
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot route to graft long-chain polymer onto silica nanoparticles and its application for high-performance poly(L-lactide) nanocomposites
Xin Wen *
*
Nanomaterials Physicochemistry Department, Faculty of Chemical Technology and Engineering, West Pomeranian University of Technology in Szczecin, al. Piastów 45, 70-311, Szczecin, Poland. E-mail: Xin.Wen@zut.edu.pl; hgwenxin@126.com
First published on 7th May 2019
Abstract
A facile “one-pot” synthetic route to modify SiO2 nanoparticles with long-chain polymer was developed. The structure and morphology of SiO2 grafted with poly(L-lactide) (SiO2-g-PLA) were characterized by FTIR, TGA, GPC, and TEM. Furthermore, a series of PLA/SiO2-g-PLA nanocomposites were prepared with different nanofiller loadings, and their related performances were investigated. As an effective nucleating agent, the SiO2-g-PLA had a positive effect to improve the crystallization rate and increase the crystallinity. Meanwhile, the PLA nanocomposites presented outstanding mechanical properties including excellent toughness and high stiffness. In addition, the PLA materials kept good transparency with less than 3 wt% nanofillers. Overall, this work provides a useful method for preparing high-performance polymer nanocomposites.
1. Introduction
Polymer nanocomposites have gathered substantial academic and industrial interest due to their remarkable improvements in thermal and mechanical properties when compared with virgin polymers or conventional micro- and macro-composites.1–3 However, the enforcement effect of nanocomposites is largely limited by the strong tendency of nanofillers to aggregate in polymer matrices. Meanwhile, the poor interfacial adhesion between inorganic nanofillers and organic polymers is also a negative factor to deteriorate the resultant properties of composite materials. Thus, it is still a great challenge to develop novel methods to achieve good dispersion and strong interfacial adhesion of nanofillers for preparing high-performance polymer nanocomposites.Hitherto much excellent research has been done on the chemical modification of nanofillers.4–6 To improve the dispersion, generally the surfaces of inorganic nanoparticles were treated with organic small molecule compounds, such as: silane coupling agents, titanate and organic isocyanates, etc.7–9 Besides, it is a more promising strategy that the nanoparticles are covered with a layer of matrix-miscible polymers. For instance, Förster group10 modified Fe3O4 and CdSe nanoparticles by coating with polystyrene of different molecular weights, and SEM image showed there was no aggregation at high nanoparticles volume fractions. Rong et al.11 prepared low nano-SiO2 loaded polypropylene composites, in which the nanoparticles were grafted by polystyrene using irradiation beforehand. A high interfacial stress transfer efficiency was demonstrated by both strengthening and toughening effects perceived in tensile tests. Recently Chen et al.12 reported that ZnO nanoparticles were activated by N,N-carbonyldiimidazole (CDI) and subsequently modified with oleic acid (OA). Results showed that the dispersibility of modified ZnO nanoparticle in the unsaturated polyester (UP) matrix improved owing to the enhanced compatibility and adhesion between the UP resin and the modified ZnO particles, resulting in the marked influence on mechanical and curing properties of the nanocomposite.
Over the past decade, biodegradable polymers have attracted considerable attention due to the increasing environmental concerns and the decreasing fossil resources.13–16 As a representative, poly(L-lactide) (PLA) possesses many advantages in terms of biodegradability, mechanical strength, transparency and compostability, so it has tremendous market in biomedical, packaging and agriculture fields.17–19 To further extend its applications, PLA based nanocomposites have been exploited by incorporating various types of nanofillers.20–23 Meanwhile, the modification of these nanoparticles was necessary to improve the dispersion and interface in PLA matrix. Hong et al.24 reported that hydroxyapatite nano-particles (n-HAP) were grafted by PLA via directly the polymerization of L-lactide, and the amount of grafted polymer was about 6 wt% in weight. The obtained PLLA/PLLA-g-HAP composite exhibited higher tensile strength and elongation at break than PLLA composites with unmodified n-HAP. Yan et al.25 also grafted PLA onto SiO2 via silanol groups initiated polymerization, and the grafted polymer was PLA oligomers. TEM and SEM results showed that grafted SiO2 (g-SiO2) nanoparticles can be comparatively uniformly dispersed in chloroform or PLLA matrix, and it can greatly improve the toughness and tensile strength of PLA. Lu et al.26 modified TiO2 nanoparticles by surface-grafting L-lactic acid oligomer, and the highest amount of grafted polymer were about 8.5% in weight. They found that the tensile strength of the PLLA/5 wt% g-TiO2 nanocomposite was 23.1% higher than that of pure PLLA, and it also exhibited better ductility.
As above described, the chemical grafting of polymer is an effective way to improve the dispersion of nanofillers and improve the mechanical properties of PLA to some extent. Nevertheless, there are still some unsatisfactory shortcomings that hinder their effectiveness on comprehensive properties. On one hand, the reactivity of these hydroxyl groups on nanofillers is limited, resulting in forming a number of free-polymerized oligomers. On the other hand, the surface hydroxyl groups had intrinsic steric hindrance for attacking the reacted groups, leading to the grafted polymer with low grafting density and low molecular weight. Therefore, to achieve highly effective overall enhancements in PLA nanocomposites with desirable performances, it is necessary to consider two aspects: (i) to increase the grafting degree of polymers, which could form isolated layer due to the increase of interparticle distance; (ii) to graft long-chain polymer, which was favourable to increase compatibility with polymer matrix via chain entanglement.
In the present study, a facile “one-pot” synthetic route was developed to modify SiO2 nanoparticles with long-chain PLA. As shown in Scheme 1, firstly SiO2 was functionalized by converting its surface silanol groups into more reactive amino groups (SiO2-g-APS); subsequently these amino groups initiated the ring-opening polymerization of L-lactide (LA) using Sn(Oct)2 as catalyst (SiO2-g-PLA). After the reaction of SiO2-g-APS, no purification was carried out and it was directly used to graft PLA. Besides, the molar ratio of L-LA/APS must be strictly controlled to obtain long-chain PLA. Moreover, the SiO2-g-PLA was employed to prepare PLA/SiO2 nanocomposites. The effect of nanofillers on the crystallization properties, mechanical properties and optical transparency of PLA was investigated.
