
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilencing of miR-486 alleviates LPS-stimulated inflammatory response of macrophages through targeting SIRT1
Jie Huanga,
Xinlei Fub,
Xue Chenc,
Shuang Xuc and
Jian Yu *c
*c
aDepartment of Severe Medicine, The Central Hospital of Wuhan, Wuhan, China
bDepartment of Severe Medicine, Dongguan Donghua Hospital, Dongguan, Guangdong, China
cDepartment of Severe Medicine, The Second Affiliated Hospital of Dalian Medical University, No. 467, Zhongshan Rd, Shahekou, 116000, Dalian, China. E-mail: tomorrow259@163.com; Tel: +86-0411-8467129
First published on 31st May 2019
Abstract
Previous studies identified that microRNAs (miRNAs) have promising diagnostic and prognostic value against sepsis. MiR-486 was demonstrated to be upregulated in sepsis. However, the detailed role and underlying mechanism of miR-486 in the inflammatory response of sepsis are still unclear. In this research, macrophages were stimulated with lipopolysaccharide (LPS) to establish a sepsis model in vitro. qRT-PCR was used to detect miR-486 expression and the mRNA levels of sirtuin (SIRT1), tumor necrosis factor (TNF)-α, interleukin (IL)-6 and IL-1β. ELISA assay was performed to measure the levels of TNF-α, IL-6 and IL-1β. SIRT1 protein expression was determined by Western blot analysis. The targeted relationship of miR-486 and SIRT1 was confirmed by dual-luciferase reporter assay. Our data supported that miR-486 was upregulated in the serum of sepsis patients. MiR-486 expression and inflammatory response were elevated by LPS stimulation in macrophages. MiR-486 silencing or SIRT1 overexpression alleviated inflammatory response in LPS-stimulated macrophages. Moreover, SIRT1 was a direct target of miR-486. Anti-miR-486-mediated anti-inflammatory response in LPS-stimulated macrophages was antagonized by SIRT1 inhibition. Our data suggested that miR-486 silencing alleviated inflammatory response in macrophages under LPS stimulation at least partly through targeting SIRT1. Targeting miR-486 may provide a novel way to protect against dysregulated inflammatory response in sepsis patients.
Introduction
Sepsis is a widespread and serious medical problem, characterized by heightened systemic inflammatory response syndrome (SIRS) following primarily bacterial infection.1 It is crucial to determine the magnitude of the inflammatory response for sepsis analysis because an excessive inflammatory response is capable of autodestruction, causing tissue injury, peritonitis, meningitis, abscesses, multiple organ failure and septic shock.1,2 Elevated serum levels of pro-inflammatory cytokines such as tumor necrosis factor (TNF)-α, interleukin (IL)-6 and IL-1β levels have been commonly found in sepsis patients.3 Although the progress of intensive care treatment has greatly improved the therapeutic outcomes for sepsis patients, it is still associated with significant mortality.4 Therefore, it is of great importance to identify novel effective targets for protection against dysregulated inflammatory response in sepsis.MicroRNAs (miRNAs), a family of small non-coding RNAs of ∼21–23 nucleotides in length, play crucial roles in almost every aspect of biology.5 MiRNAs function as the catalytic engine for miRNA-mediated post-transcriptional gene silencing by binding to the 3′-untranslated region (UTR) of target gene mRNAs, leading to translation suppression and/or the target mRNAs' degradation.5 An increasing number of studies have demonstrated that dysregulation of miRNAs is implicated in various human diseases, such as cardiovascular diseases, cancers, viral infection and inflammatory diseases.6–9 Previous studies identified that miRNAs had promising diagnostic and prognostic value against sepsis.10 It was reported that miR-486 expression was upregulated in sepsis patients compared with healthy controls.11 However, the detailed role and underlying mechanism of miR-486 in the inflammatory response of sepsis are still unclear.
Previous reports illustrated that sirtuin 1 (SIRT1) was capable of the inhibition of inflammasome activation in sepsis.12,13 Moreover, SIRT1 was demonstrated to act as a functional target of miR-486 in several other diseases.14–16 However, the effect of interplay between miR-486 and SIRT1 in sepsis remains unclear. In the present study, our data supported that miR-486 was overexpressed in the serum of sepsis patients. Moreover, miR-486 silencing alleviated the inflammatory response in macrophages under LPS stimulation at least partly through targeting SIRT1.
Materials and methods
Sample collection
A total of 41 cases of sepsis patients who were admitted to the Central Hospital of Wuhan, and 28 cases of healthy controls, were included in the study, with prior written informed consent. The inclusion criteria were: (1) diagnosed as sepsis according to the consensus definition of the American College of Chest Physicians/Society of Critical Care Medicine; (2) aged 55 years or older. The basic characteristics of the participants are shown in Table 1. No patients received any treatment before sample collection. 5 mL blood samples were collected and serum samples were obtained by centrifugation at 1200g for 10 min. All serum samples were stored at −80 °C until use. Our study was approved by the Ethics Committee of the Central Hospital of Wuhan in accordance with the principles of the Declaration of Helsinki. Informed consent was obtained from all participants.| Characteristics | Healthy controls (n = 28) | Sepsis patients (n = 41) | P-value |
|---|---|---|---|
| a Data presented as mean value ± standard deviation, median (quartile 25th to 75th). Comparison was determined by t-test or chi-square test. P value < 0.05 was considered significant. BMI, body mass index; Scr, serum creatinine; WBC, white blood cells; CRP, C-reactive protein; APACHE, acute physiology and chronic health evaluation; ICU, intensive care unit; NA, not applicable. | |||
| Age (years) | 52.3 ± 9.3 | 55.8 ± 8.2 | 0.113 |
| Sex | |||
| Male | 16 | 25 | 0.750 |
| Female | 12 | 16 | |
| BMI (kg m−2) | 24.15 ± 3.59 | 24.51 ± 3.64 | 0.652 |
| Scr (mg dL−1) | NA | 1.52 (1.02–1.91) | NA |
| Albumin (g L−1) | NA | 25.64 (22.14–35.03) | NA |
| WBC (×109/L) | NA | 13.39 (4.11–28.19) | NA |
| CRP (mg L−1) | NA | 42.12 (23.34–113.06) | NA |
| APACHE II score | NA | 15.6 ± 6.2 | NA |
| ICU stay (days) | NA | 13 ± 5 | NA |
| 28 day survival | |||
| Yes | NA | 24 | NA |
| No | NA | 17 | NA |
Cell culture and treatment
THP-1 cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in RPMI-1640 medium (Corning Cellgro, Manassas, VA, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Hyclone, Logan, UT, USA), 50 μM β-mercaptoethanol (Sigma-Aldrich, Poole, Dorset, UK), and 1% penicillin/streptomycin (Sigma-Aldrich) at 37 °C in a humidified atmosphere of 5% CO2. Phorbol-12-myristate-13-acetate (PMA) was used to induce macrophage differentiation from the monocytic THP-1 cell line as described previously.17 Briefly, in the experiments, the THP-1 cells were incubated with 100 ng mL−1 of PMA (Sigma-Aldrich) overnight for macrophage differentiation.To establish the sepsis model in vitro, macrophages were stimulated with 1 μg mL−1 of lipopolysaccharide (LPS, Escherichia coli, serotype 0111:B4, Sigma-Aldrich) for 24 h. To inhibit SIRT1, macrophages were treated with 10 μM of the SIRT1 inhibitor Selisistat (EX527, Sigma-Aldrich) for 6 h.
