
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic insight into the catalytic hydrogenation of nonactivated aldehydes with a Hantzsch ester in the presence of a series of organoboranes: NMR and DFT studies†
Go Hamasaka ab,
Hiroaki Tsujiab,
Masahiro Eharaab and
Yasuhiro Uozumi*abc
ab,
Hiroaki Tsujiab,
Masahiro Eharaab and
Yasuhiro Uozumi*abc
aInstitute for Molecular Science (IMS), Myodaiji, Okazaki 444-8787, Japan. E-mail: uo@ims.ac.jp
bSOKENDAI (The Graduate University for Advanced Studies), Myodaiji, Okazaki 444-8787, Japan
cJST-ACCEL, Myodaiji, Okazaki 444-8787, Japan
First published on 2nd April 2019
Abstract
Mechanistic studies on the organoborane-catalyzed transfer hydrogenation of nonactivated aldehydes with a Hantzsch ester (diethyl-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate) as a synthetic NADH analogue were performed by NMR experiments and DFT calculations. In the reaction of benzaldehyde with the Hantzsch ester, the catalytic activity of tris[3,5-bis(trifluoromethyl)phenyl]borane was superior to that of other borane catalysts, such as tris(pentafluorophenyl)borane, trifluoroborane etherate, or triphenylborane. Stoichiometric NMR experiments demonstrated that the hydrogenation process proceeds through activation of the aldehyde by the borane catalyst, followed by hydride transfer from the Hantzsch ester to the resulting activated aldehyde. DFT calculations for the hydrogenation of benzaldehyde with the Hantzsch ester in the presence of borane catalysts supported the reaction pathway and showed why the catalytic activity of tris[3,5-bis(trifluoromethyl)phenyl]borane is higher than that of the other boron catalysts. Association constants and Gibbs free energies in the reaction of boron catalysts with benzaldehyde or benzyl alcohol, which were investigated by 1H NMR analyses, also indicated why tris[3,5-bis(trifluoromethyl)phenyl]borane is a superior catalyst to tris(pentafluorophenyl)borane, trifluoroborane etherate, or triphenylborane in the hydrogenation reaction.
Introduction
Biological hydrogenation processes are recognized as important reactions in nature. These processes are mediated by a combination of an enzyme and an organic reduction cofactor (e.g., NADH or NADPH).1 In particular, the biological hydrogenation of acetaldehyde is a crucial final step in the fermentation of sugars to form ethanol. The reaction proceeds in the presence of alcohol dehydrogenase and NADH under anaerobic conditions. The reaction pathway of this process is known to be as follows: (1) acetaldehyde is activated by coordination of its carbonyl group to the Zn center of alcohol dehydrogenase; and (2) hydride is transferred to the activated acetaldehyde from NADH (Fig. 1).1,2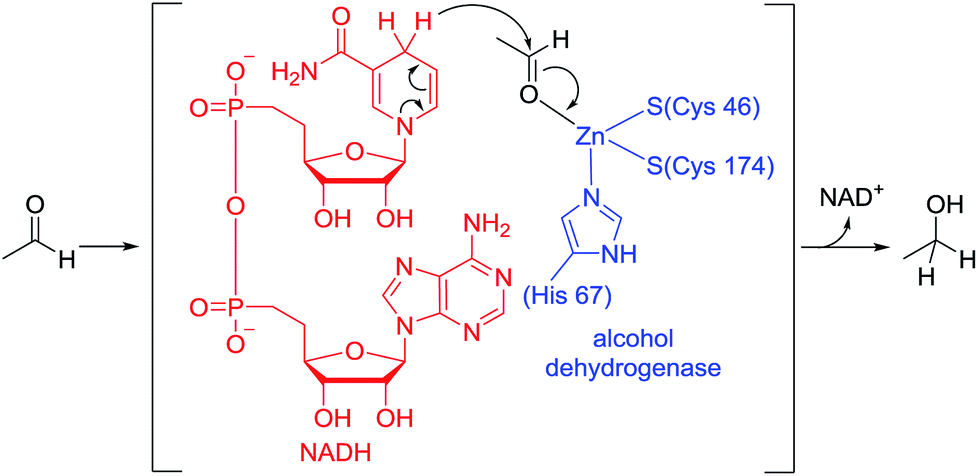 | ||
| Fig. 1 Active site of alcohol dehydrogenase for the reduction of acetaldehyde with NADH. | ||
In attempts to realize a biomimetic hydrogenation process, many organic chemists have investigated the catalytic hydrogenation of imines, α,β-unsaturated carbonyl compounds, or α-keto esters with a synthetic NADH analogue.3–7 Despite numerous investigations of Brønsted or Lewis acid-catalyzed hydrogenations of nonactivated aldehydes with synthetic NADH analogues, no efficient hydrogenation system was developed until 2015.8,9 In 2015, we reported the first example of an organoborane-catalyzed hydrogenation of nonactivated aliphatic and aromatic aldehydes with a Hantzsch ester (diethyl-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate) as a synthetic NADH analogue to give the corresponding primary alcohols (Scheme 1).10 In this reaction, tris[3,5-bis(trifluoromethyl)phenyl]borane showed superior catalytic activity to other borane catalysts. However, the details of the reaction mechanism and the origin of the superiority of tris[3,5-bis(trifluoromethyl)phenyl]borane to the other borane catalysts were not clarified.
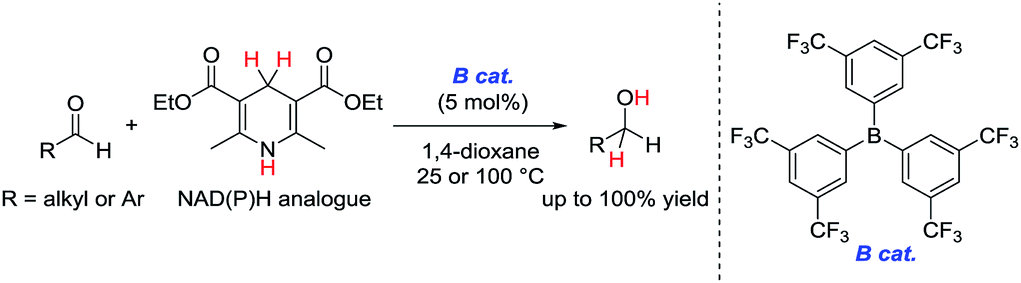 | ||
| Scheme 1 Organoborane-catalyzed hydrogenation of nonactivated aldehydes with a Hantzsch ester (our previous work). | ||
Because of their high oxophilic Lewis acidities, organoborane compounds such as tris(pentafluorophenyl)borane have been used as Lewis acid catalysts in various organic transformations including the Mukaiyama-aldol reaction and the Hosomi–Sakurai reaction.11,12 These reactions proceed through coordination of the carbonyl compound to the boron catalyst (as exemplified by the formation of intermediate Int-I in Fig. 2a) followed by attack by a nucleophile (silyl enol ether or allyl silane) on the resulting activated carbonyl compound.
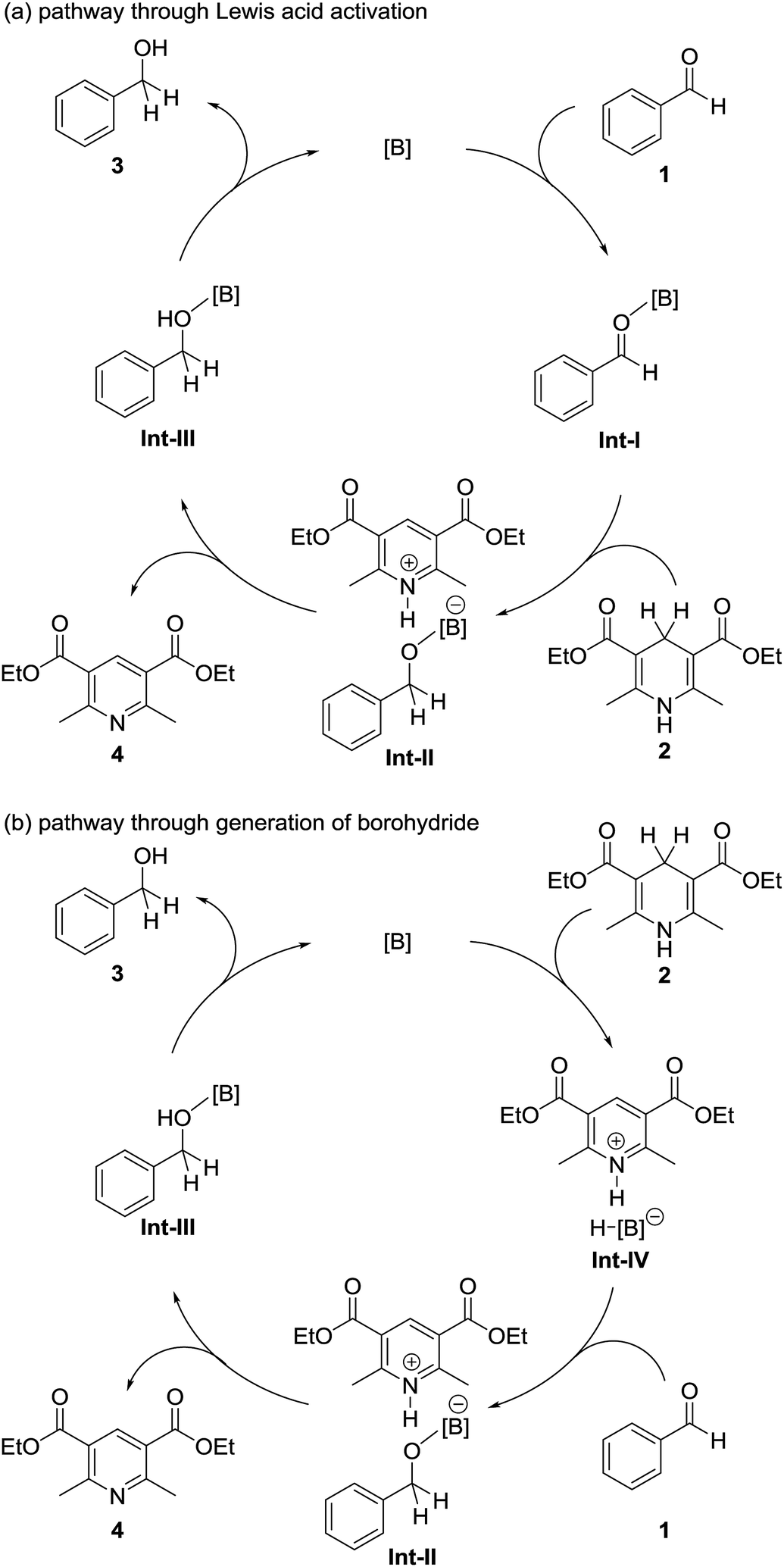 | ||
| Fig. 2 Possible reaction pathways. (a) Lewis acid activation pathway, (b) borohydride pathway. | ||
Electron-deficient borane compounds have also been used as partners in frustrated Lewis pair catalysts to promote the hydrogenation of unsaturated bonds with H2.13–15 In 2010, Stephan, Crudden, and co-workers discovered that borohydride species such as Int-IV in Fig. 2b are generated by the reaction of tris(pentafluorophenyl)borane with Hantzsch esters.16 However, catalytic applications of these borohydrides were not investigated. Recently, Oestreich and co-workers reported that tris(pentafluorophenyl)borane catalyzes the hydrogenation of imines or related N-heterocycles with cyclohexa-1,4-dienes.17,18 In this reaction, tris(pentafluorophenyl)borane abstracts hydrogen from the cyclohexa-1,4-diene to generate a borohydride species; this reaction is similar to that reported by Stephan and Crudden and co-workers.16 This process is followed by reaction with the imine or a related N-heterocycle to give the corresponding hydrogenated product.
On the basis of the reaction pathways reported in the literature,11–18 we propose two plausible reaction pathways for our organoborane-catalyzed hydrogenation of aldehydes with a Hantzsch ester, as shown in Fig. 2. The first of these proposed reaction pathways is shown in Fig. 2a. In this pathway, an aldehyde 1 is activated by a boron catalyst. Hydrogen is transferred to the activated aldehyde from the Hantzsch ester 2 to form an alcohol–boron adduct Int-III and the Hantzsch pyridine 4. Int-III then dissociates to give an alcohol 3, with the regeneration of the boron catalyst. The second possible reaction pathway is shown in Fig. 2b. This proceeds through generation of a borohydride species Int-IV by the reaction of 2 with the boron catalyst. Int-IV then reacts with 1 to give Int-III. Dissociation of Int-III to afford 3 is accompanied by regeneration of the boron catalyst.
Here, we report an investigation of the mechanism of our organoborane-catalyzed hydrogenation of aldehydes with a Hantzsch ester by means of NMR experiments and DFT calculations. We also discuss why tris[3,5-bis(trifluoromethyl)phenyl]borane shows a superior catalytic activity to that of other boron catalysts.
Results and discussion
Evaluation of the catalytic activity of borane catalysts in the hydrogenation of benzaldehyde with a Hantzsch ester
To compare the catalytic activity of borane catalysts, we examined the reaction of benzaldehyde (1) with the Hantzsch ester 2 in the presence of a catalytic amount of various borane compounds B-a to B-d (Scheme 2). When tris[3,5-bis(trifluoromethyl)phenyl]borane (B-a) was used as the catalyst, the reaction proceeded efficiently to give benzyl alcohol (3) quantitatively. Tris(pentafluorophenyl)borane (B-b) also promoted the reaction to give 3 in 88% yield. The catalytic efficiency of B-a is therefore superior to that of B-b. On the other hand, only a 7% yield of 3 was obtained from the corresponding reaction in the presence of trifluoroborane etherate (B-c), whereas triphenylborane (B-d) did not promote the reaction at all. These results indicate that the order of catalytic activity is as follows: B-a > B-b > B-c > B-d.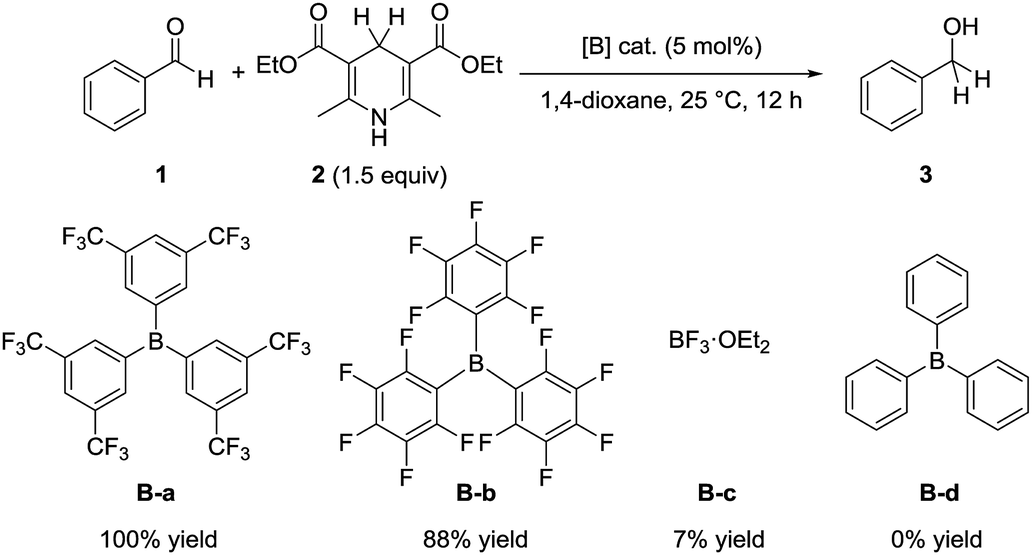 | ||
| Scheme 2 Hydrogenation of benzaldehyde (1) with Hantzsch ester 2. Reaction conditions: 1 (0.25 mmol), 2 (0.38 mmol), [B] cat. (0.013 mmol), 1,4-dioxane (1 mL), 25 °C, 12 h. | ||
Stoichiometric reaction experiments
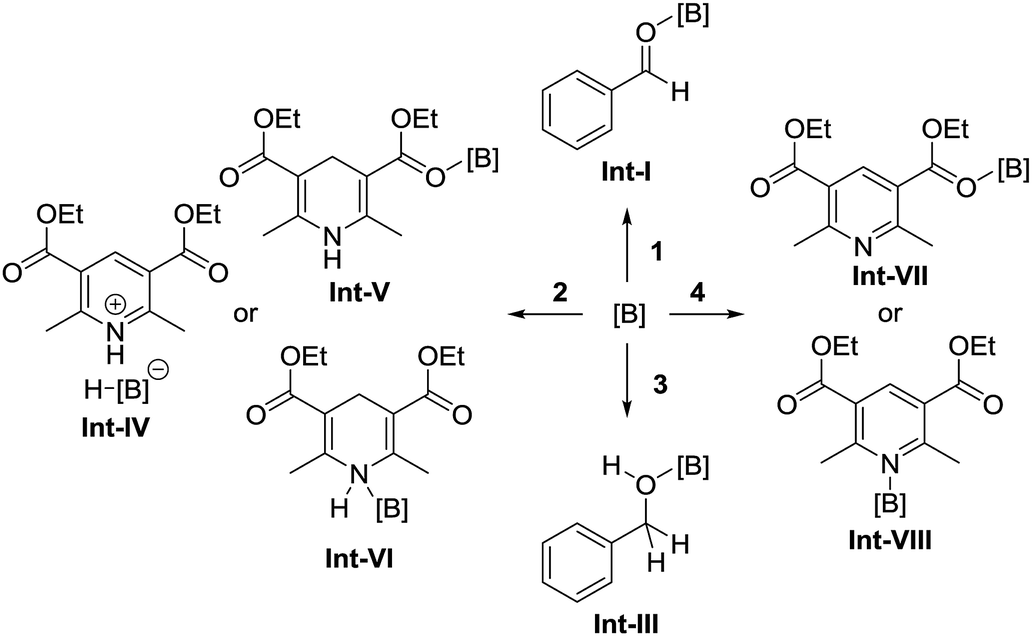
To identify the reaction pathway, we examined the stoichiometric reactions of B-a with benzaldehyde (1) and with Hantzsch ester 2 (Scheme 3). In a glove box, B-a, 1, and CD2Cl2 were introduced into a J. Young NMR tube (Scheme 3a), and the resulting sample was analyzed by 1H, 13C, and 11B NMR spectroscopy. In the 1H NMR spectrum of this sample, the peak derived from the aldehyde functionality shifted to a higher field (1: δ = 10.01 ppm; Int-Ia: δ = 9.17 ppm), whereas the peaks derived from the aromatic protons of the benzaldehyde shifted to a lower field (Fig. 3a–c). In the 13C NMR measurement, the peak derived from the aldehyde functionality shifted to a lower field (1: δ = 192.6 ppm, Int-Ia: δ = 200.2 ppm). These observations suggest that the electron density of the aldehyde functionality decreased after reaction with B-a. A high field shift was observed in the 11B NMR measurements (B-a: δ = 68.6 ppm, Int-Ia: δ = 36.0 ppm), showing that the electron density at the boron center in the resulting product had increased. These results indicated that Int-Ia was formed in this reaction. The resulting mixture was also examined by ESI-MS, and a peak corresponding to Int-Ia (m/z = 779 [M + Na]+) was observed. The addition of 2 to the reaction mixture then led to further reaction (Scheme 3a). The resulting sample was analyzed by 1H NMR spectroscopy (Fig. 3d). In the 1H NMR spectrum of the resulting mixture, the peak derived from the aldehyde functionality disappeared and a peak derived from benzylic protons appeared (Fig. 3d, inset; δ = 4.53 ppm). Int-Ia immediately reacted with 2 to give the benzyl alcohol–tris[3,5-bis(trifluoromethyl)phenyl]borane adduct Int-IIIa in 90% NMR yield. GC/MS analysis of the reaction mixture demonstrated the formation of benzyl alcohol (3) (m/z = 108 [M]+). In contrast, no peaks arising from the borohydride species Int-IVa were observed in the reaction of B-a with 2.19 If the reaction proceeds via a borohydride species (Fig. 2b), Int-IVa should have been detected. Therefore, the reaction proceeds through activation of the aldehyde by the boron catalyst, followed by hydride transfer from the Hantzsch ester (Fig. 2a).20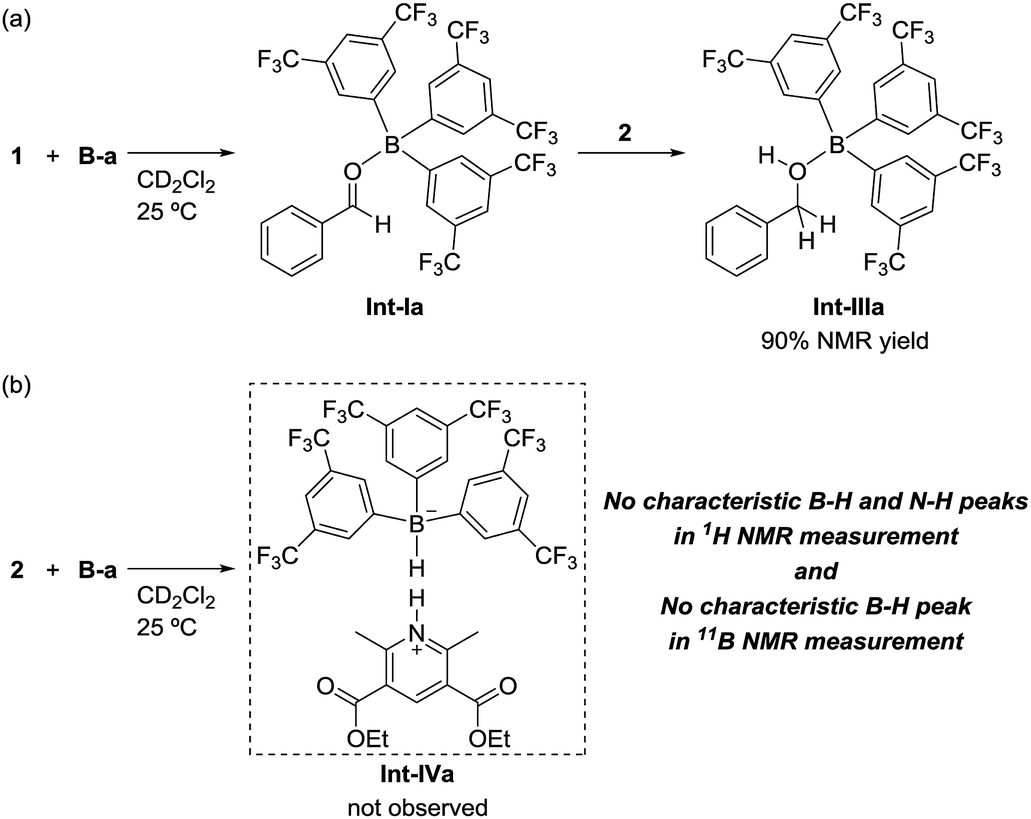 | ||
| Scheme 3 Stoichiometric experiments on (a) the reaction of benzaldehyde (1) with tris[3,5-bis(trifluoromethyl)phenyl]borane (B-a) followed by treatment with Hantzsch ester 2; (b) the reaction of benzaldehyde (1) with Hantzsch ester 2. | ||
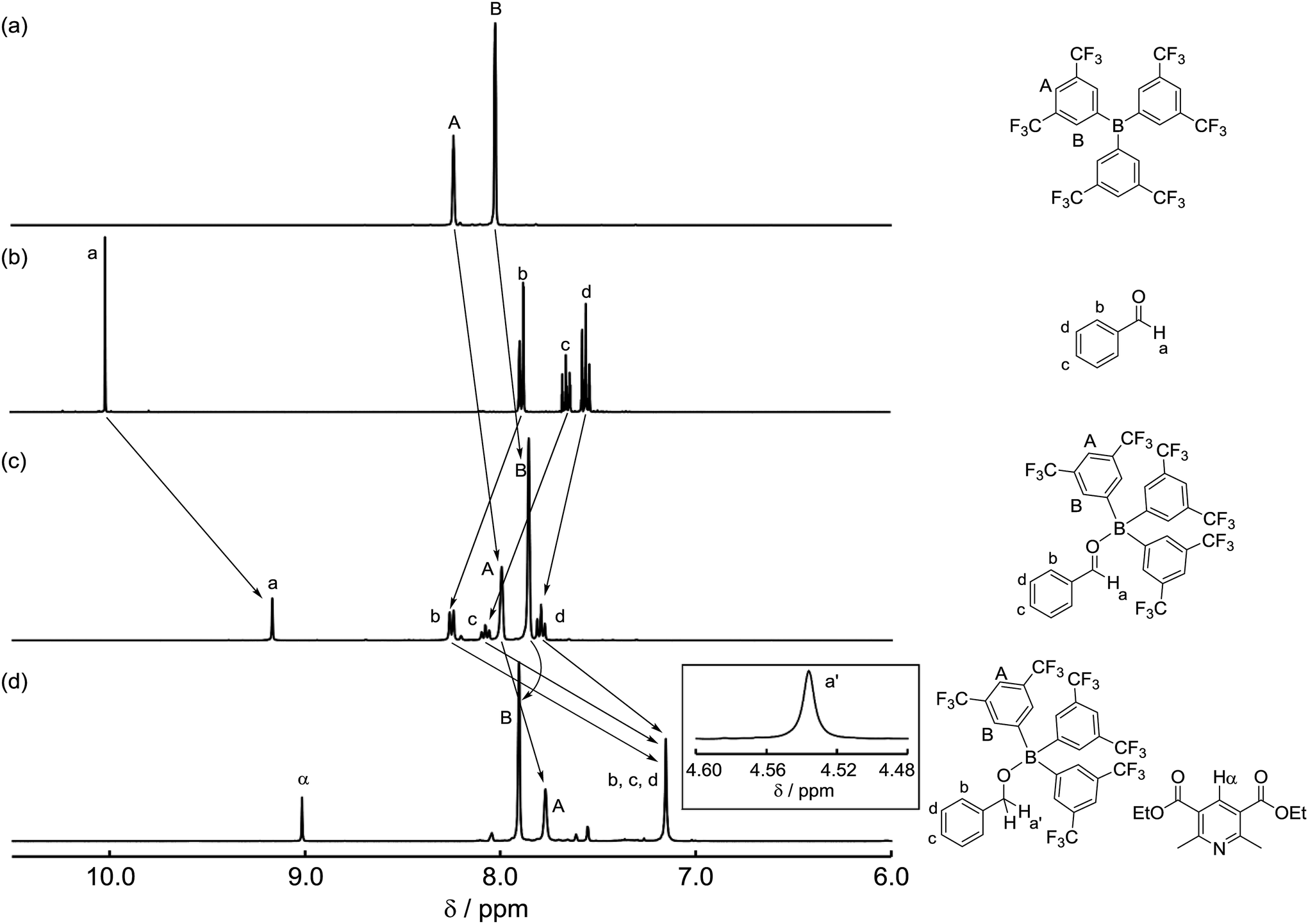 | ||
| Fig. 3 1H NMR spectra for (a) tris[3,5-bis(trifluoromethyl)phenyl]borane (B-a), (b) benzaldehyde (1), (c) a mixture of 1 and B-a, and (d) after addition of Hantzsch ester 2 in CD2Cl2. | ||
DFT calculations for the hydrogenation of benzaldehyde with a Hantzsch ester catalyzed by various organoboranes
We next performed DFT calculations to elucidate details of our catalytic reaction. All the calculations were carried out with the GAUSSIAN 09 (Revision E.01) program.21 Geometry optimizations and frequency calculations for all molecules were carried out at the M06-2x22 level of theory with the basis sets 6-31G(d,p) for C and H and 6-31+G(d,p) for B, N, O, and F. Single-point energies were obtained by calculations at the M06-2x/6-311++G(d,p) level of theory using the SMD23 solvation model (1,4-dioxane). The energies reported here includes the electronic energy, the zero-point energy, thermal correction at 298.15 K and 1 atm, and solvent-effect correction.First, we calculated the expected reaction pathway (Scheme 2a). The obtained energy diagram of this catalytic reaction and the calculated structures of transition states are shown in Fig. 4 and 5, respectively. The energy diagram for the reaction with B-a is drawn as a red line in Fig. 4. The calculated Gibbs free energy for coordination of 1 to borane B-a is −3.7 kcal mol−1, showing that this is an energetically downhill step. The activation energy of the hydride-transfer process is 9.9 kcal mol−1 relative to the initial state. The energy difference between TSa (Fig. 5a) and [Int-Ia + 2] is 13.6 kcal mol−1. After the hydride transfer, the ionic pair Int-IIa is generated and the Gibbs free energy is −8.7 kcal mol−1. Ionic pair Int-IIa is more stable than Int-Ia (ΔΔGInt-IIa–Int-Ia = −5.0 kcal mol−1). These results show that the hydride-transfer process should proceed smoothly because this step is energetically downhill and has a low activation energy. A proton is then moved to the benzyl alcohol moiety from the protonated Hantzsch pyridine (ΔGInt-IIIa = −9.2 kcal mol−1, ΔΔGInt-IIIa–Int-IIa = −0.5 kcal mol−1). Finally, B-a is liberated from Int-IIIa to give 3. The calculated stabilization energy for the formation of Int-IIIa from B-a and 3 is −1.1 kcal mol−1. The stabilization energy of the formation of Int-Ia is therefore lower than that of the formation of Int-IIIa, so the catalytic reaction should proceed smoothly.
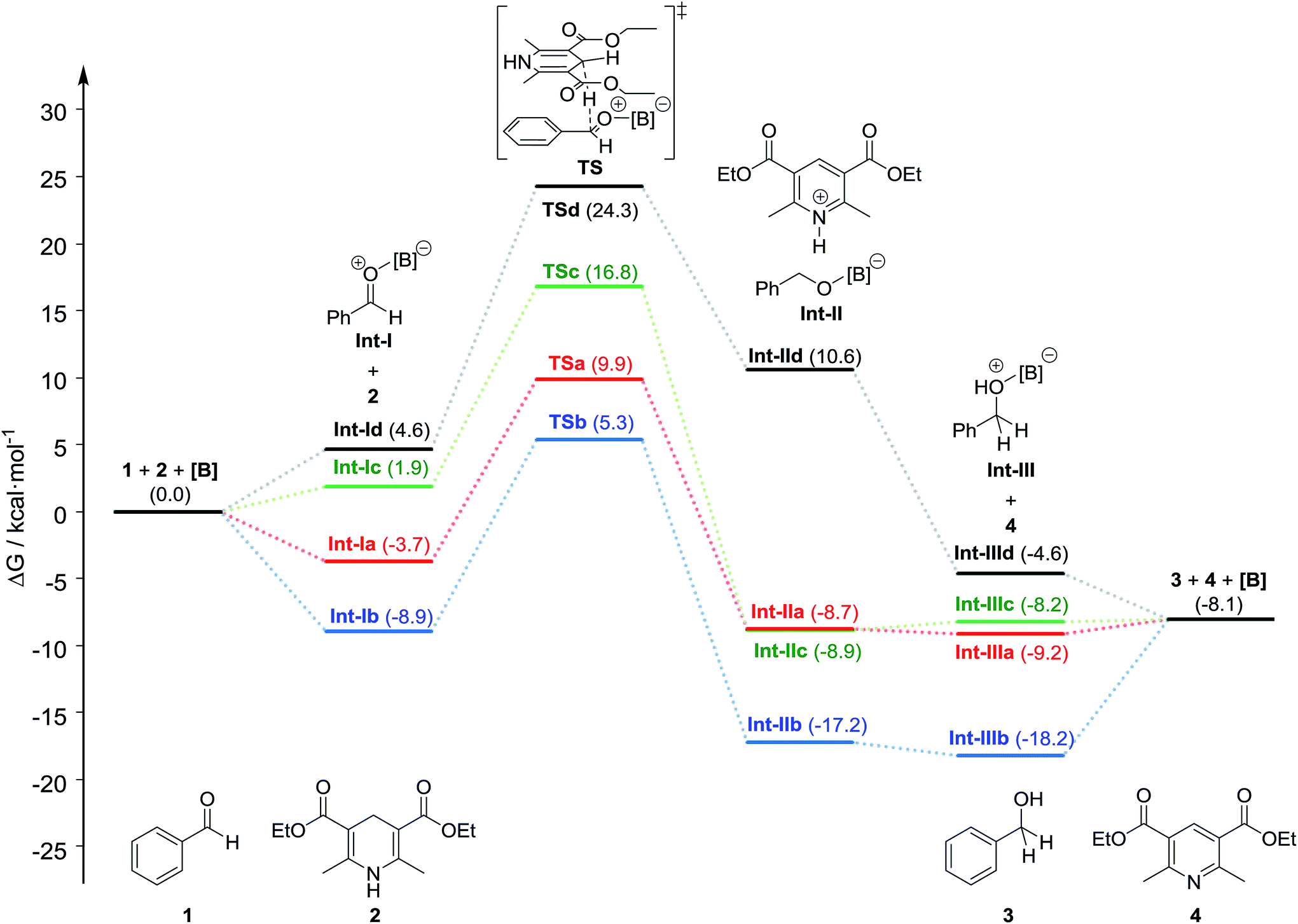 | ||
| Fig. 4 Energy diagram in the organoborane-catalyzed hydrogenation of benzaldehyde (1) with Hantzsch ester (2). Red: tris[3,5-bis(trifluoromethyl)phenyl]borane (B-a), blue: tris(pentafluorophenyl)borane (B-b), green: trifluoroborane etherate (B-c), black: triphenylborane (B-d). | ||
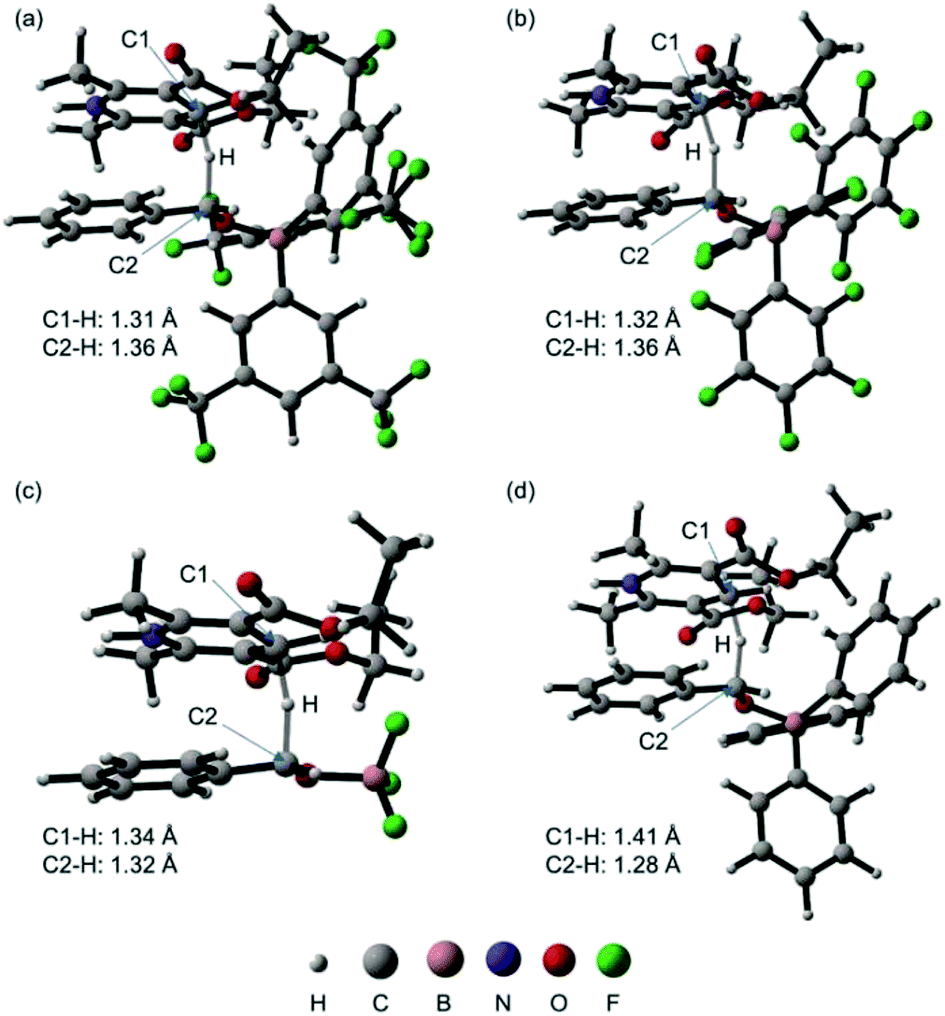 | ||
| Fig. 5 Calculated structures of transition states TSa (a), TSb (b), TSc (c), and TSd (d) and selected bond distances (the molecular structures were drawn with CYLview24). | ||
We also examined the hydrogenation with B-b as the catalyst (Fig. 4; blue line). The formation of Int-Ib is a downhill step (stabilization energy −8.9 kcal mol−1). The activation energy of the hydride-transfer process from 2 to Int-Ib is 5.3 kcal mol−1 relative to the initial state. The energy difference between TSb (Fig. 5b) and [Int-Ib + 2] is 14.2 kcal mol−1. This value is slightly greater than the value of the energy difference between TSa and [Int-Ia + 2] (13.6 kcal mol−1). After the hydride transfer, Int-IIb should be generated (ΔGInt-IIb = −17.2 kcal mol−1). The ionic intermediate Int-IIb is therefore more stable than Int-Ib (ΔΔGInt-IIb–Int-Ib = −8.3 kcal mol−1), showing that the reaction should proceed. The Gibbs free energy of Int-IIIb is −18.2 kcal mol−1, whereas that for the formation of Int-IIIb from 3 and B-b is −10.1 kcal mol−1. This value is comparable to the formation of Int-Ib: the calculated energy difference is only 1.2 kcal mol−1. These results suggest that the generated 3 suppresses regeneration of the catalyst, thereby explaining why the catalytic activity of B-b is lower than that of B-a.
The energy diagram for the reaction with B-c as the catalyst is shown by the green line in Fig. 4. In this calculations, interaction between diethyl ether and intermediates is not considered. The Gibbs free energy for the formation of Int-Ic is 1.9 kcal mol−1. Under the calculation conditions, B-c is more stable than Int-Ic. However, the energy difference is small. Therefore, formation of Int-Ic is possible. For the formation of the ionic-pair intermediate Int-IIc, the calculated Gibbs free energy is −8.9 kcal mol−1. The energy difference between Int-Ic and Int-IIc is therefore −10.8 kcal mol−1, indicating that the hydride-transfer process is energetically downhill. The activation energy for the hydride-transfer process is as high as 16.8 kcal mol−1 relative to the initial state, and the energy difference between TSc (Fig. 5c) and [Int-Ic + 2] is 14.9 kcal mol−1. These results suggest that the hydride-transfer process should proceed. The activation energy of TSc (14.9 kcal mol−1) is larger than that of TSa (13.6 kcal mol−1) and TSb (14.2 kcal mol−1). The ion-pair intermediate Int-IIc should undergo proton transfer to give Int-IIIc and 4, because this process is slightly energetically uphill (ΔΔGInt-IIIc–Int-IIc = 0.7 kcal mol−1). The formation of Int-IIIc from B-c and 3 is energetically downhill (−0.1 kcal mol−1). These results suggest that the reaction is difficult due to the higher activation energy barrier and the higher stability of the formation of Int-IIIc than that of the formation of Int-Ic by 2.0 kcal mol−1. Therefore, when B-c was used as the catalyst, 3 was obtained in only 7% yield, because the generated 3 inhibited the formation of Int-Ic (Scheme 2).
The energy diagram for the hydrogenation reaction with B-d is shown as a black line in Fig. 4. The formation of Int-Id is energetically uphill (ΔGInt-Id = 4.6 kcal mol−1), suggesting that it is difficult for 1 to coordinate to B-d. In the hydride-transfer process, the energy of the desired intermediate Int-IId is higher than that of Int-Id (ΔΔGInt-IId–Int-Id = 6.0 kcal mol−1), and the activation energy is relatively high (24.3 kcal mol−1 relative to the initial state). Therefore, the hydride-transfer process should not take place. These calculation results suggest that the reaction with B-d should not proceed at all.
In our reaction system, the boron catalysts might also react with the Hantzsch ester 2 or with the Hantzsch pyridine 4. To understand the influence of 2 or 4 on the hydrogenation reaction, we performed DFT calculations for the possible intermediates Int-IV to Int-VIII (Scheme 4). The results of these calculations are summarized in Table 1. In the case of the reaction of borane B-a with 2 and 4, the Gibbs free energies are 10.9 (Int-IVa), 1.0 (Int-Va), 18.7 (Int-VIa), 7.8 (Int-VIIa), and 7.9 kcal mol−1 (Int-VIIIa), respectively. These reactions should proceed with difficulty, because they are energetically uphill, indicating that 2 and 4 are unlikely to affect the efficiency of the hydrogenation reaction.
 | ||
| Scheme 4 Possible reactions of boron catalysts in the reaction system. | ||
| B-a | B-b | B-c | B-d | |
|---|---|---|---|---|
| a The coordinated structure Int-VId was not obtained in the DFT calculation. | ||||
| 1 | Int-Ia (−3.7) | Int-Ib (−8.9) | Int-Ic (1.9) | Int-Id (4.6) |
| 2 | Int-IVa (10.9) | Int-IVb (−4.0) | Int-IVc (32.8) | Int-IVd (29.9) |
| Int-Va (1.0) | Int-Vb (−2.5) | Int-Vc (1.2) | Int-Vd (9.1) | |
| Int-VIa (18.7) | Int-VIb (7.9) | Int-VIc (7.0) | Ind-VId (−)a | |
| 3 | Int-IIIa (−1.1) | Int-IIIb (−10.1) | Int-IIIc (−0.2) | Int-IIId (3.5) |
| 4 | Int-VIIa (7.8) | Int-VIIb (4.6) | Int-VIIc (7.0) | Int-VIId (16.4) |
| Int-VIIIa (7.9) | Int-VIIIb (3.4) | Int-VIIIc (1.9) | Int-VIIId (15.9) | |
On the other hand, in the reaction of B-b, the Gibbs free energies for the formation of Int-IVb and Int-Vb are −4.0 and −2.5 kcal mol−1, respectively. These intermediates might, therefore, be formed in the reaction of B-b with 2. The processes for the formation of Int-VIb, Int-VIIb, and Int-VIIIb are energetically uphill. From these results, the formation of Int-IIIb is the most energetically favored of these processes. Therefore, the desired hydrogenation process proceeds and the resulting hydrogenated product 3 partially inhibits the desired formation of Int-Ib. The formation of Int-IVb and Int-Vb might also disturb the formation of Int-Ib. Thus, the catalytic activity of B-b is lower than that of B-a.
In the results of our DFT calculation for the reactions of B-c with 2 or 4, the Gibbs free energies for the formation of Int-Vc and Int-VIIIc were 1.2 and 1.9 kcal mol−1, respectively. These values are comparable with the Gibbs free energy for the formation of Int-Ic (1.9 kcal mol−1). Thus, 2, 3, and 4 inhibit the desired reaction, resulting in a low yield of the desired product.
In the reaction of B-d, the formation of all intermediates is energetically uphill (by at least 3.5 kcal mol−1). Consequently, the reaction of B-d does not proceed at all.
Determination of association constants between boron reagents and benzaldehyde or benzyl alcohol
The DFT study indicated that a preferential formation of Int-I compared with the formation of Int-III is important for the catalytic hydrogenation reaction to proceed. Therefore, we next determined the association constants between boron reagents B-a to B-d and benzaldehyde (1) or benzyl alcohol (3). To obtain the association constants, boron compounds B-a to B-d and 1 or 3 were dissolved in CD2Cl2 and subjected to 1H NMR measurements. Variable-temperature (VT) 1H NMR spectra for the reaction of B-a with 1 are shown in Fig. 6. The peak derived from the aldehyde functionality separated into peaks derived from uncoordinated benzaldehyde and the coordinated benzaldehyde Int-Ia below −40 °C. By using a van't Hoff plot, the association constant was estimated to be 1.19 × 104 M−1 (Fig. 7). The Gibbs free energy for the formation of Int-Ia was calculated from the association constant to be −5.6 kcal mol−1 (Table 2).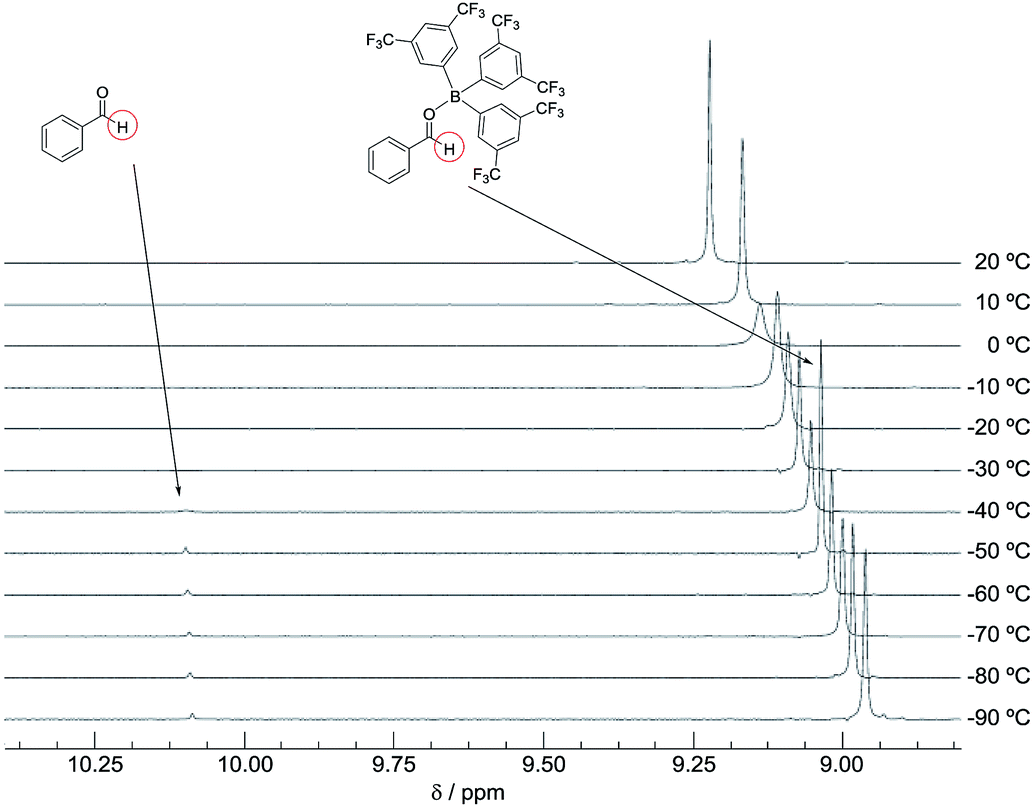 | ||
| Fig. 6 VT 1H NMR spectra of a mixture of 1 and B-a (20 °C to −90 °C). | ||
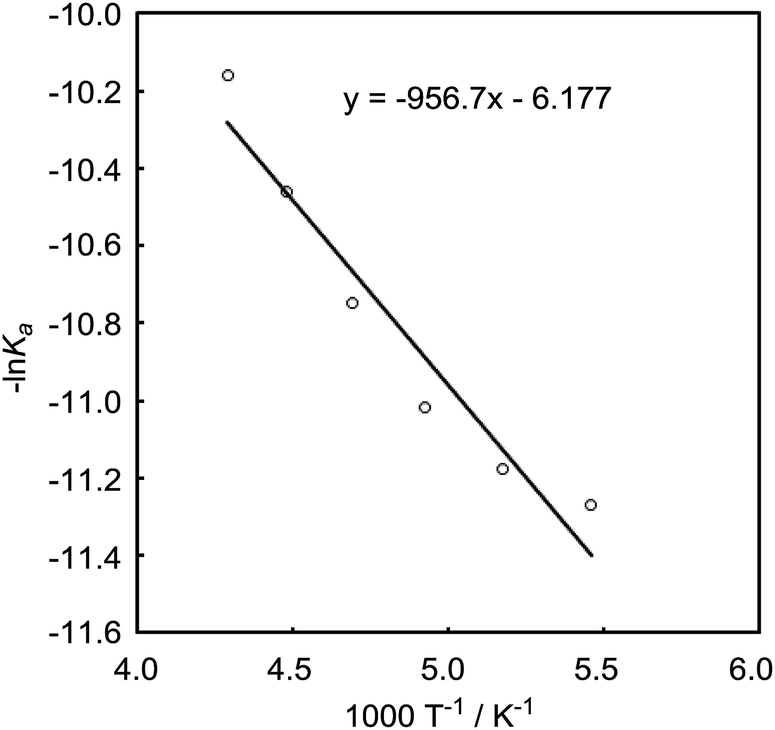 | ||
| Fig. 7 van't Hoff plot for a mixture of 1 and B-a (−40 °C to −90 °C). | ||
| Benzaldehyde (1) | Benzyl alcohol (3) | ΔΔGInt-I–Int-III, kcal mol−1 | |||
|---|---|---|---|---|---|
| Ka, M−1 | ΔG, kcal mol−1 | Ka, M−1 | ΔG, kcal mol−1 | ||
| B-a | 1.19 × 104 | -5.6 | 95.4 | -2.7 | -2.9 |
| B-b | 396 | -3.5 | 58.3 | -2.4 | -1.1 |
| B-c | 5.51 | -1.0 | 263 | -3.3 | 2.3 |
| B-d | 0.195 | 1.0 | 5.67 × 10−3 | 3.1 | -2.1 |
We also recorded VT 1H NMR spectra for a mixture of B-a and 3 in CD2Cl2 (Fig. 8). By using the van't Hoff plot (Fig. 9), the association constant and the Gibbs free energy for the formation of the borane–benzyl alcohol adduct Int-IIIa were determined to be 95.4 M−1 and -2.7 kcal mol−1, respectively (Table 2). The Gibbs free energy of the formation of Int-Ia is lower than that for the formation of Int-IIIa (ΔΔGInt-Ia–Int-IIIa = −2.9 kcal mol−1, Table 2). Therefore, coordination of B-a to 1 is preferred to that of B-a to 3, and the desired reaction should proceed smoothly (Scheme 2).
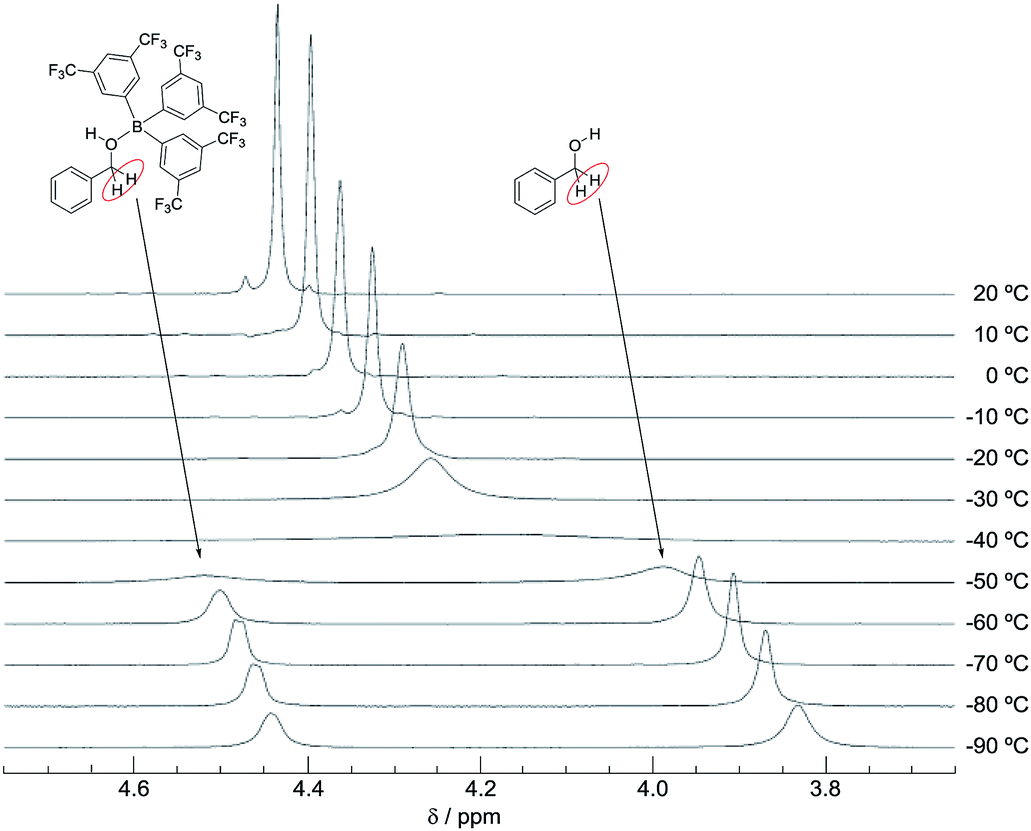 | ||
| Fig. 8 VT 1H NMR spectra for a mixture of 3 and B-a (20 °C to −90 °C). | ||
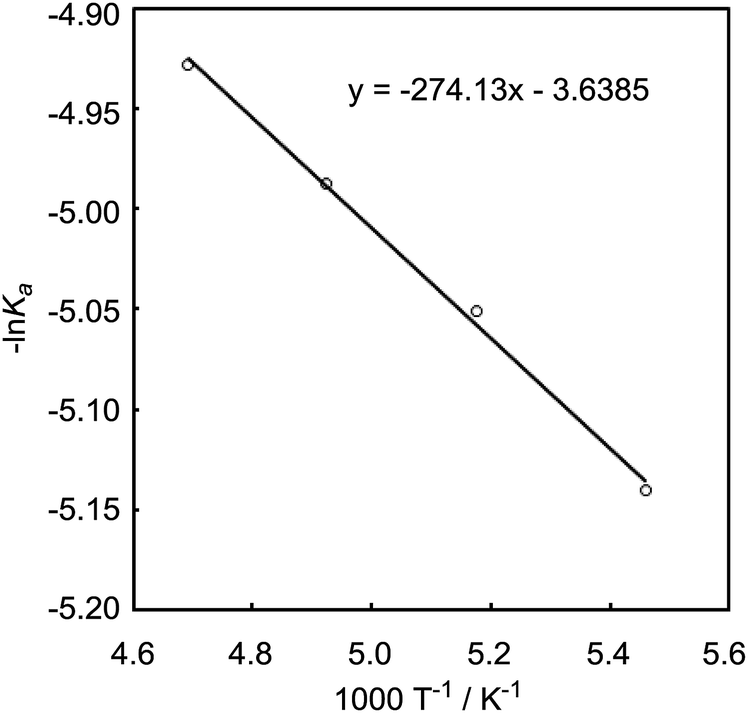 | ||
| Fig. 9 van't Hoff plot for a mixture of 3 and B-a (−60 °C to −90 °C). | ||
We also measured the VT 1H NMR spectra for the reactions of B-b with 1 or 3.25 The association constants and the Gibbs free energies for the formation of Int-Ib were 396 M−1 and -3.5 kcal mol−1, respectively, and those of Int-IIIb were 58.3 M−1 and -2.4 kcal mol−1, respectively (Table 2).26 In this case, the difference in the value of the Gibbs free energies for the formation of Int-Ib and Int-IIIb is −1.1 kcal mol−1 (Table 2), so the formation of Int-Ib competes with the formation of Int-IIIb. Consequently, the catalytic activity of B-b should be lower than that of B-a.
In the case of BF3·OEt2 (B-c), the association constant and Gibbs free energy for the formation of Int-Ic are 5.51 M−1 and -1.0 kcal mol−1, respectively, and the corresponding values for Int-IIIc are 263 M−1 and -3.3 kcal mol−1, respectively (Table 1).25 The Gibbs free energy for the formation of Int-IIIc is therefore lower than that for the formation of Int-IIIc (ΔΔGInt-Ic–Int-IIIc = 2.3 kcal mol−1, Table 2) In the hydrogenation of 1 with 2 with B-c as the catalyst, the generated 3 should therefore inhibit the catalytic reaction. Consequently, the reaction does not proceed efficiently (Scheme 2). The order of the catalytic activities of B-a, B-b, and B-c is correlated with the energy differences between the formation of Int-I and Int-III [catalytic activities: B-a > B-b > B-c (Scheme 2); energy differences between formation of Int-I and Int-III: B-a < B-b < B-c, (Table 2)].
In the reaction of B-d with 1 or 3, the association constants were 0.195 and 5.67 × 10−3 M−1, respectively, and the Gibbs free energies were 1.0 and 3.1 kcal mol−1, respectively (Table 1).25 These reactions were energetically uphill. Therefore, the coordination of 1 to B-d occurs with difficulty, explaining why the hydrogenation of 1 does not proceed at all in this case (Scheme 2).
Conclusions
We performed NMR experiments and DFT calculations to elucidate the mechanism of the organoborane-catalyzed hydrogenation of benzaldehyde with a Hantzsch ester as a synthetic NADPH analogue, and to explain the activity or lack of activity of various borane catalyst in this reaction. The reactions in the presence of tris[3,5-bis(trifluoromethyl)phenyl]borane, tris(pentafluorophenyl)borane, BF3·OEt2, and triphenylborane gave benzyl alcohol in yields of 100, 88, 4, and 0%, respectively. Tris[3,5-bis(trifluoromethyl)phenyl]borane therefore showed a superior catalytic activity to the other borane catalysts in this reaction. Stoichiometric NMR experiments demonstrated that the hydrogenation process proceeds through activation of benzaldehyde by the borane catalyst, followed by hydride transfer from the Hantzsch ester to the activated aldehyde.DFT calculations for the hydrogenation of benzaldehyde with a Hantzsch ester in the presence of borane catalysts were carried out to elucidate details of the reaction. The activation energies for the hydrogenations in the presence of tris[3,5-bis(trifluoromethyl)phenyl]borane and tris(pentafluorophenyl)borane are sufficiently low to permit the reaction to proceed. However, in the case of tris(pentafluorophenyl)borane, the benzyl alcohol–B(C6F5)3 adduct is almost as stable as the benzaldehyde–B(C6F5)3 adduct, indicating that the generated benzyl alcohol is likely to inhibit the catalytic cycle. In the case of BF3·OEt2, the formation of the BF3–benzyl alcohol adduct, the BF3–Hantzsch ester adduct, and the BF3–Hantzsch pyridine adduct should retard the formation of the BF3–benzaldehyde adduct, explaining why the catalytic reaction does not proceed efficiently. In contrast, the activation energy in the reaction with triphenylborane is relatively high and the hydrogen-transfer process is energetically uphill, explaining why the reaction proceeds with difficulty. These results explain why tris[3,5-bis(trifluoromethyl)phenyl]borane is superior to the other boron catalysts in this hydrogenation reaction.
Association constants and Gibbs free energies for the reaction of boron catalysts with benzaldehyde and benzyl alcohol were determined by 1H NMR analyses. In the case of tris[3,5-bis(trifluoromethyl)phenyl]borane, the association constant of formation of the borane–benzaldehyde adduct is greater than that for the formation of the borane–benzyl alcohol adduct, explaining why the desired catalytic hydrogenation proceeds smoothly. The association constant of tris(pentafluorophenyl)borane with benzaldehyde is comparable to that of tris(pentafluorophenyl)borane with benzyl alcohol, indicating that the formation of the benzaldehyde–B(C6F5)3 adduct competes with the formation of the benzyl alcohol–B(C6F5)3 adduct. Consequently, the catalytic activity of tris(pentafluorophenyl)borane is lower than that of tris[3,5-bis(trifluoromethyl)phenyl]borane. On the other hand, in the reaction of BF3·OEt2, the association constant for the formation of the benzaldehyde–BF3 adduct is lower than that for the BF3–benzyl alcohol adduct. The generation of benzyl alcohol therefore inhibits the hydrogenation of benzaldehyde when BF3·OEt2 is used as a catalyst. The association constants of triphenylborane with benzaldehyde and benzyl alcohol are low, suggesting the hydrogenation reaction should not proceed efficiently. These experimental results also demonstrated why tris[3,5-bis(trifluoromethyl)phenyl]borane is a superior catalyst to the other borane catalysts in the hydrogenation reaction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research project was supported by JST-ACCEL program (JPMJAC1401). G. H. appreciates funding from the JSPS-KAKENHI [Grant-in-Aid for Young Scientists (B) No. 17K14524]. The calculations were performed at the Research Center for Computational Science, Okazaki, Japan.Notes and references
- J. M. Berg, J. L. Tymoczko and L. Stryer, Biochemistry, W. H. Freeman, New York, 5th edn, 2001 Search PubMed.
- For selected recent reviews on enzymatic reduction of carbonyl compounds, see; (a) M. Kataoka, K. Kita, M. Wada, Y. Yasohara, J. Hasegawa and S. Shimizu, Appl. Microbiol. Biotechnol., 2003, 62, 437 CrossRef CAS PubMed; (b) L. R. Jarboe, Appl. Microbiol. Biotechnol., 2011, 89, 249 CrossRef CAS PubMed; (c) C. A. Rabinovitch-Deere, J. W. K. Oliver, G. M. Rodriguez and S. Atsumi, Chem. Rev., 2013, 113, 4611 CrossRef CAS PubMed.
- For selected recent reviews on transfer hydrogenation using synthetic NADH analogues as the hydrogen donors, see; (a) S.-L. You, Chem.–Asian J., 2007, 2, 820 CrossRef CAS PubMed; (b) S. G. Ouellet, A. M. Walji and D. W. C. Macmillan, Acc. Chem. Res., 2007, 40, 1327 CrossRef CAS PubMed; (c) M. Rueping, E. Sugiono and F. R. Schoepke, Synlett, 2010, 21, 852 CrossRef; (d) M. Rueping, J. Dufour and F. R. Schoepke, Green Chem., 2011, 13, 1084 RSC; (e) J. G. de Vries and N. Mršić, Catal. Sci. Technol., 2011, 1, 727 RSC; (f) C. Zheng and S.-L. You, Chem. Soc. Rev., 2012, 41, 2498 RSC; (g) A. M. F. Philips and A. J. L. Pomberio, Org. Biomol. Chem., 2017, 15, 2307 RSC.
- A. McSkimming and S. B. Colbran, Chem. Soc. Rev., 2013, 42, 5439 RSC.
- For catalytic transfer hydrogenation of imines with a Hantzsch ester, see; (a) M. Rueping, C. Azap, E. Sugiono and T. Theissmann, Synlett, 2005, 2367 CrossRef CAS; (b) M. Rueping, E. Sugiono, C. Azap, T. Theissmann and M. Bolte, Org. Lett., 2005, 7, 3781 CrossRef CAS PubMed; (c) S. Hoffmann, A. M. Seayad and B. List, Angew. Chem., Int. Ed., 2005, 44, 7424 CrossRef CAS PubMed; (d) T. B. Nguyen, H. Bousserouel, Q. Wang and F. Guéritte, Adv. Synth. Catal., 2011, 353, 257 CrossRef CAS; (e) T. B. Nguyen, Q. Wang and F. Guéritte, Chem.–Eur. J., 2011, 17, 9576 CrossRef CAS PubMed; (f) P. Bachu, C. Zhu and T. Akiyama, Tetrahedron Lett., 2013, 54, 3977 CrossRef CAS; (g) T. Akiyama, K. Saito, S. Janczak and Z. J. Lesnikowsiki, Synlett, 2014, 25, 795 CrossRef CAS; (h) V. N. Wakchaure, P. S. J. Kaib, M. Leutzsch and B. List, Angew. Chem., Int. Ed., 2015, 54, 11852 CrossRef CAS PubMed; (i) J. Chen, Z. Zhang, Z. Bao, Y. Su, H. Xing, Q. Yang and Q. Ren, ACS Appl. Mater. Interfaces, 2017, 9, 9772 CrossRef CAS PubMed; (j) Q. Wang, J. Chen, X. Feng and H. Du, Org. Biomol. Chem., 2018, 16, 1448 RSC.
- For catalytic transfer hydrogenation of α,β-unsaturated carbonyls with a Hantzsch ester, see; (a) J. W. Yang, M. T. Hechavarria Fonseca and B. List, Angew. Chem., Int. Ed., 2004, 43, 6660 CrossRef PubMed; (b) J. W. Yang, M. T. Hechavarria Fonseca, N. Vignola and B. List, Angew. Chem., Int. Ed., 2005, 44, 108 CrossRef CAS PubMed; (c) S. G. Ouellet, J. B. Tuttle and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 32 CrossRef CAS PubMed; (d) S. Mayer and B. List, Angew. Chem., Int. Ed., 2006, 45, 4193 CrossRef CAS PubMed; (e) J. B. Tuttle, S. G. Ouellet and D. W. C. MacMillan, J. Am. Chem. Soc., 2006, 128, 12662 CrossRef CAS PubMed; (f) N. J. A. Martin and B. List, J. Am. Chem. Soc., 2006, 128, 13368 CrossRef CAS PubMed; (g) G.-L. Zhao and A. Córdova, Tetrahedron Lett., 2006, 47, 7417 CrossRef CAS; (h) T. J. Hoffman, J. Dash, J. H. Rigby, S. Arseniyadis and J. Cossy, Org. Lett., 2009, 11, 2756 CrossRef CAS PubMed; (i) K. Akagawa, H. Akabane, S. Sakamoto and K. Kudo, Tetrahedron: Asymmetry, 2009, 20, 461 CrossRef CAS; (j) J. Che and Y. Lam, Synlett, 2010, 2415 CAS; (k) C. Ebner and A. Pfaltz, Tetrahedron, 2011, 67, 10287 CrossRef CAS; (l) D. B. Ramachary, R. Sakthidevi and P. S. Reddy, RSC Adv., 2013, 3, 13497 RSC.
- For catalytic transfer hydrogenations of α-keto esters with a Hantzsch ester, see; J. W. Yang and B. List, Org. Lett., 2006, 8, 5653 CrossRef CAS PubMed.
- For Brønsted acid-mediated transfer hydrogenation of nonactivated aldehydes with synthetic NADH analogues, see; (a) S. Shinkai, H. Hamada and O. Manabe, Tetrahedron Lett., 1979, 20, 1397 CrossRef; (b) S. Fukuzumi, M. Ishikawa and T. Tanaka, J. Chem. Soc., Chem. Commun., 1985, 1069 RSC; (c) S. Fukuzumi, M. Ishikawa and T. Tanaka, Tetrahedron, 1986, 42, 1021 CrossRef CAS; (d) M. Ishikawa and S. Fukuzumi, J. Chem. Soc., Chem. Commun., 1990, 1353 RSC; (e) K. Kuroda, T. Nagamatsu, R. Yanada and F. Yoneda, J. Chem. Soc., Perkin Trans. 1, 1993, 547 RSC.
- For Lewis acid-mediated hydrogenation of nonactivated aldehydes with synthetic NADH analogues, see; (a) D. J. Creighton and D. S. Sigman, J. Am. Chem. Soc., 1971, 93, 6314 CrossRef CAS (Zn); (b) M. Shirai, T. Chishina and M. Tanaka, Bull. Chem. Soc. Jpn., 1975, 48, 1079 CrossRef CAS (Zn, Pb, Cb, Cu); (c) D. J. Creighton, J. Hajdu and D. S. Sigman, J. Am. Chem. Soc., 1976, 98, 4619 CrossRef CAS PubMed (Zn). (d) Y. Ohnishi and M. Kitami, Tetrahedron Lett., 1978, 19, 4033 CrossRef (Mg); (e) M. Hughes and R. H. Prince, J. Inorg. Nucl. Chem., 1978, 40, 703 CrossRef CAS (Zn, Ni); (f) R. A. Hood and R. H. Prince, J. Chem. Soc., Chem. Commun., 1979, 163 RSC (Zn); (g) R. Mathis, G. Dupas, A. Decormeille and G. Quéguiner, Tetrahedron Lett., 1981, 22, 59 CrossRef CAS (Mg); (h) A. Ohno, Y. Ishihara, S. Ushida and S. Oka, Tetrahedron Lett., 1982, 23, 3185 CrossRef CAS (B, Ti, Al, Zr, Sb); (i) G. Dupas, J. Bourguignon, C. Ruffin and G. Quéguiner, Tetrahedron Lett., 1982, 23, 5141 CrossRef CAS (Mg); (j) H. Awano and W. Tagaki, J. Chem. Soc., Chem. Commun., 1985, 994 RSC (Zn). (k) J. Cazin, G. Dupas, J. Bourguignon and G. Quéguiner, Tetrahedron Lett., 1986, 27, 2375 CrossRef CAS (Mg); (l) J. F. J. Engbersen, A. Koudijs and H. C. van der Plas, Bioorg. Chem., 1988, 16, 215 CrossRef CAS (Zn); (m) G. Gelbard, J. Lin and N. Roques, J. Org. Chem., 1992, 57, 1789 CrossRef CAS (Mg, Zn, Nd, La, Eu, Al).
- G. Hamasaka, H. Tsuji and Y. Uozumi, Synlett, 2015, 26, 2037 CrossRef CAS.
- (a) L. Deloux and M. Srebnik, Chem. Rev., 1993, 93, 763 CrossRef CAS; (b) P. J. Duggan and E. D. Tyndall, J. Chem. Soc., Perkin Trans. 1, 2002, 1325 RSC; (c) K. Ishihara, in Boronic Acids: Preparation and Applications in Organic Synthesis and Medicine, ed. D. G. Hall, Wiley-VCH, Weinheim, ch. 10, 2005, pp. 377–409 Search PubMed; (d) E. Dimitrijević and M. S. Taylor, ACS Catal., 2013, 3, 945 CrossRef.
- (a) W. E. Piers and T. Chivers, Chem. Soc. Rev., 1997, 26, 345 RSC; (b) K. Ishihara and H. Yamamoto, Eur. J. Org. Chem., 1999, 527 CrossRef CAS; (c) G. Erker, Dalton Trans., 2005, 1883 RSC; (d) W. E. Piers, Adv. Organomet. Chem., 2005, 52, 1 CAS.
- (a) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46 CrossRef CAS PubMed; (b) D. W. Stephan and G. Erker, Top. Curr. Chem., 2013, 332, 85 CrossRef CAS PubMed; (c) J. Paradies, Synlett, 2013, 24, 777 CrossRef CAS; (d) J. Paradies, Angew. Chem., Int. Ed., 2014, 53, 3552 CrossRef CAS PubMed; (e) D. W. Stephan, Acc. Chem. Res., 2015, 48, 306 CrossRef CAS PubMed; (f) M. Oestreich, J. Hermeke and J. Mohr, Chem. Soc. Rev., 2015, 44, 2202 RSC; (g) D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed; (h) W. Meng, X. Feng and H. Du, Acc. Chem. Res., 2018, 51, 191 CrossRef CAS PubMed.
- Combinations of B(C6F5)3 and an ether (Et2O, i-Pr2O, or 1,4-dioxane) catalyze the hydrogenation of ketones and some aldehydes with H2 gas, see: (a) T. Mahdi and D. W. Stephan, J. Am. Chem. Soc., 2014, 136, 15809 CrossRef CAS PubMed; (b) D. J. Scott, M. J. Fuchter and A. E. Ashley, J. Am. Chem. Soc., 2014, 136, 15813 CrossRef CAS PubMed.
- A combination of B(C6F5)3 and cyclodextrin or molecular sieves also catalyzed the hydrogenation of ketones and aldehydes with H2 gas; see: T. Mahdi and D. W. Stephan, Angew. Chem., Int. Ed., 2015, 54, 8511 CrossRef CAS PubMed.
- J. D. Webb, V. S. Laberge, S. J. Geier, D. W. Stephan and C. M. Crudden, Chem.–Eur. J., 2010, 16, 4895 CrossRef CAS PubMed.
- I. Chatterjee and M. Oestreich, Angew. Chem., Int. Ed., 2015, 43, 1965 CrossRef PubMed.
- S. Keess and M. Oestreich, Chem. Sci., 2017, 8, 4688 RSC.
- See Fig. S1 in ESI.†.
- Very recently, Melen, Oestreich, and co-workers reported a tris[3,5-bis(trifluoromethyl)phenyl]borane-catalyzed hydroboration of imines. They also proposed a similar Lewis acid activation pathway; see; Q. Yin, Y. Soltani, R. L. Melen and M. Oestreich, Organometallics, 2017, 36, 2381 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision E.O1), Gaussian, Inc., Wallingford, 2013 Search PubMed.
- Y. Zhao and D. G. Truhalar, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS.
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, Canada, 2009, http://www.cylview.org Search PubMed.
- See ESI.†.
- Piers and co-workers determined the association constant for the formation of Int-Ib [Ka = 2.1(1) × 104] by another 1H NMR technique (not VT 1H NMR) in C6D6: see; D. J. Parks, W. E. Piers, M. Parvez, R. Atencio and M. J. Zaworotko, Organometallics, 1998, 17, 1369 CrossRef CAS . In our study, the association constant was obtained by VT 1H NMR measurements on a 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (molar) mixture of B-b and 1 in CD2Cl2. Because the solubility of B-a for C6D6 is low, we chose CD2Cl2 as the solvent for our NMR experiments. The different solvent and measurement method might have affected the result.
:1 (molar) mixture of B-b and 1 in CD2Cl2. Because the solubility of B-a for C6D6 is low, we chose CD2Cl2 as the solvent for our NMR experiments. The different solvent and measurement method might have affected the result.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details. See DOI: 10.1039/c9ra01468c |
| This journal is © The Royal Society of Chemistry 2019 |