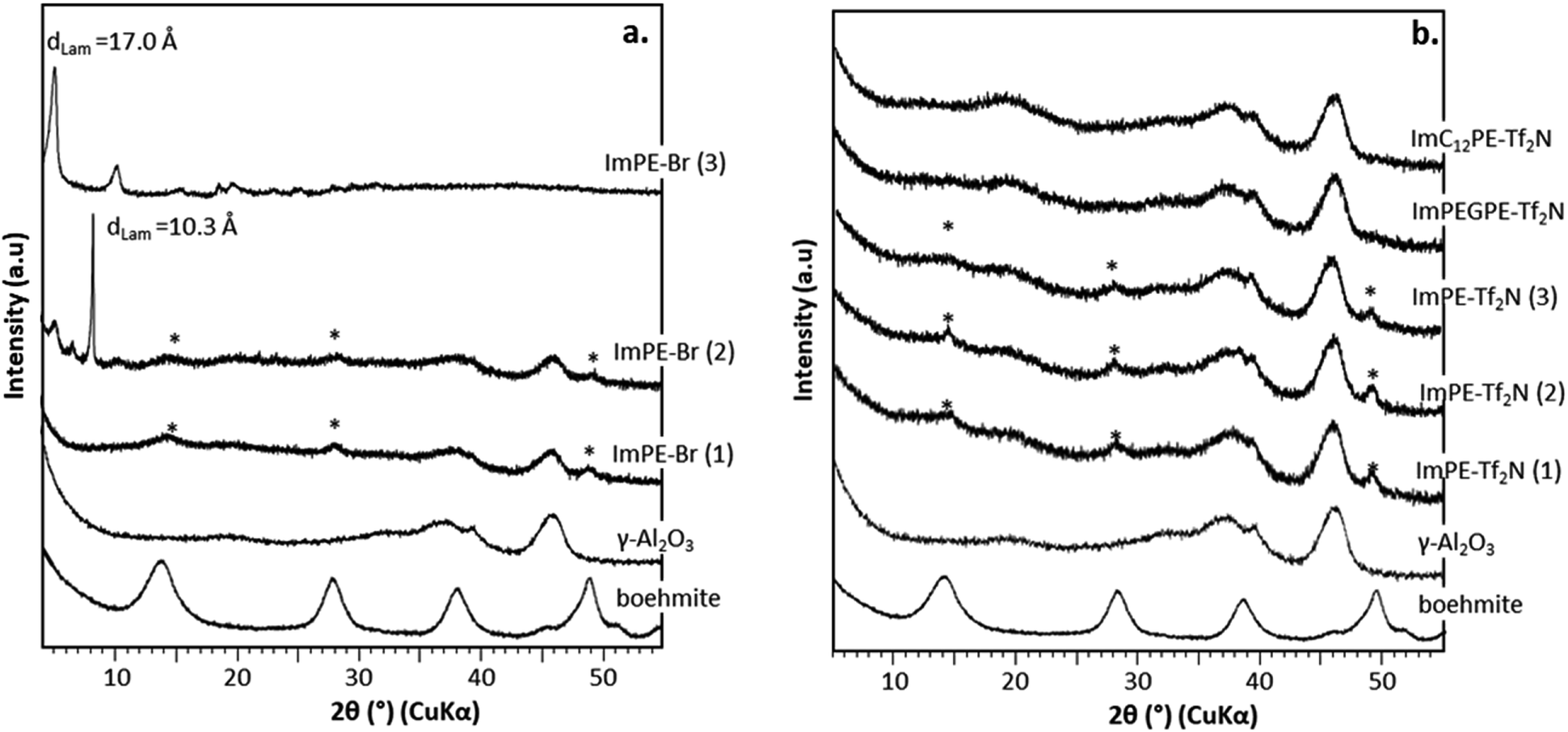 | ||
| Fig. 5 XRD diffraction patterns for pristine γ-Al2O3, boehmite (*) and grafted samples: (a) with Br− anion, (b) with Tf2N− anion. | ||
While the grafting of alumina with phosphonic acids and their parent trimethylsilyl esters may lead to bulk phosphonate aluminum phases even in soft conditions, the use of the diethylester phosphonate coupling function in organic medium only led to surface modified alumina.21,22 Indeed, the chemical nature of the coupling agent is also important to avoid the dissolution–precipitation process. The formation of phosphonic acid functions from diethyl ester parents during the grafting reaction may promote the formation of aluminum phosphonate phases. In order to better understand the formation mechanism of those lamellar phases, the stability of the diethyl ester phosphonate function toward hydrolysis in aqueous medium was evaluated. The influence of the reaction conditions has been studied on pure ILs at different reaction times (i.e., 20, 48 and 92 h). 1H and 31P liquid NMR was used respectively to confirm the molecule integrity and to reveal the evolution of the phosphonate functional groups. Details of the procedure including the 1H and 31P liquid NMR spectra are provided in the ESI.† On the basis of the chemical shifts integration in 31P liquid NMR related to the ester or acid forms of the coupling functions, it was possible to quantify the proportion of residual diethylester functions after the different grafting treatments in forcing reaction conditions. As presented in Fig. 6, after a 20 h reaction time in water, only 1% of the initial [ImPE][Br] IL is still present. The major part of the IL has been transformed to its phosphonic acid (82%) or to its monoester parents (Fig. S5†). In ethanol–water co-solvent, the hydrolysis of the coupling function is slower and only 26% or 7% of the [ImPE][Br] IL remained after 20 h and 92 h reaction time, respectively (Fig. S10–S15†). Concerning the [ImPE][Tf2N] coupling agent (only soluble in an ethanol–water co-solvent) after 20 h long reaction, 85% of the diethyl ester phosphonate IL was still present and a proportion of 15% was recovered in a monoester form (Fig. S17†). After 48 h, the conversion rate increases with only 48% of the initial ester and 15% for the monoester present in the reaction mixture (Fig. S19†). Thus, the [ImPE][Tf2N] IL is significantly more stable than [ImPE][Br] toward hydrolysis in water–ethanol media. This result confirms that the formation of bulk aluminium phosphonate phases is strongly related to the chemical nature of both the anion and the solvent used.