Cell transfection
To investigate the role of miR-486, 50 nM of modified miR-486 inhibitors (anti-miR-486, Ribobio, Guangzhou, Guangdong, China) or nontargeting control oligonucleotide (anti-miR-NC, Ribobio) were transfected into macrophages using Lipofectamine™ 3000 Transfection Reagent (Invitrogen, Carlsbad, CA, USA) referring to the guidance of the manufacturers.To observe the effect of SIRT1, macrophages were transfected with 100 ng of SIRT1 overexpression plasmids (Vector-SIRT1, Ribobio) or negative control (Vector) using Lipofectamine™ 3000 Transfection Reagent.
RNA isolation, reverse transcription and quantitative real-time PCR (qRT-PCR)
Total RNA was isolated from the serum samples and macrophages using Trizol reagent (Life Technologies, Villebon sur Yvette, France) according to the instructions of the manufacturers. The concentration and quality of the RNA extracts were determined using a NanoDrop 8000 spectrophotometer (NanoDrop, Wilmington, Delaware, USA). For mRNA detection, 1 μg of total RNA was used for reverse transcription PCR to synthesize cDNA using an iScript™ cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA, USA). qRT-PCR assay was performed using Fast SYBR™ Green Master Mix (Invitrogen) on a Stratagene Mx3000P real-time PCR system (Agilent Technologies, Santa Clara, CA, USA). The housekeeping gene GAPDH was used as an internal control and the fold changes were calculated by the 2−ΔΔCt method. For miR-486 determination, reverse transcription PCR was performed using a miScript Reverse Transcription Kit (Qiagen, Hilden, Germany) and qRT-PCR was performed using a miScript SYBR Green PCR Kit (Qiagen) with a specific primer for miR-486 (Qiagen). U6 was used as an endogenous control for miR-486 detection. The primers for the PCR analysis were as follows: miR-486: 5′-TCAGTACAGGATGCGTCAGG-3′ (forward), and 5′-AAAACCAGTGACTGCCTGCT-3′ (reverse); TNF-α mRNA: 5′-AAGCCTGTAGCCCATGTTGTA-3′ (forward), and 5′-TCAGCTCCACGCCATTG-3′ (reverse); IL-6 mRNA: 5′-AAAGAGGCACTGGCAGAAAA-3′ (forward), and 5′-AGCTCTGGCTTGTTCCTCAC-3′ (reverse); IL-1β mRNA: 5′-CACCTCTCAAGCAGAGCACAG-3′ (forward), and 5′-GGGTTCCATGGTGAAGTCAAC-3′ (reverse); SIRT1 mRNA: 5′-GCCTCACATGCAAGCTCTAGTGAC-3′ (forward), and 5′-TTCGAGGATCTGTGCCAATCATAA-3′ (reverse); GAPDH: 5′-GCACCGTCAAGGCTGAGAAC-3′ (forward), and 5′-TGGTGAAGACGCCAGTGGA-3′ (reverse); U6: 5′-GCTTCGGCAGCACATATACTAAAAT-3′ (forward), and 5′-CGCTTCACGAATTTGCGTGTCAT-3′ (reverse).Dual-luciferase reporter assay
Bioinformatic analysis for the targets of miR-486 was performed using the software TargetScan 3.1 (http://www.targetscan.org). A SIRT1 3′-UTR wild-type luciferase reporter vector (SIRT1-Wt) containing the putative binding sequence of miR-486 and its mutant-type in seed sequence (SIRT1-Mut) were designed and synthesized by Ribobio. Macrophages were cotransfected with 100 ng of SIRT1-Wt or SIRT1-Mut and 50 nM of miR-NC mimics or miR-486 mimics using Lipofectamine™ 3000 Transfection Reagent. After 48 h, the luciferase activities were analyzed using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA).ELISA assay of TNF-α, IL-6 and IL-1β measurement
The levels of TNF-α, IL-6 and IL-1β were measured with the corresponding commercially available ELISA kits (R&D Systems, Minneapolis, MN, USA) according to the instructions of the manufacturers. To be brief, serum samples or cell lysates were incubated with 100 μM of enzyme-specific substrates at 37 °C for 2 h, followed by incubation with 100 μL of HRP-conjugated detection antibody. The absorbance at 450 nm was determined using a Microplate Reader (Bio-Rad Laboratories)Western blot of SIRT1 detection
Treated macrophages were lysed in lysis buffer (50 mM Tris–HCl, pH = 7.5, 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 0.5% sodium deoxycholate) containing protease inhibitor cocktail (Calbiochem, La Jolla, CA, USA). The concentrations of protein extracts were detected by a BCA Protein Assay Kit (Sigma-Aldrich). The protein extracts were resolved on a 10% SDS polyacrylamide gel and then electronically transferred onto PVDF membranes (Bio-Rad Laboratories). After being blocked in 5% non-fat milk for 1 h, the membranes were probed with anti-SIRT1 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000, Cell Signaling Technology, Danvers, MA, USA) and anti-GAPDH (1:1000, Cell Signaling Technology) at 4 °C overnight, followed by incubation with HRP-conjugated secondary antibody (1:2000, Cell Signaling Technology). The protein bands were visualized using a chemiluminescence ECL assay kit (Life Technologies) and the images were analyzed using the ImageJ software (version 1.33).
:1000, Cell Signaling Technology, Danvers, MA, USA) and anti-GAPDH (1:1000, Cell Signaling Technology) at 4 °C overnight, followed by incubation with HRP-conjugated secondary antibody (1:2000, Cell Signaling Technology). The protein bands were visualized using a chemiluminescence ECL assay kit (Life Technologies) and the images were analyzed using the ImageJ software (version 1.33).