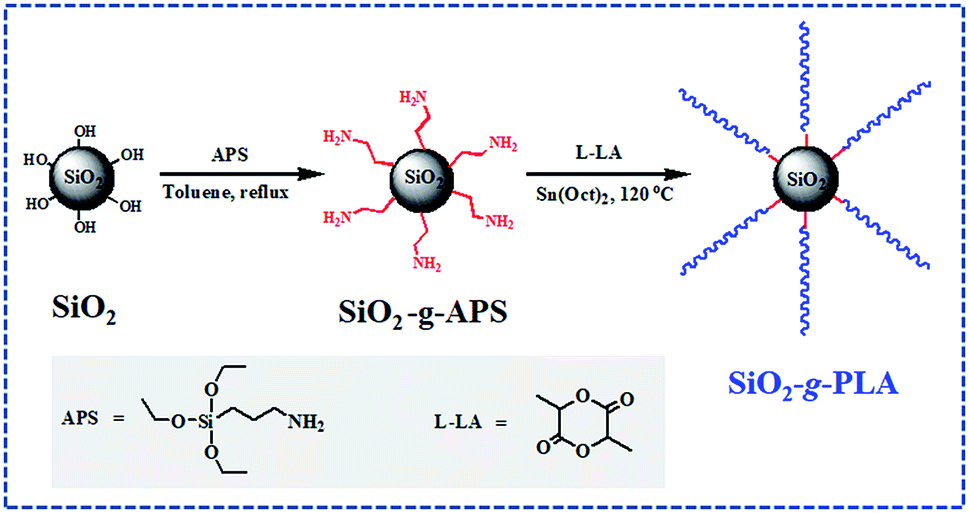 | ||
| Scheme 1 The scheme of surface modification of silica nanoparticles. | ||
2. Experimental section
2.1 Materials
The nanosilica (SiO2, AEROSIL® 200) was supplied by Degussa AG (Hanau, Germany) with average primary particle size 12 nm and a specific surface area 200 m2 g−1. L-Lactide (L-LA) was purchased from Purac. Ethylacetate and toluene were dried by refluxing over CaH2 and metal sodium, respectively. Aminopropyltriethoxysilane (APS) and Sn(Oct)2 were bought from Sigma-Aldrich without further purification. Poly(L-lactide) (PLA, 4032D) was a commercial product of Natureworks Co. Ltd., USA, and had high optical purity with about 98% L-lactide content. It had density 1.24 g cm−3, weight-average molecular weight (Mw) 250 kDa, polydispersity 1.70 (by GPC analysis).2.2 Grafting of long-chain PLA onto the surfaces of silica nanoparticles
Functionalization of silica by converting silanol groups into amino groups was carried out following a previous literature.27 To a mechanical stirred suspension of dried silica (20.0 g) in dry toluene (300 mL) under nitrogen, a solution of APS (0.123 g) in toluene (20 mL) was added dropwise over 25 min. The mixture was refluxed for 8 h under nitrogen. After cooling to room temperature, a little turbid solution from the mixture was taken out with a sucker, filtered and washed with toluene, then extracted with toluene in a Soxhlet apparatus for 8 h. The functionalized silica (SiO2-g-APS) was dried in vacuum at 100 °C for 6 h for later measurements.Subsequently, 40.0 g L-LA was added into above reaction mixture. After it completely dissolved, 4.0 mL Sn(Oct)2 solution in toluene (0.15 mol L−1) was injected into the mixture under nitrogen atmosphere with stirring. The mixture was slowly heated to 80 °C, then the vacuum was applied for about 1 h to remove the toluene. Finally the mixture was heated to 120 °C, and the bulk polymerization reaction was maintained at this temperature for 24 h. The PLA grafted silica nanoparticles (SiO2-g-PLA) were separated by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm and washed with excessive amount of chloroform for five times to completely remove the free L-lactide oligomer. Finally the separated precipitate was dried in vacuum at 60 °C for 24 h to remove the residual chloroform.
000 rpm and washed with excessive amount of chloroform for five times to completely remove the free L-lactide oligomer. Finally the separated precipitate was dried in vacuum at 60 °C for 24 h to remove the residual chloroform.
To characterize the molecular weight parameters of the grafted PLA, it is necessary to remove the silica core. As a typical procedure,28 200 mg silica-containing PLA was dissolved in 10 mL of THF, then 100 μL HF solution (30 wt%) (Caution! hydrofluoric acid is extremely corrosive) was added to this solution. After stirring at 50 °C for 4 h and then removing all the solvents, the residues were dissolved in 5 mL chloroform and precipitated into an excess of methanol. The sediments were collected and dried in a vacuum oven overnight. Additionally, the above centrifuged solution was concentrated and also precipitated into methanol, yielding small amounts of white powder. For convenience, the grafted PLA and the PLA from the centrifuged solution were denoted as PLA1 and PLA2, respectively.
2.3 Preparation of PLA/SiO2-g-PLA nanocomposites
Melt compounding of PLA with 1, 3, 5, 7 and 10 wt% of SiO2-g-PLA was conducted using a Haake batch intensive mixer (Haake Rheomix 600, Karlsruhe, Germany). The two components were mixed at 175 °C for 6 min, with screw speed 60 rpm. For comparison, neat PLA was subjected to the same mixing treatment so as to have the same thermal history as its nanocomposites. After mixing, all samples were cut into small pieces and hot compression molded into various sheets at 180 °C and 10 MPa. For convenience, the prepared PLA/SiO2-g-PLA nanocomposites were denoted as PLAGSNs in the following discussion. For example, PLAGSN1 represents PLA nanocomposites with 1 wt% SiO2-g-PLA.2.4 Characterization
Fourier Transform Infrared (FTIR) spectra were recorded on a FT/IR-460 PLUS spectrometer (JASCO, Tokyo, Japan) in the range between 4000 and 400 cm−1, with a resolution of 4 cm−1 and 64 scans. The sample of original silica (SiO2), APS functionalized silica (SiO2-g-APS), and PLA grafted silica (SiO2-g-PLA) was mixed with KBr powders and pressed into a disk suitable for FTIR measurement, respectively.The grafting amount of polymer on SiO2 was determined by Thermal Gravimetric Analysis (TGA) using a Mettler Toledo TGA/SDTA 851 (Columbus, USA). The measurements were performed under nitrogen from room temperature to 700 °C at a rate of 10 °C min−1. The amount of organic composition was determined from the weight loss percentage between 150 °C and 700 °C.
TEM (JEOL JEM-1010) was used to observe the size, shape and dispersibility of silica nanoparticles in chloroform before and after surface grafting. The TEM specimens were prepared by dripping a drop of 0.1 wt% nanoparticles/chloroform suspension onto a TEM grid covered with carbon film and evaporating the solvent completely at room temperature.
GPC measurement was carried out on a Waters 410 GPC instrument equipped with two Waters Styragel columns (HT6E and HT3) and a differential refractometer detector for calibration at 25 °C, using monodisperse polystyrene standards. Chloroform was used as solvent with a flow rate of 1.0 mL min−1. PLA1 and PLA2 were directly tested. PLA samples after melt compounding were first dissolved in chloroform and filtered off all fillers, then the solution was precipitated in an excess of methanol.
The morphologies of the fracture surfaces of neat PLA and its nanocomposites were examined by XL30 ESEM FEG (FEI Co.).The samples were frozen well in liquid nitrogen and quickly broken off to obtain a random brittle-fractured surface. A layer of gold was sputter coated uniformly over all the fractured surfaces before SEM observations.
