
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEDTA-bonded multi-connected carbon-dots and their Eu3+ complex: preparation and optical properties
Tianhao Ji *a,
Peidong Fana,
Xueli Lia,
Zhipeng Meib,
Yongyun Maob and
Yanqing Tianb
*a,
Peidong Fana,
Xueli Lia,
Zhipeng Meib,
Yongyun Maob and
Yanqing Tianb
aScience College, Beijing Technology and Business University, Beijing 100048, China. E-mail: jitianhao@th.btbu.edu.cn
bDepartment of Materials Science and Engineering, Southern University of Science and Technology, Shenzhen 518055, China
First published on 5th April 2019
Abstract
EDTA-bonded multi-connected carbon-dots (EDTA–C-dots) were prepared from carbon dot precursors and complexed with Eu3+ to give Eu3+-coordinated EDTA-bonded multi-connected carbon dots (Eu–EDTA–C-dots). Whereas EDTA–C-dots were readily soluble in DMSO, Eu–EDTA–C-dots could not be easily dissolved in DMSO, water, or other common organic solvents. The newly prepared materials were thoroughly characterized. The X-ray diffraction results showed that no crystalline phase of Eu oxides (europium oxide or europium hydroxide) could be observed in Eu–EDTA–C-dots. The infrared and UV-Vis spectra showed that coordination with Eu3+ ions did not damage the structure of the EDTA–C-dots. It was found that EDTA could be easily grafted on the surface of carbon dots and EDTA had minimal influence on the photoluminescence of the carbon dot matrix. In contrast, the existence of Eu3+ ions strongly quenched the photoluminescence of Eu–EDTA–C-dots. The measured and fitted decay lifetime indicated that Eu–EDTA–C-dots possessed two photoluminescence decay processes, i.e., radiative recombination and non-radiative recombination.
1 Introduction
Carbon dots (C-dots) include all kinds of fluorescent carbon nanoparticles having a particle diameter of <10 nm. The term was first proposed in 2006 by Sun et al.1 and received escalating attention in the subsequent decade.2–6 Carbon dots have been successfully exploited as optical materials in diverse applications, e.g., in studying cytotoxicity and biocompatibility,7–9 bioimaging,6,10,11 drug delivery,12–15 photocatalysis,16 photothermal therapy,17–19 photocurrent and photovoltaics,20,21 metal ion detection,5,22–31 H2O2 detection,32 etc.The detection of metal ions with C-dots, as a kind of promising fluorometric nanosensor, has been investigated extensively.5,22,23 For instance, the detection of Fe3+, Hg2+, and Cu2+ has been achieved based on the fluorescent quenching of the C-dots nanosensor.22–26 Since coexisting Fe3+ and Hg2+ in aqueous solution could interfere with each other, Liu et al. studied the detection of metal ions in the mixture solution of Hg2+ and Fe3+, and demonstrated that C-dots could also be effectively utilized in the presence of SCN− and P3O105− anions.27 Also, to improve the sensitivity of Hg2+ detection in water, Chen et al. prepared C-dots/Au-clusters nanocomposite and studied corresponding ratiometric fluorescence in detail.28 In addition, non-transition metal ions (e.g. K+,26 Al3+,27 Bi3+, and Sn2+) could also be detected using C-dots.31
The detection of rare earth elements with C-dots has been relatively less reported. Singhal et al. used Eu-coordinated C-dots to detect F− ions,33 and indicated that the Eu3+ ions interacted relatively loosely with the carboxylic acid moiety on the C-dots. Kailasa et al. recently reported that Eu3+ hybrid C-dots could be used as a fluorescent nanosensor to detect Hg2+ ions,34 but in their sensor the Eu3+ ions did not directly coordinate with the C-dots. Besides the application of C-dots with Eu3+ as detector, it should be also considered to extend to other field, and first of all, Eu3+ ions need be firmly bonded with C-dots.
Highly fluorescent C-dots can be synthesized from different organic precursors including citric acid, EDTA salts,35 or graphene,36,37 but it remains challenging to bond C-dots firmly with rare-earth ions. To our knowledge, nobody has obtained EDTA-bonded C-dots, which can be bonded easily with Eu3+ ions. In this work, based on the synthesis of EDTA-bonded multi-connected C-dots (referred to as EDTA–C-dots), Eu-coordinated EDTA–C-dots were successfully prepared through a simple procedure and were then characterized systemically.
2 Materials and methods
2.1 Materials
All reagents were of analytical reagent grade and used without further purification. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) and N-hydroxysulfosuccinimide sodium salt (S-NHS) were purchased from Aladdin. Eu(NO3)3·6H2O (99.9%), ethylene diamine tetraacetic acid (EDTA), citric acid, ethylenediamine, and dimethylsulfoxide (DMSO) were purchased from Sinopharm.2.2 Methods
To the solution of the obtained EDTA–C-dots (0.07 g) in DMSO (50 mL) was added Eu(NO3)3·6H2O (0.012 g) under stirring at room temperature. The mixture was then stirred at ambient temperature for one day before the addition of ethanol (100 mL), and the precipitated Eu–EDTA–C-dots suspension was collected by centrifugation and washed with ethanol for four times. After treated under vacuum, the product was stored at room temperature. The Eu3+ content in the Eu–EDTA–C-dots was analyzed by ICP-MS to be about 11.9 wt%.