Statistical analysis
All statistical analyses were performed using GraphPad Prism 7.0 software (GraphPad Software Inc., La Jolla, CA, USA) at least three times and were shown as mean ± standard deviation (SD). A Student's t-test and Mann–Whitney U test were used to compare the differences between two groups. Differences between multiple groups were analyzed by one-way ANOVA. Correlations between miR-486 and TNF-α, IL-6, and IL-1β were analyzed by Spearman's test. P < 0.05 was considered statistically significant.Results
MiR-486 was upregulated in serum of sepsis patients and associated with inflammatory response
To investigate the role of miR-486 in sepsis, we firstly quantified the expression of miR-486 in sepsis patients. The qRT-PCR results revealed that miR-486 was significantly upregulated in the serum of sepsis patients compared with healthy controls (Fig. 1A). Meanwhile, we determined the levels of TNF-α, IL-6 and IL-1β in sepsis serum. These data showed that the levels of TNF-α, IL-6 and IL-1β were higher in serum of sepsis patients than those in healthy controls (Fig. 1B–D). Intriguingly, we found that the levels of TNF-α, IL-6 and IL-1β were positively correlated with miR-486 expression in the serum of sepsis patients (Fig. 1E–G). These results suggested that upregulation of miR-486 might play an important role in sepsis.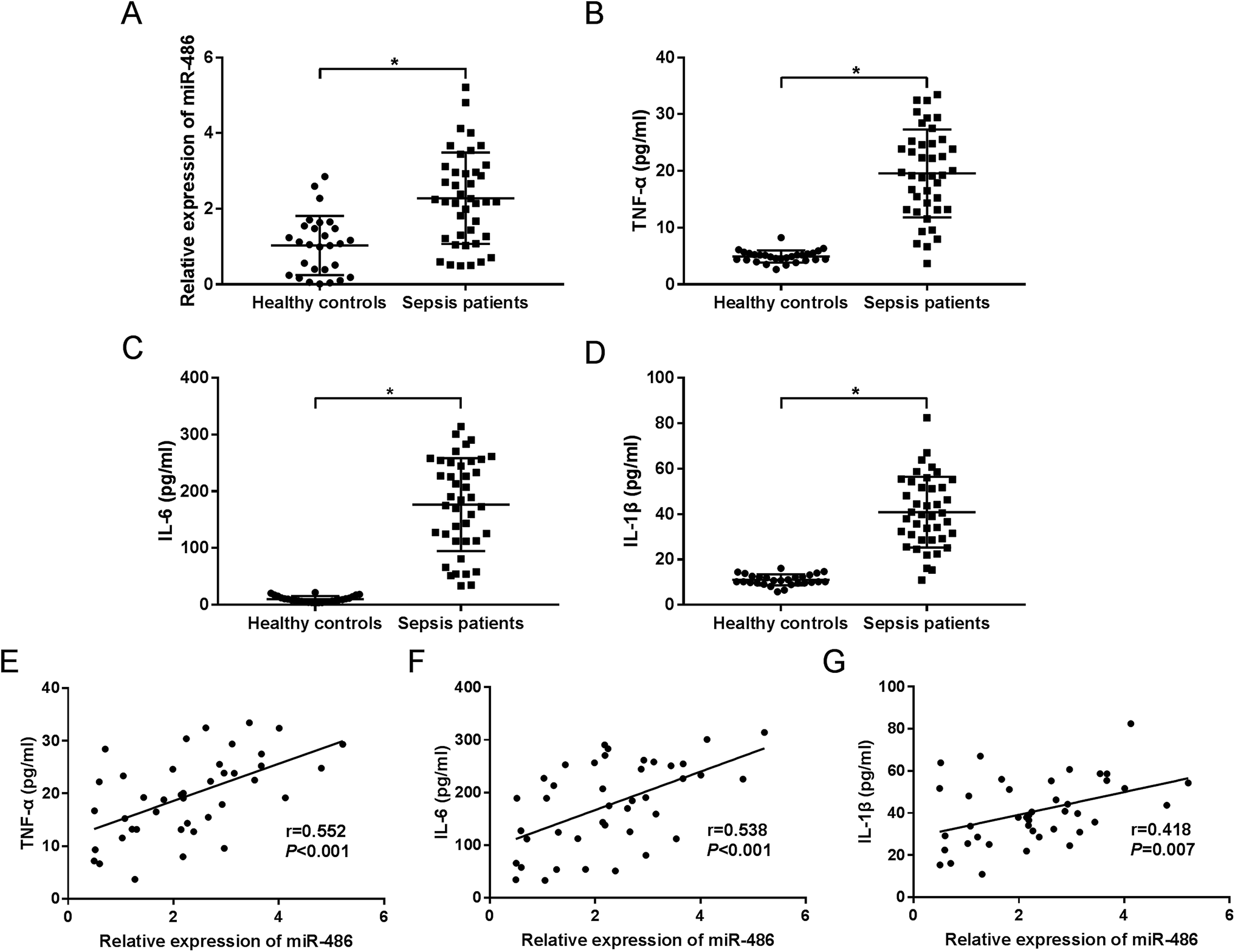 | ||
| Fig. 1 The role of miR-486 was preliminarily determined in serum of sepsis patients. MiR-486 expression was assessed by qRT-PCR assay (A), and TNF-α (B), IL-6 (C) and IL-1β (D) levels were detected by ELISA in the serum of sepsis patients (n = 41) and healthy controls (n = 28). The correlations between TNF-α (E), IL-6 (F), IL-1β (G) levels and miR-486 expression were analyzed in serum of sepsis patients. *P < 0.05 vs. healthy controls. | ||
MiR-486 expression and inflammatory response were elevated in macrophages under LPS stimulation