The thermal parameters of neat PLA and its nanocomposites were measured by DSC-7, Perkin-Elmer, under nitrogen flow. All samples were heated from 30 to 200 °C with heating rate of 50 °C min-1 and kept isothermal for 3 min to erase previous thermal history. Then they were cooled to −20 °C at 50 °C min−1 and subsequently the second scan as result curves was performed between −20 and 200 °C at the heating rate of 10 °C min−1. The crystallinity (χc) of PLA in the nanocomposites was calculated from the following formula:
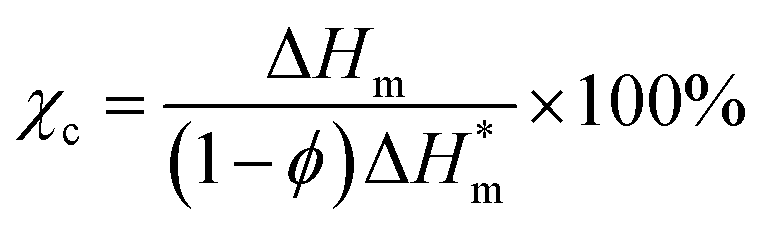
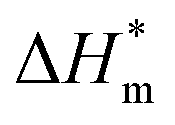 is the theoretical enthalpy of completely crystalline PLA, as 93.1 J g−1.29,30
is the theoretical enthalpy of completely crystalline PLA, as 93.1 J g−1.29,30
Polar optical microscopy studies (POM) were carried out with a Leica polarized light microscope (DM 2500P) in conjunction with a hot stage (Linkam LTS 350). The samples of PLA and PLAGSNs were prepared by cutting small pieces from prepared sheets. Each sample was melted on glass slides with coverslips to form thin films 20–50 μm thickness. The specimens were heated to 180 °C on the hot stage and held at that temperature for 3 min and then quickly cooled to 130 °C at a rate of 40 °C min−1. The POM observation was carried out as soon as the sample was cooled to 130 °C. The hot stage was calibrated with a melting point standard to 0.2 °C accuracy. In addition, photographs were taken by a digital camera.
Dynamic mechanical thermal analysis (DMTA) was performed on molded sheets with a DMA/SDTA861e apparatus (Mettler-Toledo, Switzerland) in the tensile mode. The relaxation spectrum was scanned from −80 to 150 °C at a frequency of 1 Hz and a heating rate of 3 °C min−1. The shape of the film samples was rectangular (ca. 4.0 × 9.0 × 1.0 mm3). The viscoelastic properties, such as the storage modulus (G′) and the mechanical loss factor (tanδ = G′′/G′, where G′′ is the loss modulus), were recorded as a function of the temperature.
Uniaxial tensile tests were performed at room temperature with an Instron 1121 testing machine (Canton, MA) according to GB/T1040.3-2006 (China). Specimens (20 × 4 × 1 mm3) were cut from the above compression molded sheet with a dumbbell shape. The measurements were conducted at a crosshead speed of 5 mm min−1 at room temperature. The V-notched specimens (55 × 6 × 4 mm3) were performed to measure the impact strength according to GB1843-93 procedures using an impact testing machine (CEAST, Chengde, China). All tests were carried out at room temperature and 50% relative humidity. At least five runs for each sample were measured and averaged as results.
The light transmittance and haze values of the neat PLA and PLASN sheets were measured with a photoelectric haze instrument (WGW, Suzhou, China) at room temperature. The sheets were prepared by melting and then quick quenching with a thickness of 0.50 ± 0.02 mm. At least five runs for each sample were measured and averaged value as results.
3. Results and discussion
3.1 Characterization of silica nanoparticles grafted with long-chain PLA
To prove the grafting reaction of PLA chains onto silica nanoparticles, FTIR was employed to comparatively analyse original SiO2, SiO2-g-APS and SiO2-g-PLA. As shown in Fig. 1a, original SiO2 presented its characteristic absorption peaks at 1086, 800 and 467 cm−1, which were ascribed to the stretching and bending vibrations of Si–O–Si bonds.31 For SiO2-g-APS in Fig. 1b, several weak new peaks appeared at 2936 and 1489 cm−1, 1567 and 699 cm−1, which were assigned to the vibration of C–H and N–H, respectively. In contrast, SiO2-g-PLA in Fig. 1c exhibited some different peaks. The strong peaks at 1664 cm−1 was corresponded to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in amido bond.32 Moreover, the 1758 cm−1 and 2995 cm−1 were assigned to CO in ester bond and C–H in –CH3, which are well consistent with the characteristic bands of PLA.33 These results confirmed that PLA chains were successfully grafted onto SiO2 via amino groups initiated the ring-opening polymerization of L-lactide.
O in amido bond.32 Moreover, the 1758 cm−1 and 2995 cm−1 were assigned to CO in ester bond and C–H in –CH3, which are well consistent with the characteristic bands of PLA.33 These results confirmed that PLA chains were successfully grafted onto SiO2 via amino groups initiated the ring-opening polymerization of L-lactide.
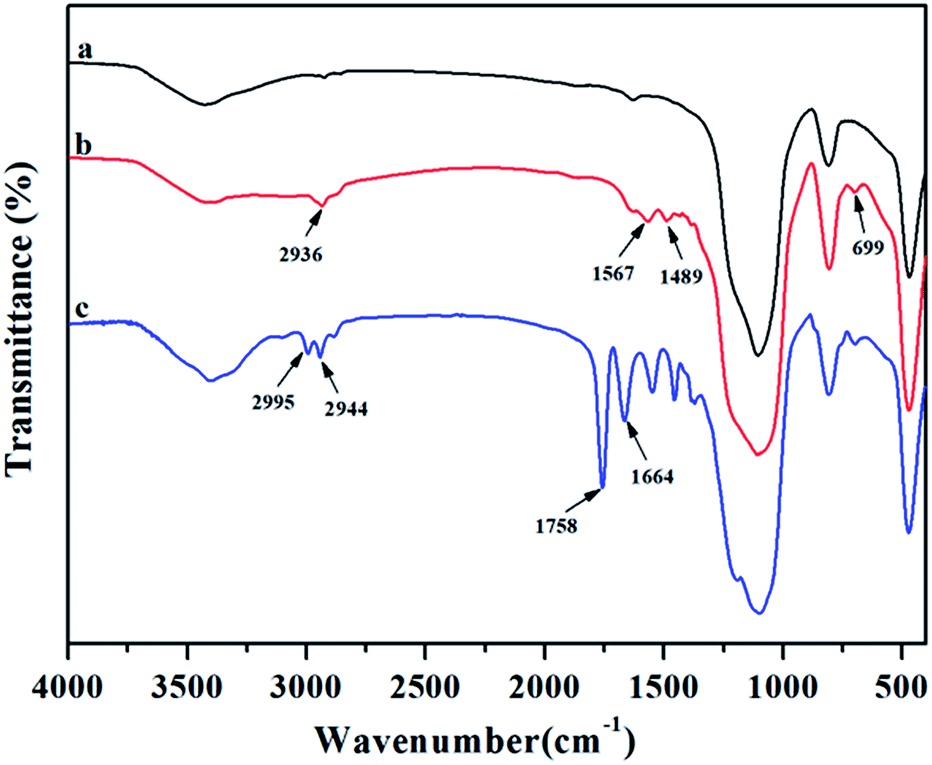 | ||
| Fig. 1 FTIR spectra of (a) original SiO2, (b) SiO2-g-APS and (c) SiO2-g-PLA. | ||
TGA was used to quantify the grafting degree of PLA on SiO2. The curves for above three types of SiO2 are plotted in Fig. 2. The weight loss of original SiO2, SiO2-g-APS and SiO2-g-PLA between 150 and 700 °C was 0.49 wt%, 1.62 wt% and 21.27 wt%, respectively. This indicated that the grafting degree of PLA was approximately 19.65 wt%.