Fig. 1 gives the schematic illustration of the preparation of EDTA–C-dots and Eu–EDTA–C-dots, along with one HRTEM image of the precursor C-dots (∼2.5 nm in particle diameter).
 | ||
| Fig. 1 Schematic illustration of the preparation process of EDTA–C-dots and Eu–EDTA–C-dots. In the inset, there includes one HRTEM image of precursor C-dots with about 2.5 nm of particle diameter. | ||
3 Results and discussion
3.1 Characterization of C-dots and x-C-dots
In the FTIR spectra of the precursor C-dots and EDTA-free x-C-dots (Fig. 2A), the amide I and amide II bands appeared at around 1690 and 1550 cm−1, which in combination with the vibration absorptions of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N–H bands39 confirmed the presence of the –NHCO– linker in the two samples.40 The absorption peak at around 1020 cm−1 could be assigned to the C–O stretching vibration.41 In addition, C-dots and x-C-dots had similar UV-Vis spectra (Fig. 2B), indicating that the x-C-dots were made of the C-dots. However, their PL spectra differed strikingly because the electron–hole state of various groups on the material surface (e.g. –COOH, –NH2, and –NHCO–) had changed after the reaction.42 The x-C-dots showed the strongest PL intensity under excitation at 340 nm, and aside from the primary fluorescent peak at 480 nm, there was also a broad fluorescent peak at about 430 nm. In contrast, the precursor C-dots had the strongest PL intensity under excitation at 365 nm, and only gave one fluorescent peak at 450 nm without broadening. It could be inferred that the surface groups with PL behaviors underwent certain changes after they were subjected to the reaction conditions (i.e., exposure to EDC and NHS).