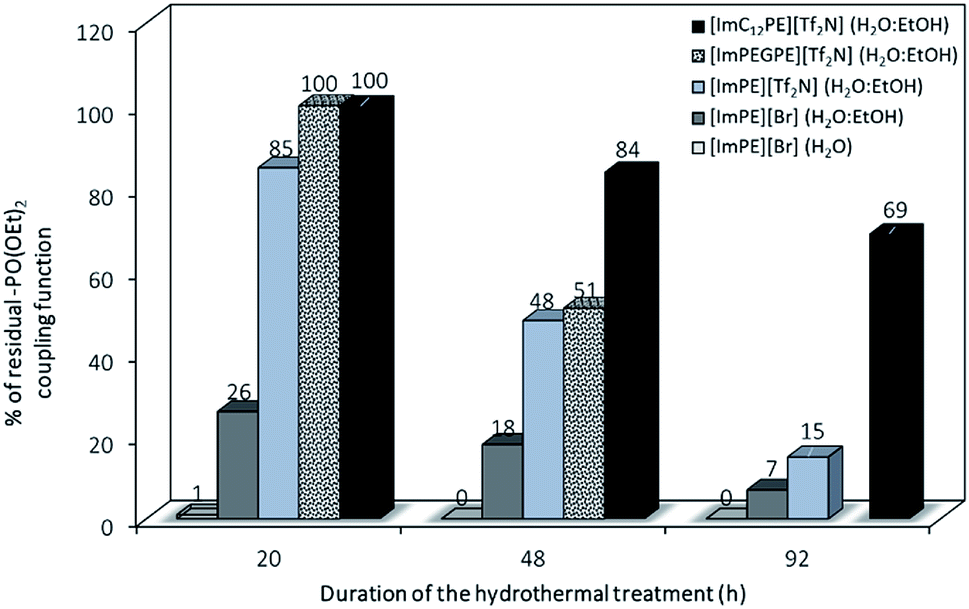 | ||
| Fig. 6 Proportion of residual-P(O)(OEt)2 coupling functions resulting from different duration of hydrothermal treatment (grafting reaction) in H2O or H2O:EtOH solvents. | ||
The stability of the coupling function was also investigated for the [ImPEGPE][Tf2N] ILs after 20 h and 48 h grafting reaction times and until 92 h for the [ImC12PE][Tf2N] IL. After 20 h, both ILs did not present any hydrolysis of the coupling function while after a 48 h treatment, only 51% and 84% of the diethylester coupling function remained unaffected for [ImPEGPE][Tf2N] and [ImC12PE][Tf2N], respectively (Fig. S22–S31†).
The long hydrophobic alkyl organic spacer seems to provide higher stability toward hydrolysis. The classical way to form phosphonic acid from diethylphosphonate functions is a strong acidic treatment (HCl 6 M) under reflux in an aqueous medium.25,26 Surprisingly, we evidenced that for the IL-based phoshonate esters studied in this work, the hydrolysis to phosphonic functions can be easily performed in either aqueous or hydroalcoholic medium without any strongly acidic conditions. Hence, the presence of phosphonic acid groups during the grafting reaction cannot be precluded. In the case of the [ImPE][Br] series, the acidic form becomes the major component during the grafting process, promoting the formation of bulk aluminum phosphonate phases when increasing the reaction time.
31P CP-MAS NMR is a useful tool to highlight the presence of phosphorus atoms in phosphonate based hybrid materials and to distinguish the grafted species from the bulk aluminum phosphonate phases.22 The latter have been largely described in the literature and are characterized by individual or multiple thin peaks in 31P solid-state NMR.21,22 The 31P CP-MAS NMR spectra of the grafted samples are shown in Fig. 7. The ImPE-Br sample obtained after 40 h of grafting (i.e., ImPE-Br(1)) presented a spectrum similar to the results obtained in our previous study.15 The simulation of ImPE-Br(1) spectrum using a minimum number of resonance lines with a Gaussian–Lorentzian shape revealed the presence of at least 3 signals at 32.4, 23.6 and 17.9 ppm (see Table 2). These sites can be attributed respectively to the monodentate, tridentate and bidentate bonding modes according to the FTIR spectra, as already discussed in our previous work concerning the grafting of [ImPE][Br] on γ-alumina powders (Fig. 3).15 After a 45 h reaction time (ImPE-Br(2)), only phosphonate units grafted in tridentate (23.6 ppm) and bidentate (16.4 ppm) bonding modes were present with an additional broad signal observed at 12.7 ppm. On the basis of the XRD patterns, this broad peak was tentatively attributed to the presence of a bulk aluminum phosphonate phase. The integrations derived from the simulated spectra indicated that the major part of the phosphonate units in a bidentate anchoring mode was converted in phosphonate sites of the lamellar phase. As expected, by increasing the reaction time up to 92 h (ImPE-Br(3)) only a sharp symmetric peak at 12.3 ppm has remained corresponding to the ordered environment in bulk aluminum phosphonate phases22 previously foreseen from EDX analysis and XRD data.
