
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An efficient 3-acylquinoline synthesis from acetophenones and anthranil via C(sp3)–H bond activation mediated by Selectfluor†
Yejun Gaoab,
Robert C. Hider c and
Yongmin Ma*ab
c and
Yongmin Ma*ab
aSchool of Pharmaceutical and Chemical Engineering, Taizhou University, Taizhou 318000, PR China. E-mail: yongmin.ma@tzc.edu.cn
bSchool of Pharmaceutical Science, Zhejiang Chinese Medical University, Hangzhou, 310053, PR China
cInstitute of Pharmaceutical Science, King's College London, Franklin-Wilkins Building, Stamford Street, London, SE1 9NH, UK
First published on 2nd April 2019
Abstract
An efficient method for the synthesis of 3-functionalized quinolines from commercially available acetophenones and anthranil has been described. Selectfluor propels the C(sp3)–H bond activation of the acetophenones and aza-Michael addition of anthranil resulting in annulated 3-acylquinolines in moderate to high yields. DMSO acts not only as a solvent but also as a one carbon donor in the reaction.
Introduction
Quinolines are one of the most ubiquitous structural units in natural products,1–5 biologically active compounds6–11 and functionalized materials (Fig. 1).12–14 The synthesis of quinolines has been an active area for many years and as a result, a number of efficient synthetic methods have been developed,15–20 such as the classical Skraup,21–23 Combes,24–26 Friedlander,27–29 Gould–Jacobs,30–32 and Doebner–von Miller reactions.33–36 More recently, alternative strategies for the quinoline synthesis such as domino cycloadditions37–41 and transition-metal mediated methods42–45 have been introduced. However, most of the existing methods necessitate the use of strong acid or base conditions, high reaction temperatures, the use of expensive/toxic metal catalysts and frequently require the use of expensive, highly functionalized starting materials. Such issues present serious problems from the environmental point of view and for industrial scale production. Therefore, the development of new synthetic protocols for the quinoline synthesis in sustainable, environmentally friendly and efficient fashion are highly desirable.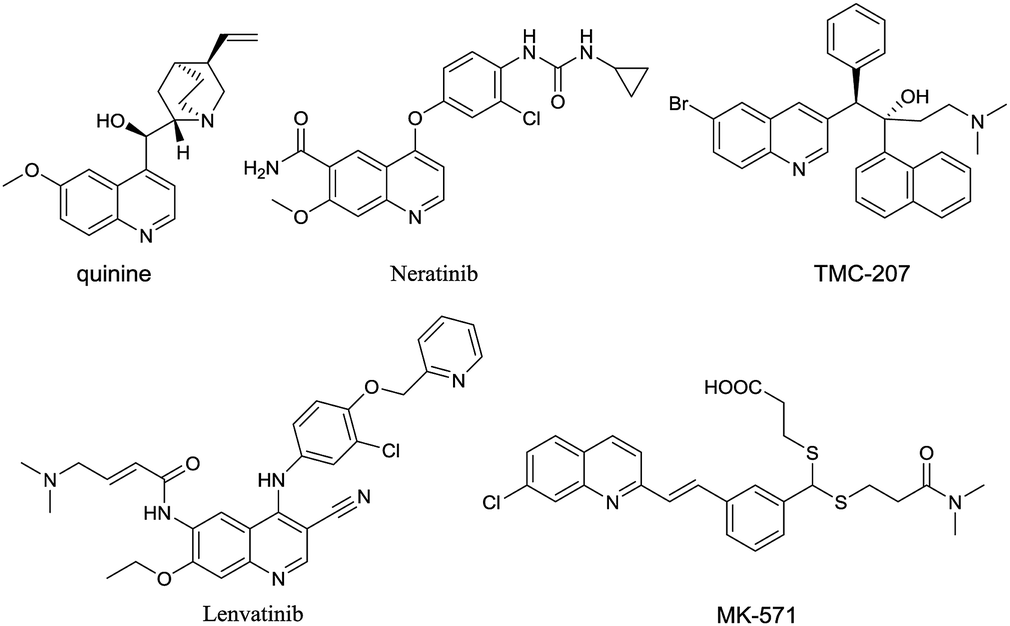 | ||
| Fig. 1 Examples of biological active quinoline derivatives. | ||
3-Acylquinolines have been reported to possess herbicidal activity46 as well as antihypertensive activity.47 Some efficient methods for the preparation of 3-acylquinolines have been developed, such as Pd-catalyzed carbonylative Suzuki cross-coupling reactions of arylboronic acid with 3-iodoquinoline,48 Pd-catalyzed coupling of aldehydes and 3-bromoquinoline49 or arylboronic acid and 3-quinolinecarbaldehyde,50 Fe-catalyzed cascade Michael addition/cyclization of o-aminoaryl aldehydes/ketones/alcohols with ynones,45 and domino reactions between N,N-dimethyl enaminones and anilines.41 In addition, dehydrogenation of saturated carbonyl compounds to afford α,β-unsaturated carbonyl derivatives has been found to be compatible with other organic transformations, leading to efficient one-pot protocols for the synthesis of functionalized molecules.51–54 Stimulated by this excellent pioneering work, Fan et al. developed a copper-catalyzed α,β functionalization of saturated ketones with 2-aminoaryl carbonyl compounds via a C(sp3)–H bond amination, enaminone formation, and enamine-carbonyl condensation process.55 Tiwari et al. reported a similar reaction starting from saturated ketones and anthranils via sequential dehydrogenation/aza-Michael addition/annulations cascade reactions in a one pot.56 The same group also reported the reaction anthranil is an isoxazole derivative which has been used for synthesis of a couple of heterocycles by cleavage of N–O bond.57–61 Inspired by these excellent pioneering work, especially Tiwari's report that quinolines can be transformed from the reaction of in situ generated α,β-unsaturated ketones from acetophenones via one-carbon homologation by DMSO followed by the aza-Michael addition of anthranils and subsequent annulation,62 we were encouraged to use Selectfluor63–68 as an alternative oxidant for the synthetic approach to 3-acylquinolines from commercially available acetophenones and anthranil. In this case, DMSO was applied as both carbon source and reaction medium. Herein, we wish to report the new protocol involving N–O bond cleavage and the formation of three C–C bonds.