O and N–H bands39 confirmed the presence of the –NHCO– linker in the two samples.40 The absorption peak at around 1020 cm−1 could be assigned to the C–O stretching vibration.41 In addition, C-dots and x-C-dots had similar UV-Vis spectra (Fig. 2B), indicating that the x-C-dots were made of the C-dots. However, their PL spectra differed strikingly because the electron–hole state of various groups on the material surface (e.g. –COOH, –NH2, and –NHCO–) had changed after the reaction.42 The x-C-dots showed the strongest PL intensity under excitation at 340 nm, and aside from the primary fluorescent peak at 480 nm, there was also a broad fluorescent peak at about 430 nm. In contrast, the precursor C-dots had the strongest PL intensity under excitation at 365 nm, and only gave one fluorescent peak at 450 nm without broadening. It could be inferred that the surface groups with PL behaviors underwent certain changes after they were subjected to the reaction conditions (i.e., exposure to EDC and NHS).
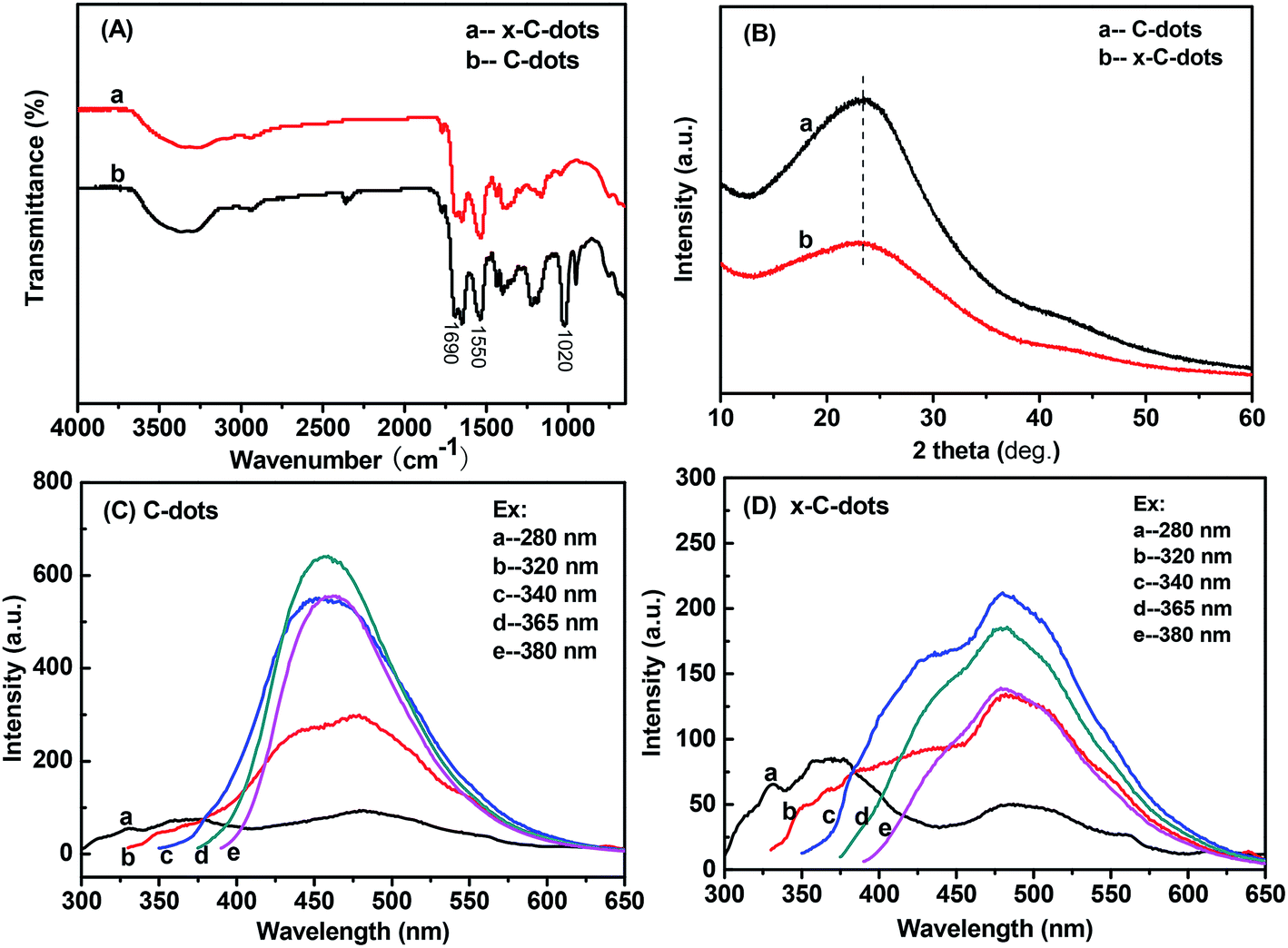 | ||
| Fig. 2 Characterizations of precursor C-dots and EDTA-free x-C-dots: (A) FTIR spectra; (B) XRD patterns; (C) PL of the C-dots; (D) PL of the x-C-dots. | ||
3.2 Characterization of EDTA–C-dots and Eu–EDTA–C-dots
Similarly, EDTA-bonded multi-connected C-dots (EDTA–C-dots) were prepared and further coordinated with Eu3+ to give Eu–EDTA–C-dots. Before the complexation produced, the EDTA–C-dots could be dissolved in DMSO, and whereas it was found that Eu–EDTA–C-dots hardly dissolved in water or other common organic solvents (e.g., DMSO, DMF, ethanol, acetone). X-ray diffraction analysis confirmed that Eu oxides did not exist in the Eu–EDTA–C-dots (Fig. 3A).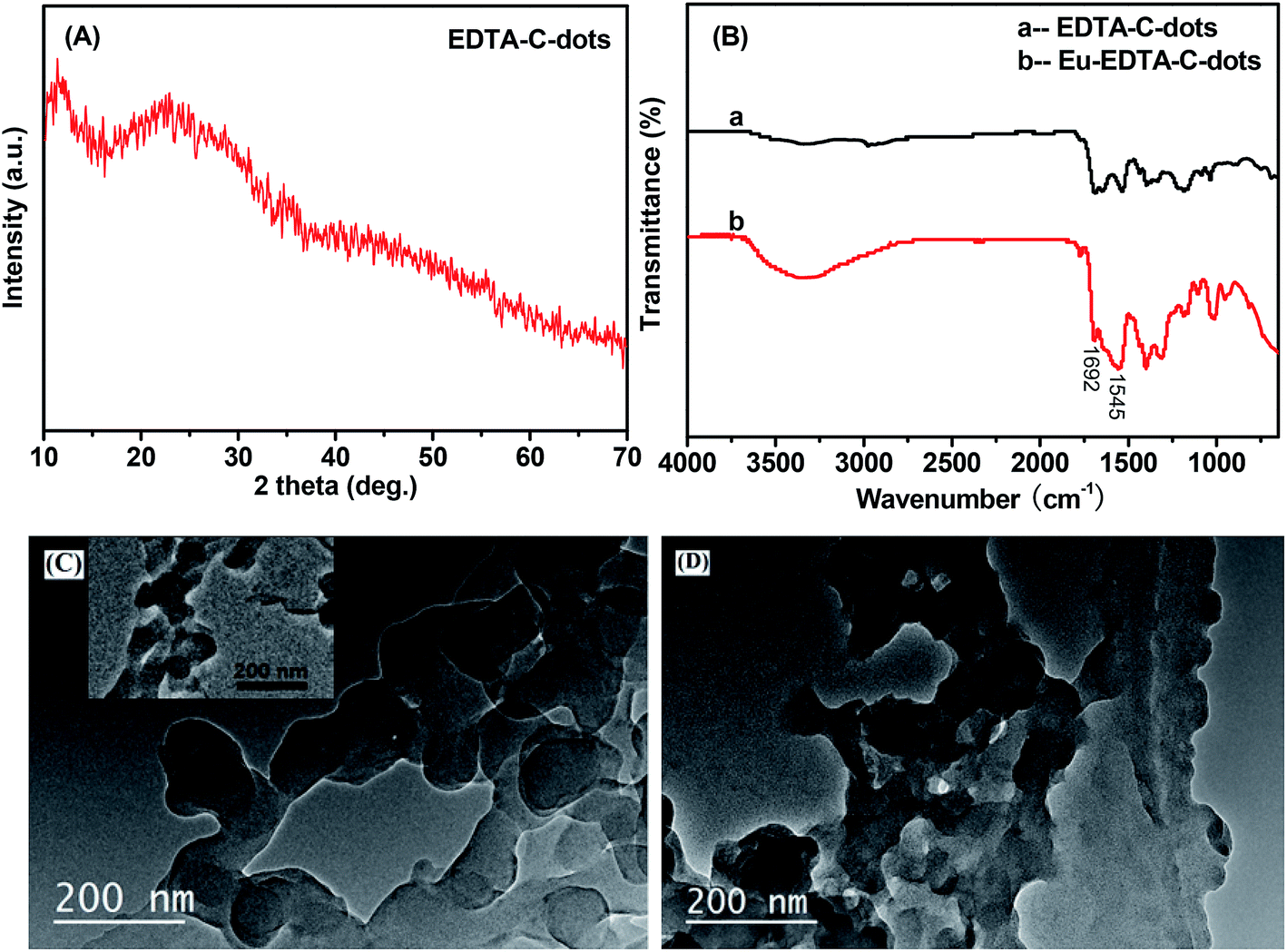 | ||
| Fig. 3 (A) XRD pattern of Eu–EDTA–C-dots; (B) FTIR spectra of EDTA–C-dots and Eu–EDTA–C-dots; (C) TEM image of EDTA–C-dots; (D) TEM image of Eu–EDTA–C-dots. Inset in (C) is the TEM image of precursor x-C-dots. | ||