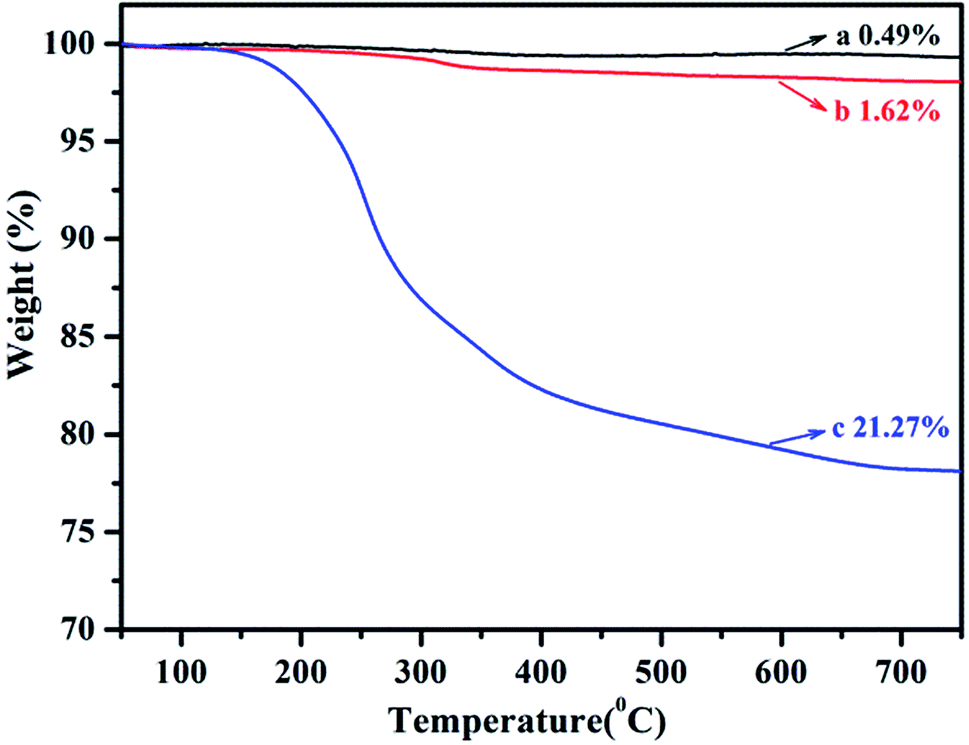 | ||
| Fig. 2 TGA curves of (a) original SiO2, (b) SiO2-g-APS and (c) SiO2-g-PLA. | ||
GPC measurement was carried out to provide information about molecular weight and its distribution for the synthesized PLAs. As listed in Table 1, the weight-average molecular weight (Mw) and molecular weight distribution for PLA1 was 109.74 kDa and 2.12. In contrast, the Mw of PLA2 from free polymerization was only 15.4 kDa, which was much smaller than that of PLA1. These data suggested that long-chain structure of PLA was successfully grafted to the surface of silica due to high reactive activity of amino groups as initiator and large molar ratio of L-LA to APS (L-LA/APS = 500/1).
| Samples | Mw (kDa) | Mn (kDa) | PDI |
|---|---|---|---|
| a PLA1 from silica surface.b PLA2 from the centrifuged solution.c PLA3 from neat PLA after melt compounding. | |||
| PLA1a | 109.74 | 51.52 | 2.13 |
| PLA2b | 15.47 | 8.50 | 1.82 |
| PLA3c | 178.24 | 96.35 | 1.85 |
| PLAGSN1 | 177.51 | 92.45 | 1.92 |
| PLAGSN3 | 174.30 | 88.03 | 1.98 |
| PLAGSN5 | 171.52 | 83.67 | 2.05 |
| PLAGSN7 | 169.19 | 80.18 | 2.11 |
| PLASN10 | 167.22 | 78.50 | 2.13 |
The morphology of original SiO2 and SiO2-g-PLA was observed by TEM. As shown in Fig. 3. It can be seen that the original SiO2 particles had a strong tendency to aggregate and it is difficult to distinguish one particle from the other (Fig. 3a). After grafted by PLA, the nanoparticles became a little larger and exhibited core–shell-like structure (dark silica core with grey polymer shell in Fig. 3b). This further proves that PLA chains were coated on SiO2 surface via chemical grafting.
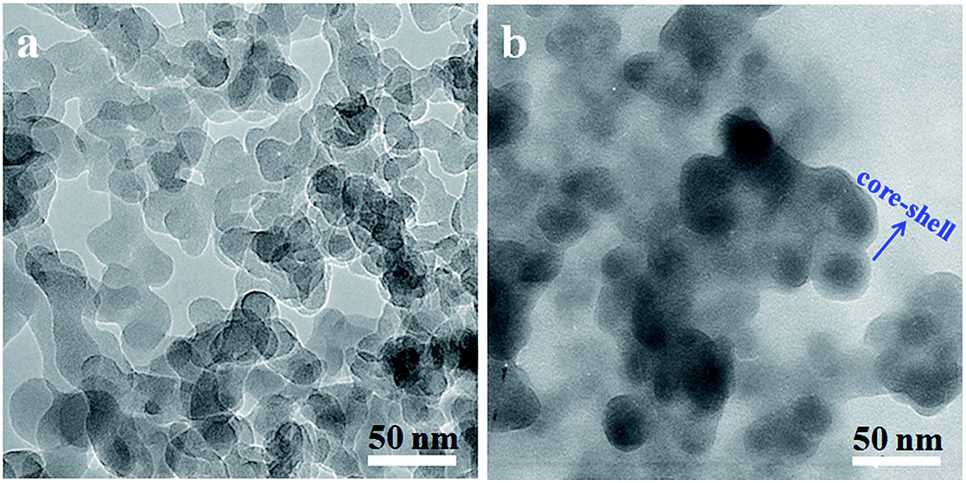 | ||
| Fig. 3 TEM micrographs of silica nanoparticles dispersed in chloroform: (a) original SiO2 and (b) SiO2-g-PLA. | ||
3.2 Effect of SiO2-g-PLA on crystallization properties of PLA
Firstly the molecular weight of PLAs after melt compounding was investigated, which is very important to understand their crystallization characteristics. As shown in Table 1, after melt compounding, all PLAs exhibited a certain level of decline on molecular weight and wider polydispersity due to essential thermal degradation of polyesters. Generally the incorporation of nanofiller will promote the degradation of polymers because of high shear viscosity. With the increase of SiO2-g-PLA content, although a gradual decease on molecular weight was present, this trend was only slight. This result indicated that the addition of SiO2 nanoparticles had no apparent influence on the thermal degradation of PLA chains.The addition of nanofiller will significantly affect the crystallization properties of polymer matrix, and further determine the resultant physical properties of polymer nanocomposites, especially for mechanical properties. Fig. 4 presents the second heating thermograms of quenched PLA samples with various filler loadings, and the detailed DSC results are summarized in Table 2. It is noted that the second heating was selected for eliminating thermal history due to the possible differences on real process condition (such as temperature fluctuation, hot-pressing time, etc.), so that the record of all PLA samples started from amorphous state. It seems that a trend with substantial increase then slow decrease in glass transition temperature (Tg) was found with the increase of nanofiller content, but the change was inconspicuous with only small differences within 1 °C. However, the SiO2-g-PLA had a great influence on the cold crystallization and melting behaviour of PLA. Neat PLA represented a tiny broad exothermic peak at 132.8 °C (ΔHcc = 21.37 J g−1), indicating a rather low cold crystallization capability. In the case of PLAGSNs, a sharper and narrower peak appeared at a much lower temperature, and correspondingly the crystallization enthalpies largely increased (Table 2). It suggested that the SiO2 played a heterogeneous nucleation role, and enhanced the crystalline ability of PLA. Similar nucleating effect from nanofillers was previously reported in PLA/clay system.34 Meanwhile, the melting peak (Tm) of PLA in the nanocomposites had a transition from single peak to two separate peaks, including a minor shoulder peak at 162 °C range. Generally, multiple melting peaks of semi-crystalline polymer are attributed to two main reasons: melt-recrystallization and dual crystal structure.35,36 During the slow DSC scans, the less perfect crystals got enough time to melt and reorganized into crystals with higher structure perfection and re-melted at a higher temperature. According to our previous work about PLA nanocomposites with direct addition of SiO2,30 the bimodal peaks might be a result of the melt-recrystallization. Furthermore, it was found that the crystallinity of PLA was greatly increased with the addition of SiO2-g-PLA. For example, with only 1 wt% SiO2-g-PLA, the crystallinity was as high as 55.6%. But the value was kept at this range (55.1–58.2%) with further increase of nanofiller loading, implying that only a small amount of SiO2-g-PLA could promote the crystallization of PLA chains to reach saturation point.