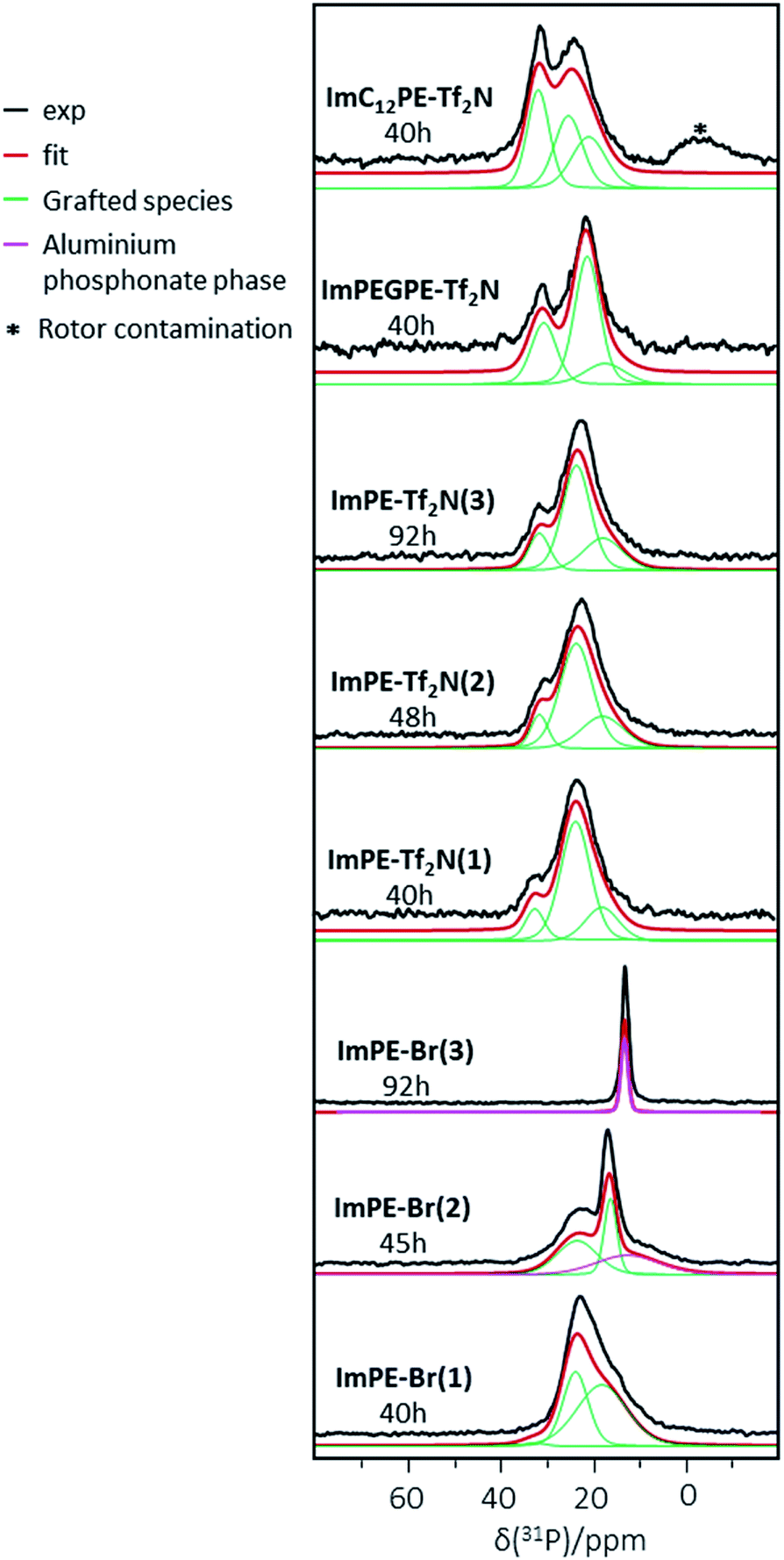 | ||
| Fig. 7 31P solid-state CP-MAS NMR spectra of grafted samples. | ||
| Sample | ImPE-Br(1) 40 h | ImPE-Br(2) 45 h | ImPE-Br(3) 92 h | ImC12PE-Tf2N 40 h | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| δ (ppm) | 32.4 | 23.6 | 17.9 | 23.6 | 16.4 | 12.7 | 12.4 | 31.7 | 25.2 | 20.8 |
| Width (ppm) | 3.2 | 41.5 | 25.5 | 22.6 | 49.8 | 12.5 | 69.2 | 65.7 | 48.1 | 34.4 |
| Integration (%) | 2 | 37 | 61 | 39 | 27 | 34 | 100 | 34 | 36 | 30 |
| Sample | ImPE-Tf2N(1) 40 h | ImPE-Tf2N (2) 48 h | ImPE-Tf2N(3) 92 h | ImPEGPE-Tf2N 40 h | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| δ (ppm) | 32.4 | 23.6 | 17.9 | 31.6 | 23.6 | 17.9 | 31.6 | 23.6 | 17.9 | 30.6 | 21.3 | 17.5 |
| Width (ppm) | 13.1 | 51 | 14.1 | 24.6 | 74.6 | 22.7 | 19.8 | 54.9 | 17.1 | 6.5 | 6.4 | 10.3 |
| Integration (%) | 11 | 68 | 21 | 11 | 64 | 25 | 16 | 58 | 26 | 28 | 57 | 15 |
31P CP-MAS NMR spectra of the sample series grafted with [ImPE][Tf2N], reveal a major broad signal centered at 23.6 ppm attributed to grafted species in a tridentate bonding mode. The absence of the thin peak at ≈12.5 ppm, even after longer reaction times, confirms that only grafting occurred. This finding highlights the influence of the anion and the solvent nature on the grafting reaction mechanism as previously discussed. The simulations of the different spectra reveal two other phosphonate sites in all the ImPE-Tf2N samples (Table 2) at the same chemical shifts than those observed for ImPE-Br(1). These two signals can be ascribed to monodentate bonding modes at 32.4 and 31.6 ppm and bidentate bonding modes at 17.9 ppm (Fig. 3) in agreement with FTIR spectra, indicating the presence of residual P–O–C bands. The 31P CP-MAS NMR spectra of the ImC12PE-Tf2N and ImPEGPE-Tf2N samples presented in Fig. 7 showed both two dominant signals respectively centered at ∼25 and 32 ppm and at 21 and 31 ppm. The simulation of the ImC12PE-Tf2N spectrum using a minimum number of resonance lines with a Gaussian–Lorentzian shape revealed that the high field resonance can be described with at least two equivalent components (at 25.2 and 20.8 ppm). The broad signal centered at ∼−0.4 ppm is attributed to impurities in the solid-state NMR rotor. In the case of ImPEGPE-Tf2N, the simulated spectrum revealed the presence of at least two signals at 21.3 and 17.5 ppm to describe the high field resonance. The overall shape of the 31P NMR signals enabled to tentatively conclude about the presence of a mixture of phosphonate units in monodentate (∼33 ppm), tridentate (∼21 and ∼25 ppm) and bidentate (∼18 ppm) bonding modes. In the case of ImC12PE-Tf2N, well-packed arrangements were expected on the alumina surface as already described for long hydrocarbon chained phosphonic acids in self-assembled monolayer (SAMs) which leads to 31P NMR spectra with thin resonances.27 The multiple broad resonances present in ImC12PE-Tf2N sample indicated that self-assembled monolayers were absent in the sample as a result of steric hindrance or disorder. This is confirmed by FTIR spectroscopy. SAMs formation usually results in a slight shift to lower wavenumbers of the symmetric and asymmetric stretching vibrations of the methylene groups of the long alkyl chain due to van der Waals interactions. In ImC12PE-Tf2N sample, no differences in the symmetric and asymmetric stretching vibration associated to the CH2 in the alkyl chain on the grafted sample can be noted compared to the pure IL (Fig. S32†).