The complexation between EDTA–C-dots and Eu3+ could also be confirmed by FTIR measurements (Fig. 3B). Since the IR spectrum of the EDTA–C-dots was similar to that of x-C-dots, it could be reasoned that the reaction of EDTA with C-dots barely affected the structure of the C-dots. The amide I and amide II bands at around 1692 cm−1 and 1545 cm−1 in the EDTA–C-dots also proved the existence of the –OCNH– linker.39 In comparison, the absorption pattern of the Eu–EDTA–C-dots in 1500–1700 cm−1 differed prominently, and the notable peak broadening in this area could be attributed to the coordination of the Eu3+ ions.43 It could be inferred that the complexation between Eu3+ ions and EDTA–C-dots changed both the state of valence electrons and the bond length in the amide linker.
The morphologies of the EDTA–C-dots and Eu–EDTA–C-dots could be clearly observed from their TEM images (Fig. 3C and D). Under the preparation condition of the EDTA–C-dots, the precursor C-dots appear obvious aggregation due to the reaction between different C-dots with each other, supported by the TEM image of x-C-dots and below PL results. Similar to the TEM image of the EDTA–C-dots, the TEM image of the x-C-dots in the inset also shows serious aggregation, inferring the morphology of the EDTA–C-dots may result from the reaction between C-dots. In fact, it should be easily understood that because the co-existence of –COOH and –NH2 groups on the surface of each carbon-dot, C-dots can also react with each other under the synthesis of EDTA–C-dots. The coordination of Eu3+ ions still remain similar aggregation state, and the solubility proves that such Eu-complex possesses much serious aggregation.
3.3 Optical properties of EDTA–C-dots and Eu–EDTA–C-dots
The UV-Vis absorptions of the EDTA–C-dots and Eu–EDTA–C-dots (Fig. 4) were similar except for the slight redshift of the absorption peak of Eu–EDTA–C-dots, indicating that the complexation of Eu3+ hardly affected the structure of the EDTA–C-dot ligands. The PL spectra of the EDTA–C-dots and Eu–EDTA–C-dots under excitation at 340 nm (Fig. 4 inset) differed obviously in 400–600 nm. After the coordination of Eu3+ with Eu–EDTA–C-dots, the PL peak at 480 nm experienced serious fluorescence quenching, due to the energy transfer of the non-radiative recombination between Eu3+ ions and C-dots.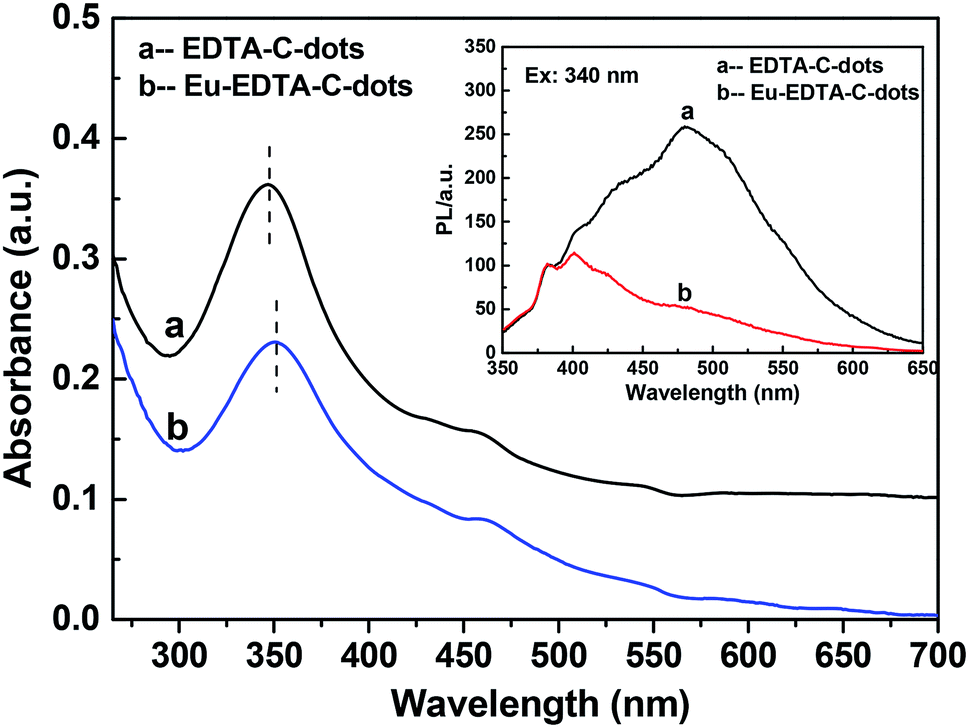 | ||