Results and discussion
To initiate the study, a reaction was carried out using acetophenone (1a) and anthranil (2a) as model substrates, the details being summarized in Table 1. We first performed the reaction with Selectfluor as an oxidant in DMSO, and a good yield (71%) was achieved when the reaction was carried out at 100 °C for 24 h (entry 1). In an attempt to improve the efficiency, the effect of various carbon donors such as DMF, N,N-dimethylacetamide (DMAC) and N-methylformamide (NMF) was investigated, but appreciably lower yields were obtained (entries 2–4). The subsequent brief investigation in varying the carbon source indicated that DMSO was a relatively ideal carbon source. Furthermore, the reaction did not proceed at all in toluene, a non-carbon donor solvent (entry 5). We then investigated the effect of oxidants on the transformation. (NH4)2S2O8 or K2S2O8 has been reported to efficiently oxidize acetophenones and other substrates,69–72 and Tiwari and co-workers declared that 37% and 76% yield of 3aa were obtained at 120 °C mediated by (NH4)2S2O8 and K2S2O8, respectively.62 However, we found that both (NH4)2S2O8 and K2S2O8 are much less efficient in our conditions as compared with Selectfluor (entries 6 and 7 vs. 1). In the absence of any oxidant, the desired product was not observed (entry 8). Increasing the reaction time to 48 h did not markedly improve the efficiency (entry 9). However, a significantly lower yield was achieved when the reaction was performed for 16 h or less, with a recovery of a large amount of the starting material (entries 10–12). Increasing or decreasing the reaction temperature away from 100 °C did not improve the product yield (entries 13–16 vs. 1). Based on these results, the optimal reaction conditions selected were acetophenone (1 mmol), anthranil (1 mmol) and Selectfluor (1.1 mmol) in DMSO (3 mL) at 100 °C in open air for 24 h.
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Oxidant | Time (h) | Temp (°C) | Yield (%) |
| a Reaction conditions: 1a (1 mmol), 2 (1 mmol), oxidant (1.1 mmol) and DMSO (3 mL) in a sealed tube at the indicated reaction conditions.b Reaction under N2.c Reaction was performed on a 10 mmol scale of 1a. | |||||
| 1 | DMSO | SF | 24 | 100 | 71, 15b, 65c |
| 2 | DMF | SF | 24 | 100 | 23 |
| 3 | DMAC | SF | 24 | 100 | Trace |
| 4 | Toluene | SF | 24 | 100 | 0 |
| 5 | NMF | SF | 24 | 100 | 18 |
| 6 | DMSO | (NH4)2S2O8 | 24 | 100 | Trace |
| 7 | DMSO | K2S2O8 | 24 | 100 | 33 |
| 8 | DMSO | — | 24 | 100 | 0 |
| 9 | DMSO | SF | 48 | 100 | 73 |
| 10 | DMSO | SF | 16 | 100 | 46 |
| 11 | DMSO | SF | 8 | 100 | 23 |
| 12 | DMSO | SF | 4 | 100 | Trace |
| 13 | DMSO | SF | 24 | 120 | 67 |
| 14 | DMSO | SF | 24 | 80 | 56 |
| 15 | DMSO | SF | 24 | 60 | 29 |
| 16 | DMSO | SF | 24 | 40 | 0 |
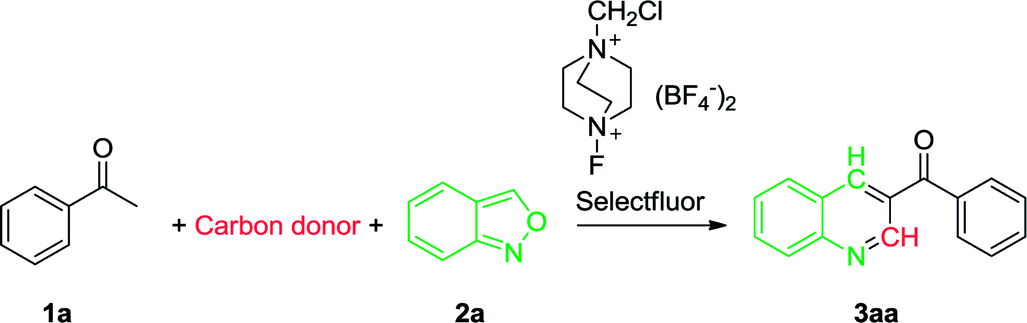
With the optimal reaction conditions in hand, we progressed to explore the substrate scope of the aryl methyl ketones and anthranils in order to investigate the reaction generality. The results are summarized in Table 2. We first examined the substituent effect of aryl ketones with anthranil 2a. A variety of aryl methyl ketones were well tolerated with both electron-donating groups (–OMe, –Me, –n-Pr) and electron-withdrawing groups (–Br, –Cl, –F) on the aromatic ring, leading to the desired products in 48–74% yield (3aa–3da, 3ga–3ka, 3ma–3ra). The electronic nature of the aryl moiety had no marked effect on the product yield. However, their steric property did influence the yield appreciably. For instance, the average yield of the ortho-substituted 3aa–3da is 57% whereas the average yields of the meta- and para-substituted analogues (3ga–3ka and 3ma–3ra) are 63% and 64%, respectively. In addition, when a hydroxyl or amino group was introduced on the aromatic ring, the corresponding product failed to form (3ea, 3fa, 3la and 3sa). This may be due to the influence of an active proton on the reaction. However, when the amino group was protected with two methyl groups (–NMe2), the corresponding 3-acylquinoline (3ta) also failed to be formed. The desired product was also not prepared when a nitro substituted acetophenone was employed as the substrate (3ua).
We also investigated the reaction generality of di-substituted acetophenones and heterocyclic ketones. In similar fashion to mono-substituted analogues, the di-substituted methyl ketones reacted smoothly to furnish the corresponding 3-acylquinolines in an average yield of 57% (3va–3b′a) (Table 2). Again, steric hindrance influences the reaction yield dramatically. As a result, 59% and 45% yield were achieved when 1-(naphthalene-2-yl)ethanone and 1-(naphthalene-1-yl)ethanone were employed respectively (3a′a and 3b′a). With the heterocyclic ketones employed, only 2-thienyl ketone (1c′a) and 2-furanyl ketone (1d′) were successfully converted into the corresponding 3-heterocyclic quinolines in moderate yields (47% and 41% respectively) and the other three (1e′–1g′) failed. Aliphatic ketones such as 1-phenylpropan-2-one (1h′) and 4-phenylbutan-2-one (1i′) failed to provide the desired products.
The scope of substituted anthranils in this novel reaction was also investigated (Table 2). Anthranils bearing either an electron-withdrawing group (EWG) such as a fluoro, chloro and bromo group, or an electron-donating group (EDG) such as dimethoxy group on the aromatic ring, underwent this reaction smoothly to provide the corresponding quinolines in moderate to good yields (3ab-3ae). In this case, the introduction of the EWG did not influence the product yield (3ab–3ad vs. 3aa) while the introduction of the EDG decreased the yield from 71% to 58% (3ae vs. 3aa).
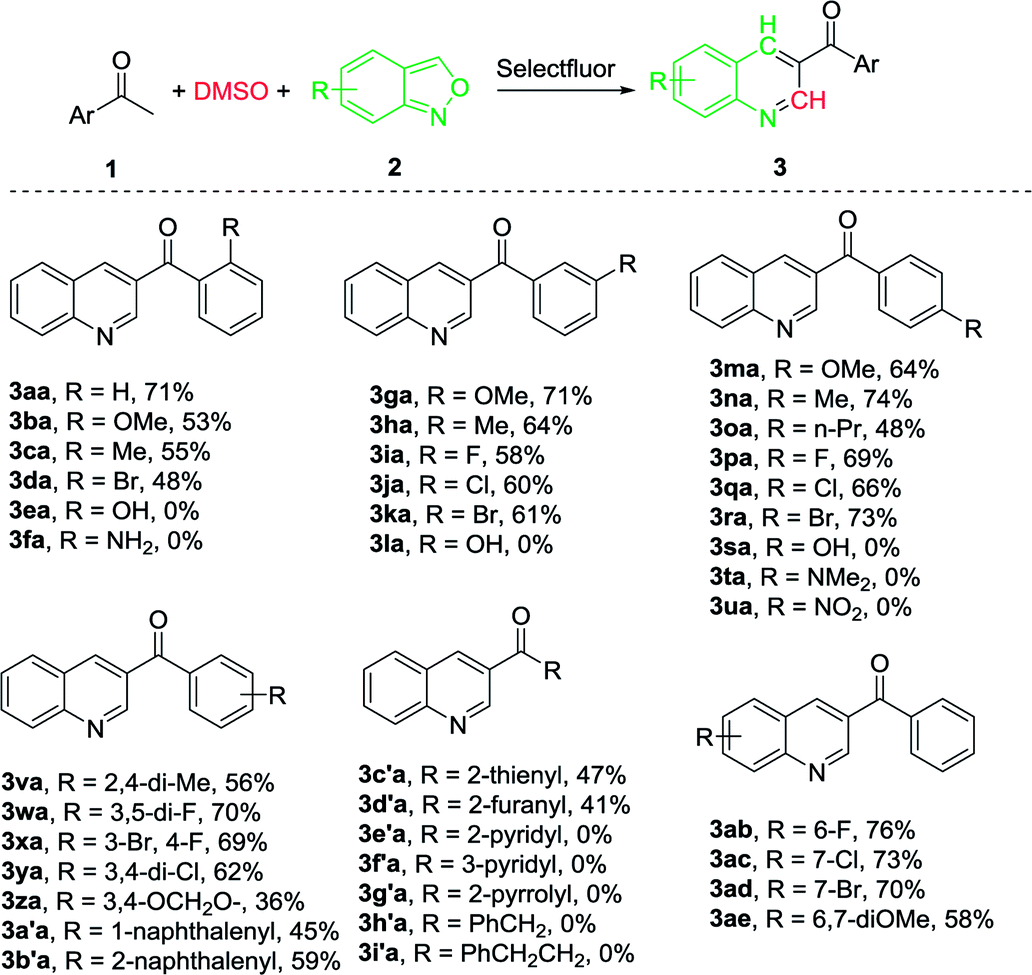
This initial success led us to further investigate the synthesis of biologically active molecules (Scheme 1). In this regard, both the ketone and halide groups are well tolerated and the method offers the chance for further diversification and amplification of the quinoline moiety. For example, the (2-bromophenyl)(quinolin-3-yl)methanone (3da) was heated in MeOH in the presence of methoxyamine to afford quinolinylmethanone oxime (4) as bactericidal agent for agricultural application.73 3da was also reported to successively synthesize 2,3-dihydro-5-(3-quinolinyl)-1H-1,4-benzodiazepine (5) as a fungicide.56,62,74
 | ||
| Scheme 1 Conversion of 3-acylquinoline into biologically active molecules. | ||
Several control experiments were performed to gain a preliminary insight into the reaction mechanism (Scheme 2). When the reaction was carried out in the presence of TEMPO (2 equiv.), a similar yield (65%) of the desired product (3aa) was afforded (71% in the absence of TEMPO), indicating that a radical process may not be involved in the reaction. When the reaction was carried out under N2 atmosphere, the desired product was obtained at 15% yield only (Table 1, entry 1), suggesting that O2 was involved in the oxidative process. On the other hand, no markedly improved yield was given when the reaction was carried out under oxygen atmosphere. To verify the carbon source in the reaction from DMSO, the reaction was performed in methyl phenyl sulfoxide and diphenyl sulfoxide, the expected product 3aa was obtained in 58% and 0% yield, respectively. Furthermore, the reaction of 1a was performed with deuterium-labelling DMSO and a C2 deuterated 3aa′ was the sole product. This result confirmed that the carbon on the 2-position of the pyridine ring originated from DMSO.
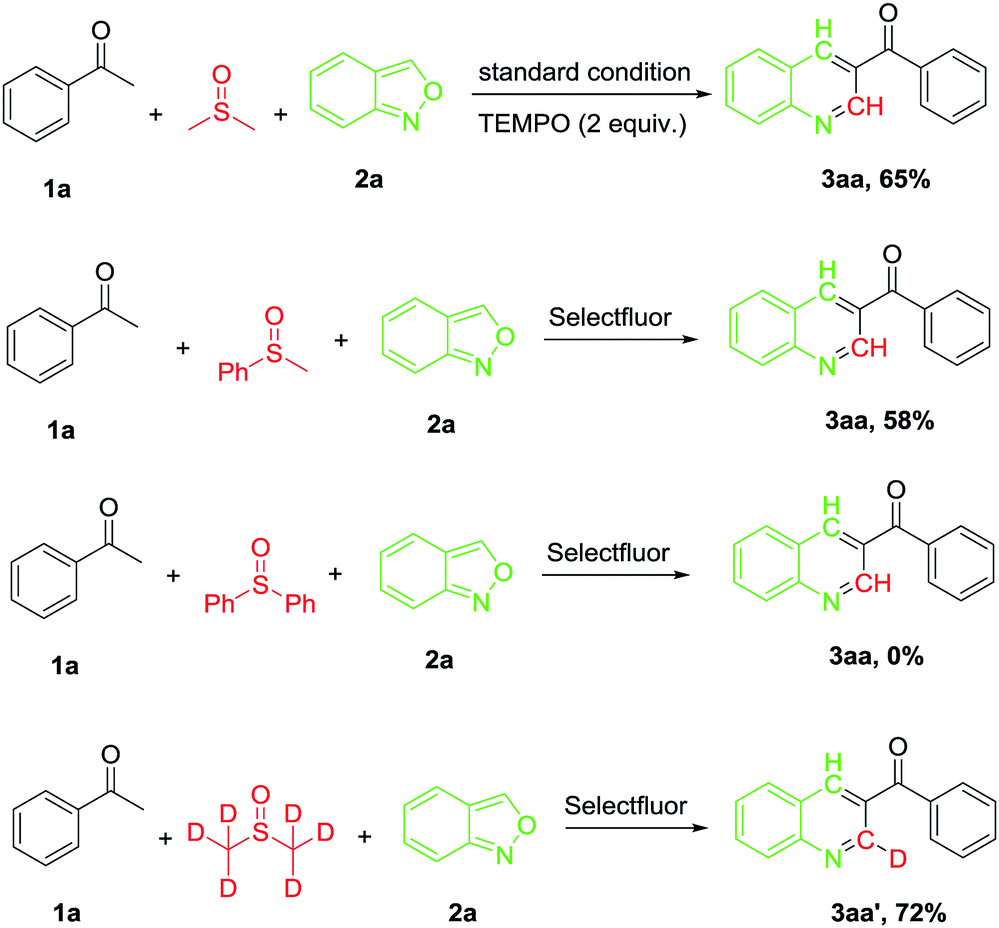 | ||
| Scheme 2 Control experiments. | ||
To illustrate the suitability of this new synthetic method on an enlarged scale, a gram-scale experiment (10 mmol) was carried out. The reaction of 1a proceeded smoothly under standard conditions to afford the corresponding product 3aa in 65% yield (Table 1, entry 1) without an appreciable loss of efficiency (1 mmol, 71%).
On the basis of these preliminary experimental results and previous literature reports,62,68 a possible mechanism is proposed (Scheme 3). DMSO is initially activated by Selectfluor to furnish α-fluorinated intermediate A or methyl(methylene)sulfonium cation A′, which on reaction with acetophenone 1 furnishes intermediate B or B′, respectively. The intermediate B or B′ undergoes spontaneous demethylsulfinylation or demethylthioation to remove MeS(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)H or MeSH, respectively. The intermediate C undergoes [4 + 2] cycloaddition with anthranil 2 to afford intermediate D which followed by double elimination to remove water to give the final product 3.
O)H or MeSH, respectively. The intermediate C undergoes [4 + 2] cycloaddition with anthranil 2 to afford intermediate D which followed by double elimination to remove water to give the final product 3.
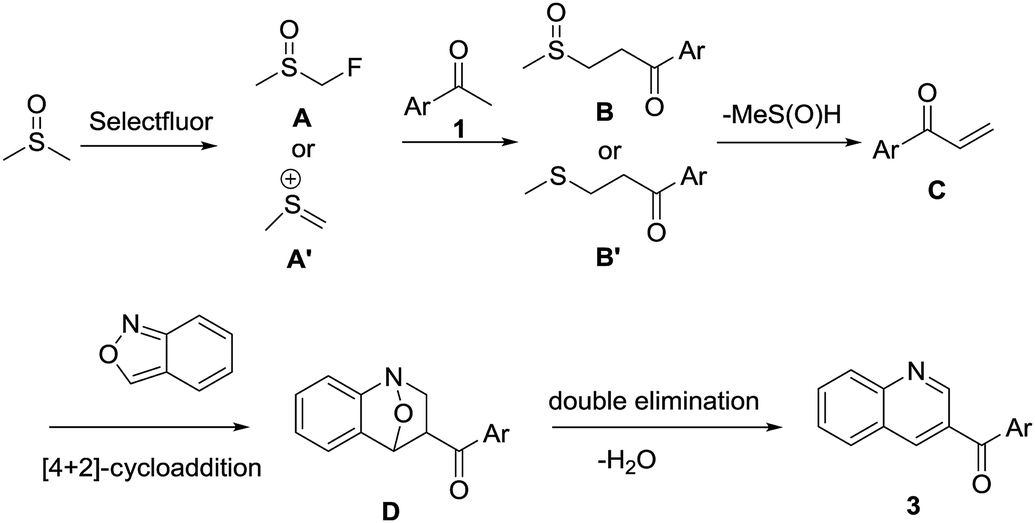 | ||
| Scheme 3 Proposed reaction mechanism. | ||
Conclusions
In conclusion, we have successfully developed a Selectfluor-promoted efficient three-component cascade reaction procedure for the synthesis of 3-acylquinolines from readily available acetophenones, anthranils and DMSO. The reaction is compatible with various ketones and anthranils and DMSO is served as both solvent and a carbon source. Considering the wide availability of the substrates, broad substrate generality and the mild reaction conditions, the present work provides an attractive novel protocol for the synthesis of the 3-acylquinolines.Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Jampilek, Curr. Org. Chem., 2017, 21, 1824–1846 CrossRef CAS.
- T. P. Nicholls, D. Leonori and A. C. Bissember, Nat. Prod. Rep., 2016, 33, 1248–1254 RSC.
- B. J. Neves, C. H. Andrade and P. V. L. Cravo, Molecules, 2015, 20, 1872–1903 CrossRef PubMed.
- N. Singh, B. B. Mishra, S. Bajpai, R. K. Singh and V. K. Tiwari, Bioorg. Med. Chem., 2014, 22, 18–45 CrossRef CAS PubMed.
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K.-D. Entian, M. A. Fischbach, J. S. Garavelli, U. Goeransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Mueller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. T. Reaney, S. Rebuffat, R. P. Ross, H.-G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Suessmuth, J. R. Tagg, G.-L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Nat. Prod. Rep., 2013, 30, 108–160 RSC.
- X.-M. Chu, C. Wang, W. Liu, L.-L. Liang, K.-K. Gong, C.-Y. Zhao and K.-L. Sun, Eur. J. Med. Chem., 2018, 161, 101–117 CrossRef PubMed.
- X.-F. Shang, S. L. Morris-Natschke, G.-Z. Yang, Y.-Q. Liu, X. Guo, X.-S. Xu, M. Goto, J.-C. Li, J.-Y. Zhang and K.-H. Lee, Med. Res. Rev., 2018, 38, 1614–1660 CrossRef PubMed.
- I. Balderas-Renteria, P. Gonzalez-Barranco, A. Garcia, B. K. Banik and G. Rivera, Curr. Med. Chem., 2012, 19, 4377–4398 CrossRef CAS PubMed.
- Y. Zhang, T. Han, Q. Ming, L. Wu, K. Rahman and L. Qin, Nat. Prod. Commun., 2012, 7, 963–968 CAS.
- C. Kloeck, Z. Herrera, M. Albertelli and C. Khosla, J. Med. Chem., 2014, 57, 9042–9064 CrossRef PubMed.
- E. S. Coimbra, L. M. R. Antinarelli, N. P. Silva, I. O. Souza, R. S. Meinel, M. N. Rocha, R. P. P. Soares and A. D. da Silva, Chem.-Biol. Interact., 2016, 260, 50–57 CrossRef CAS PubMed.
- H.-y. Chen, J.-R. Wei, J.-X. Pan, W. Zhang, F.-q. Dang, Z.-Q. Zhang and J. Zhang, Biosens. Bioelectron., 2017, 91, 328–333 CrossRef CAS PubMed.
- B.-Y. Ren, D.-K. Zhong, Y.-G. Sun, X.-H. Zhao, Q.-J. Zhang, Y. Liu, M. Jurow, M.-L. Sun, Z.-S. Zhang and Y. Zhao, Org. Electron., 2016, 36, 140–147 CrossRef CAS.
- J. G. Croissant, S. Picard, D. Aggad, M. Klausen, C. M. Jimenez, M. Maynadier, O. Mongin, G. Clermont, E. Genin, X. Cattoen, M. W. C. Man, L. Raehm, M. Garcia, M. Gary-Bobo, M. Blanchard-Desce and J.-O. Durand, J. Mater. Chem. B, 2016, 4, 5567–5574 RSC.
- F. Zhang, Q. Lai, X. Shi and Z. Song, Chin. Chem. Lett., 2019, 30, 392–394 CrossRef CAS.
- X.-F. Zhu, J. Zhang, S. Sun, Y.-C. Guo, S.-X. Cao and Y.-F. Zhao, Chin. Chem. Lett., 2017, 28, 1514–1518 CrossRef CAS.
- M. H. Muhammad, X.-L. Chen, B. Yu, L.-B. Qu and Y.-F. Zhao, Pure Appl. Chem., 2019, 91, 33–41 CAS.
- L.-Y. Xie, Y. Duan, L.-H. Lu, Y.-J. Li, S. Peng, C. Wu, K.-J. Liu, Z. Wang and W.-M. He, ACS Sustainable Chem. Eng., 2017, 5, 10407–10412 CrossRef CAS.
- L.-Y. Xie, S. Peng, F. Liu, Y.-F. Liu, M. Sun, Z.-L. Tang, S. Jiang, Z. Cao and W.-M. He, ACS Sustainable Chem. Eng., 2019 DOI:10.1021/acssuschemeng.9b00200.
- L.-Y. Xie, S. Peng, T.-G. Fan, Y.-F. Liu, M. Sun, L.-L. Jiang, X.-X. Wang, Z. Cao and W.-M. He, Sci. China: Chem., 2019 DOI:10.1007/s11426-018-9446-1.
- J. Jin, S. Guidi, Z. Abada, Z. Amara, M. Selva, M. W. George and M. Poliakoff, Green Chem., 2017, 19, 2439–2447 RSC.
- C. Luedtke, A. Haupt, M. Wozniak and N. Kulak, J. Fluorine Chem., 2017, 193, 98–105 CrossRef CAS.
- G. A. Ramann and B. J. Cowen, Tetrahedron Lett., 2015, 56, 6436–6439 CrossRef CAS.
- F. Aribi, E. Schmitt, A. Panossian, J.-P. Vors, S. Pazenok and F. R. Leroux, Org. Chem. Front., 2016, 3, 1392–1415 RSC.
- T. Selvi and S. Velmathi, J. Org. Chem., 2018, 83, 4087–4091 CrossRef CAS PubMed.
- S. El Kharrat, P. Laurent and H. Blancou, Tetrahedron, 2014, 70, 1252–1266 CrossRef CAS.
- M. K. Barman, A. Jana and B. Maji, Adv. Synth. Catal., 2018, 360, 3233–3238 CrossRef CAS.
- B. Schaefer, M. Schmidtmann and J. Christoffers, Eur. J. Org. Chem., 2018, 4490–4497, DOI:10.1002/ejoc.201800931.
- R. Rubio-Presa, S. Suarez-Pantiga, M. R. Pedrosa and R. Sanz, Adv. Synth. Catal., 2018, 360, 2216–2220 CrossRef CAS.
- M. Bella, M. Schultz and V. Milata, ARKIVOC, 2012, 242–251 Search PubMed.
- L. Lengyel, T. Z. Nagy, G. Sipos, R. Jones, G. Dorman, L. Uerge and F. Darvas, Tetrahedron Lett., 2012, 53, 738–743 CrossRef CAS.
- S. Trah and C. Lamberth, Tetrahedron Lett., 2017, 58, 794–796 CrossRef CAS.
- M. M. Heravi, S. Asadi and F. Azarakhshi, Curr. Org. Synth., 2014, 11, 701–731 CrossRef CAS.
- J. Fotie, H. V. K. Wangun, F. R. Fronczek, N. Massawe, B. T. Bhattarai, J. L. Rhodus, T. A. Singleton and D. S. Bohle, J. Org. Chem., 2012, 77, 2784–2790 CrossRef CAS PubMed.
- J. Horn, S. P. Marsden, A. Nelson, D. House and G. G. Weingarten, Org. Lett., 2008, 10, 4117–4120 CrossRef CAS PubMed.
- R. N. Monrad and R. Madsen, Org. Biomol. Chem., 2011, 9, 610–615 RSC.
- C. M. Melendez Gomez, M. Marsiglia, R. Escarsena, E. del Olmo and V. V. Kouznetsov, Tetrahedron Lett., 2018, 59, 22–25 CrossRef CAS.
- R. Yi, X. Li and B. Wan, Adv. Synth. Catal., 2018, 360, 875–880 CrossRef CAS.
- I. N. C. Kiran, R. S. Reddy, C. Lagishetti, H. Xu, Z. Wang and Y. He, J. Org. Chem., 2017, 82, 1823–1832 CrossRef CAS PubMed.
- E. Sanchez-Diez, D. L. Vesga, E. Reyes, U. Uria, L. Carrillo and J. L. Vicario, Org. Lett., 2016, 18, 1270–1273 CrossRef CAS PubMed.
- J.-P. Wan, Y. Jing and L. Wei, Asian J. Org. Chem., 2017, 6, 666–668 CrossRef CAS.
- B. Orwat, M.-j. O. Oh, M. Kubicki and I. Kownacki, Adv. Synth. Catal., 2018, 360, 3331–3344 CrossRef CAS.
- F. Xiao, C. Liu, S. Yuan, H. Huang and G.-J. Deng, J. Org. Chem., 2018, 83, 10420–10429 CrossRef CAS PubMed.
- L.-Y. Xi, R.-Y. Zhang, L. Zhang, S.-Y. Chen and X.-Q. Yu, Org. Biomol. Chem., 2015, 13, 3924–3930 RSC.
- H. Li, X. Xu, J. Yang, X. Xie, H. Huang and Y. Li, Tetrahedron Lett., 2011, 52, 530–533 CrossRef CAS.
- D.-W. Wang, H.-Y. Lin, R.-J. Cao, T. Chen, F.-X. Wu, G.-F. Hao, Q. Chen, W.-C. Yang and G.-F. Yang, J. Agric. Food Chem., 2015, 63, 5587–5596 CrossRef CAS PubMed.
- H. Kumar, V. Devaraji, R. Joshi, M. Jadhao, P. Ahirkar, R. Prasath, P. Bhavana and S. K. Ghosh, RSC Adv., 2015, 5, 65496–65513 RSC.
- Z. S. Qureshi, K. M. Deshmukh, P. J. Tambade and B. M. Bhanage, Synthesis, 2011, 243–250 CAS.
- T. Wakaki, T. Togo, D. Yoshidome, Y. Kuninobu and M. Kanai, ACS Catal., 2018, 8, 3123–3128 CrossRef CAS.
- M. Kuriyama, N. Hamaguchi, K. Sakata and O. Onomura, Eur. J. Org. Chem., 2013, 3378–3385 CrossRef CAS.
- Y. Shang, X. Jie, J. Zhou, P. Hu, S. Huang and W. Su, Angew. Chem., Int. Ed., 2013, 52, 1299–1303 CrossRef CAS PubMed.
- X. Jie, Y. Shang, X. Zhang and W. Su, J. Am. Chem. Soc., 2016, 138, 5623–5633 CrossRef CAS PubMed.
- J.-L. Zhan, M.-W. Wu, F. Chen and B. Han, J. Org. Chem., 2016, 81, 11994–12000 CrossRef CAS PubMed.
- Y. Chen, J. P. Romaire and T. R. Newhouse, J. Am. Chem. Soc., 2015, 137, 5875–5878 CrossRef CAS PubMed.
- Z. Wang, G. Chen, X. Zhang and X. Fan, Org. Chem. Front., 2017, 4, 612–616 RSC.
- D. K. Tiwari, M. Phanindrudu, S. B. Wakade, J. B. Nanubolu and D. K. Tiwari, Chem. Commun., 2017, 53, 5302–5305 RSC.
- Y. B. Pandit, R. L. Sahani and R.-S. Liu, Org. Lett., 2018, 20, 6655–6658 CrossRef CAS PubMed.
- Z. Zeng, H. Jin, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2018, 57, 16549–16553 CrossRef CAS PubMed.
- Z. Zeng, H. Jin, K. Sekine, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2018, 57, 6935–6939 CrossRef CAS PubMed.
- R. L. Sahani and R.-S. Liu, Angew. Chem., Int. Ed., 2017, 56, 12736–12740 CrossRef CAS PubMed.
- Z. Wang, Z. Yin and X.-F. Wu, Chem.–Eur. J., 2017, 23, 15026–15029 CrossRef CAS PubMed.
- S. B. Wakade, D. K. Tiwari, P. S. K. P. Ganesh, M. Phanindrudu, P. R. Likhar and D. K. Tiwari, Org. Lett., 2017, 19, 4948–4951 CrossRef CAS PubMed.
- Y. Lin, L. Zhu, Y. Lan and Y. Rao, Chem.–Eur. J., 2015, 21, 14937–14942 CrossRef CAS PubMed.
- X. Gao, X. Pan, J. Gao, H. Jiang, G. Yuan and Y. Li, Org. Lett., 2015, 17, 1038–1041 CrossRef CAS PubMed.
- H. Bao, Z. Xu, D. Wu, H. Zhang, H. Jin and Y. Liu, J. Org. Chem., 2017, 82, 109–118 CrossRef CAS PubMed.
- X. Li, X. Fu, Y. Huang and Z. Yan, J. Chem. Res., 2018, 42, 202–205 CrossRef CAS.
- L.-Y. Xie, J. Qu, S. Peng, K.-J. Liu, Z. Wang, M.-H. Ding, Y. Wang, Z. Cao and W.-M. He, Green Chem., 2018, 20, 760–764 RSC.
- Y. Gao, D. Zhou and Y. Ma, ChemistrySelect, 2018, 3, 9374–9377 CrossRef CAS.
- S. D. Jadhav and A. Singh, Org. Lett., 2017, 19, 5673–5676 CrossRef CAS PubMed.
- M. Phanindrudu, S. B. Wakade, D. K. Tiwari, P. R. Likhar and D. K. Tiwari, J. Org. Chem., 2018, 83, 9137–9143 CrossRef CAS PubMed.
- Y.-F. Liu, P.-Y. Ji, J.-W. Xu, Y.-Q. Hu, Q. Liu, W.-P. Luo and C.-C. Guo, J. Org. Chem., 2017, 82, 7159–7164 CrossRef CAS PubMed.
- P. S. Mahajan, S. D. Tanpure, N. A. More, J. M. Gajbhiye and S. B. Mhaske, RSC Adv., 2015, 5, 101641–101646 RSC.
- A. Mi-Tani, WO2010/026771A1, 2010.
- J. I. A. Mitani, R. Kuwahara and M. Sato, US2012/0184732A1, 2012.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra01481k |
| This journal is © The Royal Society of Chemistry 2019 |