
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEco-friendly and facile one-step synthesis of a three dimensional net-like magnetic mesoporous carbon derived from wastepaper as a renewable adsorbent†
Jing Yua,
Donghuan Zhangb,
Shukui Zhu *a,
Pin Chena,
Gang-Tian Zhuc,
Xiangtao Jianga and
Siyuan Dia
*a,
Pin Chena,
Gang-Tian Zhuc,
Xiangtao Jianga and
Siyuan Dia
aState Key Laboratory of Biogeology and Environmental Geology, China University of Geosciences, 388 Lumo Road, Wuhan 430074, P. R. China. E-mail: shukuizhu@126.com; Tel: +86-27-67883452
bSchool of Environmental Studies, China University of Geosciences, Wuhan 430075, P. R. China
cKey Laboratory of Tectonics and Petroleum Resources, Ministry of Education, China University of Geosciences, Wuhan 430075, P. R. China
First published on 23rd April 2019
Abstract
Millions of tons of paper and its derivatives are annually wasted without being recycled and reused. To promote the comprehensive utilization of resources and eco-friendly preparation, waste filter paper, printer paper, and napkins were chosen as carbon sources to one-step synthesize three types of three dimensional (3D) net-like magnetic mesoporous carbon (MMC) by an eco-friendly and low-cost method. These mesoporous (3.90–7.68 nm) composites have a high specific surface area (287–423 m2 g−1), well-developed porosity (0.24–0.74 cm3 g−1) and abundant oxygen-containing functional groups. Compared to the other two composites, the adsorbent derived from filter paper showed the highest adsorption capacity towards methylene blue (MB) (qmax = 332.03 mg g−1) and rhodamine B (RhB) (qmax = 389.59 mg g−1) with a high adsorption rate (<5 min). According to the effect of pH value on adsorption capacity, and combining the analysis of Fourier transform infrared spectrometry and X-ray photoelectron spectroscopy, the main adsorption mechanisms can be summarized as hydrogen bonds, electrostatic interactions, and π–π interaction. Besides, the occurrence of redox reactions between Fe2+/Fe0 and dye cannot be ignored. Finally, experiments on reusability were performed. They showed that the 3D net-like MMC could be easily regenerated and still maintained a removal efficiency of above 80% for RhB and 90% for MB after five cycles.
1. Introduction
Wastepaper is ubiquitously present in our daily life. Millions of tons of paper and its derivatives are annually wasted without being recycled and reused.1 Such an enormous amount of paper waste mostly originates from wood pulp or cotton, which consist of 90–99% cellulose fibers with a network-frame morphology.2,3 This structure is conducive to form 3D interconnected carbon architectures with wastepaper after carbonization.4 The cellulose fibers are rich in carbon, pore structure and functional groups, such as carboxylic groups and hydroxyl groups.5 Therefore, given the considerably high availability, the large quantities of by-products, and significantly high ratio of paper-to-paper recycling efficiency, low-cost wastepaper is suitable to provide a recyclable carbon source.6,7 Recently, several types of mesoporous carbon derived from print paper and filter paper were developed into functional materials for supercapacitors and lithium-ion batteries.4,8 The unique properties of these carbon materials, including high specific surface area, 3D network with high porosity and good stability endow the lithium-ion batteries with low-cost and high-performance energy systems. However, as far as we know, current researchers have paid little attention to the application of wastepaper in the field of environmental remediation.Many methods including adsorption,9 chemical oxidation,10 membrane separation,11 solvent extraction,12 and biological processes13 have been widely used for removal of environmental pollutants from wastewater. Among them, adsorption is generally regarded as the most effective and frequently used techniques, due to its easy operation, high efficiency, economical advantages, and no harmful by-products. So far, all kinds of adsorbents have sprung up and parts of them have been commercialized. Polymeric ionic liquids (PLL),14 clay minerals,15 metal–organic frameworks,16 and cellulosic materials,17,18 etc. have been widely used as adsorption/separation materials for environmental pollutant treatment. Besides, carbon materials also have recently drawn broad attention for adsorption.19 In order to design a “smart” carbon material with high specific surface area, pores structure, suitable pore size, numerous methods have been used, such as soft template, hard template, chemical/physical activation and halogen erosion method.20–22 However, these current prevailing strategies need a complex synthetic procedure or costly ingredients, as well as the utilization of toxic and erosion solutions, such as sodium and potassium hydroxide (NaOH and KOH) or hydrofluoric acid (HF).22–24 In addition, previous efforts on the transformation of precursors into carbon material almost relied on harsh synthesis conditions, for instance, chemical vapor deposition,25 pyrolysis in sealed reactors,26 or salt melt-based ionothermal carbonization.27 Therefore, an eco-friendly, facile and scalable method is indispensable for the preparation of carbon materials.
From a practical point of view, easy operation is also a very essential property for an ideal adsorbent. Magnetic separation technology, in comparison with other methods such as centrifugation, precipitation, and filtration, has advantages of reduced energy consumption and higher separation efficiency.28 For the sake of enhancing stability as well as the adsorption performance of magnetic nanoparticles (MNPs), encapsulating these MNPs in a shell with selective extraction properties is a widely used method.29 However, in this method, MNPs need to be synthesized at first and then modified by functional materials, which is complex and time-consuming. Therefore, a one-step, cost-effective synthesis, as well as eco-friendly preparation method towards obtaining a magnetic nano-adsorbent is highly imperative.
In this study, some wastepaper including filter paper, printer paper and napkins were chosen as carbon sources, and Fe(NO3)3 was chosen as magnetism source to one-step calcine 3D net-like MMC as an adsorbent for the removal of two typical pollutants, methylene blue (MB) and rhodamine B (RhB). This process requires no toxic and erosion ingredients with almost no environmental pollutants, can produce large amounts of adsorbents only by one step. Therefore, the synthetic method has many significant advantages, such as eco-friendly, easy operation, low-cost, and high yield. As a result, the produced materials had targeted mesoporous size, effectively attracted the MB and RhB with high adsorption capacity and possess superior physical and chemical stabilities. Moreover, in this study, the reuse of theses low-cost wastepaper not only promotes the comprehensive utilization of resources, but also produces large quantities of adsorbents with high efficiency for environmental pollutants removal from water samples.
2. Materials and methods
2.1. Materials
Fe(NO3)3·9H2O (≥98%), ethanol, methylene blue (MB), rhodamine B (RhB) were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Some almost unpolluted scraps of paper were collected and were treated as waste papers. These waste papers came from filter papers (Hangzhou Xinhua Paper Industry Co., Ltd., China), printer papers (Shanghai Deli stationery Co., Ltd., China) and napkins (Hubei Weida Paper Industry Co., Ltd., China), which were the necessity of daily experiments, office, and living, respectively. A Heal Force Water Purification System (Shanghai Canrex Analytic Instrument Co., Ltd) was used throughout to obtain ultrapure water.2.2. Synthesis of 3D net-like MMC
3D net-like MMC (FeOx/C) was prepared by a dry-production method,8 and the carbon sources used were waste filter paper in the laboratory, waste printer paper, and waste napkin. Firstly, the waste paper was immersed in ethanol, and placed into the 300 Hz ultrasonic system for 30 min. After washing for 3 times, the treated paper was dried in a 60 °C oven. Then, 0.28 g of Fe(NO3)3·9H2O was dissolved in 2 mL of deionized water and stirred until the iron nitrate solid was completely dissolved. The dried paper was immersed in the above ferric nitrate solution for 2 h at room temperature. Finally, the excess iron nitrate solution on the surface of the waste paper was sucked out with the tissue paper, and then the paper was dried in a 60 °C oven for 12 h. The dried filter paper, printer paper, and napkin were calcined at 750 °C in a tubular furnace under N2 atmosphere with a heating rate 3 °C min−1 to the final products FeOx/C, separately, named FC1, FC2, FC3. Additionally, filter paper without adsorption of ferric nitrate was also calcined at 750 °C under the same condition to obtain carbon, named C.2.3. Instruments and sample preparation
SB-5200DTN Ultrasonic Device (Ningbo scientz biotechnology Co., Ltd.) was used for the dispersion of FeOx/C in the dye aqueous solution and carried out the adsorption experiments. The morphologies of FeOx/C and dye-loaded adsorbent were observed using a HITACHI-SU8010 Field Emission Scanning Electron Microscope (FESEM). Prior to the FESEM-image observation, samples were immersed in ethyl alcohol and dispersed by sonication for one hour. Next, the sample suspensions were spotted in the object stage. Finally, after volatilization of ethyl alcohol, the dried samples were carefully gold-coated by sputtering before SEM observation. Transmission electron microscopy (TEM) characterization was carried out after 1-methyl-2-pyrrolidone (NMP) dispersion. Briefly, about 1 mg sample was dispersed in NMP by sonication for 30 min and obtain a suspension. TEM observation (Philips CM12 Transmission Electron Microscope) was then made under 120 kV operation after dropping the suspension onto carbon-film-coated copper microgrids. Further observation was carried out by High-resolution Transmission Electron Microscope (HR-TEM, FEI Tecnai G2 F20 S-TWIN), Selected Area Electron Diffraction (SAED) and Energy Dispersive Spectrometer (EDS) element mapping. In order to obtain an atomic resolution for carbon, about 1 mg sample was placed into 50 mL plastic centrifuge tubes with 30 mL 1.0 mol L−1 HCl and shaken for 4 h. Then, the supernatant was removed after 1000g centrifugation for 10 min. This washing process was repeated four times, and then the washed samples were washed by ultrapure water until reaching a neutral pH. After oven drying overnight at 80 °C, the sample was prepared by the method mentioned above. Finally, HR-TEM (FEI Titan G2 60-300) with image corrector was used to investigate carbon at atomic resolution.X-ray Diffraction (XRD) patterns were obtained on a D8-FOCUS (Bruker AXS, Germany) instrument with Cu-Kα radiation source at 30 kV and 20 mA. The scanning range was 10–55° with a scanning rate of 1° min−1. Fourier Transform Infrared (FT-IR) spectra were recorded in a Nicolet 6700 Spectrometer (Thermo Fisher Scientific, USA) operating in the spectral region of 4000–400 cm−1 with a resolution of 4 cm−1. The samples were ground with spectroscopic grade KBr and pressed into disks. The Raman spectroscopy was detected on a Raman system (Renishaw 2000). The valence states of the element on the surface of materials were analyzed by X-ray Photoelectron Spectroscope with Al Kα X-ray radiation (XPS, Thermo Fisher, ESCALAB 250XI). Charge neutralization was achieved using both low-energy electrons (<5 eV) and argon ions. The binding energy peaks were corrected to the CHx band in the C 1s spectra at 284.4 eV. Measurements were carried out at a takeoff angle of 45° with respect to the sample surface. This resulted in a typical sampling depth of 3–6 nm (95% of the signal originated from this depth or shallower).
The zeta potential analysis was carried out to determine the surface charge distribution of each sample in an aqueous solution. A Malvern Zetasizer (ZS) was employed for electrophoretic light scattering. Each example was first dispersed in ultrapure water at a concentration of 0.1 mg mL−1 and sonicated for 1 h until a stable dispersion was achieved. Subsequently, the pH value of the solution was adjusted by a minimum amount (below 500 μL) of 0.1 M HCl or 0.1 M NaOH, and again the resulting solution was sonicated for 1 h before carrying out the analysis.
The specific surface area and pore size distribution of the synthesized material were measured by N2 adsorption isotherm at 77 K using the surface area and pore size analyzer (MicroActive for ASAP 2460 2.01) employing the Brunauer–Emmett–Teller (BET) method. The Barrett–Joyner–Halenda (BJH) method was used to calculate the pore distribution for FeOx/C.
Magnetic data for FeOx/C were detected by a Physical Property Measurement System (PPMS-9T) with the VSM (Vibrating Sample Magnetometer) option (USA, Quantum Design).
2.4. Batch adsorption test
The FeOx/C was dispersed in the ultrapure water via sonication to form a suspension with a concentration of 5 g L−1. The batch mode adsorption experiments were carried out by adding the FeOx/C aqueous solution into 20 mL of MB aqueous solution (C0 10–60 mg L−1) and RhB aqueous solution (C0 50–100 mg L−1), respectively. The mixture was dispersed uniformly in solution after 60 min sonication. At each predetermined time point, the adsorbent was separated by an external magnetic field, and then the supernatant was measured on a UV-vis spectrophotometer at a wavelength of 664 nm and 554 nm for MB and RhB, respectively. The adsorption capacity of MB and RhB was calculated by the following equation:
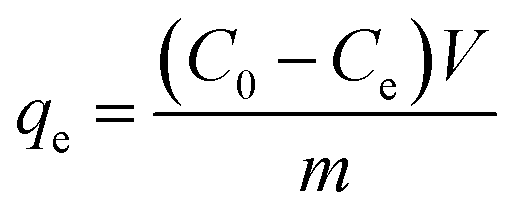 | (1) |
2.5. Desorption and reusability test
FC1 aqueous solution (4 mL) was added into 20 mL of MB or RhB aqueous solution with a concentration of 50 mg L−1 under the ultrasonic for 60 min. After collecting dye-loaded adsorbents by the external magnetic field, MB and RhB were eluted from FC1 with ethanol until no blue or pink color was observed in the ethanol solution. Finally, the recovered adsorbents were washed with ultrapure water and dried under vacuum at 100 °C overnight, which can be reused for another adsorption in succeeding cycles.3. Results and discussion
3.1. Characterization
Fig. S1† shows the SEM and TEM images of waste filter paper, printer paper, and napkins before and after calcination with a different magnification, respectively. The fibers in the filter paper are finer and thinner than the two others (Fig. S1a–c†), and a lot of micropores exist in the fibers of filter paper. This special structure is more beneficial to crimping and coating on the MNPs during the calcination process. As a result, the pores in the filter paper are well developed with high pore size and good connectivity, and a perfect 3D net-like structure is formed. The above conclusions are further confirmed by the SEM (Fig. S1d–f†) and TEM (Fig. S1g–i†) images of waste filter paper, printer paper, and napkins after calcination. A large amount of small MNPs, ranging from 50 to 100 nm, are embedded in the 3D net-like carbon material of calcined filter paper, printer paper, and napkin. However, compared with printer paper and napkin, the filter paper is more porous and has a more obvious 3D net-like structure. In order to evaluate the effect of ferric nitrate on 3D net-like structure, the calcination process of the filter paper without immersion of ferric nitrate was repeated. There is hardly any pore existed in filter paper after calcination (Fig. S2†). In addition, the proportion of filter paper and ferric nitrate were optimized (Fig. S3†). When the paper![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe(NO3)3 (m:m) ratio was 7:1, the product had the highest adsorption capacity. These results indicate that the ferric nitrate was involved in the carbonization process of filter paper and facilitated the production of pores and 3D net-like structure. This porous structure is mainly attributed to the decomposition of ferric nitrate and paper fiber during the calcination process, with the release of some volatiles like CO, CO2, NO2 and H2O (eqn (2)–(5)) resulting in the formation of pore and 3D net-like structure in the carbon matrix.30–32
:Fe(NO3)3 (m:m) ratio was 7:1, the product had the highest adsorption capacity. These results indicate that the ferric nitrate was involved in the carbonization process of filter paper and facilitated the production of pores and 3D net-like structure. This porous structure is mainly attributed to the decomposition of ferric nitrate and paper fiber during the calcination process, with the release of some volatiles like CO, CO2, NO2 and H2O (eqn (2)–(5)) resulting in the formation of pore and 3D net-like structure in the carbon matrix.30–32| 4Fe(NO3)3·9H2O → 2Fe2O3 + 12NO2↑ + 3O2↑ + 36H2O↑ | (2) |
| 3Fe2O3 + CO → 2Fe3O4 + CO2↑ | (3) |
| Fe3O4 + CO → 3FeO + CO2↑ | (4) |
| FeO + CO → Fe + CO2↑ | (5) |
However, when there is too much Fe(NO3)3 participated in the calcination process, the carbon content was reduced, which would cause a decrease of adsorption capacity. Therefore, the proportion of filter paper and ferric nitrate is a key factor for the adsorption capacity of the product.
In order to further reveal the morphology and 3D net-like structure of FeOx/C nanoparticles, the study of HRTEM, EDS element mapping and SAED of product FC1 were also carried out. The HRTEM images and EDS element mapping are shown in Fig. 1. According to HRTEM images in Fig. 1a and b, 3D net-like structure of the product can be clearly observed. The SAED pattern inserted in Fig. 1a indicates the polycrystalline property of the products. Upon further magnification, the HRTEM image revealed the highly graphitic structures of the prepared FeOx/C, which was predominantly made up of randomly orientated graphitic layers. The highly graphitic structures (Fig. 1c) may result from the graphitization catalyst of ferric nitrate and would be crucial for practical applications in terms of selectivity and chemical stability.33,34 The regular crystalline stripes appeared on MNPs are directional growth (Fig. 1d–e). Two kinds of lattice spacing about 0.25 nm and 0.3 nm are indexed to the (311) and (220) crystal planes of Fe3O4, Furthermore, EDS element mapping (Fig. 1f) disclosed the distribution of C, Fe, and O. It can be seen that Fe and O intensively distribute in MNPs. Meanwhile, C and O uniformly distribute in the part of carbon, indicating that a large amount of oxygen-containing functional groups are still retained on the surface of MMC after calcination.
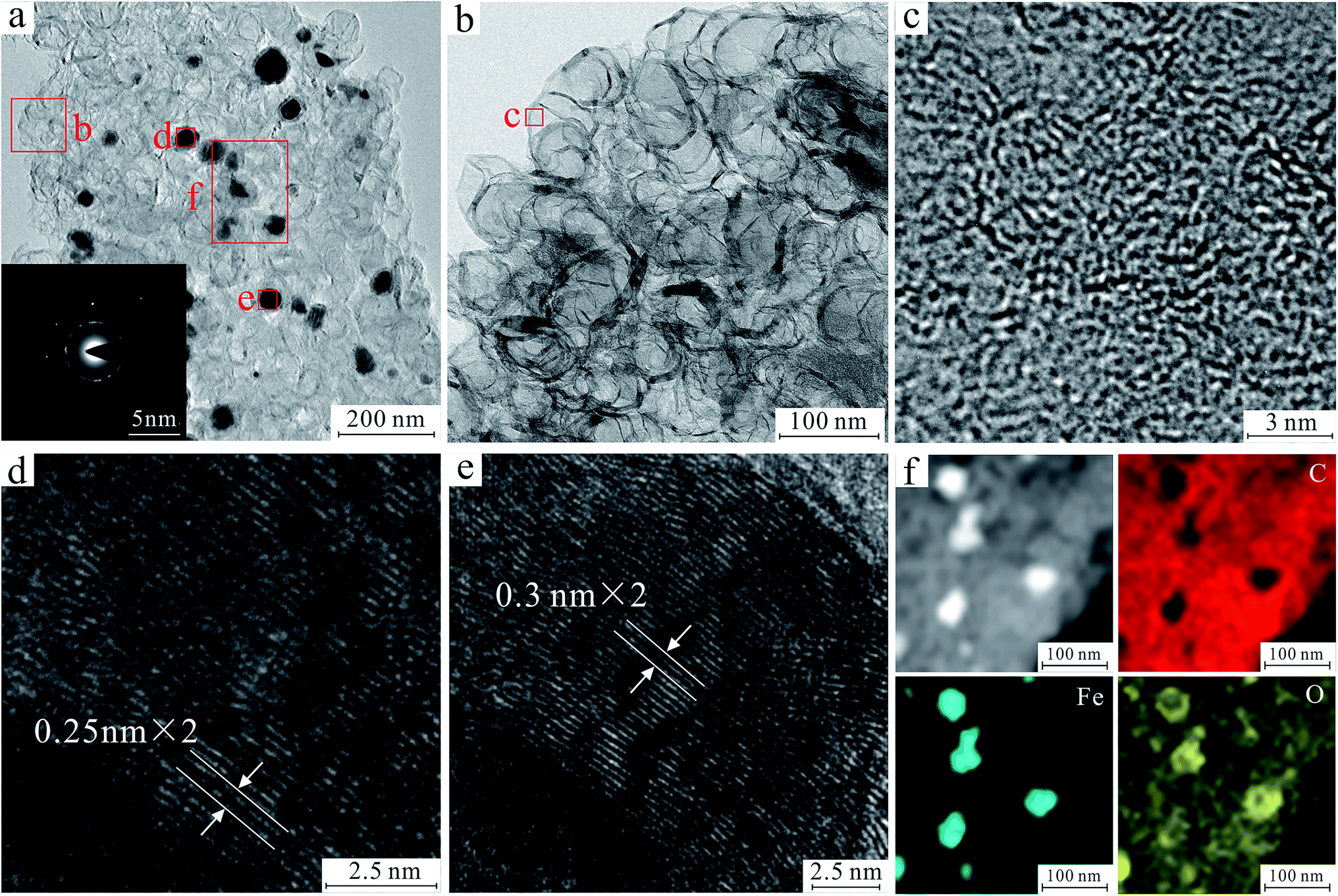 | ||
| Fig. 1 HRTEM image and SAED of 3D net-like MMC (a). HRTEM image of the marked area in a (b–e). Corresponding mapping of Fe, O and C for 3D net-like MMC (f). | ||
The specific surface area and pore size distribution of sample FC1, FC2, FC3, and C were measured by the BET and BJH methods. The nitrogen adsorption–desorption isotherm curves and pore size distribution curves are shown in Fig. 2 and the results are listed in Table 1. According to Fig. 2a, the N2 adsorption isotherm curves of FC1, FC2, and FC3 can be classified as type III isotherm with hysteresis loops, but no hysteresis loop is found in curves of C. In Table 1, the specific surface areas of FC1, FC2, FC3 and C are 287 m2 g−1, 294 m2 g−1, 422 m2 g−1 and 36 m2 g−1, respectively. The specific surface area of C is much smaller than the other three. It indicates that the porous structure of FeOx/C is mainly attributed to the decomposition of Fe(NO3)3·9H2O during the calcination process of cellulose, with the release of volatile matters like CO, CO2, NO2 and H2O (eqn (2)–(5)). The pore size distribution of these four types of materials shown in Fig. 2b and Table 1 is in the range of 3.90–7.68 nm, indicating that these four products are mesoporous materials. However, the average pore sizes (Dpore), mesoporous volume (Vmeso) and total volume (Vcum) of FC1 are larger than those of FC2 and FC3. Since micropores have a higher surface-area-to-volume ratio than mesopores and macropores, FC2 and FC3 exhibited higher SSA values than FC1.
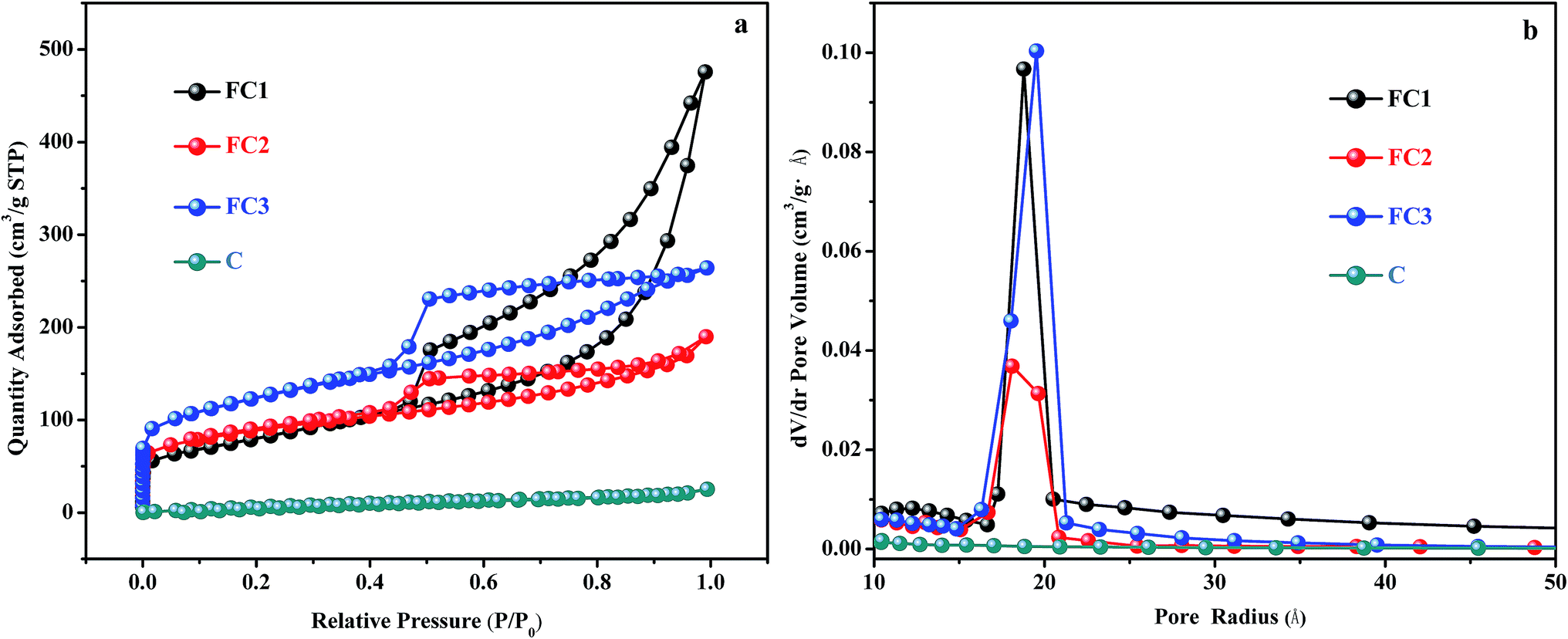 | ||
| Fig. 2 Nitrogen adsorption–desorption isotherm curves (a) and pore size distribution curves (b) of FC1, FC2, FC3 and C. | ||
| Adsorbent | Raw materialsa | Pore structureb | ||||
|---|---|---|---|---|---|---|
| SSA (m2 g−1) | Dpore (nm) | Vmicro (cm3 g−1) | Vmeso (cm3 g−1) | Vtotal (cm3 g−1) | ||
| a From the supplier.b Specific surface area (SSA) was determined with nitrogen by multi-point Brunauer–Emmett–Teller (BET) method. Average pore diameter (Dpore), mesopore volume (Vmeso), and cumulative volume of pores were determined by Barrett–Joyner–Halenda (BJH) desorption method. Micropore volume (Vmicro) was measured by t-plot micropore volume. The pores were divided into micropores (Dpore < 2 nm) and mesopores (2 < Dpore < 50 nm). | ||||||
| FC1 | Filter paper | 287 | 7.68 | 0.01 | 0.53 | 0.74 |
| FC2 | DIN A4 paper | 294 | 4.42 | 0.05 | 0.21 | 0.24 |
| FC3 | Napkin | 422 | 3.90 | 0.05 | 0.35 | 0.36 |
| C | Filter paper | 36 | 5.88 | 0.01 | 0.02 | 0.03 |
Fig. S4† shows the FTIR spectra of FC1, FC2, and FC3 for characterizing their functional groups. The adsorption bands at 2865–2973, 1630–1658, 1383–1384 and 1045–1048 cm−1 are attributed to the skeletal vibration of –CH3, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, OC–O and C–O bonds, respectively.35,36 However, the adsorption intensity of the OC–O vibration in FC2 and FC3 are higher than that in FC1, while the vibration of CC in FC1 is more intense than that in FC2 and FC3. Therefore, compared with FC1, there are more OC–O and fewer CC bonds in FC2 and FC3. The presence of relatively weak adsorption band at 508–584 cm−1 for Fe–O,37 confirms that a large number of FeOx particles were successfully embedded in the 3D net-like carbon material.
C, OC–O and C–O bonds, respectively.35,36 However, the adsorption intensity of the OC–O vibration in FC2 and FC3 are higher than that in FC1, while the vibration of CC in FC1 is more intense than that in FC2 and FC3. Therefore, compared with FC1, there are more OC–O and fewer CC bonds in FC2 and FC3. The presence of relatively weak adsorption band at 508–584 cm−1 for Fe–O,37 confirms that a large number of FeOx particles were successfully embedded in the 3D net-like carbon material.
The composites were also investigated by XRD patterns (Fig. S5†). It can be seen that all the three types of products consist of graphite, Fe3O4, and Fe0. This result indicates that the simultaneous decomposition of ferric nitrate and paper fiber happened during the calcination process. Furthermore, the signal of carbon could be testified in the Raman spectra of FC1 shown in Fig. S6.† The characteristic peaks at 1332 cm−1 and 1569 cm−1 are attributed to the D- and G-bands of C, respectively. The intensity ratio of D band and G band (ID/IG) is 0.89, indicating the feature of amorphous graphite.38
To further confirm the composition of FC1, FC2, FC3, and C, XPS patterns were obtained in Fig. S7.† Fig. S7a† shows three characteristic peaks at 709.9 eV, 528.9 eV, and 283.1 eV, which are indexed to Fe2p, O1s and C1s, respectively. In the patterns of FC1, FC2, FC3, peaks of Fe2p, O1s and C1s are obvious. However, there is hardly any Fe2p peak in the pattern of C. The peaks at about 709.8 eV and 719.18 eV are attributed to Fe2p3/2 and Fe2p1/2 in Fe2+ (Fig. S7b†). The 713.4 eV and 723.9 eV peaks are indexed to Fe2p3/2 and Fe2p1/2 in Fe3+. The 714.9 eV and 726.5 eV peaks correspond to Fe2p3/2 and Fe2p1/2 in Fe0. These peaks also indicate that the Fe content in FC1, FC2, and FC3 is a mixture of Fe3O4 and Fe0.39 Due to the existence of these magnetic nanoparticles, the maximum saturation magnetization value of FC1 is 21.18 emu g−1 at room temperature (Fig. S8†), which is enough high for a magnetic response.
3.2. Adsorption performance
The removal efficiency of MB and RhB, as a function of contact time for the four adsorbent–adsorbate systems, are shown in Fig. 3. It can be seen that very high adsorption rates were obtained. Generally, within only 5 min, 62.4% of MB and 94.2% of RhB were adsorbed by FC1 from aqueous solution, about 53% removal of MB and 76% removal of RhB were achieved by FC2 and FC3, and 4.5% of MB and 7.5% of RhB were removed by C. Therefore, for a given adsorbate, FC1 has much higher removal efficiency than FC2, FC3, and C under the same conditions. The adsorption efficiency of FC2 is nearly equal to that of FC3, and about 1.2 times lower than that of FC1. C has little adsorption capacity compared with the other three.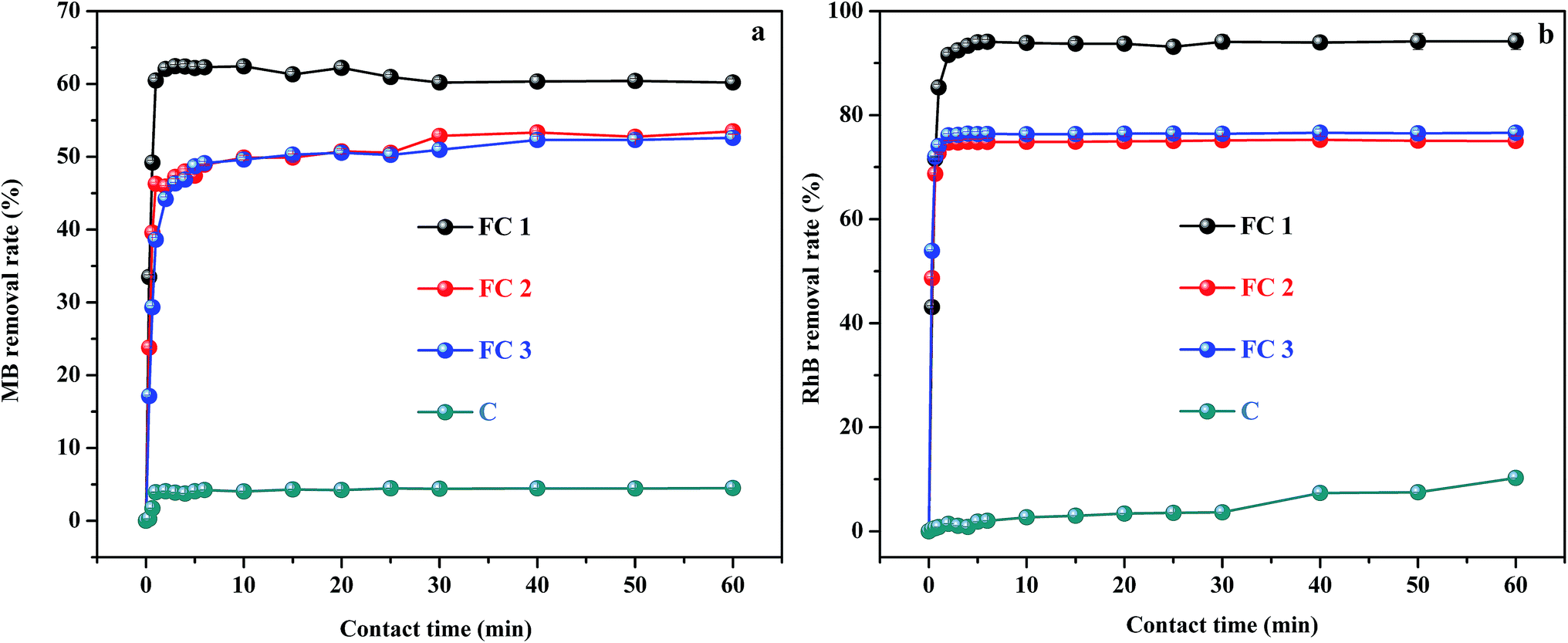 | ||
| Fig. 3 Adsorption performance of FC1, FC2, FC3 and C for MB (a) and RhB (b) in an aqueous solution. (Conditions: initial concentration of MB and RhB: 50 mg L−1; amount of adsorbents for MB and RhB: 8 mg; sample volume of MB and RhB: 20 mL; pH of MB and RhB: 7; temperature: 25 °C.) | ||
The adsorption capacity among the four adsorbents is closely related to their SSA and pore structures. C has a very poor adsorption performance due to the lowest SSA (36 cm2 g−1). Whereas, FC3 with the highest SSA (422 cm2 g−1) did not show the best adsorption performance. At the same time, FC2 also showed a relatively lower removal efficiency than FC1, even though the SSA of FC2 (294 cm2 g−1) is a little higher than that of FC1 (286 cm2 g−1). According to the “structure–performance relationship” of mesoporous materials, SSA is not the only factor determining adsorption performance.40 Large enough and interconnected mesopores are also critical, which can facilitate the diffusion of adsorbate, thereby increasing the accessibility of inner active sites and shortening the response time.41,42 Therefore, the poor connectivity and lower Dpore, Vmeso, and Vtotal of FC2 and FC3 compared with that of FC1 (Table 1) decreased the adsorption capacity of FC2 and FC3, even though the pore sizes of FC2 and FC3 were suitable for the molecule sizes of MB and RhB (Table S1†). By contrast, the mesopores of FC1 with the highest Dpore, Vmeso, and Vtotal are more suitable for the adsorption of molecules, and so shows satisfactory removal efficiency.
For a given adsorbate, the removal efficiency of RhB by FC1, FC2, and FC3 was higher than that of MB. It can be attributed to that the carboxylic group in RhB (Table S1†) endowed the molecule with high hydrophilicity and good affinity to MMC.
The adsorption kinetics was analyzed by two common semi-empirical models respectively, i.e. the pseudo-first-order (eqn (S1)) and the pseudo-second-order (eqn (S2)) (Fig. S9).† The constants k1 and k2 were calculated and listed in Table S2.† Evidently, the pseudo-second-order model provided the better correlation to the MB and RhB adsorption data with correlation coefficients (R2 > 0.99), and good agreement between experimental (qe,cal) and calculated (qe,exp) values. Thus it could be concluded that adsorption interaction of FC1, FC2, FC3, and C to MB and RhB were determined by electron transfer or electron sharing interactions between dye molecule and FeOx/C.24
The interaction mechanisms of MB and RhB molecule on FC1, FC2, FC3 and C were investigated using Langmuir, Freundlich, Sips and Redlich-Peterson isotherm models (the linear forms of Langmuir (eqn (S3) and (S4)), Freundlich (eqn (S5)), Sips (eqn (S6)) and Redlich-Peterson (eqn (S7)) isotherms were given in Fig. S10).† The calculated parameters from the isotherms and R2 values were summarized in Table S3.† The R2 values of the Redlich-Peterson isotherm model for the MB adsorption onto FC1, FC2, and FC3 were higher as compared to those obtained from Langmuir, Freundlich and Sips isotherm models. However, the values of β were already close to 1, and the R2 values were almost equal to the obtained by the Langmuir model. Therefore, the isotherm can be reduced the Langmuir isotherm form, which designates that the adsorption process was driven by the monolayer formation on MB and the surfaces of FC1, FC2, and FC3. In addition, the favorability and feasibility of the MB adsorption can be delivered in terms of a dimensionless constant known as separation ration factor (RL) that was calculated by the Langmuir isotherm model (eqn (S4)†). The RL values for all the systems of MB onto FC1, FC2 and FC3 were found to be in the range of 0–1 (Table S4†), which confirms that the adsorption processes of MB on FC1, FC2, and FC3 were favorable under the set experimental conditions.17 The equilibrium data of the MB adsorption onto C fitted well to Freundlich isotherm, indicating that the interaction between MB and C belong to multilayer adsorption. For RhB adsorption, the R2 values for the Sips model were all higher than that of the other three models. Additionally, based on the Sips isotherm model, theoretical isotherm saturation adsorption capacities (qm) were more close to the experimental results. Those indicated that Sips model describes well the adsorption performance of FC1, FC2, FC3, and C for RhB. The heterogeneity factors ns−1 for both four adsorbents were close to 1.0, illustrating that the binding force between the adsorbent and adsorbate was chemical force instead of physical force.43
As we know, the adsorption removal capacity depends on the adsorbent surface properties and adsorbate structure, which are greatly affected by pH. Fig. 4 presents two sets of the adsorption data measured at different solution pH for FC1 and the two adsorbates. As shown in Fig. 4, generally, the adsorption capacity for MB decreased slowly with the increase of pH from 2.0 to 6.0, remained constant from 6.0 to 10.0, and increased slowly from 10.0 to 12.0. However, the adsorption capacity for RhB increased with the increase of pH from 2.0 to 8.0 and decreased with the increase of pH from 8.0 to 12.0. Apparently, the effect of solution pH should be illustrated by both the speciation of the adsorbates and the surface charge density of adsorbent under various pH values.44 First of all, MB and RhB are typical cationic dyes and stably exist in solution as positively charged ions in the studied pH range. On the other hand, the isoelectric point of the FC1 is identified to be 5.0. This value indicates a positively charged surface of FC1 with pH below 5.0 and a negatively charged surface with pH above 5.0. Therefore, electrostatic repulsion exists between MB and FC1, RhB and FC1 at pH below 5.0, while electrostatic attraction arises between MB and FC1, RhB and FC1 when pH above 5.0. However, there is a slight increase of adsorption capacity of MB with the decreasing of pH from 5.0 to 2.0. It can be explained by the protonation of the amino group in the molecule of MB, which intensifies the hydrogen bond interaction between the amino of MB and carboxyl of FC1.45 In addition, the deprotonation of the carboxyl of RhB also exists in the solution at the alkaline condition. It results in the electrostatic repulsion between the negative carboxylic group of RhB and FC1 with a negatively charged surface and reduces the interaction between RhB and FC1.43
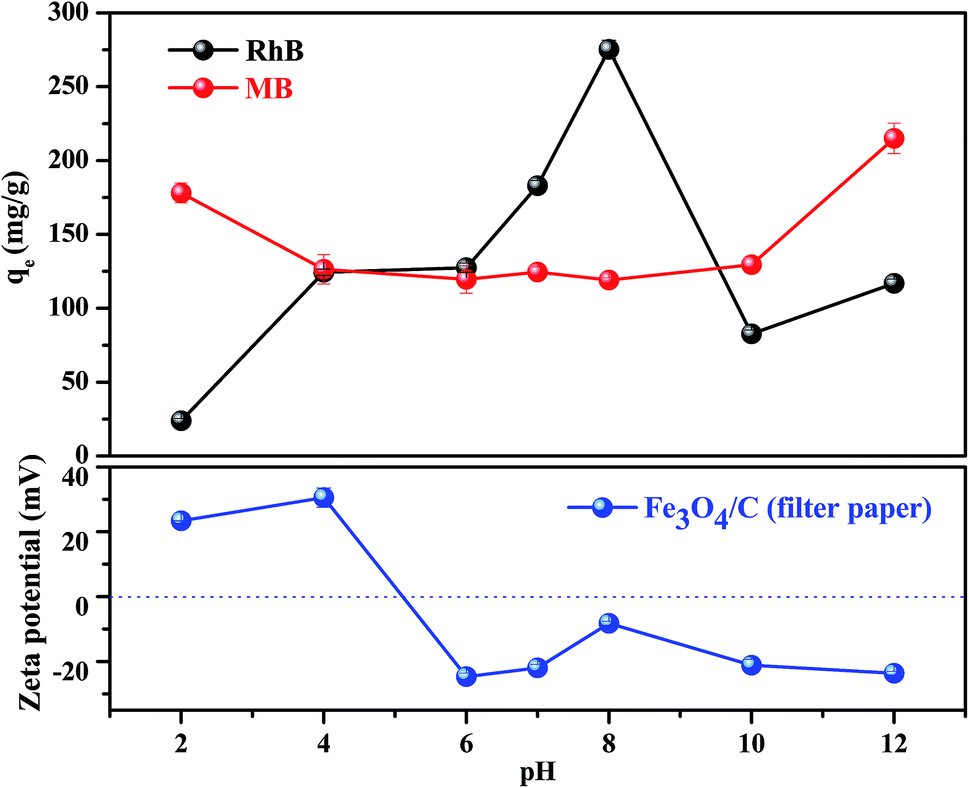 | ||
| Fig. 4 Equilibrium adsorption capacities of FC1 in MB and RhB aqueous solutions with different pH. (Conditions: amount of adsorbents: 2 mg; sample volume: 20 mL; initial concentration of MB: 60 mg L−1; initial concentration of RhB: 100 mg L−1; temperature: 25 °C.) | ||
The effect of temperature on the equilibrium adsorption capacity of FC1 to MB and RhB was also investigated (Fig. 5). It is shown that the MB and RhB adsorption increased obviously as the temperature increased from 20 to 50 °C, suggesting that both MB and RhB adsorption onto FC1 were endothermic processes.46 The highest adsorption capacity of MB (332.03 mg g−1) and RhB (389.59 mg g−1) were both obtained at 50 °C. Thermodynamic parameters were calculated (eqn (S8)–(S11)†) and were used to investigate the adsorption processes further. Results of standard Gibbs free energy (ΔG0), enthalpy (ΔH0) and entropy (ΔS0) were displayed in Table 2. It is demonstrated that the ΔG0 values calculated for the MB and RhB adsorption on FC1 were negative for all tested temperature, indicating that the adsorption processes were spontaneous. ΔH0 and ΔS0 were calculated from the linearized vanít Hoff plot (eqn (S11)†). Positive values of ΔH0 also indicate that the adsorption processes were endothermic, and positive values of ΔS0 suggest some increase in the randomness of the FC1/solution interface.
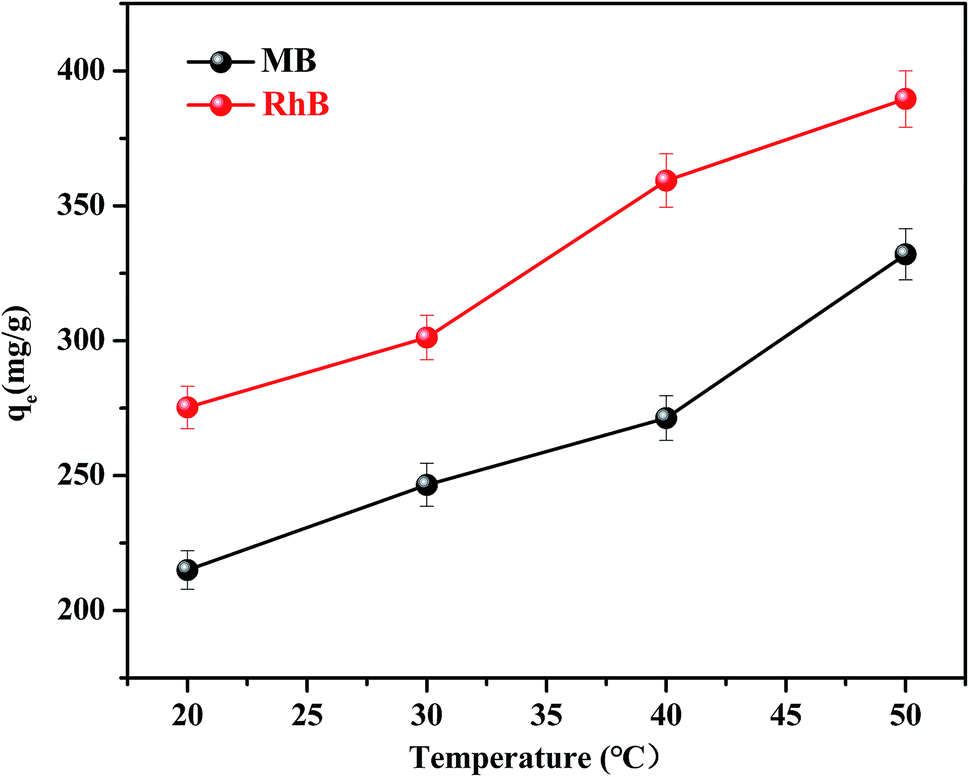 | ||
| Fig. 5 Equilibrium adsorption capacities of FC1 in MB and RhB aqueous solutions with different temperature. (Conditions: amount of adsorbents: 2 mg; sample volume: 20 mL; initial concentration of MB: 60 mg L−1; initial concentration of RhB: 100 mg L−1; pH of MB aqueous solution: 12; pH of RhB aqueous solution: 8.) | ||
| Temp. (K) | MB | RhB | ||||
|---|---|---|---|---|---|---|
| ΔG0 (kJ mol−1) | ΔH0 (kJ mol−1) | ΔS0 (kJ mol−1) | ΔG0 (kJ mol−1) | ΔH0 (kJ mol−1) | ΔS0 (kJ mol−1) | |
| 293 | −1.36 | 20.03 | 0.06 | −0.93 | 14.31 | 0.04 |
| 303 | −1.76 | −1.09 | ||||
| 313 | −2.15 | −1.46 | ||||
3.3. Desorption studies
Desorption and reusability of adsorbents are essentially important for commercial feasibility. The adsorption and desorption cycles were conducted five times using the same batch of adsorbent. As shown in Fig. S11,† the removal rate of MB and RhB by the regenerated pure FC1 slightly decreased after five cycles, and the removal rate remained higher than 80% and 90% for MB and RhB, respectively. It indicates that FC1 is a kind of reusable nanocomposite with high adsorption performance.4. Conclusion
Three types of 3D net-like MMC have been one-step synthesized by an eco-friendly method. Waste filter paper, printer paper, and napkins were chosen as three types of carbon source, and Fe(NO3)3 as magnetism source. During the calcination process, some MNPs including Fe3O4 and Fe0 were formed. These iron oxide not only brings magnetic response for adsorbent, but also play an important role for the formation of pores. The pores are directly accessible without extra steps to remove templates or organic solvents in the pores. The synthesis method is simple, fast, low-cost and non-toxic. The adsorbent derived from filter paper showed higher adsorption performance than those from printer paper and napkin. Additionally, the adsorption mechanism was investigated by FT-IR and XPS analysis. It was confirmed that the functional groups including CC, O–CO, C–O on the surface of FC1 participated in the adsorption process, and the occurrence of redox reactions between Fe2+/Fe0 and dye cannot be ignored. Furthermore, the adsorbents could be regenerated by a single elution step. It is worthwhile to point out that the 3D net-like MMC is not limited to dye adsorption, and shows enormous potential to removal of other pollutants.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The study was supported by grants from Environmental Protection Department of Hubei Province (No. 2017HB04) and the Fundamental Research Funds for the Central Universities, China University of Geosciences (Wuhan) (No. CUG170102).References
- H. Argun and G. Onaran, Int. J. Hydrogen Energy, 2016, 41, 8057–8066 CrossRef CAS.
- H. T. Sahin and M. B. Arslan, Int. J. Mol. Sci., 2008, 9, 78–88 CrossRef CAS PubMed.
- L. K. Voon, S. C. Pang and S. F. Chin, Carbohydr. Polym., 2016, 142, 31–37 CrossRef CAS PubMed.
- P. Dai, Y. Xue, S. Zhang, L. Cao, D. Tang, X. Gu, L. Li, X. Wang, X. Jiang, D. Liu, L. Kong, Y. Bando, D. Golberg and X. Zhao, ACS Appl. Mater. Interfaces, 2018, 10, 37046–37056 CrossRef CAS PubMed.
- M. Shahrousvand, F. A. Tabar, E. Shahrousvand, A. Babaei, M. M. Hasani-Sadrabadi, G. M. M. Sadeghi, H. Jafari and A. Salimi, Carbohydr. Polym., 2017, 175, 293–302 CrossRef CAS PubMed.
- W. H. Danial, Z. A. Majid, M. N. M. Muhid, S. Triwahyono, M. B. Bakar and Z. Ramli, Carbohydr. Polym., 2015, 118, 165–169 CrossRef CAS PubMed.
- M. A. Mohamed, W. N. W. Salleh, J. Jaafar, S. E. A. M. Asri and A. F. Ismail, RSC Adv., 2015, 5, 29842–29849 RSC.
- M. Li, H. Du, L. Kuai, K. Huang, Y. Xia and B. Geng, Angew. Chem., Int. Ed., 2017, 56, 12649–12653 CrossRef CAS.
- K. Yang, W. Wu, Q. Jing and L. Zhu, Environ. Sci. Technol., 2008, 42, 7931–7936 CrossRef CAS PubMed.
- M. A. Oturan and J. Aaron, Crit. Rev. Environ. Sci. Technol., 2014, 44, 2577–2641 CrossRef CAS.
- D. P. Zagklis and C. A. Paraskeva, Sep. Purif. Technol., 2015, 156, 328–335 CrossRef CAS.
- M. Ruesgas-Ramón, M. C. Figueroa-Espinoza and E. Durand, J. Agric. Food Chem., 2017, 65, 3591–3601 CrossRef PubMed.
- A. E. Navarro, N. A. Cuizano, J. C. Lazo, M. R. Sun-Kou and B. P. Llanos, J. Hazard. Mater., 2009, 164, 1439–1446 CrossRef CAS PubMed.
- L. Chen, J. Pei, X. Huang and M. Lu, J. Chromatogr. A, 2018, 1564, 34–41 CrossRef CAS PubMed.
- M. S. U. Rehman, M. Munir, M. Ashfaq, N. Rashid, M. F. Nazar, M. Danish and J. Han, Chem. Eng. J., 2013, 228, 54–62 CrossRef CAS.
- J. Qin, P. Tan, Y. Jiang, X. Liu, Q. He and L. Sun, Green Chem., 2016, 18, 3210–3215 RSC.
- E. F. Lessa, M. S. Gularte, E. S. Garcia and A. R. Fajardo, Carbohydr. Polym., 2017, 157, 660–668 CrossRef CAS PubMed.
- B. C. Melo, F. A. A. Paulino, V. A. Cardoso, A. G. B. Pereira, A. R. Fajardo and F. H. A. Rodrigues, Carbohydr. Polym., 2018, 181, 358–367 CrossRef CAS PubMed.
- H. Li, Y. Li, Y. Chen, M. Yin, T. Jia, S. He, Q. Deng and S. Wang, ACS Sustainable Chem. Eng., 2018, 7, 858–866 CrossRef.
- D. Jia, X. Yu, H. Tan, X. Li, F. Han, L. Li and H. Liu, J. Mater. Chem. A, 2017, 5, 1516–1525 RSC.
- M. Inagaki, M. Toyoda, Y. Soneda, S. Tsujimura and T. Morishita, Carbon, 2016, 107, 448–473 CrossRef CAS.
- Y. Boyjoo, Y. Cheng, H. Zhong, H. Tian, J. Pan, V. K. Pareek, S. P. Jiang, J. Lamonier, M. Jaroniec and J. Liu, Carbon, 2017, 116, 490–499 CrossRef CAS.
- Y. Li, C. Yang and X. Yan, Chem. Commun., 2017, 53, 2511–2514 RSC.
- Y. Xie, W. Yang, M. Wang and X. Ge, Chem. Eng. J., 2017, 323, 224–232 CrossRef CAS.
- Z. Chen, W. Ren, L. Gao, B. Liu, S. Pei and H. Cheng, Nat. Mater., 2011, 10, 424–428 CrossRef CAS PubMed.
- Y. Ito, C. Christodoulou, M. V. Nardi, N. Koch, M. Klaeui, H. Sachdev and K. Muellen, J. Am. Chem. Soc., 2015, 137, 7678–7685 CrossRef CAS PubMed.
- Y. Chang, M. Antonietti and T. Fellinger, Angew. Chem., Int. Ed., 2015, 54, 5507–5512 CrossRef CAS PubMed.
- J. Yu, S. Zhu, L. Pang, P. Chen and G. Zhu, J. Chromatogr. A, 2018, 1540, 1–10 CrossRef CAS PubMed.
- J. Li, J. Wang, Y. Ling, Z. Chen, M. Gao, X. Zhang and Y. Zhou, Chem. Commun., 2017, 53, 4018–4021 RSC.
- K. Wieczorek-Ciurowa and A. J. Kozak, J. Therm. Anal. Calorim., 1999, 58, 647–651 CrossRef CAS.
- F. Gong, T. Ye, L. Yuan, T. Kan, Y. Torimoto, M. Yamamoto and Q. Li, Green Chem., 2009, 11, 2001–2012 RSC.
- P. R. Patwardhan, R. C. Brown and B. H. Shanks, ChemSusChem, 2011, 4, 636–643 CrossRef CAS PubMed.
- Z. Yu, S. Xin, Y. You, L. Yu, Y. Lin, D. Xu, C. Qiao, Z. Huang, N. Yang, S. Yu and J. B. Goodenough, J. Am. Chem. Soc., 2016, 138, 14915–14922 CrossRef CAS PubMed.
- K. Zhou, M. Hu, Y. He, L. Yang, C. Han, R. Lv, F. Kang and B. Li, Carbon, 2018, 129, 667–673 CrossRef CAS.
- Y. Yan, S. Sun, Y. Song, X. Yan, W. Guan, X. Liu and W. Shi, J. Hazard. Mater., 2013, 250–251, 106–114 CrossRef CAS PubMed.
- L. Jiang, Y. Liu, G. Zeng, F. Xiao, X. Hu, X. Hu, H. Wang, T. Li, L. Zhou and X. Tan, Chem. Eng. J., 2016, 284, 93–102 CrossRef CAS.
- L. Jiang, Y. Liu, S. Liu, X. Hu, G. Zeng, X. Hu, S. Liu, S. Liu, B. Huang and M. Li, Chem. Eng. J., 2017, 308, 597–605 CrossRef CAS.
- X. Chen, X. Lai, J. Hu and L. Wan, RSC Adv., 2015, 5, 62913–66292 RSC.
- Y. Jiang, Z. Jiang, L. Yang, S. Cheng and M. Liu, J. Mater. Chem. A, 2015, 3, 11847–11856 RSC.
- J. Wang, Q. Ma, Y. Wang, Z. Li, Z. Li and Q. Yuan, Chem. Soc. Rev., 2018, 47, 8766–8803 RSC.
- T. Wagner, S. Haffer, C. Weinberger, D. Klaus and M. Tiemann, Chem. Soc. Rev., 2013, 42, 4036–4053 RSC.
- W. Luo, T. Zhao, Y. Li, J. Wei, P. Xu, X. Li, Y. Wang, W. Zhang, A. A. Elzatahry, A. Alghamdi, Y. Deng, L. Wang, W. Jiang, Y. Liu, B. Kong and D. Zhao, J. Am. Chem. Soc., 2016, 138, 12586–12595 CrossRef CAS PubMed.
- A. B. Albadarin, M. N. Collins, M. Naushad, S. Shirazian, G. Walker and C. Mangwandi, Chem. Eng. J., 2017, 307, 264–272 CrossRef CAS.
- Q. Yang, R. Lu, S. Ren, C. Chen, Z. Chen and X. Yang, Chem. Eng. J., 2018, 348, 202–211 CrossRef CAS.
- Y. Zhou, Y. Hu, W. Huang, G. Cheng, C. Cui and J. Lu, Chem. Eng. J., 2018, 341, 47–57 CrossRef CAS.
- F. Zhao, E. Repo, D. Yin, Y. Meng, S. Jafari and M. Sillanpää, Environ. Sci. Technol., 2015, 49, 10570–10580 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra01332f |
| This journal is © The Royal Society of Chemistry 2019 |