LPS has been widely acknowledged to trigger SIRS and identified as a crucial mediator of sepsis.18 Hence, to validate whether miR-486 was involved in sepsis, we stimulated macrophages with 1 μg mL−1 of LPS or PBS. These results revealed that LPS stimulation in macrophages, in contrast to PBS, substantially elevated the expression of TNF-α, IL-6 and IL-1β (Fig. 2A–C). Moreover, their mRNA expressions were markedly enhanced by LPS stimulation (Fig. 2D). More importantly, LPS stimulation resulted in a dramatic increase of miR-486 expression in macrophages (Fig. 2E).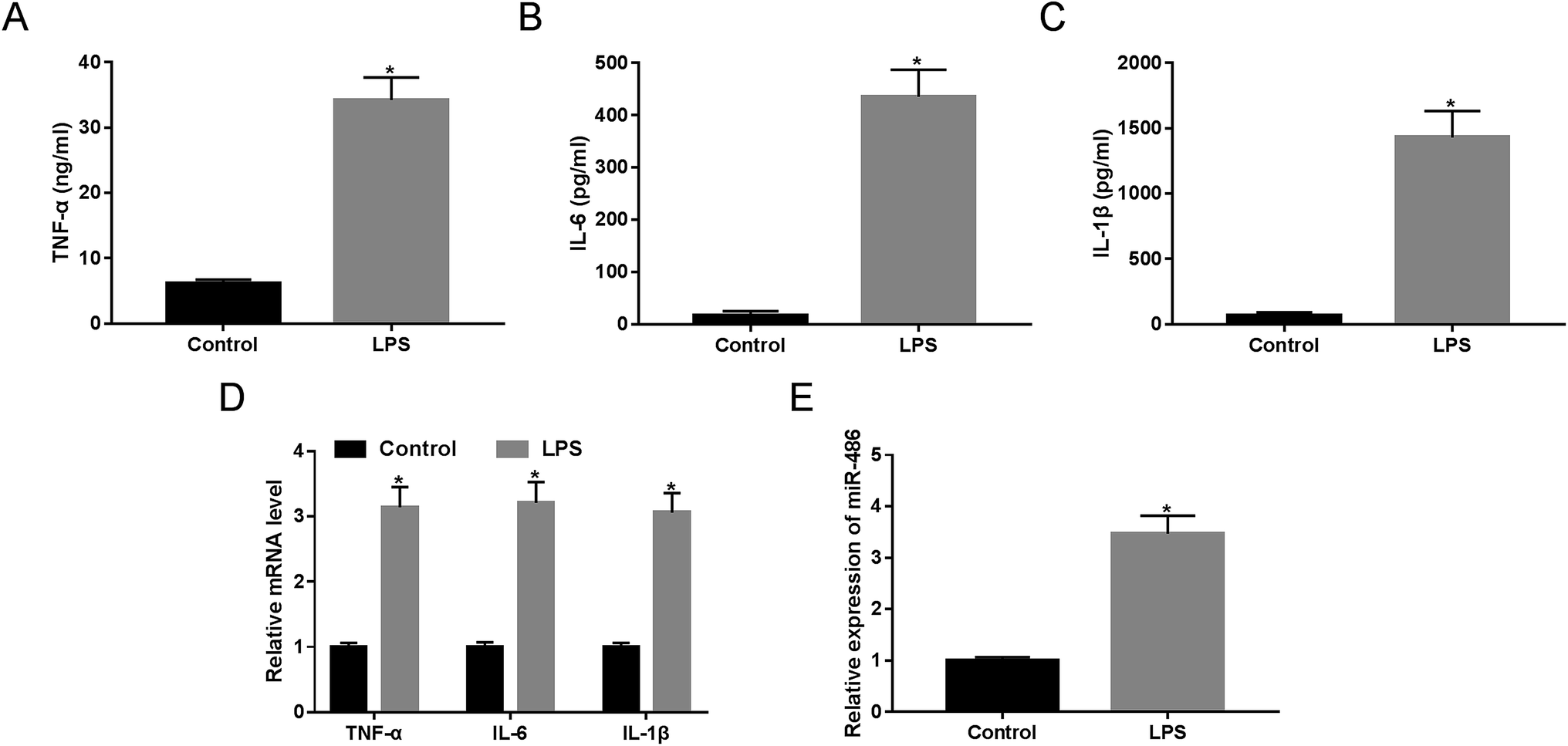 | ||
| Fig. 2 The expression of miR-486 and the inflammatory response were detected in macrophages under LPS stimulation. Macrophages were stimulated with 1 μg mL−1 of LPS or PBS. (A–C) After 24 h stimulation, the TNF-α, IL-6 and IL-1β levels were detected by ELISA. (D) After 6 h stimulation, the TNF-α, IL-6 and IL-1β mRNA levels were assessed by qRT-PCR assay. (E) After 24 h stimulation, miR-486 expression was determined by qRT-PCR assay. *P < 0.05 vs. control. | ||
MiR-486 silencing alleviated inflammatory response in macrophages under LPS stimulation
Then, to further explore the function of miR-486 in the inflammatory response in sepsis, macrophages were transfected with anti-miR-486 or anti-miR-NC prior to LPS stimulation. These results showed that transfection of anti-miR-486, but not anti-miR-NC, significantly reduced the expression of miR-486 in LPS-stimulated macrophages (Fig. 3A). Moreover, miR-486 silencing markedly inhibited the expression of TNF-α, IL-6, IL-1β and their respective mRNA (Fig. 3B–E), indicating that miR-486 silencing repressed the inflammatory response of LPS-stimulated macrophages.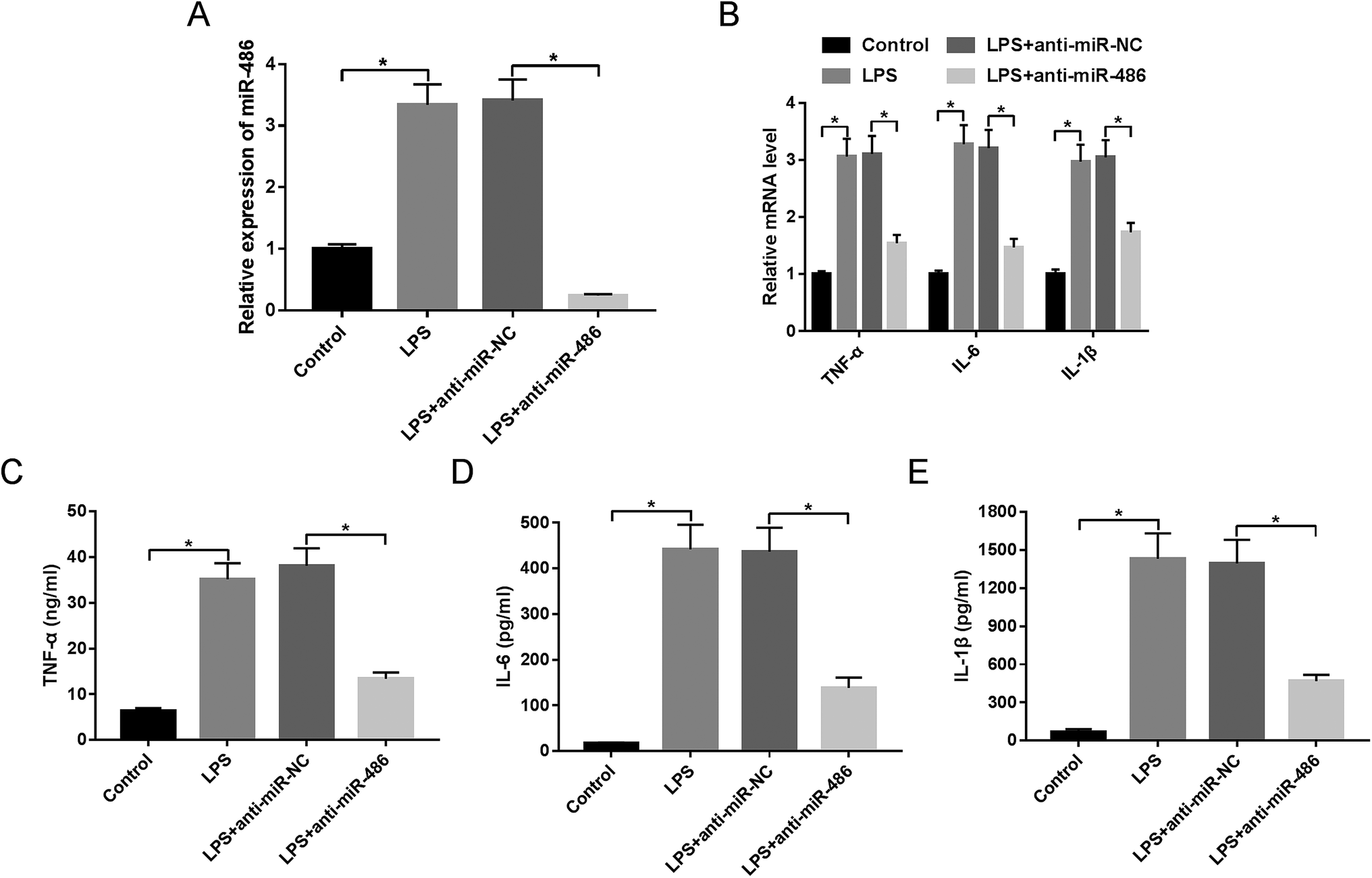 | ||
| Fig. 3 MiR-486 silencing repressed the inflammatory response in LPS-stimulated macrophages. Macrophages were transfected with anti-miR-486 or anti-miR-NC prior to LPS stimulation, followed by the detection of miR-486 expression by qRT-PCR assay (A), the mRNA levels of TNF-α, IL-6 and IL-1β by qRT-PCR assay (B), TNF-α (C), IL-6 (D) and IL-1β (E) levels by ELISA. *P < 0.05 vs. control or LPS + anti-miR-NC. | ||
SIRT1 was a direct target of miR-486
Next, to identify the molecular mechanism by which miR-486 regulated the inflammatory response, we carried out a detailed analysis of its targets. The data predicted by TargetScan software revealed that SIRT1 mRNA contained a potentially complementary sequence for miR-486 in its 3′-UTR (Fig. 4A). To verify whether SIRT1 was a target of miR-486, we performed luciferase reporter assays with SIRT1 3′-UTR wild-type luciferase reporter vector (SIRT1-Wt) containing the putative binding sequence of miR-486 and its mutant-type in seed sequence (SIRT1-Mut). Transfection of the miR-486 mimics, but not the negative control, reduced the luciferase activity of SIRT1-Wt (Fig. 4B). In contrast, SIRT1-Mut abolished the effect of the miR-486 mimics on luciferase activity under the same conditions (Fig. 4B).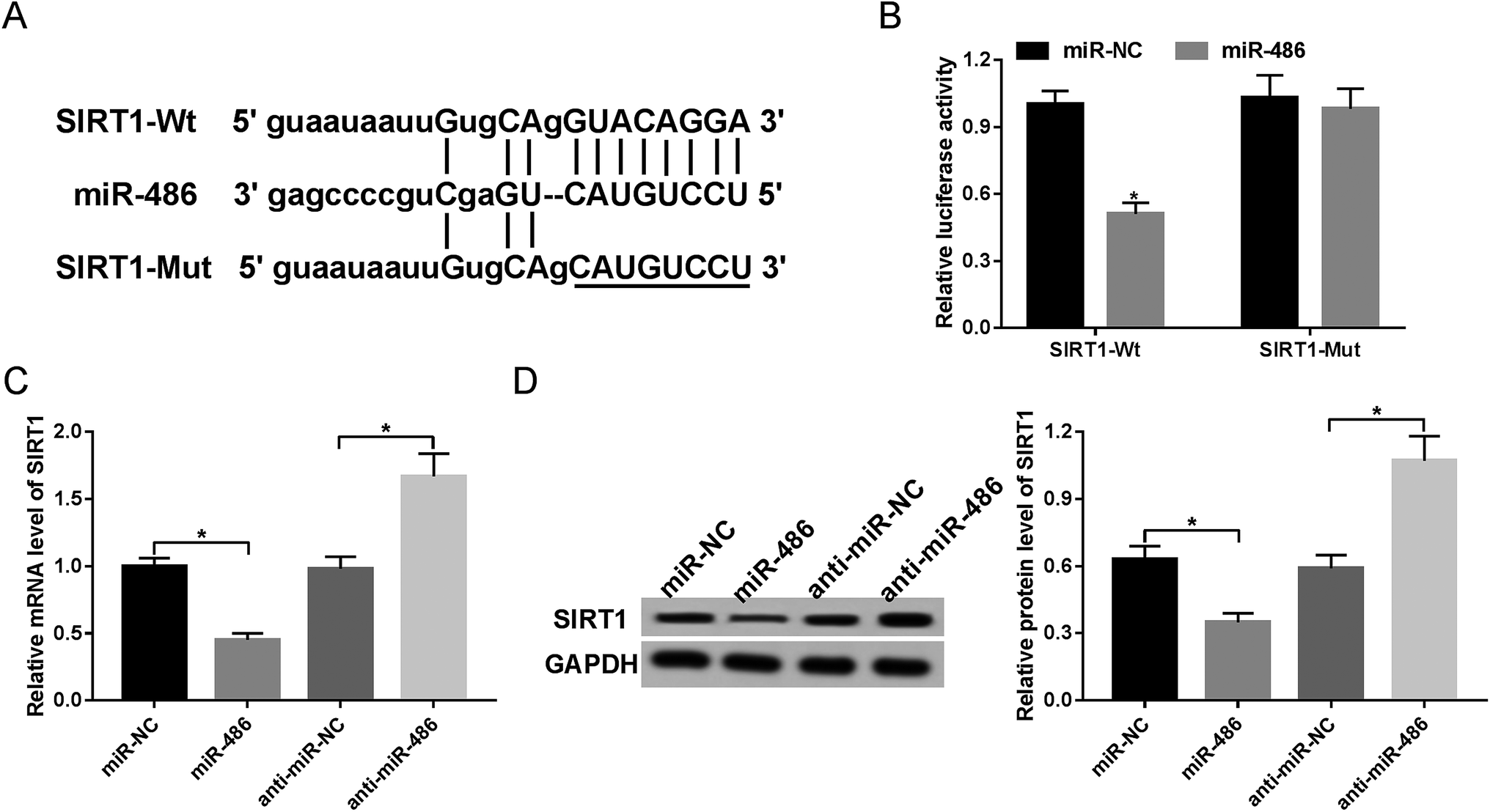 | ||
| Fig. 4 SIRT1 was a direct target of miR-486. (A) Nucleotide resolution of the predicted miR-486 binding site in 3′-UTR of SIRT1 mRNA: seed sequence and the mutated seed sequence. (B) Dual-luciferase reporter assay was performed to verify whether SIRT1 was a target of miR-486 by cotransfecting SIRT1-Wt or SIRT1-Mut into macrophages and miR-486 mimics or miR-NC mimics. Macrophages were transfected with miR-NC mimics, miR-486 mimics, anti-miR-NC or anti-miR-486, followed by the detection of SIRT1 mRNA (C) and protein (D) levels. *P < 0.05 vs. miR-NC mimics or anti-miR-NC. | ||
Then, we investigated whether miR-486 affected SIRT1 expression in macrophages. These results demonstrated that in comparison to their counterparts, the expression levels of SIRT1 mRNA and protein were markedly inhibited by transfection of miR-486 mimics, while they were highly promoted by miR-486 silencing (Fig. 4C and D), indicating that miR-486 negatively regulated SIRT1 expression.
Restoration of SIRT1 expression attenuated inflammatory response in macrophages under LPS stimulation
To observe the expression of SIRT1 in sepsis, macrophages were stimulated with 1 μg mL−1 of LPS or PBS. The data showed that SIRT1 expression was strikingly decreased by LPS stimulation (Fig. 5A and B). Then, we transfected Vector-SIRT1 or Vector (control) into macrophages before LPS stimulation. These results revealed that in contrast to the control, transfection of Vector-SIRT1 significantly abrogated the inhibitory effect of LPS on SIRT1 expression (Fig. 5A and B). Moreover, SIRT1 expression restoration strongly reversed the promotional effect of LPS on the expression of TNF-α, IL-6, IL-1β and their mRNAs (Fig. 5C–F), suggesting that restoration of SIRT1 expression attenuated the inflammatory response in macrophages under LPS stimulation.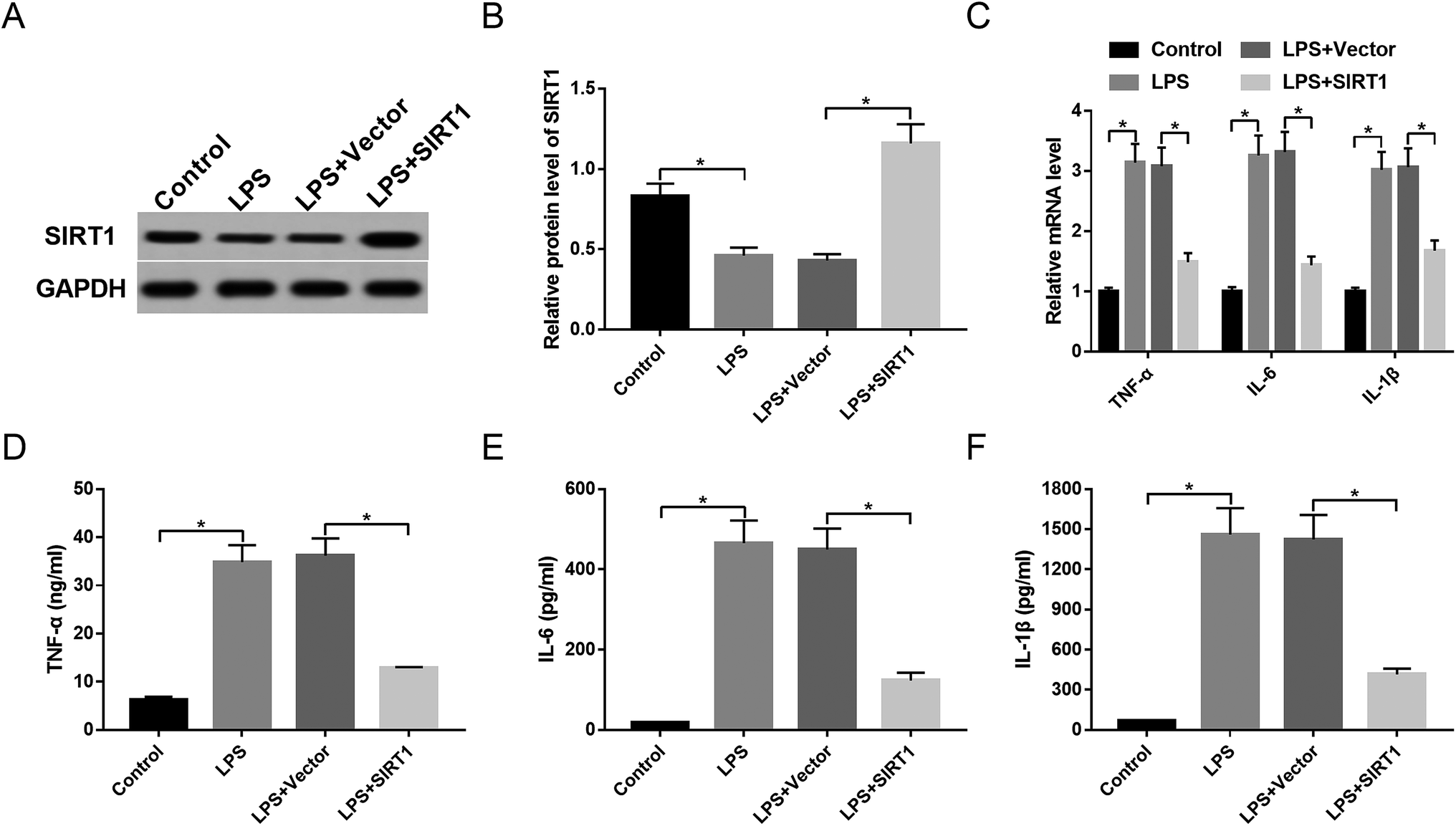 | ||
| Fig. 5 SIRT1 affected inflammatory response in LPS-stimulated macrophages. Macrophages were transfected with Vector or Vector-SIRT1 and then were stimulated with 1 μg mL−1 of LPS or PBS, followed by the determination of SIRT1 protein expression by Western blot (A and B), the mRNA levels of TNF-α, IL-6 and IL-1β by qRT-PCR assay (C), TNF-α (D), IL-6 (E) and IL-1β (F) levels by ELISA. *P < 0.05 vs. control or LPS + vector. | ||
SIRT1 inhibition antagonized the effect of anti-miR-486-mediated anti-inflammatory response in macrophages under LPS stimulation
To provide further mechanistic insight into the link between miR-486 and SIRT1 in the inflammatory response of macrophages in sepsis, macrophages were transfected with anti-miR-486 and then were treated with the SIRT1 inhibitor EX527 prior to LPS stimulation. These data revealed that anti-miR-486-mediated inhibition of TNF-α, IL-6, IL-1β and their respective mRNA expression was significantly abrogated by EX527 treatment in LPS-stimulated macrophages (Fig. 6A–F).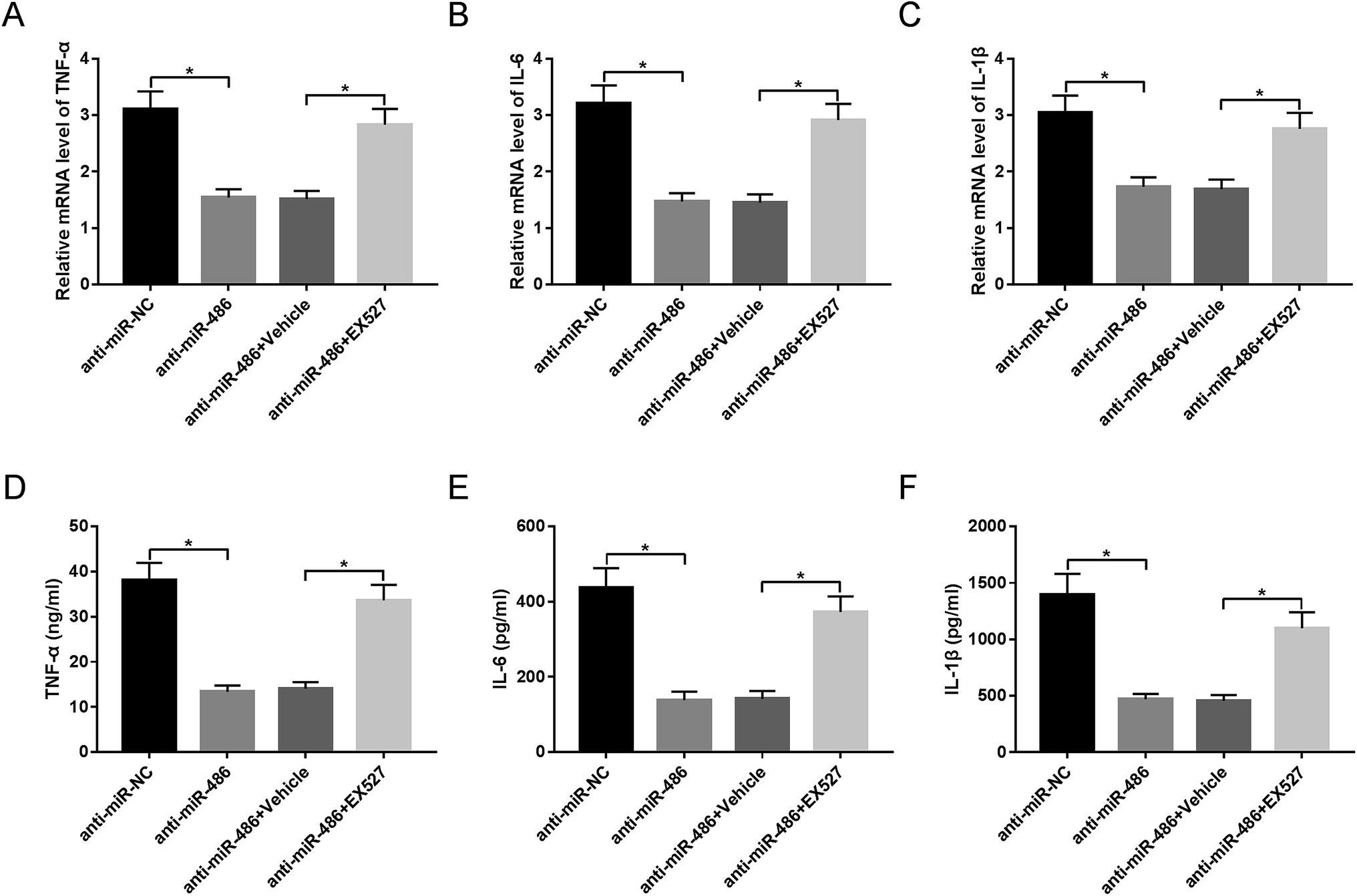 | ||
| Fig. 6 SIRT1 moderated the effect of miR-486 on inflammatory response in LPS-stimulated macrophages. Macrophages were transfected with anti-miR-486 or anti-miR-NC and then were treated with 10 μM of EX527 or vehicle, followed by stimulation with LPS. (A–C) After 6 h stimulation, the TNF-α, IL-6 and IL-1β mRNA levels were detected by qRT-PCR assay. (D–F) After 24 h stimulation, the TNF-α, IL-6 and IL-1β levels were determined by ELISA. *P < 0.05 vs. anti-miR-NC or anti-miR-486 + vehicle. | ||
Discussion
To date, a large number of miRNAs have been identified to have promising diagnostic, therapeutic and prognostic value against sepsis. For instance, some miRNAs were detected to be upregulated in sepsis patients, such as miR-143, miR-150, miR-15a and miR-16.11,19,20 Other miRNAs were demonstrated to be downregulated, including miR-146a, miR-223, miR-122 and miR-499-5p.21,22 Moreover, high miR-27a expression was found to enhance the inflammatory response in sepsis.23 The simultaneous silencing of miR-21 and miR-181b in vivo was found to enhance bacterial clearance and reduce the late sepsis mortality by 74%.24 In the present study, our data revealed a high upregulation of miR-486 expression in serum of sepsis patients, in accordance with previous work.11 Additionally, we found that the levels of TNF-α, IL-6 and IL-1β were positively correlated with miR-486 expression. Moreover, we verified that miR-486 expression was elevated in macrophages under LPS stimulation. In a word, miR-486 might play an important role in the inflammatory response of sepsis.MiR-486 has been identified as a tumor suppressor in a series of human cancers, such as lung cancer,25 gastric cancer26 and hepatocellular carcinoma.27 MiR-486 also was reported to be a potential novel target for the therapy of spinal cord injury.28 Also, miR-486 protected against muscle protein degradation by inhibiting the expression of forkhead transcription factors (FoxOs) and phosphatase and tensin homolog (PTEN) in chronic kidney disease.29 In addition, circulating miR-486 was found to be associated with acute myocardial infarction.30 Moreover, miR-486 directly repressed NF-κB-negative regulators, thus sustaining NF-κB activity.31 The aberrant expression of miR-486 was discovered in sepsis patients, suggesting miR-486 might function as a marker of early sepsis.11 In the present study, we validated for the first time that miR-486 silencing repressed the inflammatory response of LPS-stimulated macrophages.
MiRNAs play crucial roles in many human diseases by negatively regulating gene expression.5 Therefore, we carried out a detailed analysis of the molecular targets of miR-486 using TargetScan software. Among the candidates found in this study, SIRT1 was a particularly attractive target, considering its important role as a negative regulator of inflammatory response.32 Further, we confirmed that SIRT1 was a direct target of miR-486.
SIRT1, the mammalian SIR2 homolog, is a NAD+-dependent class III histone deacetylase and plays a negative regulatory role in inflammation progression by transcriptional inhibition of various inflammation-related genes.32 This effect is attributed to multiple SIRT1 activities, such as deacetylation of NF-κB and chromatin remodeling at the promoters of inflammation-related genes.33 It was previously reported that the inhibition of SIRT1 by the specific inhibitor EX527 facilitated immunity during the hypoinflammatory phenotype of sepsis.34 SIRT1 repressed acute lung inflammatory response in a murine model of sepsis through negatively regulating the inflammasome activation pathway.12 In the present study, we found that restoration of SIRT1 expression attenuated the inflammatory response of LPS-stimulated macrophages. Similar to our findings, Li and colleagues35 demonstrated that SIRT1 had an anti-inflammatory role in an LPS-induced TC-1 cell line and that SIRT1 activation relieved acute lung injury in an LPS-induced sepsis mouse model. Moreover, we demonstrated that the anti-miR-486-mediated anti-inflammatory response in LPS-stimulated macrophages was antagonized by SIRT1 inhibition. Similar to our work, SIRT1 was earlier demonstrated to be a direct functional target of miR-486 in erythroleukemia cells,14 liver cancer stem-like cells15 and human adipose tissue-derived mesenchymal stem cells.16 Additionally, recent studies elucidated that several other miRNAs regulated the inflammatory responses of macrophages through targeting SIRT1, such as miR-138 and miR-199a.36,37
In conclusion, our data indicated that miR-486 expression was elevated in serum of sepsis patients and LPS-stimulated macrophages. Furthermore, miR-486 silencing alleviated the inflammatory response at least partly through targeting SIRT1 in LPS-stimulated macrophages. Targeting miR-486 may provide a novel way to protect against dysregulated inflammatory response in sepsis patients.
Ethical statement
A total of 41 cases of sepsis patients who were admitted to the Central Hospital of Wuhan, and 28 cases of healthy controls, were included in the study, with prior written and informed consent. The study was approved by the Ethics Committee of the Central Hospital of Wuhan.Conflicts of interest
There are no conflicts of interest to declare.References
- K. M. Kaukonen, M. Bailey, D. Pilcher, D. J. Cooper and R. Bellomo, N. Engl. J. Med., 2015, 372, 1629–1638 CrossRef CAS PubMed.
- J. Cohen, Nature, 2002, 420, 885 CrossRef CAS.
- S. Mera, D. Tatulescu, C. Cismaru, C. Bondor, A. Slavcovici, V. Zanc, D. Carstina and M. Oltean, APMIS, 2011, 119, 155–163 CrossRef CAS PubMed.
- C. Fleischmann, A. Scherag, N. K. Adhikari, C. S. Hartog, T. Tsaganos, P. Schlattmann, D. C. Angus and K. Reinhart, Am. J. Respir. Crit. Care Med., 2016, 193, 259–272 CrossRef CAS PubMed.
- Z. Li and T. M. Rana, Nat. Rev. Drug Discov., 2014, 13, 622–638 CrossRef CAS.
- A. S. Sayed, K. Xia, U. Salma, T. Yang and J. Peng, Heart Lung Circ., 2014, 23, 503–510 CrossRef PubMed.
- S. Shrivastava, A. Mukherjee and R. B. Ray, World J. Hepatol., 2013, 5, 479–486 CrossRef PubMed.
- A. Kishore, J. Borucka, J. Petrkova and M. Petrek, Mediat. Inflamm., 2014, 2014, 259131 Search PubMed.
- J. Hayes, P. P. Peruzzi and S. Lawler, Trends Mol. Med., 2014, 20, 460–469 CrossRef CAS PubMed.
- K. Essandoh and G. C. Fan, Biochim. Biophys. Acta, 2014, 1842, 2155–2162 CrossRef CAS PubMed.
- C. Vasilescu, S. Rossi, M. Shimizu, S. Tudor, A. Veronese, M. Ferracin, M. S. Nicoloso, E. Barbarotto, M. Popa, O. Stanciulea, M. H. Fernandez, D. Tulbure, C. E. Bueso-Ramos, M. Negrini and G. A. Calin, PLoS One, 2009, 4, e7405 CrossRef PubMed.
- R. Gao, Z. Ma, Y. Hu, J. Chen, S. Shetty and J. Fu, Am. J. Physiol. Lung Cell Mol. Physiol., 2015, 308, L847–L853 CrossRef CAS PubMed.
- V. T. Vachharajani, T. Liu, C. M. Brown, X. Wang, N. L. Buechler, J. D. Wells, B. K. Yoza and C. E. McCall, J. Leukoc. Biol., 2014, 96, 785–796 CrossRef PubMed.
- X. F. Shi, H. Wang, F. X. Kong, Q. Q. Xu, F. J. Xiao, Y. F. Yang, R. L. Ge and L. S. Wang, Exp. Cell Res., 2017, 351, 74–81 CrossRef CAS PubMed.
- X. Yan, X. Liu, Z. Wang, Q. Cheng, G. Ji, H. Yang, L. Wan, C. Ge and Q. Zeng, Oncol. Rep., 2019, 41, 1938–1948 Search PubMed.
- Y. J. Kim, S. H. Hwang, S. Y. Lee, K. K. Shin, H. H. Cho, Y. C. Bae and J. S. Jung, Stem Cell. Dev., 2012, 21, 1749–1760 CrossRef CAS PubMed.
- M. Daigneault, J. A. Preston, H. M. Marriott, M. K. Whyte and D. H. Dockrell, PLoS One, 2010, 5, e8668 CrossRef PubMed.
- J. Shi, Y. Zhao, Y. Wang, W. Gao, J. Ding, P. Li, L. Hu and F. Shao, Nature, 2014, 514, 187–192 CrossRef CAS PubMed.
- Y. Han, Q. C. Dai, H. L. Shen and X. W. Zhang, J. Int. Med. Res., 2016, 44, 875–881 CrossRef CAS PubMed.
- H. Wang, P. Zhang, W. Chen, D. Feng, Y. Jia and L. X. Xie, Clin. Chem. Lab. Med., 2012, 50, 1423–1428 CAS.
- J. F. Wang, M. L. Yu, G. Yu, J. J. Bian, X. M. Deng, X. J. Wan and K. M. Zhu, Biochem. Biophys. Res. Commun., 2010, 394, 184–188 CrossRef CAS PubMed.
- H. J. Wang, P. J. Zhang, W. J. Chen, D. Feng, Y. H. Jia and L. X. Xie, J. Trauma Acute Care Surg., 2012, 73, 850–854 CrossRef CAS PubMed.
- Z. Wang, Z. Ruan, Y. Mao, W. Dong, Y. Zhang, N. Yin and L. Jiang, Cell. Immunol., 2014, 290, 190–195 CrossRef CAS PubMed.
- C. McClure, L. Brudecki, D. A. Ferguson, Z. Q. Yao, J. P. Moorman, C. E. McCall and M. El Gazzar, Infect. Immun., 2014, 82, 3816–3825 CrossRef PubMed.
- J. Wang, X. Tian, R. Han, X. Zhang, X. Wang, H. Shen, L. Xue, Y. Liu, X. Yan, J. Shen, K. Mannoor, J. Deepak, J. M. Donahue, S. A. Stass and L. Xing, et al., Oncogene, 2014, 33, 1181–1189 CrossRef CAS PubMed.
- H. K. Oh, A. L. Tan, K. Das, C. H. Ooi, N. T. Deng, I. B. Tan, E. Beillard, J. Lee, K. Ramnarayanan, S. Y. Rha, N. Palanisamy, P. M. Voorhoeve and P. Tan, Clin. Cancer Res., 2011, 17, 2657–2667 CrossRef CAS PubMed.
- X. P. Huang, J. Hou, X. Y. Shen, C. Y. Huang, X. H. Zhang, Y. A. Xie and X. L. Luo, FEBS J., 2015, 282, 579–594 CrossRef CAS PubMed.
- M. K. Jee, J. S. Jung, J. I. Choi, J. A. Jang, K. S. Kang, Y. B. Im and S. K. Kang, Brain, 2012, 135, 1237–1252 CrossRef PubMed.
- J. Xu, R. Li, B. Workeneh, Y. Dong, X. Wang and Z. Hu, Kidney Int., 2012, 82, 401–411 CrossRef CAS PubMed.
- R. Zhang, C. Lan, H. Pei, G. Duan, L. Huang and L. Li, BMC Cardiovasc. Disord., 2015, 15, 51 CrossRef PubMed.
- L. Song, C. Lin, H. Gong, C. Wang, L. Liu, J. Wu, S. Tao, B. Hu, S. Y. Cheng, M. Li and J. Li, Cell Res., 2013, 23, 274–289 CrossRef CAS PubMed.
- J. Xie, X. Zhang and L. Zhang, Pharmacol. Res., 2013, 67, 60–67 CrossRef CAS PubMed.
- M. E. Kotas, M. C. Gorecki and M. P. Gillum, Adipocyte, 2013, 2, 113–118 CrossRef.
- V. T. Vachharajani, T. Liu, C. M. Brown, X. Wang, N. L. Buechler, J. D. Wells, B. K. Yoza and C. E. McCall, J. Leukoc. Biol., 2014, 96, 785–796 CrossRef PubMed.
- T. Li, J. Zhang, J. Feng, Q. Li, L. Wu, Q. Ye, J. Sun, Y. Lin, M. Zhang, R. Huang, J. Cheng, Y. Cao, G. Xiang, J. Zhang and Q. Wu, Mol. Med. Rep., 2013, 7, 1889–1895 CrossRef CAS PubMed.
- Y. Liu, H. Guan, J. L. Zhang, Z. Zheng, H. T. Wang, K. Tao, S. C. Han, L. L. Su and D. Hu, Am. J. Physiol. Cell Physiol., 2018, 314, C449–C455 CrossRef CAS PubMed.
- X. Z. Bai, J. L. Zhang, Y. Liu, W. Zhang, X. Q. Li, K. J. Wang, M. Y. Cao, J. N. Zhang, F. Han, J. H. Shi and D. H. Hu, Cell. Physiol. Biochem., 2018, 49, 489–500 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |