DOI:
10.1039/C9RA01408J
(Paper)
RSC Adv., 2019,
9, 20314-20322
Platinum on 2-aminoethanethiol functionalized MIL-101 as a catalyst for alkene hydrosilylation†
Received
24th February 2019
, Accepted 16th April 2019
First published on 28th June 2019
Abstract
Hydrosilylation is one of the largest-scale applications for homogeneous catalysis and is widely used to enable the commercial manufacture of silicon products. In this paper, a bifunctional heterogeneous catalyst, Ptδ+/AET-MIL-101 (AET = 2-aminoethanethiol) with a partially positively charged Ptδ+ electronic structure is reported, which was successfully prepared using post-synthesis modification with AET and a platinum precursor. The catalysts were characterized using X-ray diffraction (XRD), nitrogen (N2) adsorption–desorption, transmission electron microscopy (TEM), scanning electron microscopy (SEM) and X-ray photoelectron spectroscopy (XPS) techniques which showed that the synergy of AET-MIL-101 provides a good dispersion of Ptδ+ in the channels, which can efficiently catalyze the hydrosilylation reaction with almost complete conversion and produce a unique adduct. In addition, the synthetic heterogeneous catalyst Ptδ+/AET-MIL-101 achieves reasonable use of Pt in terms of number cycles and atomic utilization efficiency, indicating the potential to achieve a green hydrosilylation industry.
1. Introduction
1Hydrosilylation of alkene is an important synthetic method for the preparation of organo-silicon compounds in the silicone industrial process.1–6 In particular, silanes containing various functional groups have been extensively used in coatings,7,8 adhesives, vulcanized rubbers, and fine chemicals, and it is one of the most used industrial processes in silicon chemistry.9 Although many transition metal catalysts have been used for a large number of organic synthesis reactions,10,11 there are fewer catalysts used for hydrosilylation reactions. Among which platinum (Pt) catalysts,12 such as hexachloroplatinic acid (Speier catalyst)13 and Karstedt catalyst,14 are the most effective in the hydrosilylation reaction.15,16 Despite widespread application, homogeneous Pt-based catalysts still suffer from considerable problems: (i) the decomposition of catalysts causes undesirable side reactions and colored products, which have significant effects on the quality of the products and limits their applications,17 and (ii) the consumption of Pt not only leads to high catalyst cost but is also a waste of Pt resources. These disadvantages push the development of heterogeneous catalysts, which have been extensively studied because of their ease of separation and their recyclability.18–20 For example, Wawrzyńczak and co-workers succeeded in loading Pt and rhodium (Rh) on a styrene-divinylbenzene copolymer (SDB) for the hydrosilylation of various alkenes. The results show that the Rh is poorly active in the reaction studied, whereas Pt/SDB exhibits excellent catalytic activity. At the same time, by comparing the catalyst carriers: divinylbenzene, activated carbon RX3 extra and carbon black XC72, it showed that the high hydrophobicity and specific surface area of SDB are the focus of the high activity of the catalyst.21 The team further compared the effects of bimetallic Pt–Cu/SDB and Pt/SDB on the hydrosilylation of allyl chloride, 1-octene and 1-butene. The results show that the bimetallic Pt–copper (Cu) catalyst on the polymer support appears to be much less active than the single metal catalyst. Compared to the Pt/carbon (C) catalyst, SDB seems to be a better carrier material.22 In addition, based on the specificity of the structure of silsesquioxane (HSiMe2O)(i-Bu)7Si8O12, Walczak et al. decided to examine the hydrosilylation of a wide range of functional alkenes and alkynes (terminal and internal) using (HSiMe2O)(i-Bu)7Si8O12, catalyzed by Pt supported on SDB. The results show that Pt/SDB is a more effective catalyst than commercial and industrially applied Pt/C catalysts. The heterogeneous character of the Pt/SDB catalyst allows for repeated use up to 20 times and still maintains a high turnover number (TON). Thus, use of the Pt/SDB produced one of the most versatile heterogeneous systems in hydrosilylation reactions.23 Chen et al. synthesized a heterogeneous, partially charged single atom Pt supported on an anatase titanium dioxide (Pt1δ+/TiO2) catalyst using an electrostatic induction in an exchange two-dimensional confinement strategy. As a result of this they discovering that a partially charged single atom Pt can catalyze the hydrosilylation reaction with almost complete conversion and produce an exclusive adduct.24 Hu et al. reported the synthesis of a super-microporous silica-supported Pt catalyst using n-dodecylamine as a surfactant template which was applied for the hydrosilylation of octene with dichloromethylsilane. The catalyst has a conversion of up to 98% for dichloromethylsilane (99% selectivity to the β-adduct). Furthermore, the catalyst can be recycled seven times without significant loss of activity.25 However, currently most heterogeneous catalysis still have the disadvantages of low atomic utilization and leaching of active substances.26 It is still challenging to develop a heterogeneous catalyst with a combination of the advantages of homogeneous and heterogeneous catalysts.
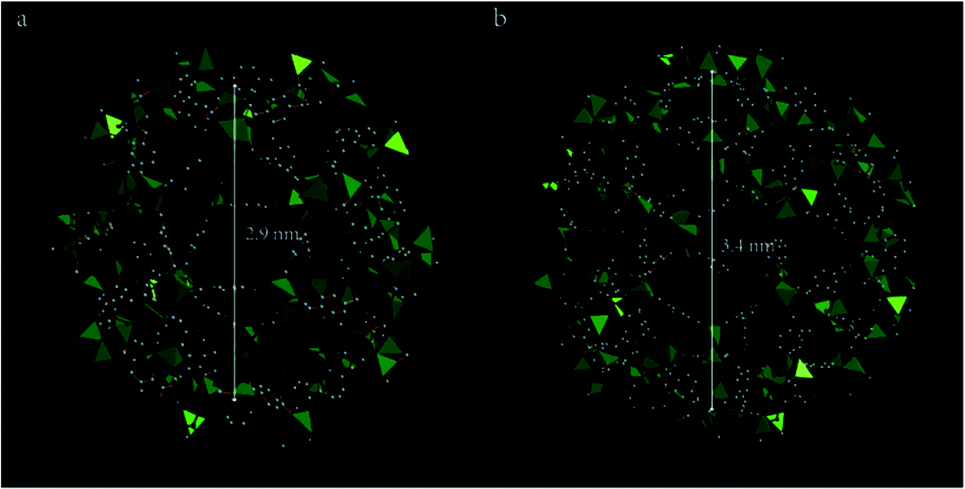
In recent years, the development of metal–organic frameworks (MOFs) has provided a good carrier for gas storage,27 separation,28 chemical sensing29 and catalysis.30 The MOFs are a new type of inorganic–organic porous material coordination polymers.31,32 Because MOFs have a larger specific surface area (up to several thousand m2 g−1) and higher volume (up to 90% of the crystal volume), more controllable pore structure, and so on,33–35 the MOFs have been widely used in the field of catalysis. The MOFs have a controlled size distribution and show the tremendous advantage of limiting metal nanoparticles (NPs). When the metal or metal oxide particles are confined in the cavities or the porous channels of MOFs, the prepared catalytic materials had uniform particle dispersion, which is beneficial for the heterogeneous catalyst.36,37 However, the absence of functional groups and selective sites in most of the stable MOFs has limited their uses in catalysis.38,39 In particular, the MIL-101 type MOF is a one-dimensional porous material formed by an octahedron chrome atom and a terephthalic acid connection (Fig. 1), the interior of which consists of two types of pores [2.9 nm (a) and 3.4 nm (b), respectively]. It has a large surface area, multiple unsaturated metal sites and good thermal stability which provides a good site for functionalization.40–42 Zhang et al.43 reported a highly efficient bifunctional catalyst palladium/sulfonic acid-MIL-101 (chromium) [Pd/SO3H-MIL-101(Cr)], in which Pd NPs are uniformly dispersed on a mesoporous SO3H functionalized MIL-101(Cr) and exhibit high catalytic performance in the hydrodeoxygenation of vanillin. Xu et al.44 successfully encapsulated Pd NPs in cages of ethylenediamine grafted MIL-101 (ED-MIL-101), giving highly efficient catalysts (Pd/ED-MIL-101) for the racemization of primary amines. Furthermore, the catalyst has excellent stability and can be reused at least eight times without significantly reducing efficiency.
 |
| | Fig. 1 Ball-and-stick view of MIL-101 showing two types of cages. | |
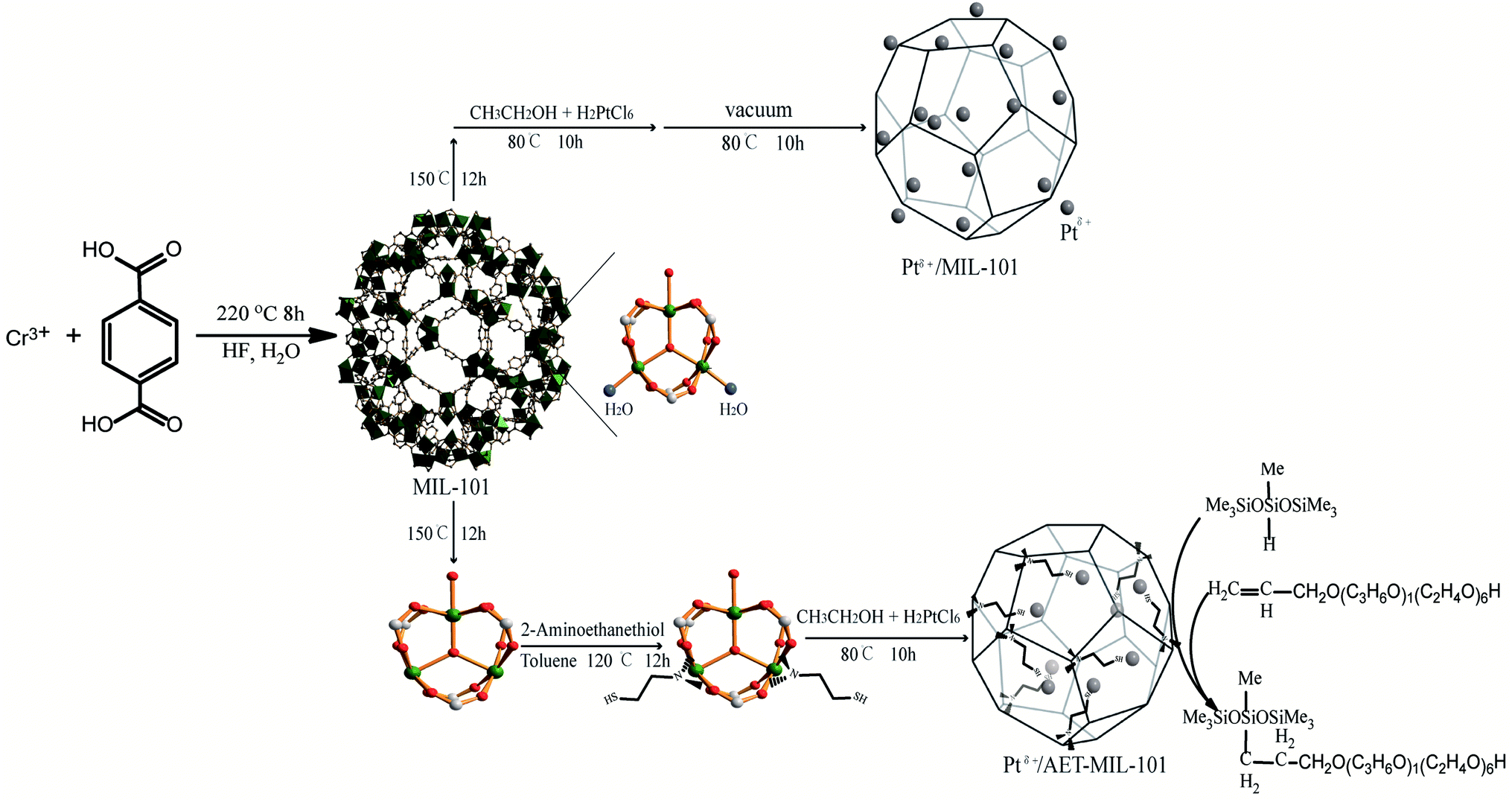
In the present research, 2-aminoethanethiol (AET)-MIL-101 was successfully synthesized using AET functionalized MIL-101. Taking advantage of the unique properties of AET-MIL-101, Ptδ+ was uniformly dispersed in the mesoporous cages of AET-MIL-101 to develop a highly efficient catalyst for the catalytic hydrosilylation reaction (Scheme 1). In addition, this paper also explores the commercial value of the catalytic product, and discusses the development prospects for green industrial catalysis.
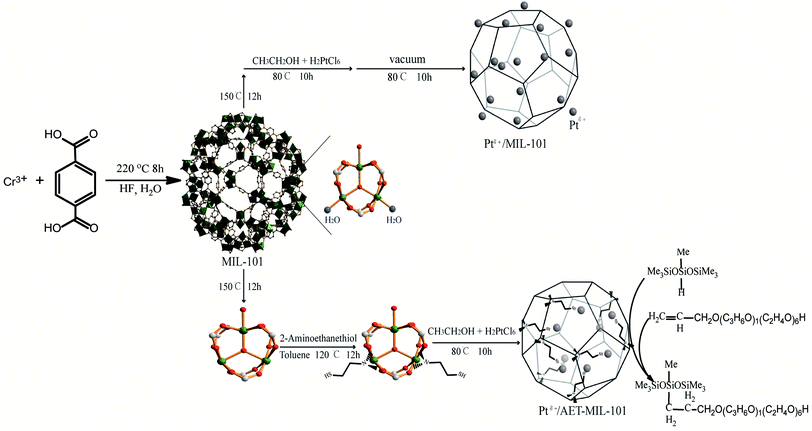 |
| | Scheme 1 The strategy for the synthesis of Ptδ+/MIL-101 and Ptδ+/AET-MIL-101 catalysts using bi-functionalized MIL-101 and their application in the hydrosilylation process. | |
2. Experimental
2.1. Reagents
All reagents of analytical or technical grade were purchased and used as received without further purification. Terephthalic acid (H2BDC), chromic nitrate nonahydrate [Cr(NO3)3·9H2O], styrene, 1-dodecene, 1-tetradecene and AET were purchased from Macklin Ltd. Hexachloroplatinic acid hydrate (H2PtCl6·6H2O) were obtained from Sigma Aldrich Ltd. N,N-Dimethylformamide (DMF), hydrofluoric acid (HF), ethanol, and toluene were purchased from Tianjin Damao Chemical Reagent Factory, Ltd. Heptamethyltrisiloxane (MDHM, MW = 222 g mol−1), acrylic polyether (APEG, MW = 200 g mol−1) and allyl alcohol polyether (F-6, MW = 380 g mol−1) was kindly provided by Zhejiang Huangma Technology Co., Ltd. Deionized water was obtained from a pure water machine produced by Sichuan Youpu Chaochun Technology Co., Ltd.
2.2. Preparation of catalyst
2.2.1 Synthesis of MIL-101. MIL-101 was synthesized according to a method previously described in the literature.36 The Cr(NO3)3·9H2O (10 mmol), H2BDC (10 mmol) and 40 wt% of HF (10 mmol) were added to deionized water (2650 mmol). The resulting suspension was stirred for 30 min at room temperature then heated under autogenous pressure at 220 °C for 8 h in a Teflon-lined autoclave. After cooling to room temperature, the green precipitate was collected using centrifugation and washed several times with deionized water and DMF to remove unreacted H2BDC. The sample was then immersed in ethanol, heated and stirred at 100 °C for 24 h for further purification. Finally, the green solid was separated using filtration, and then dried in a vacuum at 80 °C overnight.