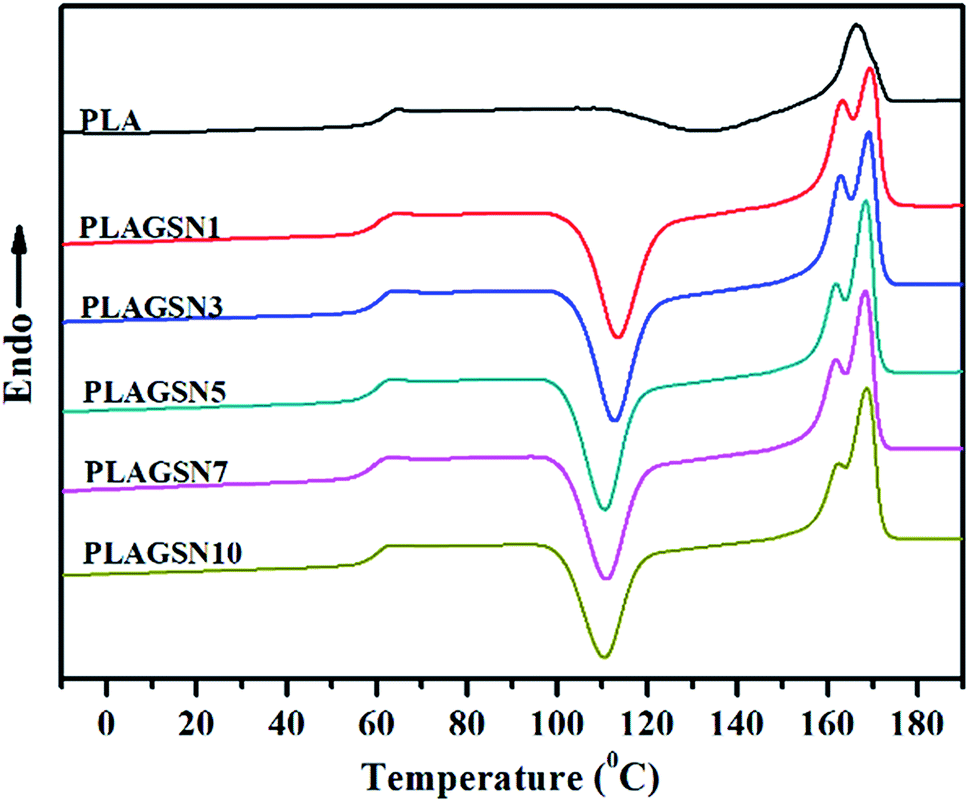 | ||
| Fig. 4 DSC curves from the second heating for neat PLA and PLA/SiO2-g-PLA nanocomposites. | ||
| Samples | Tg (°C) | Tcc (°C) | Tm1 (°C) | Tm2 (°C) | ΔHcca (J g−1) | ΔHma (J g−1) | Crystallinity (%) |
|---|---|---|---|---|---|---|---|
| a Data calculated from the peaks in DSC curves. | |||||||
| PLA | 60.1 | 132.8 | 166.6 | 21.4 | 24.1 | 25.9 | |
| PLAGSN1 | 60.3 | 113.4 | 163.3 | 169.4 | 45.0 | 51.2 | 55.6 |
| PLAGSN3 | 60.9 | 112.7 | 162.9 | 169.1 | 44.5 | 50.9 | 56.3 |
| PLAGSN5 | 60.4 | 110.5 | 161.9 | 168.5 | 43.7 | 50.8 | 57.4 |
| PLAGSN7 | 59.8 | 110.9 | 161.8 | 168.4 | 43.1 | 50.4 | 58.2 |
| PLAGSN10 | 59.7 | 110.5 | 162.1 | 168.7 | 40.4 | 46.2 | 55.1 |
To further investigate the heterogeneous nucleation of nanofiller, polar optical microscopy (POM) was used to illustrate the difference of their crystal morphologies. As shown in Fig. 5, the spherulites of PLA samples were formed after isothermal crystallization at 130 °C for 5 min, 10 min and 20 min, respectively. For neat PLA in Fig. 5(a1–a3), it exhibited bright spherules and its size systematically increased with the increase of crystallization time. For PLAGSN1, the spherule size significantly became smaller and its amount increased largely at the same record time (shown in Fig. 5(b1–b3)). In the case of PLAGSN3, the nucleus density of crystallites further increased and the diameter of spherules dramatically reduced, but these spherules became dim and showed unclear boundary, indicating the formation of less ordered spherules (shown in Fig. 5(c1–c3)). Similar results were also reported by Wang et al. in PLA/rectorite nanocomposites.37 As an effective heterogeneous nucleating agent, a minor weight percentage of SiO2-g-PLA could increase the overall crystallization rate, reduce the nucleation induction period and increase the number of primary nucleation sites.38 As a result, the PLA matrix presented a decrease in Tm and an increase in crystallinity. These changes for spherules were in good accordance with above DSC results.