The γ-alumina 27Al NMR spectrum presented in Fig. 8I is composed of two large peaks, at 10.6 and 69.2 ppm corresponding respectively to aluminum atoms in octahedral (AlVI) and tetrahedral (AlIV) coordination modes.28 27Al MAS NMR spectra of the grafted samples ImPE-Br(1) and ImPE-Tf2N(1) are presented in Fig. 8I. The spectra revealed only the presence of signals observed for pristine γ-Al2O3 and did not present any additional upfield sharp resonance which could be attributed to the formation of bulk aluminum phosphonate phases by dissolution/precipitation phenomena. Same observations were made from the spectra of ImPEGPE-Tf2N and ImC12PE-Tf2N (Fig. S33†). The spectra of ImPE-Br(2) and ImPE-Br(3) were different. The progressive formation of a bulk aluminum phosphonate phase after a 45 h grafting duration is indicated by a new weak resonance at ∼−17 ppm (Fig. 8I). After a 92 h grafting time, a thin peak centered at ∼−8 ppm attributed to AlVI atoms in aluminum imidazolium-based phosphonate phase has appeared with the progressive disappearance of the AlIV atoms coordination mode.29 The occurrence of this signal fits with the conclusions derived from 31P MAS NMR, XRD and EDX analysis.
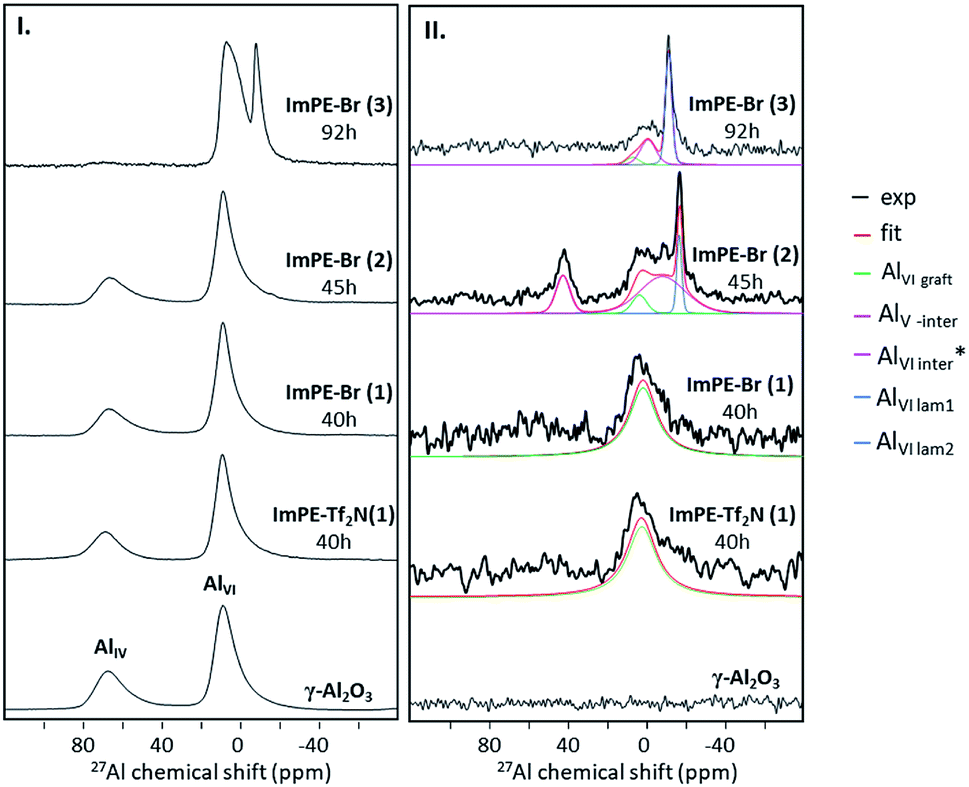 | ||
| Fig. 8 (I) 27Al MAS NMR spectra of γ-Al2O3, ImPE-Br(1), ImPE-Br(2), ImPE-Br(3) and ImPE-Tf2N(1) samples, (II) 1D 27Al NMR spectra issued from 31P–27Al D-HMQC experiments for γ-Al2O3, ImPE-Br(1), ImPE-Br(2), ImPE-Br(3) and ImPE-Tf2N(1) samples. | ||
Furthermore, spatial proximity between 31P nuclei of phosphonate-based ILs and 27Al nuclei on γ-Al2O3 surface could be attractively investigated by double resonance NMR methods to differentiate the aluminum nuclei involved in the grafting process from those present in the aluminum imidazolium-based phosphonate phase.30 Only few NMR methods can be applied to our systems. As an example, the basic CP (cross-polarisation) experiment is difficult to be carried out when the system contains quadrupolar nuclei such as 27Al, due to quadrupolar interactions.
Trébosc and co-workers,31 proposed the use of 2D D-HMQC method to demonstrate the spatial proximity between phosphorus and quadrupolar nuclei in mixed phosphate network materials. This technique is significantly more robust for the correlation between spin ½ and quadrupolar nuclei and applicable for low phosphorus content as ours. The 31P–27Al D-HMQC method can be used to observe only the phosphorus and aluminum nuclei which have a spatial proximity due to the grafting reaction. The 31P–27Al D-HMQC method can be used to observe only the phosphorus and aluminum nuclei which have a spatial proximity due to the grafting reaction. The 27Al and 31P MAS NMR spectra, the 1D 31P–27Al D-HMQC MAS NMR projections and 2D 31P–27Al D-HMQC of ImPE-Br(3) sample are shown in Fig. 9. The 1D 27Al NMR projection spectra issued from the 31P–27Al D-HMQC experiment for ImPE-Tf2N(1), ImPE-Br(1), ImPE-Br(2) and ImPE-Br(3) samples are shown in Fig. 8II.
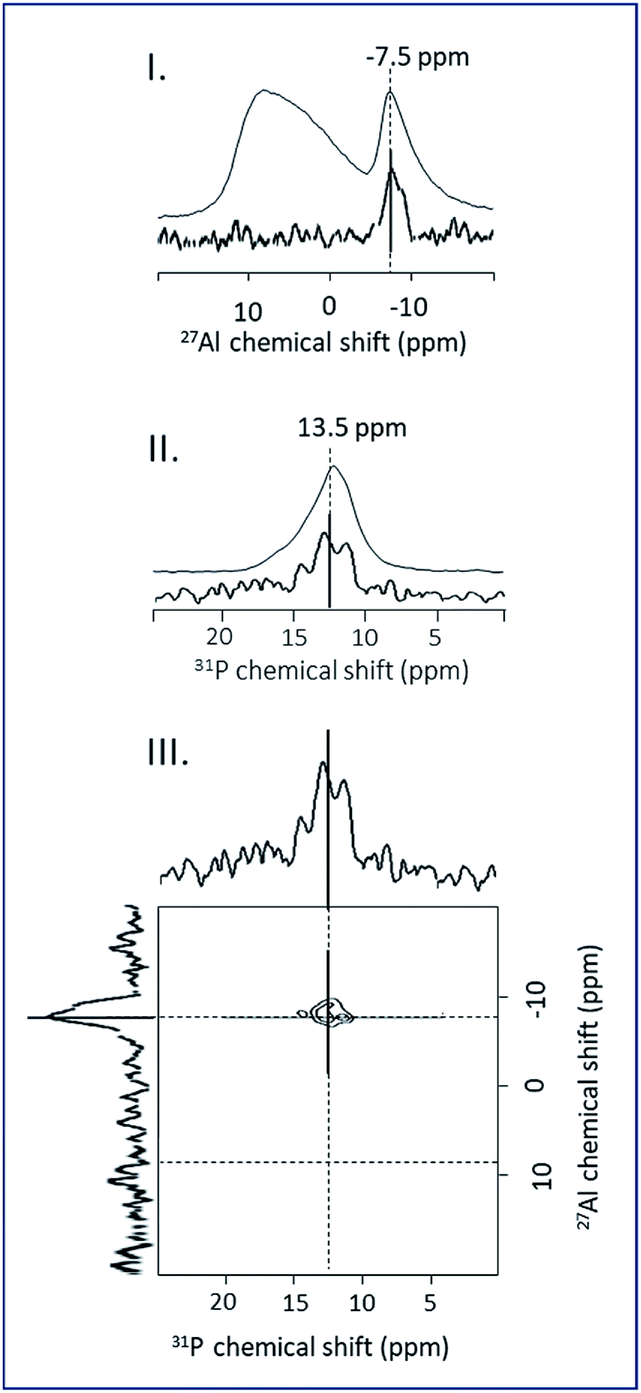 | ||
| Fig. 9 (I) 27Al MAS NMR spectrum and 1D 31P–27Al D-HMQC MAS NMR projection, (II) 31P MAS NMR spectrum and 1D 31P–27Al D-HMQC MAS NMR projection, (III) 2D 31P–27Al D-HMQC MAS NMR spectra of ImPE-Br(3) sample. | ||
A comparison between the 27Al NMR spectra from one-pulse experiment and 1D 31P–27Al D-HMQC immediately evidences the nature of the 27Al nuclei involved in grafting reactions. Simulation of the 1D 27Al NMR projection spectra using a minimum number of signals with Gaussian–Lorentzian shape reveals the presence of multiple sites listed in Table 3. 27Al nuclei involved in the grafting can be distinguished from those present in bulk lamellar aluminum phosphonate or alumina phases. In the 2D 31P–27Al D-HMQC spectra of ImPE-Br(3) (Fig. 9III), 27Al nuclei corresponding to bulk aluminum phosphonate phase appears as a sharp signal at −7.86 ppm and are assigned to hexacoordinated 27Al nuclei in the crystalline phase (AlVI lam1). In the 1D 31P–27Al D-HMQC spectra of ImPE-Br samples (Fig. 8II), a broad signal centered at 3.33 ppm is present in all samples and predominant in ImPE-Br(1).
| Grafted samples | ImPE-Tf2N(1) | ImPE-Br(1) | ImPE-Br(2) | ImPE-Br(3) |
|---|---|---|---|---|
| Grafting reaction duration (h) | 40 | 40 | 45 | 92 |
| AlX type | AlVI graft | AlVI graft | AlV inter | AlVI graft |
| AlVI graft | AlVI inter* | |||
| AlVI inter* | AlVI lam1 | |||
| AlVI lam2 | — | |||
| 27Al [ppm] | 3.33 | 3.33 | 42.4 | 3.33 |
| 3.33 | −0.51 | |||
| −8.46 | −7.86 | |||
| −16.9 | — | |||
| Signal width (ppm) | 19.02 | 19.02 | 8.53 | 3.94 |
| 8.83 | 4.62 | |||
| 27.36 | 1.9 | |||
| 2.99 | — | |||
| Integration (%) | 100 | 100 | 18 | 8 |
| 10 | 32 | |||
| 58 | 60 | |||
| 14 | — |
The same broad signal centered at 3.33 ppm is predominant in the 1D 31P–27Al D-HMQC spectrum of ImPE-Tf2N(1). As ImPE-Br(1) and ImPE-Tf2N(1) do not contain any bulk aluminum phosphonate phase, this signal is attributed to hexacoordinated 27Al nuclei involved in the grafting process (AlVI graft). The 1D 31P–27Al D-HMQC spectrum of ImPE-Br(2) sample reveals the presence of pentacoordinated aluminum centers at ∼43 ppm (AlV inter) which were not present in the 27Al one-pulse spectra of all samples. We assume that this resonance may be attributed to aluminum nuclei in spatial proximity with phosphorus nuclei during the transitory dissolution precipitation process. Also in this sample, the hexacoordinated aluminum nuclei in bulk aluminum phosphonate phase appeared as a sharp signal centered at −16.9 ppm (AlVI lam2). Spectra simulation allows to identify two more signals at −8.46 ppm for ImPE-Br(2) and −0.51 ppm for ImPE-Br(3). The signals were too broad to be attributed to well-ordered lamellar phases and were not in the range of expected chemical shifts for grafted species. Thus, we classified these resonances (AlVI inter*) as characteristic of the formed species during the transitions/rearrangements.
D-HMQC techniques provided experimental evidence on the 27Al nuclei involved in the grafting processes from those involved in structural transformations and those present in lamellar phases. We demonstrated for the first time that all the relevant aluminum nuclei involved in the grafting of phosphonate-based molecules were hexacoordinated. These results coupled with 31P solid-state NMR and FTIR results provide more complete informations about the bonding configurations in the phosphonate-based ILs/alumina system.
Gas sorption studies
As discussed in the introduction, ionic liquids are known to interact strongly and reversibly with CO2.1 To check the absence of undesirable (irreversible) chemisorption interactions, CO2 sorption experiments were conducted on the pristine γ-Al2O3 and on the grafted ImPE-Tf2N(1), ImC12PE-Tf2N and ImPEGPE-Tf2N samples. Only samples composed of a Tf2N− anion were chosen because of the chemical stability and the ability to easily solubilize CO2 of this anion by comparison with Br−. In fact, the [Tf2N]− anion is known to yield a very high CO2 solubility and this anion is a common choice when designing ILs for CO2 separation from N2 or CH4.33–36 The CO2 adsorption isotherm obtained at 298 K for the γ-Al2O3 powder (Fig. 10) is similar to the isotherm obtained at 315 K in the literature.32 With the help of both FTIR spectroscopy and previously published results, it was possible to clearly distinguish the chemisorbed CO2 species (i.e., bicarbonate, monocarbonate) and physisorbed ones (Fig. S34†). The chemisorbed species react with the high adsorption energy sites and are adsorbed at negligible equilibrium pressure such as the initial uptake at ∼130 μmol g−1 as observed from the γ-Al2O3 adsorption isotherm. The physisorbed species are energetically weak and therefore they require a higher gas phase pressure to be effectively adsorbed.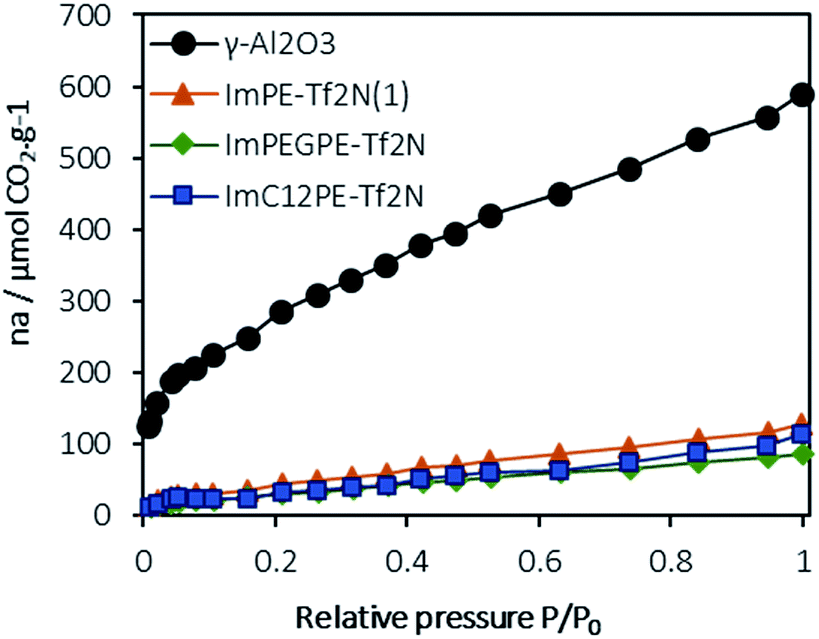 | ||
| Fig. 10 Adsorption isotherms of CO2 (μmol of CO2 per g of powder) on the pristine γ-Al2O3, ImPE-Tf2N(1), ImC12PE-Tf2N and ImPEGPE-Tf2N grafted samples (measured at 298 K, with P0 = 1 atm). | ||
In the case of ImPE-Tf2N(1) sample, the initial CO2 uptake was ∼30 μmol g−1, respectively and correspond to chemisorption interactions (acid–base type). Values in the same range were found for the ImPEGPE-Tf2N and ImC12PE-Tf2N samples, respectively with 12 and 11 μmol g−1. As major part of the γ-Al2O3 hydroxyl surface groups are involved in the grafting with the phosphonate-based ILs, the possible formation of carbonate or bicarbonate species is low. Hence, the small CO2 uptake at the beginning of the experiment could be caused by anion/CO2 interactions. In addition, the amount of adsorbed CO2 increases almost linearly with the relative pressure, thus suggesting the dominating interactions of physisorbed CO2 with the ILs.
To compare the sorption properties of the ILs before and after grafting, CO2 sorption measurements were conducted on the pure ILs ([ImPE][Tf2N], [ImPEGPE][Tf2N] and [ImC12PE][Tf2N]) at 30 °C. The molar quantity of CO2 per mol of IL (molCO2/molIL) are shown in Fig. 11. Similar values were obtained for all the ILs featuring comparable sorption capacities with conventional ILs (i.e., [emim][Tf2N]: 0.03 molCO2/molIL at 20 °C).1 It should be noted that in the most performable conventional ILs, only 0.05 molCO2/molIL can be sorbed at a partial pressure of ∼0.15 bar.1 Concerning the grafted powders, at the pressure of 1 atm, the quantity of CO2 adsorbed per gram of the sample was 128, 113 and 87 μmol g−1 for ImPE-Tf2N(1), ImC12PE-Tf2N and ImPEGPE-Tf2N, respectively. To determine the actual sorption capacity of IL without the contribution of the support, the quantity of physisorbed CO2 was expressed per mol of IL grafted on γ-Al2O3 powder (the amount of grafted IL was estimated from EDX measurements). The as-defined sorption value obtained for the grafted ImPE-Tf2N(1) sample indicated in Fig. 11 was in the same range as those obtained for the pure IL ([ImPE][Tf2N]) (∼0.03 mol CO2/mol IL). These results confirm that CO2 physisorption properties of ILs do not alter upon grafting. On the other hand, different CO2 absorption values between the grafted samples ImPEGPE-Tf2N or ImC12PE-Tf2N and corresponding pure ILs ([ImPEGPE][Tf2N]; [ImC12PE][Tf2N]) were observed. A particularly high sorption capacity of CO2 was found for the ImC12PE-Tf2N grafted sample. This result could be explained by the steric hindrance or disorder evidenced in the sample which could induce a special arrangement of the different domains (ionic and nonpolar) and thus enhance the CO2 sorption properties of the grafted IL. Whereas, a low sorption capacity of CO2 was found for the ImPEGPE-Tf2N grafted sample composed of the ethylene glycol organic spacer, suggesting that the arrangement of the IL on the support surface does not favor the CO2 sorption. As published by Bara et al.,37 ILs with oligo(ethylene glycol) chain present a higher CO2/N2 and CO2/CH4 ideal solubility selectivity compare to the same ILs without any functional group and with an alkyl chain. The results obtained for the hybrid materials are in contradiction with those obtained for pure ILs. In fact they suggests that not only the organic spacer play a key role in the CO2 sorption properties but also other factors such as the orientation of the anion and cation after grafting, the nature of the bonding modes (predominantly tridentate or a mixture of mono, bi and tridentate) influence in the final material properties.
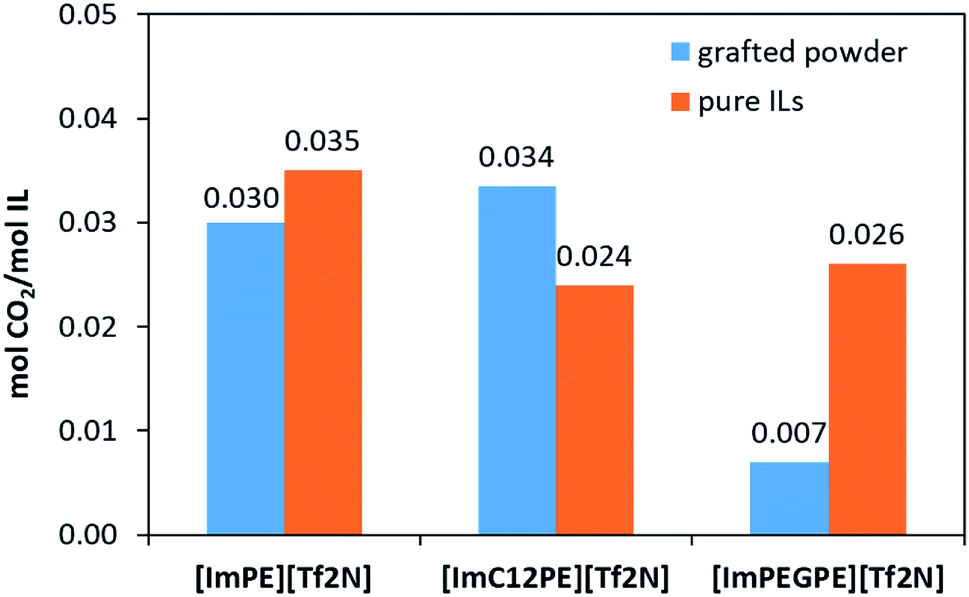 | ||
| Fig. 11 Molar quantity of adsorbed CO2 per mol of IL (mol CO2/mol IL) for the pure ILs ([ImPE][Tf2N], [ImPEGPE][Tf2N] and [ImC12PE][Tf2N]) at 30 °C and for the grafted powders ImPE-Tf2N(1), ImC12PE-Tf2N and ImPEGPE-Tf2N at 25 °C. | ||
Conclusions
In this study we have demonstrated how the grafting density and the bonding configuration of phosphonate-based ILs anchored to γ-alumina powder are influenced both by the nature of the anion and organic spacers composition in IL molecules. It has been assessed that the anion in the IL molecule and the reaction time must be carefully selected to avoid/minimize the hydrolysis reaction and thus the formation of bulk aluminum phosphonate phases. A particular attention has been devoted to D-HMQC experiments in order to determine the nature of the aluminum nuclei involved in the grafting, together with the different types of hydroxyl surface groups on the γ-Al2O3 support. Such detailed characterizations are of an utmost importance to establish a suitable procedure for the grafting reactions, enabling to reach a substantial quantity of grafted species in a minimum reaction time. The sorption experiments conducted on pure ILs and their grafted counterparts revealed virtually the same values, thus confirming that CO2 physisorption properties of the studied ILs do not alter upon grafting. Indeed, this finding confirms the huge potential of such hybrid systems for the preparation of IL-based materials for CO2 separation applications in either static (adsorbers) or dynamic (membranes) mode. It must be emphasized that the findings assessed in this work could be possibly extended to other metal oxides supports such as TiO2, ZrO2 or ZnO but also to Layered Double Hydroxide (LDH) compounds, zeolites, Metal Organic Frameworks (MOFs), etc. Other porous ceramic supports with different pore sizes/porous structures and geometries could be considered as well. The prospects of this exploratory research work are wide, whether as a direct complement to the work already started, as complementary approaches (e.g., modeling) or as an extension of the strategy to other systems and applications. Apart from the acidic gas separation, the developed IL-grafted layers could be also considered for other applications in relation with the specific properties of both the selected support and the grafted IL, e.g. for antimicrobial, hydrophilic–hydrophobic surfaces, ionic conductors, hybrid electronic devices or catalysis (metallic carbens).Conflicts of interest
The authors declare no conflict of interest.Abbreviations
| ImPE | 1-Methyl-3-(3-(diethylphosphinyl)propyl)-imidazolium |
| ImPEGPE | 1-Methyl-3-(3-(diethylphosphinyl)2-(2-(2-(2-ethoxy)ethoxy)ethoxy)ethyl)-imidazolium |
| ImC12PE | 1-Methyl-3-(3-(diethylphosphinyl)dodecyl)-imidazolium |
| Tf2N | Bis(trifluoromethanesulfonimide) |
Acknowledgements
P. Gaveau (Ingénieur de Recherche CNRS) from the Institute Charles Gerhardt in Montpellier, France is sincerely acknowledged for his helpful contribution in NMR analysis. Authors also thank L. Brun and T. Delage for their assistance in experiments related to ILs grafting. Franck Martin and Guillaume Gracy from SIKEMIA are sincerely acknowledged for their advice in ILs synthesis.Notes and references
- M. Ramdin, T. W. De Loos and T. J. H. Vlugt, Ind. Eng. Chem. Res., 2012, 51(24), 8149–8177 CrossRef CAS
.
- A. V. Perdikaki, O. C. Vangeli, G. N. Karanikolos, K. L. Stefanopoulos, K. G. Beltsios, P. Alexandridis, N. K. Kanellopoulos and G. E. Romanos, J. Phys. Chem. C, 2012, 116(31), 16398–16411 CrossRef CAS
- O. C. Vangeli, G. E. Romanos, K. G. Beltsios, D. Fokas, E. P. Kouvelos, K. L. Stefanopoulos and N. K. Kanellopoulos, J. Phys. Chem. B, 2010, 114(19), 6480–6491 CrossRef CAS PubMed
- M. A. Pizzoccaro-Zilamy, M. Drobek, E. Petit, C. Totee, G. Silly, G. Guerrero, M. G. Cowan, A. Ayral and A. Julbe, Ind. Eng. Chem. Res., 2018, 57, 16027–16040 CrossRef CAS
- B. Xin and J. Hao, Chem. Soc. Rev., 2014, 43(20), 7171–7187 RSC
- F. Nkinahamira, T. Su, Y. Xie, G. Ma, H. Wang and J. Li, Chem. Eng. J., 2017, 326, 831–838 CrossRef CAS
- J. M. Zhu, F. Xin, Y. C. Sun and X. C. Dong, Theor. Found. Chem. Eng., 2014, 48(6), 787–792 CrossRef CAS
- P. Bollini, S. A. Didas and C. W. Jones, J. Mater. Chem., 2011, 21, 15100–15120 RSC
- R. Fehrmann, M. Haumann and A. Riisager, Introduction, in Supported Ionic Liquids: Fundamentals and Applications, ed. R. Fehrmann, A. Riisager and M. Haumann, Wiley-VCH Verlag GmbH & Co. KGaA, Publisher, Weinheim, Germany, 1st edn, 2014, pp. 1–9 Search PubMed
- Z. Dai, R. D. Noble, D. L. Gin, X. Zhang and L. Deng, J. Membr. Sci., 2016, 497, 1–20 CrossRef CAS
- L. C. Tomé and I. M. Marrucho, Chem. Soc. Rev., 2016, 45(10), 2785–2824 RSC
- S. D. Hojniak, I. P. Silverwood, A. L. Khan, I. F. J. Vankelecom, W. Dehaen, S. G. Kazarian and K. Binnemans, J. Phys. Chem. B, 2014, 118(26), 7440–7749 CrossRef CAS PubMed
- S. D. Hojniak, A. L. Khan, O. Hollo, B. Kirchner, I. F. J. Vankelecom, W. Dehaen and K. Binnemans, J. Phys. Chem. B, 2013, 117(14), 15131–15140 CrossRef CAS PubMed
- J. J. Close, K. Farmer, S. S. Moganty and R. E. Baltus, J. Membr. Sci., 2012, 390–391, 201–210 CrossRef CAS
- M. A. Pizzoccaro, M. Drobek, E. Petit, G. Guerrero, P. Hesemann and A. Julbe, Int. J. Mol. Sci., 2016, 17, 1212 CrossRef PubMed
- A. Rout, K. A. Venkatesan, T. G. Srinivasan and P. R. Vasudeva Rao, Radiochim. Acta, 2010, 98(8), 459–466 CAS
- A. Cattani-Scholz, ACS Appl. Mater. Interfaces, 2017, 9(31), 25643–25655 CrossRef CAS PubMed
- F. Brodard-Severac, G. Guerrero, J. Maquet, P. Florian, C. Gervais and P. H. Mutin, Chem. Mater., 2008, 20(16), 5191–5196 CrossRef CAS
- O. Höfft, S. Bahr and V. Kempter, Langmuir, 2008, 24(20), 11562–11566 CrossRef PubMed
- L. D. Freedman and G. O. Doak, Chem. Rev., 1957, 57(3), 479–523 CrossRef CAS
- G. Guerrero, P. H. Mutin and A. Vioux, Chem. Mater., 2001, 13(11), 4367–4373 CrossRef CAS
- G. Guerrero, P. H. Mutin and A. Vioux, J. Mater. Chem., 2001, 11(12), 3161–3165 RSC
- L. Raki and C. Detellier, Chem. Commun., 1996,(21), 2475–2476 RSC
- G. Alberti, M. Casciola, U. Costantino and R. Vivani, Adv. Mater., 1996, 8(4), 291–303 CrossRef CAS
- Z. C. Duan, X. P. Hu, C. Zhang and Z. Zheng, J. Org. Chem., 2010, 75(9), 8319–8321 CrossRef CAS PubMed
- C. E. McKenna, B. A. Kashemirov, K. M. Błazewska, I. Mallard-Favier, C. A. Stewart, J. Rojas, M. W. Lundy, F. H. Ebetino, R. A. Baron, J. E. Dunford, M. L. Kirsten, M. C. Seabra, J. L. Bala, M. S. Marma, M. J. Rogers and F. P. Coxon, J. Med. Chem., 2010, 53, 3454–3464 CrossRef CAS PubMed
- W. Gao and L. Reven, Langmuir, 1995, 11, 1860–1863 CrossRef CAS
- L. A. O'Dell, S. L. P. Savin, A. V. Chadwick and M. E. Smith, Solid State Nucl. Magn. Reson., 2007, 31(4), 169–173 CrossRef PubMed
- G. Chaplais, J. Le Bideau, D. Leclercq, H. Mutin and A. Vioux, J. Mater. Chem., 2000, 10, 1593–1601 RSC
- G. Tricot, O. Lafon, J. Trébosc, L. Delevoye, F. Méar, L. Montagne and J.-P. Amoureux, Phys. Chem. Chem. Phys., 2011, 13, 16786–16794 RSC
- J. Trébosc, B. Hu, J. P. Amoureux and Z. Gan, J. Magn. Reson., 2007, 186, 220–227 CrossRef PubMed
- M. Cabrejas Manchado, J. M. Guil, A. Pérez Masiá, A. Ruiz Paniego and J. M. Trejo Menayo, Langmuir, 1994, 10, 685–691 CrossRef CAS
- J. E. Bara, T. K. Carlisle, C. J. Gabriel, D. Camper, A. Finotello, D. L. Gin and R. D. Noble, Ind. Eng. Chem. Res., 2009, 48(6), 2739–2751 CrossRef CAS
- J. Jacquemin, M. F. Costa Gomes, P. Husson and V. J. Majer, Chem. Thermodyn., 2006, 38(4), 490–502 CrossRef CAS
- J. Jacquemin, P. Husson, V. Majer and M. F. C. Gomes, Fluid Phase Equilib., 2006, 240(1), 87–95 CrossRef CAS
- Z. Lei, C. Dai and B. Chen, Chem. Rev., 2014, 114(2), 1289–1326 CrossRef CAS PubMed
- J. E. Bara, C. J. Gabriel, S. Lessmann, T. K. Carlisle, A. Finotello, D. L. Gin and R. D. Noble, Ind. Eng. Chem. Res., 2007, 46(16), 5380–5386 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: γ-Al2O3 powder synthesis, ionic liquid synthesis and characterization details, FTIR and DFT calculated spectra of the [ImPE][Tf2N] IL, physisorbed sample, 19F NMR spectra, stability of ILs phosphonate functions toward hydrolysis under forcing reaction conditions, FTIR spectra of the pure ionic liquid [ImC12PE][Tf2N] and grafted sample ImC12PE-Tf2N between 2600 and 4000 cm−1, 27Al MAS NMR spectra, schematic representation of the chemisorbed and physisorbed CO2 on a pristine γ-Al2O3. See DOI: 10.1039/c9ra01265f |
| ‡ Current address: Inorganic Membranes, MESA+ Institute for Nanotechnology, University of Twente, P. O. Box 217, 7500 AE Enschede, The Netherlands |
| This journal is © The Royal Society of Chemistry 2019 |