2.2.2 Preparation of AET-MIL-101. The activated MIL-101 (0.3 g) was dehydrated at 150 °C in a vacuum oven for 12 h. Then, the dehydrated MIL-101 was dispersed in anhydrous toluene (30 mL). An appropriate amount of AET (1.125 mmol) was added and the mixture was stirred and refluxed for 12 h at 110 °C. Finally, the solid product was separated using filtration, washed extensively with excess ethanol and dried at 80 °C in a vacuum for 12 h. The final product was named AET-MIL-101.
2.2.3 Preparation of 1.5% Ptδ+/AET-MIL-101. First, 0.3 g of vacuum dried AET-MIL-101 was dispersed in 30 mL of absolute ethanol, and 0.66 mL of H2PtCl6–ethanol (0.035 mol L−1) was added into the suspension. The mixture was mixed ultrasonically for 5 min in an ultrasonic bath (100 W) and then stirred vigorously at 80 °C for 10 h. The solid product was washed with absolute ethanol, and dried at 80 °C in a vacuum. The as-prepared samples were designated as 1.5% Ptδ+/AET-MIL-101, where 1.5% denotes the nominal Pt loading. For comparison, reference 1.0% Ptδ+/AET-MIL-101, 2.0% Ptδ+/AET-MIL-101 and 1.5% Ptδ+/MIL-101 catalysts were prepared using the same method.
2.3. Catalyst characterization
The crystalline structure and phase composition were investigated using X-ray powder diffraction on a D8 ADVANCE (XRD; Bruker) with Cu Kα radiation operated at 35 kV and 35 mA, λ = 0.154 nm, scanning step 0.02°, scanning range 2–15°. Fourier transform-infrared (FT-IR) spectra were recorded on a Spectrum-100 FT-IR spectrometer (PerkinElmer) using a potassium bromide wafer technique with scanning from 4000 to 450 cm−1. The nitrogen (N2) adsorption–desorption isotherms and pore size distribution were obtained at 77 K using a Quadrasorb SI instrument (Quantachrome Instruments). After the sample was evacuated at 150 °C for 10 h, the Brunauer–Emmett–Teller (BET) surface area was calculated using the multiple point BET method in the relative pressure range of P/P0 = 0.05–0.2, and the pore diameter distribution curves were obtained using the density functional theory (DFT) calculations method. The morphology of the material and the presence and distribution of Pt was observed using both a S4800 scanning electron microscope (SEM; Hitachi) and a JEM-2100F transmission electron microscopy (TEM; Jeol). The surface electronic states were determined using an ESCALAB 250Xi X-ray photoelectron spectrometer (XPS; ThermoFisher Scientific), and the XPS data were internally calibrated, fixing the binding energy of C 1s at 284.6 eV. The product conversion of various substrates was determined using a 6890N gas chromatography-mass spectrometer (GC-MS; Agilent), equipped with an HP-5MS capillary column (30 m × 0.25 mm, 0.25 μm). An auto-injector was used to inject the samples at 250 °C and was set to a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 split ratio. The carrier gas flow rate of helium was 1 mL min−1. The warming procedure was as follows: held at 50 °C for 2 min, ramped to 280 °C at a rate of 15 °C min−1 and then held for 10 min. The ionization mode was electron ionisation (70 eV, 300 μA) and the ion source of the MS was set to 230 °C. Proton nuclear magnetic spectrometry (1H-NMR) spectra were recorded on an Ascend 400 (400 MHz, Bruker) NMR spectrometer using deuterated chloroform as solvent. The Pt content of the catalyst was determined using an iCAP 6000 Series inductively coupled plasma emission spectrometer (ICP; ThermoScientific). The water or diluted catalytic product (Vproduct:Vwater = 1:1000) droplet contact angles and surface tension were measured at room temperature on a self-supporting pressed sample disc using a Attention Theta contact angle measuring system (Biolin Scientific). By slowly dripping the liquid on to the surface of cucumber leaves, the contact angle was determined from images captured within 7 s using a high-performance coupled device camera.
:1 split ratio. The carrier gas flow rate of helium was 1 mL min−1. The warming procedure was as follows: held at 50 °C for 2 min, ramped to 280 °C at a rate of 15 °C min−1 and then held for 10 min. The ionization mode was electron ionisation (70 eV, 300 μA) and the ion source of the MS was set to 230 °C. Proton nuclear magnetic spectrometry (1H-NMR) spectra were recorded on an Ascend 400 (400 MHz, Bruker) NMR spectrometer using deuterated chloroform as solvent. The Pt content of the catalyst was determined using an iCAP 6000 Series inductively coupled plasma emission spectrometer (ICP; ThermoScientific). The water or diluted catalytic product (Vproduct:Vwater = 1:1000) droplet contact angles and surface tension were measured at room temperature on a self-supporting pressed sample disc using a Attention Theta contact angle measuring system (Biolin Scientific). By slowly dripping the liquid on to the surface of cucumber leaves, the contact angle was determined from images captured within 7 s using a high-performance coupled device camera.
2.4. Catalytic performance evaluation
Typical hydrosilylation reaction procedures were as follows. A mixture of the catalyst (Ptδ+/AET-MIL-101 or Ptδ+/MIL-101) and varying amounts of F-6 were added into 50 mL three-necked flasks vigorously stirred at 80 °C for 30 min. Then, 0.035 mol of MDHM was added dropwise under stirring and the reaction mixture was heated up to a certain temperatures. After cooling to room temperature, the product was separated using centrifugation (6000 rpm, 20 min, 25 °C) and further characterized using GC-MS and 1H-NMR (see S2 and S3, ESI†). Finally, catalytic recycling was studied using the recovered catalysts.
3. Results and discussion
3.1. Catalyst characterization
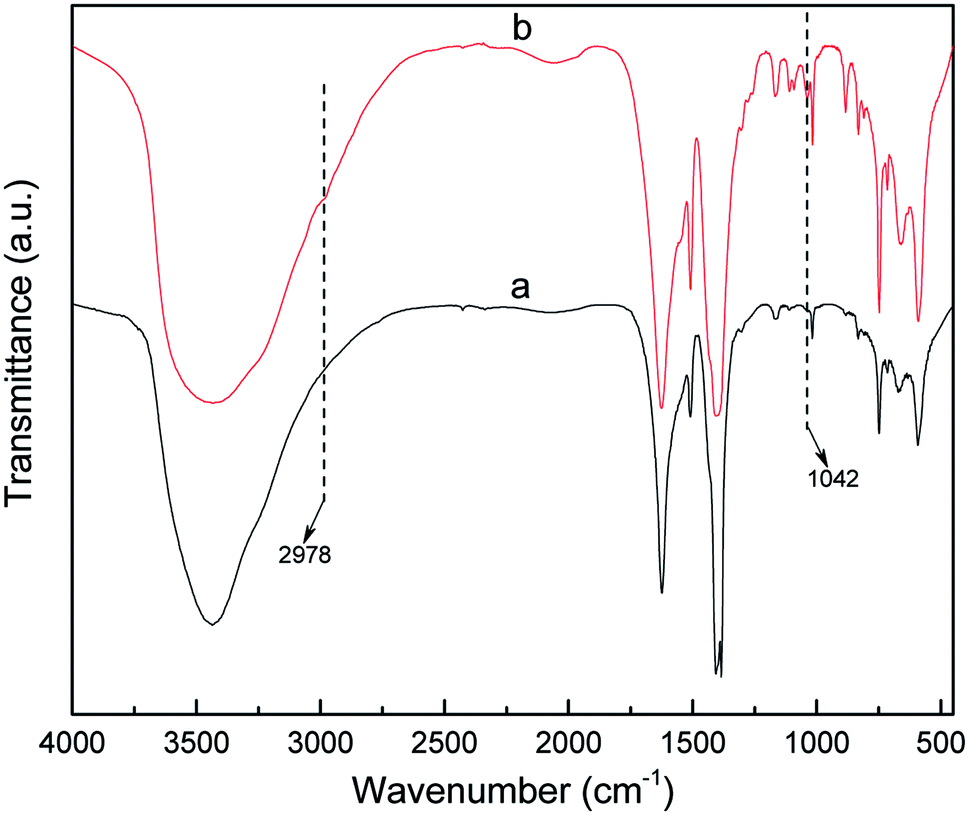
The powder XRD pattern of the synthesized MIL-101 (Fig. 2) matched well with the powder XRD pattern diffraction peak of MIL-101 in the single crystal data simulation pattern, and there was no apparent loss, the main characteristic diffraction peaks appeared at 2θ = 2.96°. 3.44°, 4.46°, 5.28°, 6.01°, 8.55°, and 9.18°, which indicated good phase purity of the synthesized MIL-101 carrier. After the introduction of AET on the backbone of MIL-101 and the incorporation of Pt, no significant change in peak position was observed, indicating that the crystal structure of MIL-101 was well maintained during grafting and doping. Additionally, the peak intensity at 3.44° dramatically decreased, which was because the MIL-101 channels were partially filled with AET and Pt.45 The FT-IR spectra of MIL-101 and AET-MIL-101 are shown in Fig. 3. Comparing the two spectra, it can be observed that two new characteristic peaks appeared in MIL-101 modified with AET. Among them, weak absorption peak appeared at 2978 cm−1, which belong to the –CH2–CH2– stretching vibration, whereas the sharp peak at about 1043 cm−1 indicated the stretching vibration of the C–N bond. However, no characteristic stretching vibration peak of the –NH2 group was observed, possibly because of the combination with Cr3+ being shielded.
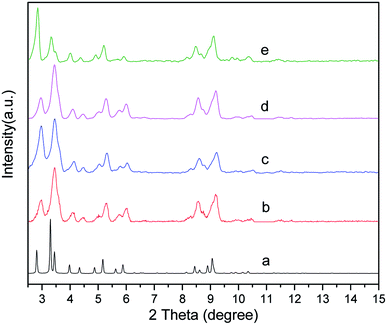 |
| | Fig. 2 XRD patterns of MIL-101 simulated from crystal structure data (a), MIL-101 (b), 1.5% Ptδ+/MIL-101 (c), AET-MIL-101 (d) and 1.5% Ptδ+/AET-MIL-101 (e). | |
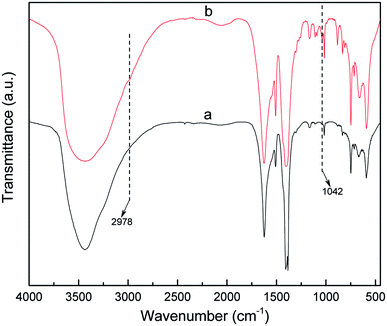 |
| | Fig. 3 FT-IR spectra of MIL-101 (a) and AET-MIL-101 (b). | |
The surface morphology of unmodified and modified MIL-101 samples was characterized using SEM and TEM (Fig. 4). The MIL-101 mainly exhibited uniformly sized octahedron morphology (Fig. 4a) and a good lattice structure (Fig. 4b), indicating that the material had good crystallinity. The MIL-101 modified by AET clearly shows a small amount of aggregate formation (Fig. 4c), which could be because of the formation of aggregates of AET crystals. After Pt was incorporated into the carrier AET-MIL-101, only a few Ptδ+ appear on the outside surface of the parent carrier (Fig. 4d), whereas most of the metal Ptδ+ was confined to the cages of the carrier (Fig. 4e). This demonstrated that the Ptδ+ was well dispersed in the AET-MIL-101 guest molecule, had a narrow size distribution between 1 nm and 4 nm (Fig. 4f), and was concentrated around 2 nm. This may be attributed to the stability of the structure of the crystal itself and the presence of functional groups attached to the framework.
 |
| | Fig. 4 SEM images of MIL-101 (a), TEM images of MIL-101 (b), AET-MIL-101 (c), 1.5% Ptδ+/AET-MIL-101 (e and d) and the corresponding size distribution plot of Ptδ+ (f). | |
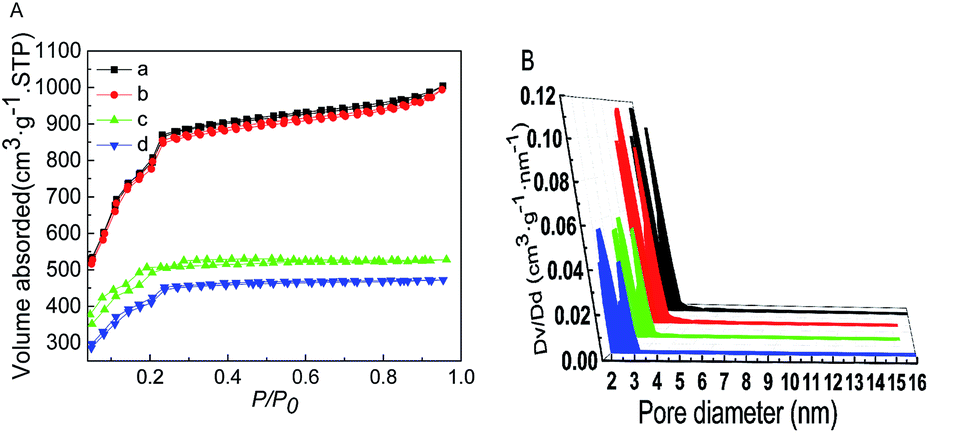
The N2 adsorption–desorption isotherms and the pore size distribution profiles are shown in Fig. 5. The BET surface area and pore volume of MIL-101 loaded with Ptδ+ did not change significantly (Table 1), and this was mainly because Ptδ+ were well dispersed on the support or most of the Ptδ+ was concentrated on the outer surface. After modification with AET, the N2 adsorption of the AET-MIL-101 samples decreased significantly, and the corresponding BET surface area was reduced to 1603 m2 g−1. The pore volume was reduced to 0.756 cm3 g−1, and the average pore diameter also decreased rapidly. However, the pore sizes and pore volume of AET-MIL-101 loaded with Ptδ+ were also reduced compared to those of the AET-MIL-101 samples, which were 1454 m2 g−1 and 0.661 cm3 g−1, respectively, indicating that most of the AET and Ptδ+ were dispersed inside the pores instead of being distributed on the outer surface.
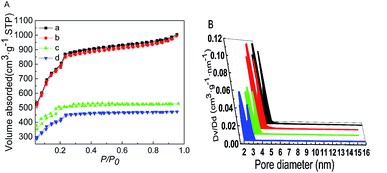 |
| | Fig. 5 The N2 isotherms at −196 °C (A) and DFT pore size distributions (B) of MIL-101 (a), 1.5% Ptδ+/MIL-101 (b), AET-MIL-101 (c) and 1.5% Ptδ+/AET-MIL-101 (d). | |
Table 1 Selected structural properties of the catalysts
| Sample |
SBET (m2 g−1) |
Pore volume (cm3 g−1) |
| MIL-101 |
2877 |
1.39 |
| 1.5% Ptδ+/MIL-101 |
2823 |
1.36 |
| AET-MIL-101 |
1603 |
0.76 |
| 1.5% Ptδ+/AET-MIL-101 |
1454 |
0.66 |
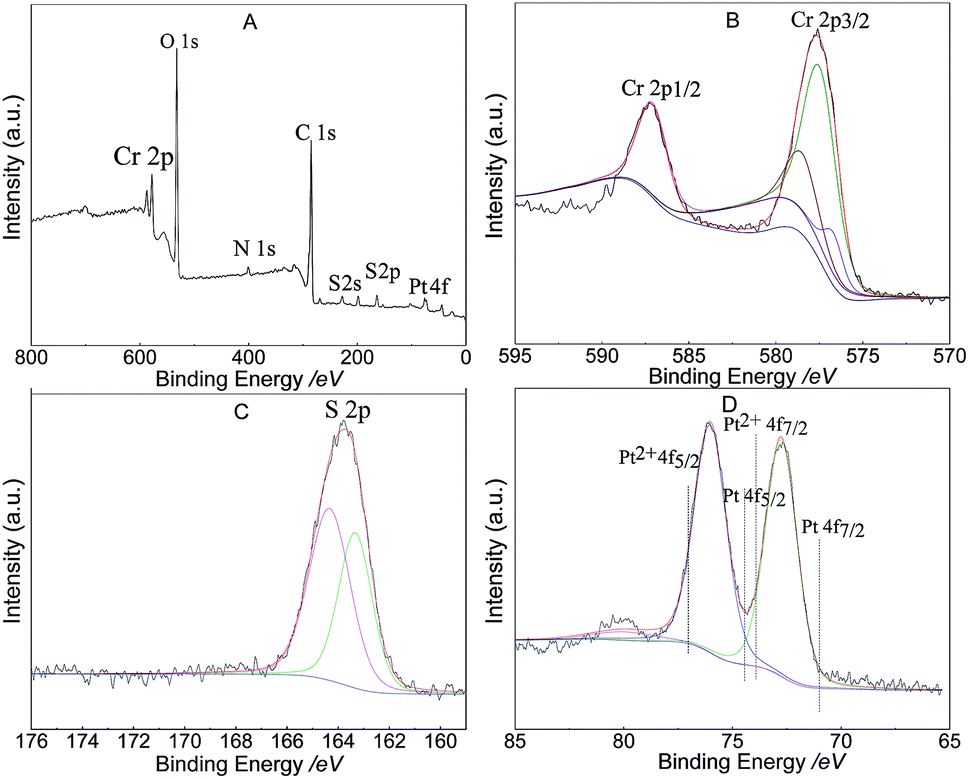
Fig. 6A shows the XPS survey data and indicates that 1.5% Ptδ+/AET-MIL-101 contained six elements C, Cr, N, O, Pt, and, S indicating that AET was successfully modified to MIL-101. The S 2p spectrum of the catalyst showed two different peaks (Fig. 6C), which can be attributed to the S–H (163.7 eV) and Pt–S bonds (164.1 eV). The oxidation state of Pt in Ptδ+/AET-MIL-101 was determined using the data in Fig. 6D. The Pt 4f spectrum could be deconvolved into two peaks at binding energies of 76.0 eV and 72.8 eV, corresponding to the 4f5/2 and 4f7/2 levels, respectively. The peak positions are between Pt(II) and Pt(0), which indicated that the Pt atom carried a portion of the positive charge from the electron transfers between the metal and the support because of the enhanced metal-support interaction.24 In terms of kinetics, because the partially charged Ptδ+ electronic structure of Ptδ+/AET-MIL-101 was produced before the catalytic reaction proceeded, it did not need to undergo additional formation to initiate the hydrosilylation reaction compared to the other forms. Thus, the Ptδ+/AET-MIL-101 catalyst has a higher catalytic performance for the hydrosilylation of alkenes.46
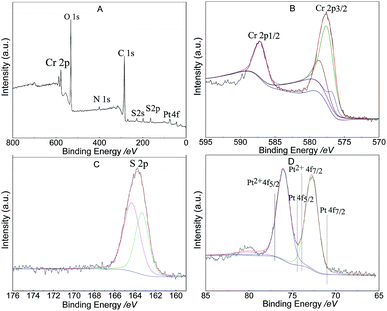 |
| | Fig. 6 XPS spectra of 1.5% Ptδ+/AET-MIL-101: survey spectrum (A), high resolution Cr spectrum (B), high resolution S spectrum (C) and high resolution Pt spectrum (D). | |
3.2. Catalytic hydrosilylation of alkenes
A number of related reports have demonstrated that Pt was an efficient catalyst for the hydrosilylation of alkenes. However, because of its environmentally friendly nature and ecological protection, the above catalyst was applied to catalyze the hydrosilylation of F-6 as a representative substrate for MDHM. There was no need to add other organic solvents or additives, and the product is environmentally friendly and non-toxic, with excellent wettability, ductility and permeability. Therefore, it has a certain use in industrial production.15
The effects of different loadings of Ptδ+/AET-MIL-101 and separate factors on the catalytic activity of F-6 and MDHM hydrosilylation reactions were investigated. The catalytic results are summarized in Table 2. Under the same catalytic conditions, the catalytic effect of the 1.5% Ptδ+/AET-MIL-101 catalyst was better than that of the 1% and 2% catalysts (Table 2, entries 6, 13, 14). However, using the pure AET-MIL-101 support or MIL-101, virtually no reaction took place (only about 3% conversion), implying that the use of Pt is inevitable for catalyzing alkene hydrosilylation (Table 2, entries 1 and 2).
Table 2 The effect of various factors on the hydrosilylation conversion of F-6
| Entry |
Catalyst |
Catalyst amount (mmol%) |
T (°C) |
F-6 (mol) |
Conv. (%) |
| Reaction conditions: MDHM = 0.035 mol, reaction time: 180 min, feeding method: MDHM is added dropwise to alkenes. Reaction conditions: MDHM = 0.035 mol, reaction time: 30 min, feeding method: MDHM is added dropwise to alkenes. |
| 1a |
MIL-101 |
— |
90 |
0.035 |
3.3 |
| 2a |
AET-MIL-101 |
— |
90 |
0.035 |
3.1 |
| 3a |
1.5% Ptδ+/AET-MIL-101 |
2.86 |
90 |
0.035 |
60 |
| 4a |
1.5% Ptδ+/AET-MIL-101 |
4.29 |
90 |
0.035 |
75 |
| 5a |
1.5% Ptδ+/AET-MIL-101 |
5.71 |
90 |
0.035 |
87 |
| 6a |
1.5% Ptδ+/AET-MIL-101 |
8.57 |
90 |
0.035 |
90 |
| 7a |
1.5% Ptδ+/AET-MIL-101 |
10.00 |
90 |
0.035 |
91 |
| 8a |
1.5% Ptδ+/AET-MIL-101 |
8.57 |
80 |
0.035 |
46 |
| 9a |
1.5% Ptδ+/AET-MIL-101 |
8.57 |
100 |
0.035 |
91 |
| 10a |
1.5% Ptδ+/AET-MIL-101 |
8.57 |
110 |
0.035 |
90 |
| 11a |
1.5% Ptδ+/AET-MIL-101 |
8.57 |
90 |
0.0315 |
86 |
| 12a |
1.5% Ptδ+/AET-MIL-101 |
8.57 |
90 |
0.0385 |
90 |
| 13a |
1.0% Ptδ+/AET-MIL-101 |
8.57 |
90 |
0.035 |
76 |
| 14a |
2.0% Ptδ+/AET-MIL-101 |
8.57 |
90 |
0.035 |
89 |
| 15b |
1.5% Ptδ+/MIL-101 |
8.57 |
90 |
0.035 |
93 |
The hydrosilylation reaction was carried out at 90 °C with different low doses (relative to the amount of Pt in MDHM) of Ptδ+/AET-MIL-101 (1.5% Pt loading) catalyst. It was found that as the dose increased, the conversion rate of the substrate also showed an upward trend, and the dosages of 8.57 mmol% and 10.00 mmol% showed better conversion (Table 2, entries 3–7). This was because the unit catalyst and the unit substrate used for the catalytic reaction at a dosage of 8.57 mmol% forms a saturated state. Thus, further research was conducted with a catalyst amount of 8.57 mmol%. The reaction temperature acts as an important factor affecting the activity of the catalyst. The reaction at 80 °C for 180 min only had a conversion of 46%, and further increasing the reaction temperature led to a remarkable increase in the reaction rate (Table 2, entries 6, 8–10), which could reach 90% when the temperature was raised to 90 °C.
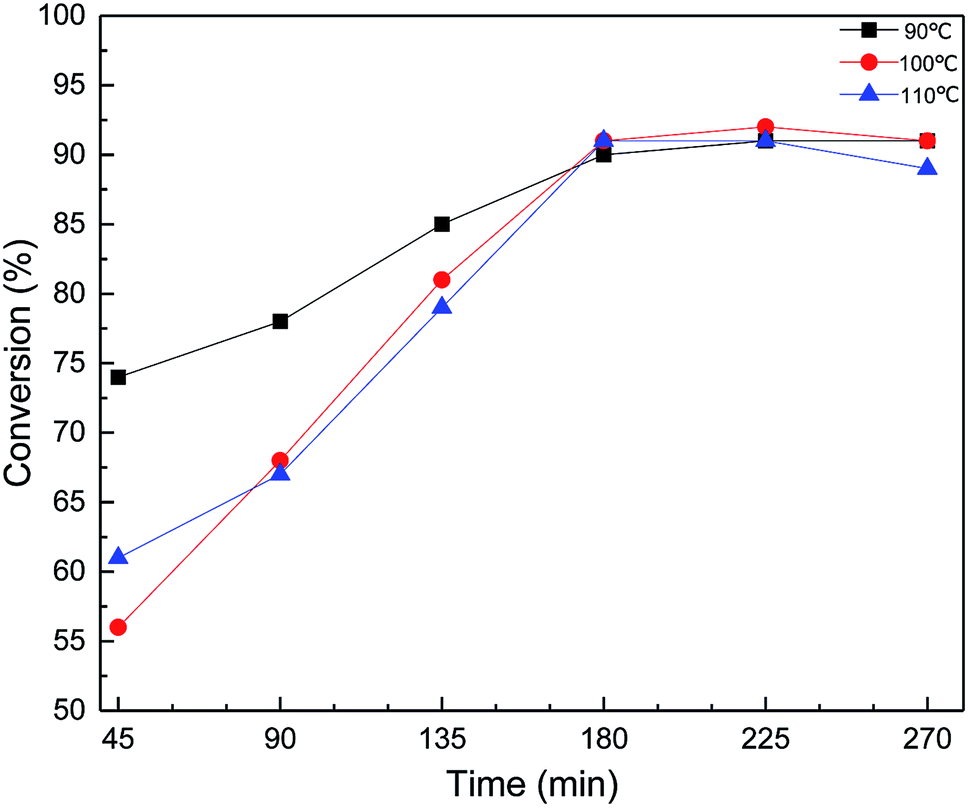
The conversion of MDMA was further investigated by sampling every 45 min at each temperature (Fig. 7). The reaction was accompanied by a time increase, each curve showed a tendency to rise first and then stabilize. As shown in Fig. 7, the MDHM conversion at 90 °C was significantly higher than 100 °C and 110 °C in a relatively short period of time. This may be because the temperature was too high causing some of the MDHM to be vaporized and reflux re-reacted. This is due to the excessive temperature causing a portion of the MDHM to be refluxed into the flask to participate in the reaction again. As the reaction progressed, most of the MDHM participates in the reaction, and the reflux rate gradually decreases. There was almost no reflux of MDHM after 180 min. Then the conversion of the reaction was compared by adding different molar ratios of the substrates. Different molar ratios of the substrates show good catalytic efficiency (Table 2, entries 6, 11 and 12). According to the mechanism of the hydrosilylation reaction, the optimum conversion could be achieved when the molar ratio was increased to 1:1 (90% conversion).
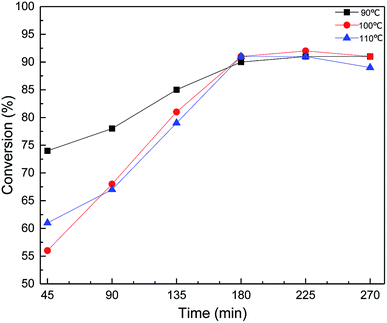 |
| | Fig. 7 Effect of Ptδ+/AET-MIL-101 on MDHM conversion at different temperatures. Reaction conditions: MDHM: 0.035 mol, F-6: 0.035 mol, amount of catalyst: 39 mg, feeding method: MDHM is added dropwise to the alkenes. | |
Finally, the catalytic activities of the different catalysts Ptδ+/MIL-101 were compared (Table 2, entries 6 and 15),47 although the Ptδ+/MIL-101 catalyst has better catalytic activity (conversion: 93%). However, in the experiment, it was found that its catalytic rate was too fast during the reaction, because of a sharp rise in temperature, and difficulty in controlling the reaction rate. This was a reaction phenomenon similar to homogeneous catalysis, which is unfavorable for the protection of industrial equipment.
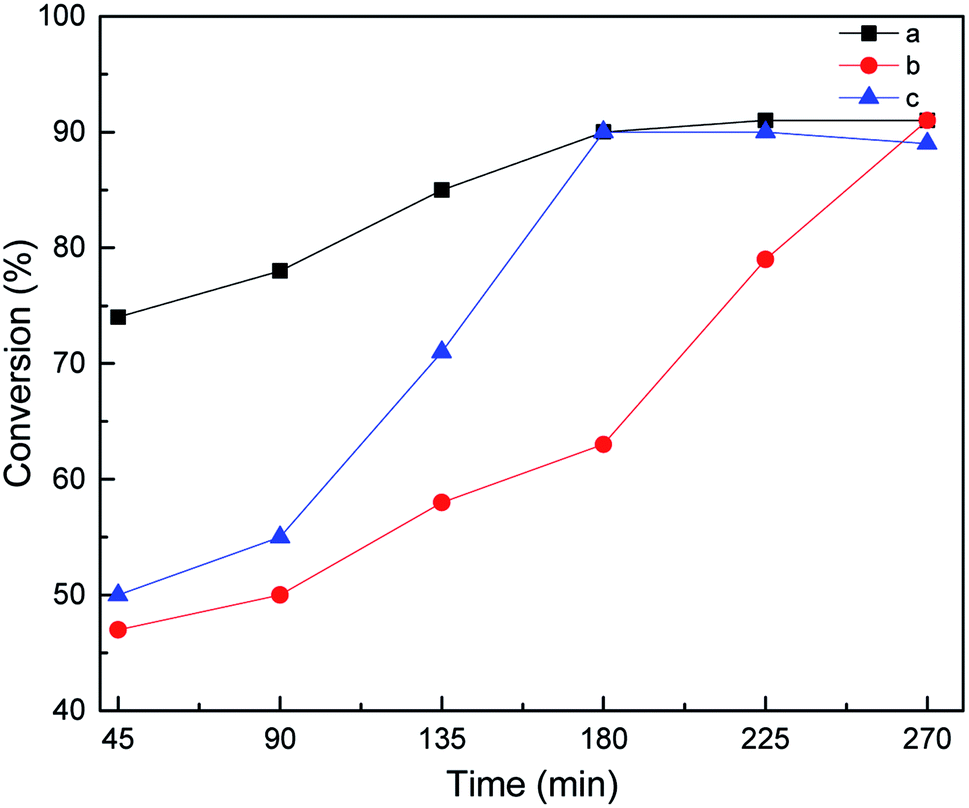
In general, according to the Chalk–Harrod theory,48 hydrosilylation can proceed by the following four steps: (i) activation by Pt, (ii) hydrosilane addition, (iii) alkenes are inserted into the Pt–H bond, and (iv) product shedding. The activation priority of hydrosilylation was studied by changing the addition order of the substrates. The results show that although the three substrate addition methods can achieve excellent conversion, the conversion rate of different addition methods follows this trend: a > c > b. Thus, it is believed that the activation before the reaction is beneficial to the smooth progress of step (i). It can increase the reaction rate and reduce the reaction time. According to the a and c curves in Fig. 8, it can be seen that both reactants can be activated by Ptδ+, but under the same reaction conditions, the alkene will participate in step (i) earlier than MDHM.
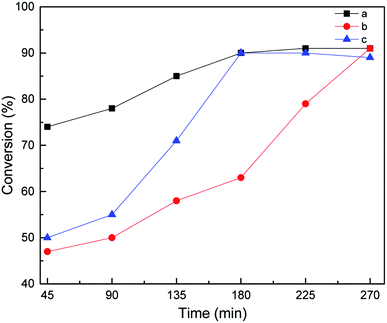 |
| | Fig. 8 Effect of feeding mode on MDHM conversion, feeding method: MDHM is added dropwise to alkenes (a), mixed alkenes are added together with MDHM (b), alkenes are added dropwise to MDHM (c). | |
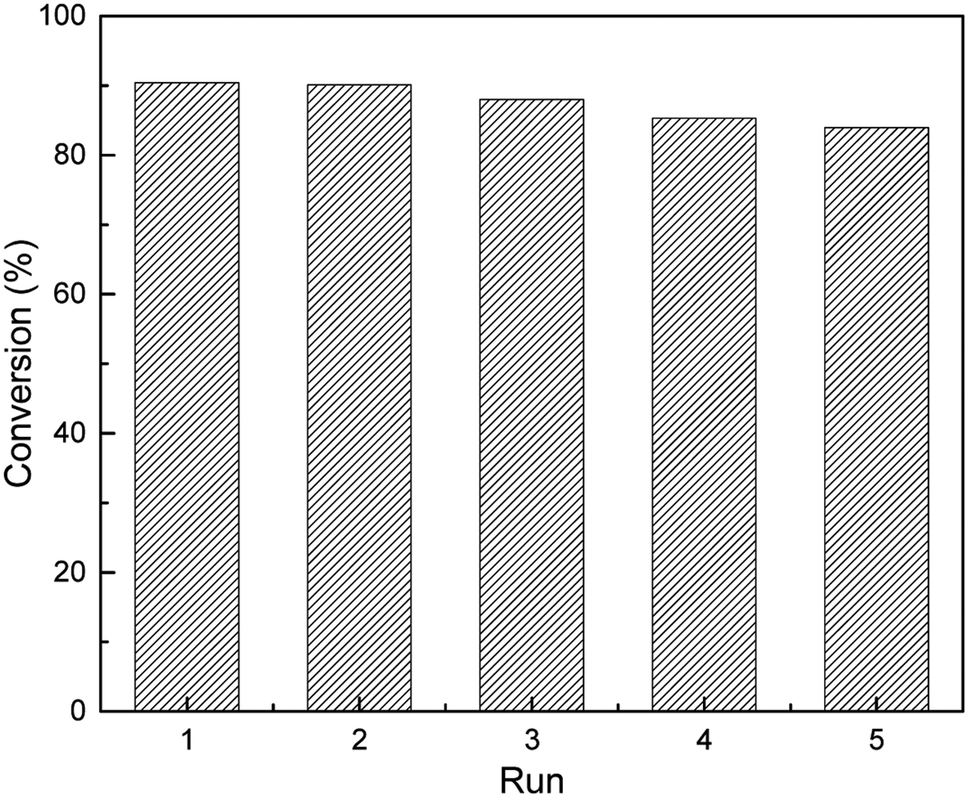
The reusability and stability of the heterogeneous catalysts was of significant importance in terms of industrial applications and green chemistry. To address the concerns, the catalyst can easily be separated from the reaction solution by a simple centrifugal separation operation, followed by washing it several times with ethanol and then using it for cyclic experiments after vacuum drying. In a series of consecutive runs, the catalyst was found to be highly stable and catalytic activity could still be maintained at about 84% after repeated use for five cycles as shown in Fig. 9. In addition, the recovered catalyst was characterized using XRD, ICP and XPS analysis. The XRD analysis of the recovered catalyst (S1(b), ESI†) showed that its crystallinity and the crystallinity of the fresh catalyst remained substantially unchanged, and the loading of Pt was reduced to 1.18% from the original 1.47%. The XPS of the recovered catalyst showed that the catalyst retained the Ptδ+ morphology (S2, ESI†) after several cycles. Finally, in order to explore the versatility of this catalyst, the hydrosilylation reactions were carried out under the previously mentioned optimal conditions using various alkenes (Table 3). Although the conversion changed as the molecular chain grew, the catalytic effect was satisfactory.
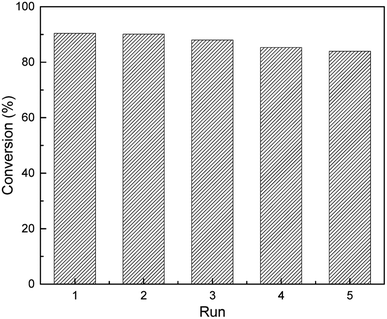 |
| | Fig. 9 Reusability of 1.5% Ptδ+/AET-MIL-101 in the catalytic hydrosilylation of F-6. Reaction conditions: MDHM: 0.035 mol, F-6: 0.035 mol, amount of catalyst: 39 mg, reaction temperature: 90 °C, reaction time: 180 min, feeding method: MDHM is added dropwise to alkenes. | |
Table 3 Hydrosilylation of various alkenes and MDHM using the Ptδ+/AET-MIL-101 catalysta
| Entry |
Substrate |
Conv. (%) |
TOF (h−1) |
| Reaction conditions: amount of catalyst: 39 mg (Pt 8.57 mmol%), MDHM: 0.035 mol, reaction time: 180 min, temperature: 90 °C, MDHM:alkene molar ratio: 1:1, feeding method: MDHM is added dropwise to alkene. Data in parentheses represent the molecular weight of the alkene. |
| 1 |
Ally alcohol polyether (380) |
90 |
8.3 × 103 |
| 2 |
Acrylic polyethers (200) |
90 |
7.6 × 103 |
| 3 |
1-Dodecene |
99 |
1.1 × 104 |
| 4 |
1-Tetradecene |
95 |
1.0 × 104 |
| 5 |
Styrene |
89 |
5.0 × 103 |
3.3. Performance test of hydrosilylation products
The contact angles of water and the catalytic products on cucumber leaves are shown in Fig. 10. Because the cucumber itself is highly hydrophobic, the angles observed remain unchanged after adding water droplets to the cucumber leaves for 7 s (Fig. 10a–c). However, by adding a small amount of the catalytic product, the contact area of water on the cucumber leaf surface can be greatly increased, and only 0.58 s was required to completely spread the water on the leaf surface (Fig. 10d–f). This was because of the surface properties of the catalytic product are imparted by the crosslinking of the functional silanes with the polymer, and thus, good wettability was obtained. Therefore, the product shows good prospects for future applications in agriculture and fine chemicals.
 |
| | Fig. 10 Contact angles of water droplets on cucumber leaves (a), stationary 1 s (b), stationary 7 s (c), adding a small amount of the catalytic products to water (d), stationary 0.07 s (e) and stationary 0.58 s (f). | |
At the same time, the same treatment method was used to test the surface tension of the product. The test results are shown in Table 4. The hydrosilylation product synthesized from Ptδ+/AET-MIL-101 had an average surface tension of 19.32 mN m−1 and the surface tension of water was 67.77 mN m−1, indicating that the product synthesized by the catalyst could significantly reduce the surface tension of water.
Table 4 Comparison of the surface tension of various products in water
| Substance |
Surface tension (mN m−1) |
| No added catalytic product |
68.05 |
67.58 |
68.68 |
| Catalytic product added |
19.26 |
19.33 |
19.37 |
| Homogeneous catalytic product added |
19.29 |
19.34 |
19.33 |
4. Conclusion
In summary, a new and robust heterogeneous catalyst was developed by encapsulating Pt into MIL-101 cages modified with AET. Pt as highly catalytically active sites for AET-MIL-101 for the alkenes hydrosilylation under mild reaction conditions and exhibits excellent reusability. The unexpected property of Ptδ+/AET-MIL-101 can be attributed to unique partially positive valence electronic structure and the highly dispersible properties of the Pt species. The stable performance of Ptδ+/AET-MIL-101 achieves a reasonable use of the catalysts by reducing consumption of the precious platinum by increased recycling and greatly increases the atomic utilization efficiency in the form of highly dispersed Ptδ+. Meanwhile, the findings highlight the fact that the addition of modifiers on the surface of the cages in MIL-101 give the material better stability and controllability of the catalytic rate compared to the unmodified MIL-101. In addition, it was shown through this research that the catalytic products have excellent interfacial properties, which will help to achieve the green hydrosilylation for agricultural and industrial applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The project was supported by the National Natural Science Foundation of China (No. 21476272), Provincial Department of Agriculture Project of Guangdong (KB1814324, KB1814325), Science and Technology Planning Project of Guangdong Province of China (2017A050506055) and the Graduate Education Innovation Program (KA180582712).
References
- J. S. Rhee, R. M. Sneeringer and A. S. Penzias, Coord. Chem. Rev., 2011, 255, 1440–1459 CrossRef.
- H. Wu, C. Zheng, N. Chen, Z. Jie and D. Gao, Tetrahedron Lett., 2017, 58, 1576–1578 CrossRef CAS.
- B. Marciniec, Hydrosilylation: a comprehensive review on recent advances, Springer Science & Business Media, 2008 Search PubMed.
- R. Hofmann, M. Vlatković and F. Wiesbrock, Polymers, 2017, 9, 534 CrossRef PubMed.
- X. Du and Z. Huang, ACS Catal., 2017, 7, 1227–1243 CrossRef CAS.
- M. Oestreich, Angew. Chem., Int. Ed., 2016, 55, 494–499 CrossRef CAS PubMed.
- Y. Nakajima and S. Shimada, RSC Adv., 2015, 5, 20603–20616 RSC.
- T. K. Mukhopadhyay, F. Marco, T. L. Groy and R. J. Trovitch, J. Am. Chem. Soc., 2014, 136, 882–885 CrossRef CAS PubMed.
- Z. Yan, G. Uo Caihong and W. Haishun, Prog. Chem., 2014, 26, 345–357 Search PubMed.
- F. Hoffmann, J. Wagler, U. Böhme and G. Roewer, J. Organomet. Chem., 2012, 705, 59–69 CrossRef CAS.
- J. Sun and L. Deng, ACS Catal., 2015, 6, 290–300 CrossRef.
- H. Zai, Y. Zhao, S. Chen, L. Ge, C. Chen, Q. Chen and Y. Li, Nano Res., 2018, 1–9 Search PubMed.
- J. L. Speier, J. A. Webster and G. H. Barnes, J. Am. Chem. Soc., 1957, 79, 974–979 CrossRef CAS.
- L. N. Lewis, J. Stein, Y. Gao, R. E. Colborn and G. Hutchins, Platin. Met. Rev., 1997, 41, 66–75 CAS.
- H. Xie, H. Yue, W. Zhang, W. Hu, X. Zhou, P. Prinsen and R. Luque, Catal. Commun., 2018, 104, 118–122 CrossRef CAS.
- A. K. Roy, Adv. Organomet. Chem., 2007, 55, 1–59 CrossRef.
- S. R. Arepalli, B. Shewit and B. S Lori, J. Long Term Eff. Med. Implant., 2002, 12, 299 CrossRef CAS.
- F. Li and Y. Li, J. Mol. Catal. A Chem., 2016, 420, 254–263 CrossRef CAS.
- D. Troegel and J. Stohrer, Coord. Chem. Rev., 2011, 255, 1440–1459 CrossRef CAS.
- M. Pagliaro, R. Ciriminna, V. Pandarus and F. Béland, Eur. J. Org. Chem., 2013, 2013, 6227–6235 CrossRef CAS.
- H. Maciejewski, A. Wawrzyńczak, M. Dutkiewicz and R. Fiedorow, J. Mol. Catal. A: Chem., 2006, 257, 141–148 CrossRef CAS.
- A. Wawrzyńczak, M. Dutkiewicz, J. Guliński, H. Maciejewski, B. Marciniec and R. Fiedorow, Catal. Today, 2011, 169, 69–74 CrossRef.
- M. Walczak, K. Stefanowska, A. Franczyk, J. Walkowiak, A. Wawrzyńczak and B. Marciniec, J. Catal., 2018, 367, 1–6 CrossRef CAS.
- Y. Chen, S. Ji, W. Sun, W. Chen, J. Dong, J. Wen, J. Zhang, Z. Li, L. Zheng and C. Chen, J. Am. Chem. Soc., 2018, 140, 8b–3121b Search PubMed.
- W. Hu, H. Xie, H. Yue, P. Prinsen and R. Luque, Catal. Commun., 2017, 97, 51–55 CrossRef CAS.
- H. Zai, Y. Zhao, S. Chen, R. Wang, L. Ge, C. Chen and Y. Li, RSC Adv., 2016, 6, 98520–98527 RSC.
- W. T. Koo, J. S. Jang, S. J. Choi, H. J. Cho and I. D. Kim, ACS Appl. Mater. Interfaces, 2017, 9, 18069–18077 CrossRef CAS PubMed.
- L. Jian-Rong, S. Julian and Z. Hong-Cai, Chem. Rev., 2012, 112, 869–932 CrossRef PubMed.
- L. Ye, Y. Bing and L. Jin-Liang, Chem. Commun., 2014, 50, 9969–9972 RSC.
- C. K. Brozek, B. Luca, S. Tomohiro, T. V. Clark, N. López and D. Mircea, Chemistry, 2014, 20, 6871–6874 CrossRef CAS PubMed.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef PubMed.
- R. Ricco, L. Malfatti, M. Takahashi, A. J. Hill and P. Falcaro, J. Mater. Chem. A, 2013, 1, 13033–13045 RSC.
- A. Corma, H. García and F. X. Llabrés I Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed.
- G. Felipe, F. Hiroyasu, L. Seungkyu and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 5271 CrossRef PubMed.
- L. T. L. Nguyen, K. K. A. Le, H. X. Truong and N. T. S. Phan, Catal. Sci. Technol., 2012, 2, 521–528 RSC.
- P. Falcaro, R. Ricco, A. Yazdi, I. Imaz, S. Furukawa, D. Maspoch, R. Ameloot, J. D. Evans and C. J. Doonan, Coord. Chem. Rev., 2016, 307, 237–254 CrossRef CAS.
- L. Ning, S. Liao, H. Cui, L. Yu and X. Tong, ACS Sustain. Chem. Eng., 2017, 6, 135–142 CrossRef.
- S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS PubMed.
- D. Jiang, L. L. Keenan, A. D. Burrows and K. J. Edler, Chem. Commun., 2012, 48, 12053–12055 RSC.
- W. Zhenqiang, K. K. Tanabe and S. M. Cohen, Inorg. Chem., 2009, 48, 296–306 CrossRef PubMed.
- F. David, A. Sonia and P. Catherine, ChemInform, 2010, 40, 7502–7513 Search PubMed.
- K. K. Tanabe and S. M. Cohen, Inorg. Chem., 2010, 49, 6766 CrossRef CAS PubMed.
- F. Zhang, Y. Jin, Y. Fu, Y. Zhong, W. Zhu, A. A. Ibrahim and M. S. El-Shall, J. Mater. Chem. A, 2015, 3, 17008–17015 RSC.
- S. Xu, W. Meng, F. Bo, X. Han, Z. Lan, H. Gu, H. Li and L. Hui, J. Catal., 2018, 363, 9–17 CrossRef CAS.
- X. Wang, H. Li and X. J. Hou, J. Phys. Chem. C, 2012, 116, 19814–19821 CrossRef CAS.
- R. Bandari and M. R. Buchmeiser, Catal. Sci. Technol., 2011, 2, 220–226 RSC.
- L. Ji, C. Yang, Z. Lei and T. Ma, J. Organomet. Chem., 2011, 696, 1845–1849 CrossRef.
- J. F. Harrod and A. J. Chalk, J. Am. Chem. Soc., 1965, 87, 1133 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra01408j |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Weiwen Chen,
Xiuying Chen,
Xinhua Zhou,
Wenbin Hu* and
Xugang Shu*
,
Weiwen Chen,
Xiuying Chen,
Xinhua Zhou,
Wenbin Hu* and
Xugang Shu*