 | ||
| Fig. 5 Polarized optical microscopy images of neat PLA, PLAGSN1 and PLAGSN3 via isothermal crystallization at 130 °C for (a1, b1, c1) 5 min, (a2, b2, c2) 10 min and (a3, b3, c3) 20 min, respectively. | ||
3.3 Effect of SiO2-g-PLA on mechanical properties of PLA
In order to evaluate the enhancement effect of SiO2-g-PLA, mechanical properties of PLA samples were measured by tensile and notched-impact tests, respectively. Stress–strain curves of neat PLA and PLAGSNs are shown in Fig. 6, the detailed data for Young's modulus, tensile strength, elongation at break and notched-impact strength are summarized in Table 3. It was found the Young's modulus gradually increased with the increasing nanofiller loading, which can be attributed to the reinforcing effect of rigid inorganic filler and the increase of crystalline degree of PLA. Meanwhile, the tensile strength gradually increased for SiO2-g-PLA content up to 3 wt%, and then slightly decreased with further increase of nanofiller content. The elongation at break is usually used to evaluate the toughness of polymer materials. In our PLA system, the PLAGSN3 exhibited the highest value for elongation at break, which reached to 118.6%, then greatly decreased to 8.4% for PLAGSN10. Besides the tensile tests, the notched impact is also useful to examine the toughness of polymers. Similar to the trend of elongation at break, PLASN3 presented the maximum value for impact strength, which was nearly 3.5 times higher than that of neat PLA (14.5 kJ m−2 vs. 4.2 kJ m−2). Based on above results, a good balance on mechanical properties was achieved in PLASN3, which not only displayed excellent toughness, but also kept fairly high stiffness.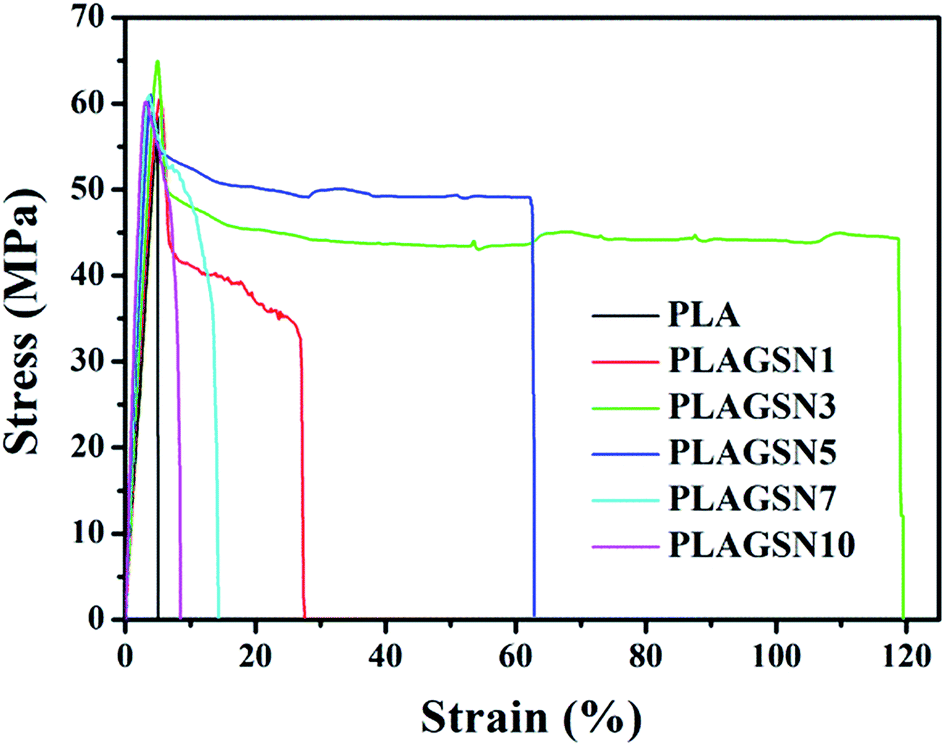 | ||
| Fig. 6 Stress–strain curves of neat PLA and PLAGSNs. | ||
| Samples | Young's modulus (GPa) | Tensile strength (MPa) | Elongation at break (%) | Impact strength (kJ m−2) |
|---|---|---|---|---|
| PLA | 1.58 ± 0.08 | 58.5 ± 1.6 | 4.9 ± 1.2 | 4.2 ± 0.3 |
| PLAGSN1 | 1.65 ± 0.16 | 60.5 ± 0.9 | 26.6 ± 3.6 | 9.6 ± 0.5 |
| PLAGSN3 | 1.72 ± 0.12 | 65.1 ± 2.2 | 118.6 ± 9.8 | 14.5 ± 0.9 |
| PLAGSN5 | 1.78 ± 0.09 | 62.8 ± 1.3 | 62.2 ± 3.2 | 11.2 ± 0.7 |
| PLAGSN7 | 1.82 ± 0.13 | 61.4 ± 0.8 | 13.8 ± 1.6 | 7.5 ± 0.3 |
| PLAGSN10 | 1.85 ± 0.11 | 60.9 ± 0.7 | 8.4 ± 0.8 | 6.6 ± 0.4 |
In polymer nanocomposites, it has been well-accepted that the dispersion of nanofillers and matrix–nanoparticle interaction are the most important factors to determine their resultant mechanical performances. Fig. 7a–d shows micrographs of fracture surfaces of PLAGSNs with nanofiller from 3 to 10 wt%. The SiO2 nanoparticles in PLASNs were detected as bright dots and were uniformly distributed in the PLA matrix. When the SiO2-g-PLA loading was beyond 5 wt%, some small aggregates with 3–5 nanoparticles were present but their size did not exceed 100 nm in diameter. Furthermore, it was seen that all nanofillers looked a little dark, and adhered firmly to the polymer matrix without interfacial debonding. These results indicated that there was a strong interfacial adhesion between polymer matrix and nanofillers due to the grafting of long-chain PLA, which was perfectly compatible with PLA matrix.
 | ||
| Fig. 7 SEM micrographs of the brittle-fractured surface of: (a) PLAGSN3, (b) PLAGSN5, (c) PLAGSN7 and (d) PLAGSN10. | ||
DMTA was employed to evaluate the compatibility between nanofillers and polymer matrix. The mechanical relaxation curves of PLA sample are depicted in Fig. 8, in the form of loss tangent (tanδ) and storage modulus (G′), respectively. A sharp tanδ peak was observed at 64.85 °C for neat PLA, corresponding to its glass transition temperature. In the case of PLASNs, the tanδ peak gradually increased to 68.80 °C for PLAGSN3, then decreased slightly with further increase of the filler concentration (Fig. 7a). The trend for Tg shift was in good agreement with the above mentioned DSC results, but the change was more apparent in the DMA due to its higher sensitive than DSC technique. As is well-known, the Tg of polymer matrix can be greatly influenced by the polymer–nanofiller interactions. In this study, the silica grafted with long-chain PLA had strong chain entanglement with PLA matrix, which resulted in the decrease of free volume for PLA chain, so the Tg shifted to a higher temperature. Meanwhile, the chain entanglement was also influenced by the nanofiller size. Once some aggregates were present, this interaction will be weakened due to the decrease of interface area, leading to the shift of Tg to a lower temperature. Thus, the optimal ratio of SiO2-g-PLA was 3 wt% in our PLA system, which corresponded to the best dispersion and compatibility to PLA matrix. Moreover, Fig. 7b shows the variations of storage modulus (G′) as a function of temperature for neat PLA and PLAGSNs. Comparing the storage modulus in the low temperature zone (−60 °C ∼ Tg), one can find that the G′ gradually increase with the increasing SiO2 content due to the enhancement effect of nanofiller, which is similar to the trend for Young's modulus. The storage modulus decreased sharply at Tg, then increased gradually with the increase of temperature due to the cold crystallization of PLA. Meanwhile, it can be seen that the cold crystallization region became narrower and shifted to a lower temperature range with the addition of silica. This result further confirmed that the grafted silica was an effective nucleation agent to accelerate the crystallization rate of PLA.