| Fig. 4 UV-Vis absorption spectra of EDTA–C-dots and Eu–EDTA–C-dots. Inset is the PL spectra of the EDTA–C-dots and Eu–EDTA–C-dots under 340 nm of excitation wavelength. | ||
The PL behaviors of EDTA–C-dots and Eu–EDTA–C-dots upon different excitations were further inspected (Fig. 5). The PL spectrum of the EDTA–C-dots was similar to that of x-C-dots (Fig. 5A), further demonstrating that grafting EDTA on the surface of x-C-dots did not destroy the surface fluorescent groups. The strongest fluorescent peak (at ∼480 nm) of EDTA–C-dots was observed when excitation was applied at 340 nm. In contrast, the addition of Eu3+ obviously quenched the strong PL emission at ∼480 nm, probably due to energy transfer quenching via non-radiative recombination between Eu3+ ions and C-dots (elaborated below further in the lifetime measurements). Similar quenching was previously observed for Au@C-dots or C-dots/ZnO44,45 in the presence of Fe3+, Hg2+, or Pb2+ ions.46–48
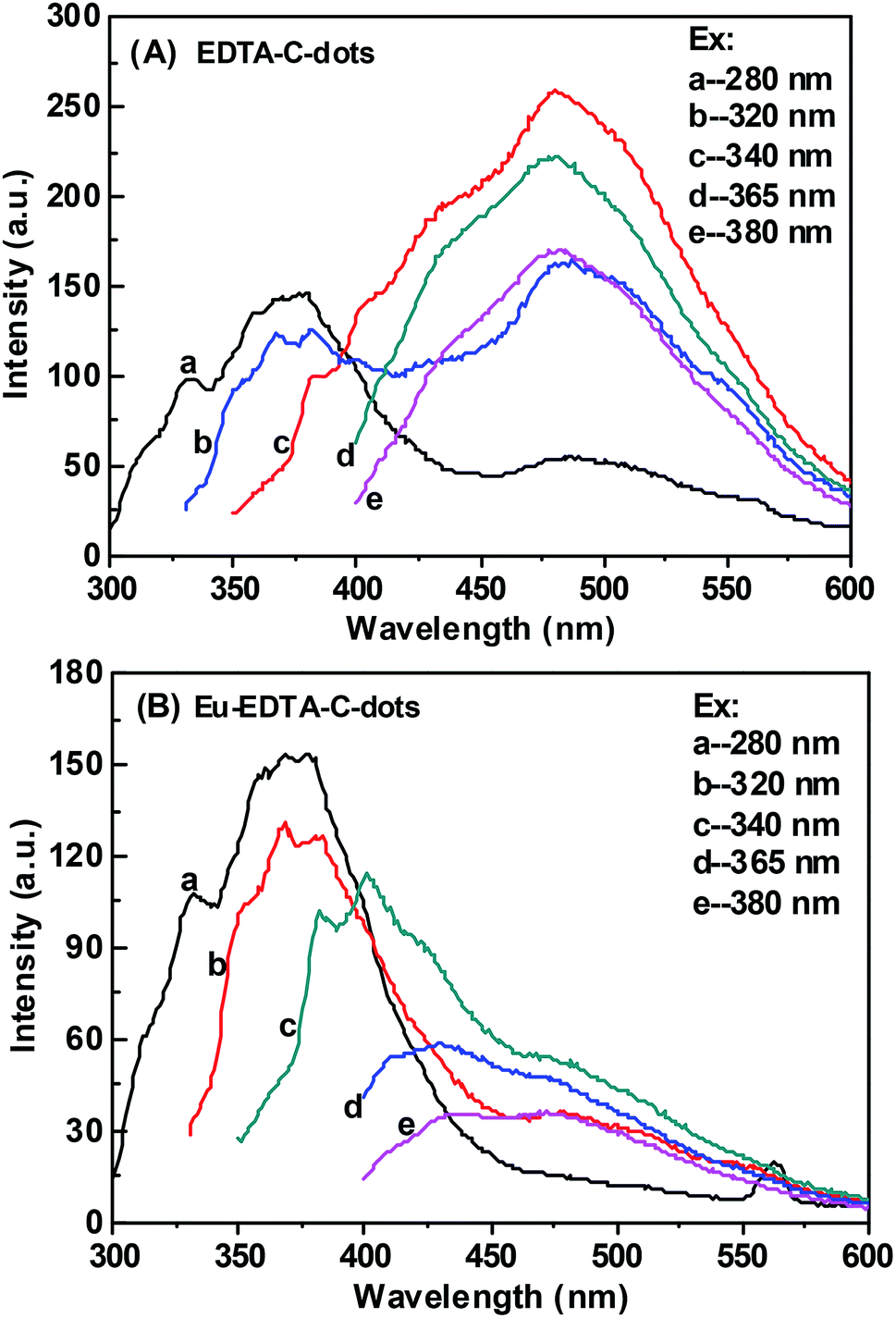 | ||
| Fig. 5 Photoluminescence spectra of EDTA–C-dots and Eu–EDTA–C-dots under different excitation wavelength. | ||
3.4 Luminescent decay spectra of EDTA–C-dots and Eu–EDTA–C-dots
The luminescent decay spectra of EDTA–C-dots and Eu–EDTA–C-dots subjected to excitation at 340 nm revealed the transfer properties of the photo-induced electron (Fig. 6). The PL decay (exciton lifetime) of EDTA–C-dots was measured to be 9.47 ns and could be single-exponentially fitted to be 9.83 ns. In contrast, Eu–EDTA–C-dots had a measured PL decay of 2.47 and 9.86 ns that could be double-exponentially fitted to be 0.85 and 7.13 ns, respectively. Since the C-dot precursor was reported to have a short PL decay of about 3.6 ns,49 the longer exciton lifetime of EDTA–C-dots probably resulted from the radiative recombination between linked C-dots. In contrast, Eu–EDTA–C-dots showed two obviously distinct exciton lifetimes, probably because two kinds of processes were involved in the decay, i.e., radiative recombination between linked C-dots and non-radiative recombination between C-dot and Eu3+.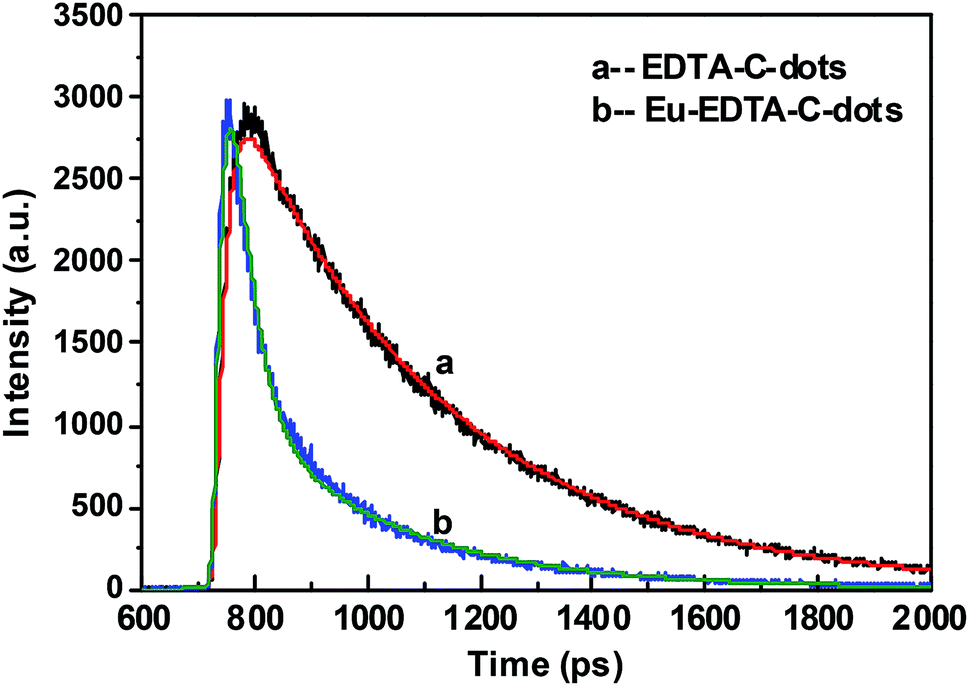 | ||
| Fig. 6 Luminescence decay spectra of EDTA–C-dots (a) and Eu–EDTA–C-dots (b) with 340 nm of excitation wavelength. | ||
4 Conclusions
In this work, we prepared and characterized EDTA-bonded multi-connected carbon dots (EDTA–C-dots) and Eu3+-coordinated EDTA-bonded multi-connected carbon dots (Eu–EDTA–C-dots). We found that EDTA–C-dots readily dissolved in DMSO but Eu–EDTA–C-dots could not be easily dissolved in DMSO, water, or other common organic solvents. In preparing EDTA–C-dots, the –COOH and –NH2 groups on the surface of the precursor C-dots not only reacted with the added EDTA but also reacted with each other. The attachment of EDTA barely affected the photoluminescence (PL) properties of the carbon dots, but the existence of Eu3+ ions in Eu–EDTA–C-dots notably quenched the fluorescence, which suggested the occurrence of non-radiative processes between the carbon dots and the Eu3+ ions. The measured and fitted PL decay of Eu–EDTA–C-dots showed two decay lifetimes, which corresponded to two kinds of electron transfer processes, i.e., radiative recombination and non-radiative recombination.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the funding of the Agricultural Environmental Monitoring Station of Beijing (No. 19000551347) and the National Natural Science Foundation of China (No. 21774054). Authors are grateful to Prof. H. Yang and J. Sun, working in the Engineering College of Peking University, for the measurement of PL lifetime.References
- Y. P. Sun, B. Zhou, Y. Lin, W. Wang, K. A. S. Fernando, P. Pathak, M. J. Meziani, B. A. Harruff, X. Wang, H. Wang, P. G. Luo, H. Yang, M. E. Kose, B. Chen, L. M. Veca and S. Y. Xie, J. Am. Chem. Soc., 2006, 128, 7756–7757 CrossRef CAS PubMed.
- X. Wang, L. Cao, F. S. Lu, M. J. Meziani, H. T. Li, G. Qi, B. Zhou, B. A. Harruff, F. Kermarrec and Y. P. Sun, Chem. Commun., 2009, 3774–3776 RSC.
- R. L. Liu, D. Q. Wu, S. H. Liu, K. Koynov, W. Knoll and Q. Li, Angew. Chem., Int. Ed., 2009, 48, 4598–4601 CrossRef CAS PubMed.
- P. Anilkumar, X. Wang, L. Cao, S. Sahu, J. H. Liu, P. Wang, K. Korch, K. N. Tackett, A. Parenzan and Y. P. Sun, Nanoscale, 2011, 3, 2023–2027 RSC.
- Q. Song, Y. S. Ma, X. Wang, T. Tang, Y. Y. Song, Y. J. Ma, G. H. Xu, F. D. Wei, Y. Cen and Q. Hu, J. Colloid Interface Sci., 2018, 516, 522–528 CrossRef CAS PubMed.
- Y. Y. Yang, X. F. Wang, G. C. Liao, X. Q. Liu, Q. L. Chen, H. M. Li, L. Lu, P. Zhao and Z. Q. Yu, J. Colloid Interface Sci., 2018, 509, 515–521 CrossRef CAS PubMed.
- E. J. Goh, K. S. Kim, Y. R. Kim, H. S. Jung, S. Beack, W. H. Kong, G. Scarcelli, S. H. Yun and S. K. Hahn, Biomacromolecules, 2012, 13, 2554–2561 CrossRef CAS PubMed.
- W. Wang, L. Cheng and W. G. Liu, Sci. China: Chem., 2014, 57, 522–539 CrossRef CAS.
- N. Zhou, Z. Y. Hao, X. H. Zhao, S. Maharjan, S. J. Zhu, Y. B. Song, B. Yang and L. Lu, Nanoscale, 2015, 7, 15635–15642 RSC.
- V. B. Kumar, J. Sheinberger, Z. Porat, Y. Shav-Tal and A. Gedanken, J. Mater. Chem. B, 2016, 4, 2913–2920 RSC.
- R. Y. Li, X. Wang, Z. J. Li, H. Y. Zhu and J. K. Li, New J. Chem., 2018, 42, 4352–4360 RSC.
- S. H. Li, D. Amat, Z. L. Peng, S. Vanni, S. Raskin, G. D. Angulo, A. M. Othman, R. M. Graham and R. M. Leblanc, Nanoscale, 2016, 8, 16662–16669 RSC.
- B. B. Wang, S. J. Wang, Y. F. Wang, Y. Lv, H. Wu, X. J. Ma and M. Q. Tan, Biotechnol. Lett., 2016, 38, 191–201 CrossRef CAS PubMed.
- R. K. Singh, K. D. Patel, C. Mahapatra, M. S. Kang and H. W. Kim, ACS Appl. Mater. Interfaces, 2016, 8, 24433–24444 CrossRef CAS PubMed.
- Z. L. Peng, E. H. Miyanji, Y. Q. Zhou, J. Pardo, S. D. Hettiarachchi, S. H. Li, P. L. Blackwelder, I. Skromne and R. M. Leblanc, Nanoscale, 2017, 9, 17533–17543 RSC.
- R. Wang, K. Q. Lu, Z. R. Tang and Y. J. Xu, J. Mater. Chem. A, 2017, 5, 3717–3734 RSC.
- C. S. Hou, S. Q. Chen and M. Q. Wang, Dalton Trans., 2018, 47, 1777–1781 RSC.
- Z. Liu, Q. Xu, Y. H. Li and W. Chen, Mater. Chem. Front., 2017, 1, 538–541 RSC.
- M. Thakur, M. K. Kumawat and R. Srivastava, RSC Adv., 2017, 7, 5251–5261 RSC.
- M. K. Barman, P. Mitra, R. Bera, S. Das, A. Pramanik and A. Parta, Nanoscale, 2017, 9, 6791–6799 RSC.
- J. B. Essner and G. A. Baker, Environ. Sci.: Nano, 2017, 4, 1216–1263 RSC.
- X. H. Gao, C. Du, Z. H. Zhuang and W. Chen, J. Mater. Chem. C, 2016, 4, 6927–6945 RSC.
- V. Sharma, P. Tiwari and S. M. Mobin, J. Mater. Chem. B, 2017, 5, 8904–8924 RSC.
- S. N. Qu, H. Chen, X. M. Zheng, J. S. Cao and X. Y. Liu, Nanoscale, 2013, 5, 5514–5518 RSC.
- C. Yuan, B. H. Liu, F. Liu, M. Y. Han and Z. P. Zhang, Anal. Chem., 2014, 86, 1123–1130 CrossRef CAS PubMed.
- A. Salinas-Castillo, M. Ariza-Avidad, C. Pritz, M. Camprubi-Robles, B. Fernandez, M. J. Ruedas-Rama, A. Megia-Fernández, A. Lapresta-Fernández, F. Santoyo-Gonzalez, A. Schrott-Fischer and L. F. Capitan-Vallve, Chem. Commun., 2013, 49, 1103–1105 RSC.
- Y. Liu, Y. N. Liu, J. P. Lee, J. H. Lee, M. Park and H. Y. Kim, Analyst, 2017, 142, 1149–1156 RSC.
- W. Liu, X. Y. Wang, Y. Q. Wang, J. H. Li, D. Z. Shen, Q. Kang and L. X. Chen, Sens. Actuators, B, 2018, 262, 810–817 CrossRef CAS.
- L. Y. Zhang, S. N. Chen, Q. Zhao and H. W. Huang, Anal. Chim. Acta, 2015, 880, 130–135 CrossRef CAS PubMed.
- F. Qu, S. Wang, D. Y. Liu and J. M. You, RSC Adv., 2015, 5, 82570–82575 RSC.
- S. N. A. M. Yazid, S. F. Chin, S. C. Pang and S. M. Ng, Microchim. Acta, 2013, 180, 137–143 CrossRef.
- D. D. Chen, X. M. Zhuang, J. Zhai, Y. Y. Zheng, H. Lu and L. X. Chen, Sens. Actuators, B, 2018, 255, 1500–1506 CrossRef CAS.
- P. Singhal, B. G. Vats, S. K. Jha and S. Neogy, ACS Appl. Mater. Interfaces, 2017, 9, 20536–20544 CrossRef CAS PubMed.
- M. L. Desai, S. Jha, H. Basu, R. K. Singhal, P. K. Sharma and S. K. Kailasa, New J. Chem., 2018, 42, 6125–6133 RSC.
- D. Y. Pan, J. C. Zhang, Z. Li, C. Wu, X. M. Yan and M. H. Wu, Chem. Commun., 2010, 46, 3681–3683 RSC.
- L. A. Ponomarenko, F. Schedin, M. I. Katsnelson, R. Yang, E. W. Hill, K. S. Novoselov and A. K. Geim, Science, 2008, 320, 356–358 CrossRef CAS PubMed.
- K. A. Ritter and J. W. Lyding, Nat. Mater., 2009, 8, 235–242 CrossRef CAS PubMed.
- S. J. Zhu, Q. N. Meng, L. Wang, J. H. Zhang, Y. B. Song, H. Jin, K. Zhang, H. C. Sun, H. Y. Wang and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 3953–3957 CrossRef CAS PubMed.
- Y. P. Shi, Y. Pan, H. Zhang, Z. M. Zhang, M. J. Li, C. Q. Yi and M. S. Yang, Biosens. Bioelectron., 2014, 56, 39–45 CrossRef CAS PubMed.
- W. J. Liu, C. Li, Y. J. Ren, X. B. Sun, W. Pan, Y. H. Li, J. P. Wang and W. J. Wang, J. Mater. Chem. B, 2016, 4, 5772–5788 RSC.
- X. Cui, L. Zhu, J. Wu, Y. Hou, P. Y. Wang, Z. Wang and M. Yang, Biosens. Bioelectron., 2015, 63, 506–512 CrossRef CAS PubMed.
- L. Cao, M. J. Meziani, S. Sahu and Y. P. Sun, Acc. Chem. Res., 2012, 46, 171–180 CrossRef PubMed.
- S. Y. Lu, J. Sun, Y. Wang, W. S. Yu, M. T. Sun and S. Y. Cui, J. Rare Earths, 2018, 36, 669–674 CrossRef CAS.
- E. Priyadarshini and K. Rawat, J. Mater. Chem. B, 2017, 5, 5425–5432 RSC.
- M. K. Barman, P. Mitra, R. Bera, S. Das, A. Pramanik and A. Parta, Nanoscale, 2017, 9, 6791–6799 RSC.
- H. J. Zhang, Y. L. Chen, M. J. Liang, L. F. Xu, S. D. Qi, H. L. Chen and X. G. Chen, Anal. Chem., 2014, 86, 9846–9852 CrossRef CAS PubMed.
- R. H. Liu, H. T. Li, W. Q. Kong, J. Liu, Y. Liu, C. Y. Tong, X. Zhang and Z. H. Kang, Mater. Res. Bull., 2013, 48, 2529–2534 CrossRef CAS.
- S. S. Wee, Y. H. Ng and S. M. Ng, Talanta, 2013, 116, 71–76 CrossRef CAS PubMed.
- P. A. Sajid, S. S. Chetty, S. Praneetha, A. V. Murugan, Y. Kumar and L. Periyasamy, RSC Adv., 2016, 6, 103482–103490 RSC.
| This journal is © The Royal Society of Chemistry 2019 |