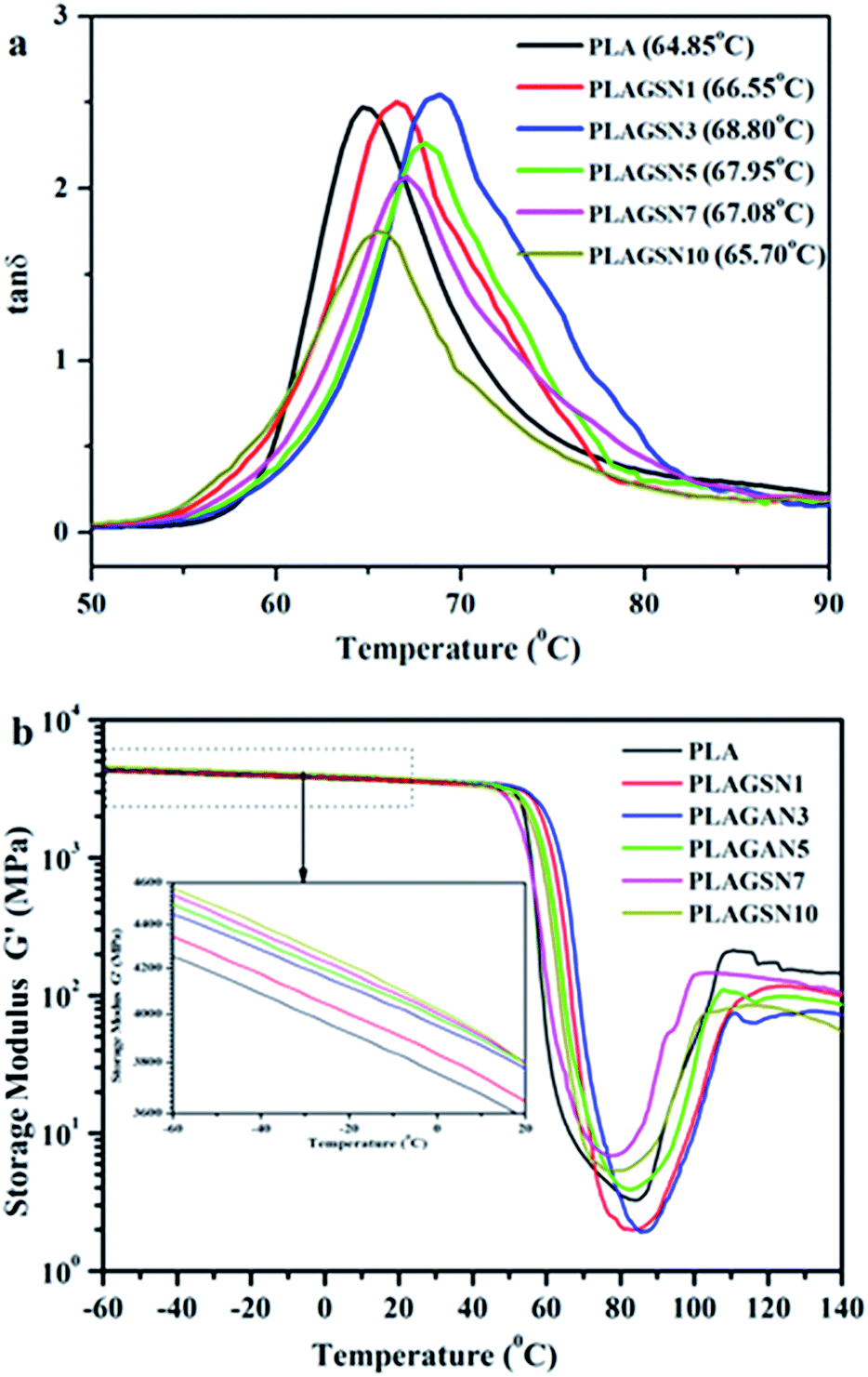 | ||
| Fig. 8 DMTA curves for neat PLA and PLAGSNs: (a) tanδ vs. temperature, (b) storage modulus (G′) vs. temperature. | ||
3.4 Effect of SiO2-g-PLA on optical transparency of PLA
As an advantage of PLA, it has good transparency due to its low crystallinity degree. Some excellent work has been done to improve the mechanical properties of PLA, but in most case it will sacrifice its optical transparency. It has been reported that silica as an isotropic spherical nanoparticles, played a critical role in preparing transparent nanomaterials based on semi-crystalline polymers.39 As shown in Fig. 9, the visible light transmittance and haze value of neat PLA and PLASNs are plotted. When the SiO2-g-PLA content was less than 3 wt%, the transmittance of obtained PLA nanocomposites was almost at the same level of neat PLA, which is related to the good dispersion and intrinsic refractive index of SiO2. Correspondingly, the haze values for PLAGSNs were almost unaffected with less than 3 wt% nanofiller.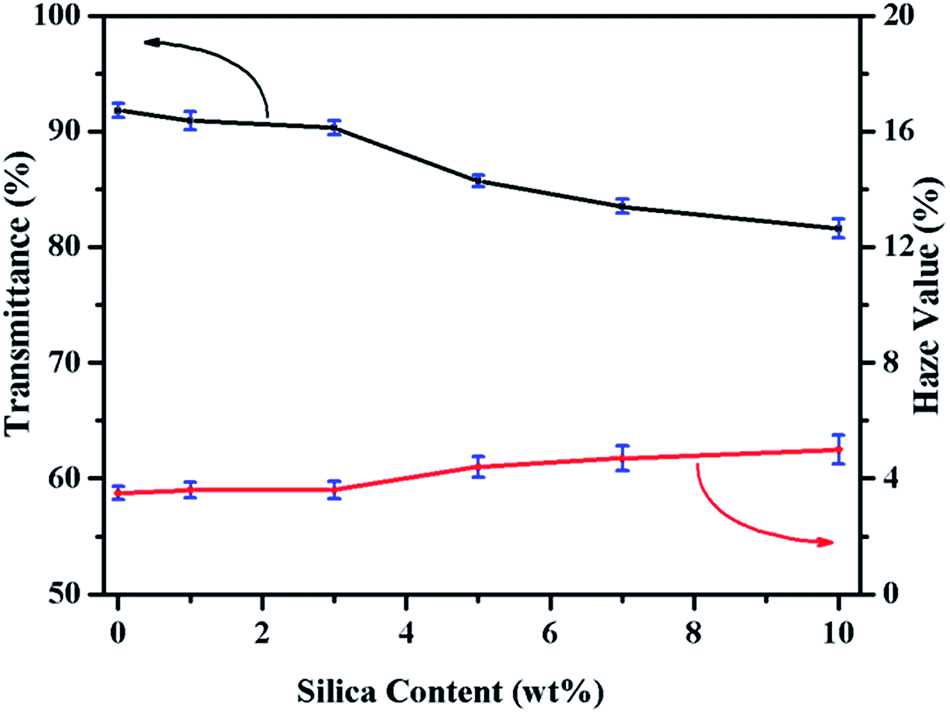 | ||
| Fig. 9 Effect of SiO2-g-PLA on the transparence and haze value of PLA. | ||
To visually display the transparency of PLA nanocomposites, the photographs of neat PLA and PLASN sheets covered on the same graphic pattern and collected by a digital camera were shown in Fig. 10. The PLASNs with less than 3 wt% nanofillers exhibited almost the same transparency as neat PLA, and it decreased slightly with higher SiO2 content. This is well consistent with above trends on transmittance and haze value. Therefore, the PLAGSN3 displayed the best comprehensive performances, including high crystallinity, balanced mechanical properties and good transparence.
 | ||
| Fig. 10 Digital images of neat PLA and PLASNs: (a) neat PLA, (b) PLAGSN1, (c) PLAGSN3, (d) PLAGSN5, (e) PLAGSN7, and (f) PLAGSN10. | ||
4. Conclusions
We proposed a “one-pot” route to graft long-chain PLA onto silica nanoparticles. Results showed that the grafting degree of PLA was 19.65 wt%, the Mw of PLA was about 109 kDa and it presented a core–shell structure. Moreover, the grafted silica was used to prepare PLA/SiO2-g-PLA nanocomposites by melt compounding. With 3 wt% SiO2-g-PLA, the PLA nanocomposites showed the optimal comprehensive performances. Its crystallinity was as high as 56.3%; its elongation at break and impact strength were improved to 118.6% and 14.5 kJ m−2, respectively. In addition, it had good transparency with high transmittance to 90.3%. We believe that this strategy will be applicable to modify not only silica nanoparticles, but also various other inorganic nanofillers. It will open up a door for fabricating high-performance and functional polymer nanocomposites.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Science Centre Poland OPUS UMO-2018/29/B/ST8/01265.References
- J. George and H. Ishida, Prog. Polym. Sci., 2018, 86, 1–39 CrossRef CAS.
- J. J. Chen, B. F. Liu, X. H. Gao and D. G. Xu, RSC Adv., 2018, 8, 28048–28085 RSC.
- Z. Q. Liu, Z. Li, Y. X. Yang, Y. L. Zhang, X. Wen, N. Li, C. Fu, R. K. Jian, L. J. Li and D. Y. Wang, Polymers, 2018, 10, 14 Search PubMed.
- S. Srivastava, S. Choudhury, A. Agrawal and L. A. Archer, Curr. Opin. Chem. Eng., 2017, 16, 92–101 CrossRef.
- G. K. Nie, G. Z. Li, L. Wang and X. W. Zhang, Polym. Chem., 2016, 7, 753–769 RSC.
- S. Y. Chen, X. Dong and L. S. Zha, Prog. Chem., 2015, 27, 831–840 Search PubMed.
- Y. F. Hou, Y. Xu, H. P. Li, Y. Y. Li and Q. J. Niu, Chem. Eng. Technol., 2019, 42, 65–72 CrossRef CAS.
- H. N. Minh, N. T. Chinh, T. T. T. Van, D. X. Thang and T. Hoang, J. Nanosci. Nanotechnol., 2018, 18, 3624–3630 CrossRef CAS PubMed.
- Q. Liu, J. R. de Wijn, D. Bakker, M. van Toledo and C. A. van Blitterswijk, J. Mater. Sci.: Mater. Med., 1998, 9, 23–30 CrossRef CAS PubMed.
- S. Fischer, A. Salcher, A. Kornowski, H. Weller and S. Förster, Angew. Chem., Int. Ed., 2011, 50, 7811–7814 CrossRef CAS PubMed.
- M. Z. Rong, M. Q. Zhang, Y. X. Zheng, H. M. Zeng and K. Friedrich, Polymer, 2001, 42, 3301–3304 CrossRef CAS.
- H. Z. Chen, X. X. Tian and J. Liu, Polymers, 2018, 10, 14 Search PubMed.
- F. Wu, M. Misra and A. K. Mohanty, RSC Adv., 2019, 9, 2836–2847 RSC.
- N. Iqbal, A. S. Khan, A. Asif, M. Yar, J. W. Haycock and I. U. Rehman, Int. Mater. Rev., 2019, 64, 91–126 CrossRef CAS.
- R. Muthuraj, M. Misra and A. K. Mohanty, J. Appl. Polym. Sci., 2018, 135, 35 CrossRef.
- S. S. Bari, A. Chatterjee and S. Mishra, Polym. Rev., 2016, 56, 287–328 CrossRef CAS.
- L. K. Kian, N. Saba, M. Jawaid and M. T. H. Sultan, Int. J. Biol. Macromol., 2019, 121, 1314–1328 CrossRef CAS PubMed.
- K. Hamad, M. Kaseem, M. Ayyoob, J. Joo and F. Deri, Prog. Polym. Sci., 2018, 85, 83–127 CrossRef CAS.
- B. P. Chang, A. K. Mohanty and M. Misra, RSC Adv., 2018, 8, 27709–27724 RSC.
- T. F. da Silva, F. Menezes, L. S. Montagna, A. P. Lemes and F. R. Passador, J. Appl. Polym. Sci., 2019, 136, 8 Search PubMed.
- D. L. Wu, H. Guo, R. L. Zhang and H. Z. Cui, ECS J. Solid State Sci. Technol., 2018, 7, 139–144 CrossRef.
- Y. T. Liu, L. M. Cao, D. S. Yuan and Y. K. Chen, Compos. Sci. Technol., 2018, 165, 231–239 CrossRef CAS.
- M. Kodal, H. Sirin and G. Ozkoc, Polym. Compos., 2018, 39, 2674–2684 CrossRef CAS.
- Z. K. Hong, X. Y. Qiu, J. R. Sun, M. X. Deng, X. S. Chen and X. B. Jing, Polymer, 2004, 45, 6699–6706 CrossRef CAS.
- S. F. Yan, J. B. Yin, Y. Yang, Z. Z. Dai, J. Ma and X. S. Chen, Polymer, 2007, 48, 1688–1694 CrossRef CAS.
- X. L. Lu, X. Q. Lv, Z. J. Sun and Y. F. Zheng, Eur. Polym. J., 2008, 44, 2476–2481 CrossRef CAS.
- Z. X. Guo and J. Yu, J. Mater. Chem., 2002, 12, 468–472 RSC.
- Z. S. Ge, D. Wang, Y. M. Zhou, H. W. Liu and S. Y. Liu, Macromolecules, 2009, 42, 2903–2910 CrossRef CAS.
- S. Aslan, L. Calandrelli, P. Laurienzo, M. Malinconico and C. Migliaresi, J. Mater. Sci., 2000, 35, 1615–1622 CrossRef.
- X. Wen, Y. Lin, C. Y. Han, K. Y. Zhang, X. H. Ran, Y. S. Li and L. S. Dong, J. Appl. Polym. Sci., 2009, 114, 3379–3388 CrossRef CAS.
- S. F. Yan, J. X. Geng, J. F. Chen, L. Yin, Y. C. Zhou and E. L. Zhou, J. Cryst. Growth, 2004, 262, 415–419 CrossRef CAS.
- K. N. Liang, M. D. Weir, M. A. Reynolds, X. D. Zhou, J. Y. Li and H. H. K. Xu, Mater. Sci. Eng., C, 2017, 72, 7–17 CrossRef PubMed.
- D. Garlotta, J. Polym. Environ., 2001, 9, 63–84 CrossRef CAS.
- Y. W. Di, S. Iannace, E. Di Maio and L. Nicolais, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 689–698 CrossRef CAS.
- L. Jiang, M. P. Wolcott and J. W. Zhang, Biomacromolecules, 2006, 7, 199–207 CrossRef CAS PubMed.
- Y. Lin, K. Y. Zhang, Z. M. Dong, L. S. Dong and Y. S. Li, Macromolecules, 2007, 40, 6257–6267 CrossRef CAS.
- B. Li, F. X. Dong, X. L. Wang, J. Yang, D. Y. Wang and Y. Z. Wang, Eur. Polym. J., 2009, 45, 2996–3003 CrossRef CAS.
- S. C. Schmidt and M. A. Hillmyer, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 300–313 CrossRef CAS.
- K. Asuka, B. P. Liu, M. Terano and K. H. Nitta, Macromol. Rapid Commun., 2006, 27, 910–913 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |