
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExplaining the influence of the introduced base sites into alkali oxide modified CsX towards side-chain alkylation of toluene with methanol†
Peidong Liabc,
Qiao Hanabc,
Xiaomin Zhangb,
Yangyang Yuanb,
Yanfei Zhangabc,
Li Xub,
Hongchen Guo*a and
Lei Xu *b
*b
aState Key Laboratory of Fine Chemicals, School of Chemical Engineering, Dalian University of Technology, Dalian 116024, People's Republic of China. E-mail: hongchenguo@dlut.edu.cn
bNational Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, People's Republic of China. E-mail: leixu@dicp.ac.cn
cUniversity of Chinese Academy of Sciences, Beijing 100049, People's Republic of China
First published on 30th April 2019
Abstract
The effect of the introduced base sites in CsX with alkali oxide modification towards side-chain alkylation of toluene with methanol was systematically investigated in the present work. A series of alkali oxide modified CsX with a base properties (base strength and amount) gradient were prepared by impregnating CsX with alkali metal hydroxide. Here, CsX with different base strengths were selected as the parent zeolite, which is favorable to clarify the different types of base site in the modified CsX. The base properties of the samples were elucidated based on the CO2-TPD results as well as with characterization by XPS and FT-IR of adsorbed CO2. It was revealed that the base strength of the oxygen atoms with negative charge (Oδ−) in the zeolite framework can be strengthened by the electron donating function of the alkali oxides in the supercage of CsX. Detailed analysis of the correlation between the reaction behaviors of side-chain alkylation reaction and the base properties of the indicated catalysts was carried out. It was found that the dehydrogenation of methanol to formaldehyde step was promoted by the increasing base properties of Oδ− in the CsX framework and that was the main reason for the promotion of the whole side-chain alkylation reaction. However, it was also found that the stronger base properties of the alkali oxide modified CsX would enhance the formation of active hydrogen atoms, which aggravated the unwanted styrene conversion to ethylbenzene.
1. Introduction
Styrene synthesis by side-chain alkylation of toluene with methanol was first reported by Sidorenko et al. in 1967.1 Since then, this reaction has attracted widespread attention and was deemed as a sustainable one-step alternative for the current two-step route through the alkylation of benzene with ethylene and sequential dehydrogenation of the ethylbenzene product.2,3 In spite of many studies in the past decades, there are still lots of unsolved problems, among which the relatively low catalytic conversion of reactants is the most urgent one as it is the main restriction for realizing industrial application of this potential and sustainable technology.Side-chain alkylation of toluene requires the catalysts possessing base sites to activate the carbon atom of the methyl group in toluene and to dehydrogenate methanol to formaldehyde which acts as the actual alkylating reagent, and the suitable base properties of catalysts are the key point for efficient conversion of toluene to styrene.4–6 Base faujasite zeolite (X or Y), which can be obtained through exchanging the hydrogen proton of the parent zeolite with alkali metal ions (Li+, Na+, K+, Ru+, Cs+), is generally regarded as a potential catalyst for side-chain alkylation of toluene with methanol.7–9 The alkali cations act as Lewis acid sites and the oxygen atoms with negative charge (Oδ−) in the zeolite framework act as Lewis base sites.10–12 According to a current consensus, cesium ion exchanged zeolite X (CsX) is the most promising catalyst for side-chain alkylation of toluene with methanol.2,4,13 It was found that further modifying CsX with cesium oxide can greatly improve its catalytic activity for side-chain alkylation of toluene with methanol.5,8,14,15 Archier et al. found that modifying CsX with cesium oxides can promote the side-chain alkylation of toluene with methanol, and suggested it was attributed to the promotion for the generation of formaldehyde from methanol on the alkali oxides particles.15 And Han et al. gave a similar speculation after study of side-chain alkylation of toluene with methanol over KX or CsX modified with potassium oxide or cesium oxide.8 However, Alabi et al. thought that the generation of new strong basic sites was helpful in activating the methyl group of toluene, which was responsible for the promotion effect.5 As mentioned above, the introduction of alkali oxides can promote the side-chain alkylation of toluene with methanol, but its promotion mechanism is still controversial, and few studies are carried out to investigate the concrete effects on side-chain alkylation of toluene with methanol through varying the base properties of the alkali oxides modified CsX.
It is widely accepted that modifying alkali ion exchanged zeolite with alkali oxides can greatly improve its base properties which is owing to the introduction of strong base sites.10,16–18 Davis et al. found that modified CsX with cesium oxide exhibited higher CO2 adsorption capacities and higher heats than the parent zeolite, but the adsorption heats was significantly lower than bulk cesium oxide.19,20 They perceived that the cesium oxide loaded in the zeolite was not stoichiometric oxides, and the Cs–O bond in the loaded cesium oxides was more ionic than in the bulk-phase.10,11 Regarding the introduced base sites in alkali oxides modified zeolite, Hattori et al. considered that the alkali oxides located in the zeolite cavities were the introduced strong base sites which accounted for the promotion of 1-butene isomerization.21,22 Hunger et al. confirmed the existence of strong base sites on the guest alkali oxides by 13C CP/MAS NMR with methyl iodide as a probe, and found that the chemical bonds of the parent zeolite can be affected by the loaded cesium oxide which would lead to the enhancement of base strength of Oδ− in the host zeolite framework.23,24 It can be found that the promoting effects for base properties of the base zeolite by further modification with alkali oxides were investigated widely. However, scarce attentions have been given to the correlation of the introduced base sites in alkali oxides modified CsX with the promotion for side-chain alkylation of toluene with methanol, especially for the mechanism understanding.
We recently discovered that side-chain alkylation of toluene with methanol is extraordinarily sensitive to the base properties of the catalysts.4 The investigation of the effects on side-chain alkylation of toluene with methanol by the introduced base sites in alkali oxides modified CsX will be of great importance to interpret the promoting effect for side-chain alkylation reaction over CsX by modification with alkali oxides.
In the present work, we further studied the influence of base properties gradient of the alkali oxides modified CsX and the type of introduced base sites to side-chain alkylation of toluene with methanol. A series of alkali oxides modified CsX with base properties gradient were prepared by the impregnation of CsX with indicated alkali metal hydroxide: CsX modified with different cesium oxide loading amount, with different kinds of alkali oxide; and CsX with different base strength (different cesium ion exchange degree) modified with same cesium oxide loading amount. And pure silica zeolite β, modified with cesium oxide, was also prepared as the comparison sample. The base properties of the catalyst samples were characterized by temperature-program desorption of CO2 (CO2-TPD), XPS and FT-IR of adsorbed CO2. Furthermore, detailed analysis of the correlation between the reaction behaviors of side-chain alkylation of toluene with methanol and the base properties and base site type of the indicated catalysts was carried out. Base on above studies, the promoting reasons for side-chain alkylation of toluene with methanol by modifying CsX with alkali oxides was figured out, and the cause for the aggravated conversion of styrene to ethylbenzene in the products over the alkali oxides modified CsX was also revealed.
2. Experimental
2.1. Catalyst preparation
Zeolite NaX (Catalyst Plant of Nankai University, Si/Al = 1.2) was ion-exchanged with aqueous solutions of cesium chloride (Aladdin, 99%) to prepare CsX. CsX-1, CsX-2 and CsX-4 with exchange degrees of 21.8%, 32.3% and 45.9% shown in our preceding paper were chosen as the parent zeolites, which is denoted as CsX-a, CsX-b and CsX-c respectively in this work.4 And the XRD, CO2-TPD and catalytic activity for reaction between toluene and methanol of them were also taken from our preceding work. The pure silica zeolite β was synthesized according to the reported procedure.25The alkali oxides modified CsX were prepared by incipient wetness impregnation of CsX with alkali metal hydroxides aqueous solutions at room temperature. After addition of impregnation precursor solution, the mixtures were stood in ceramic crucible for 12 h at room temperature, followed by drying at 110 °C for 4 h, and then calcined at 550 °C in air atmosphere for 4 h. The loaded amount of alkali oxides was adjusted by the concentration of alkali metal hydroxides in the aqueous solutions. The alkali oxides supported on the zeolite were denoted as MOx (M represents Na, K or Cs) after calcination. And the catalysts were named as AMOx/CsX-B, where A and B referred to the alkali oxides amount and the code for CsX with different cesium ion exchange degree, respectively. For example, 8CsOx/CsX-c was CsX-c loaded with 8CsOx per unit cell. Similarly, 1.72CsOx/β was prepared by the same method, which had the same loading weight as 8CsOx/CsX-c.
2.2. Catalyst characterization
Powder X-ray diffraction (XRD) patterns were obtained on a PANalytical X'Pert PRO X-ray diffractometer with Cu Kα radiation, operating at 40 kV and 40 mA. The patterns were recorded in the 2θ range of 5 to 50°.The compositions of the samples were measured by X-ray fluorescence (XRF) spectroscopy with a Philips Magix-601 X-ray fluorescence spectrometer.
N2 adsorption–desorption isotherms were measured at −196 °C on a Quantachrome Autosorb iQ2 physical adsorption analyzer system after the samples were outgassed in vacuum for 10 h at 300 °C.
Temperature-program desorption (TPD) of adsorbed CO2 and NH3 were performed on Micromeritics Autochem 2920. After the sample was pretreated in helium stream at 550 °C for 1.5 h and cooled down to 80 °C (100 °C for NH3-TPD), it was exposed to pure CO2 or 8% NH3/He (vol/vol) for 30 min and swept with helium at a flow rate of 30 mL min−1 (STP) for 30 min, then, the temperature elevated at a rate of 10 °C min−1. For the CO2-TPD, a cold trap (−47 °C) with a mixture of liquid nitrogen and m-xylene was installed to avoid errors caused by water in thermal conductivity detector (TCD).
X-ray photoelectron spectroscopy (XPS) measurements were performed on Thermo Fisher ESCALAB 250Xi, and the based pressure was 1 × 10−8 Pa. An Al Kα radiation (hν = 1486.6 eV) was used as the X-ray source, and operated at 15 kV and 10.8 mA. The C 1s peak at 284.6 eV in the background was used as the reference energy for the binding energy calibration.
Fourier transform infrared (FT-IR) spectra of adsorbed CO2 and NH3 were recorded at a spectral resolution of 4 cm−1 on Bruker Tensor 27 instrument. The samples was pressed into a self-supporting wafer (10 mg, R = 0.65 cm), and placed in an in situ quartz IR cell with KBr windows. After pretreatment under vacuum (10−2 Pa) for 2 h at 500 °C (450 °C for FT-IR of adsorbed NH3), the sample was exposed to 650 Pa CO2 at 80 °C or 8% NH3/He (vol/vol) at 100 °C for 30 min. For the FT-IR of adsorbed CO2, the samples was then swept with 40 mL min−1 N2 (STP) at the indicated temperature for 30 min. And for FT-IR of adsorbed NH3, the samples were evacuated under vacuum (10−2 Pa) at 100 °C for 30 min. All the spectra for FT-IR of adsorbed CO2 were recorded at 80 °C, and 100 °C for FT-IR of adsorbed NH3.
2.3. Catalyst testing
1.0 g of the catalysts (20–40 mesh) was used in the catalyst testing on a fixed bed reactor (quartz, 9 mm i.d.). The samples were activated under a flow of air (40 mL min−1) for 1.5 h at 550 °C. After the sample was cooled to the reaction temperature of 430 °C and swept with helium for 30 min at a flow rate of 40 mL min−1 (STP), a liquid mixture of toluene and methanol (molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) was fed into the reactor by a ram pump at a weight hourly space velocity (WHSV) of 2 h−1 with helium at a flow rate of 10 mL min−1 (STP). For investigating the influence for catalytic activity of 8CsOx/CsX-c by pretreating with CO2, 40 mL min−1 (STP) CO2 was given at 80 °C for 30 min, and then swept the catalysts with 40 mL min−1 (STP) helium for 30 min at 400 °C before introducing the reactants.
:2) was fed into the reactor by a ram pump at a weight hourly space velocity (WHSV) of 2 h−1 with helium at a flow rate of 10 mL min−1 (STP). For investigating the influence for catalytic activity of 8CsOx/CsX-c by pretreating with CO2, 40 mL min−1 (STP) CO2 was given at 80 °C for 30 min, and then swept the catalysts with 40 mL min−1 (STP) helium for 30 min at 400 °C before introducing the reactants.
An on-line gas chromatograph (Agilent GC7890A) was used for the analysis of products. Hydrocarbons, methanol and dimethyl ether were analyzed by a flame ionization detector (FID) with Agilent CP-WAX (25 m × 32 μm × 1.2 μm) capillary column, and CO, CO2 and formaldehyde was analyzed by thermal conductivity detector (TCD) with a Porapak Q (4 m × 1/8′′) packed column, the activity results are presented as conversion of methanol (Xmethanol), toluene (Xtoluene), yield of styrene and ethylbenzene (YST+EB), xylene (Yxylene), CO and CO2 (YCO+CO2) and molar ratio of styrene to ethylbenzene in products (ST/EB), which are defined as follows (quantities are molar flow rates):
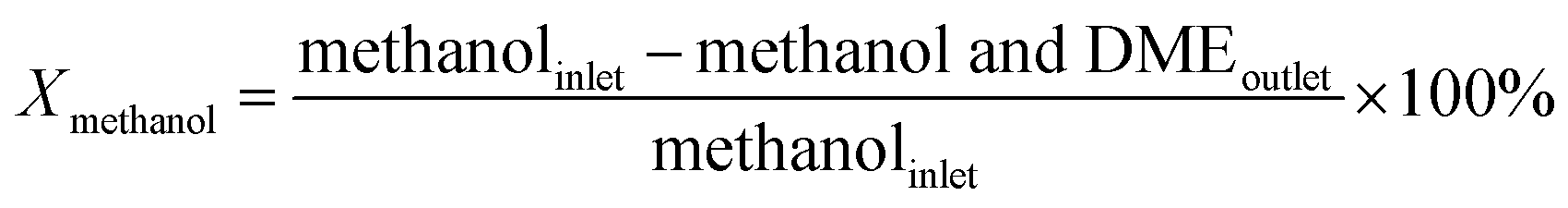
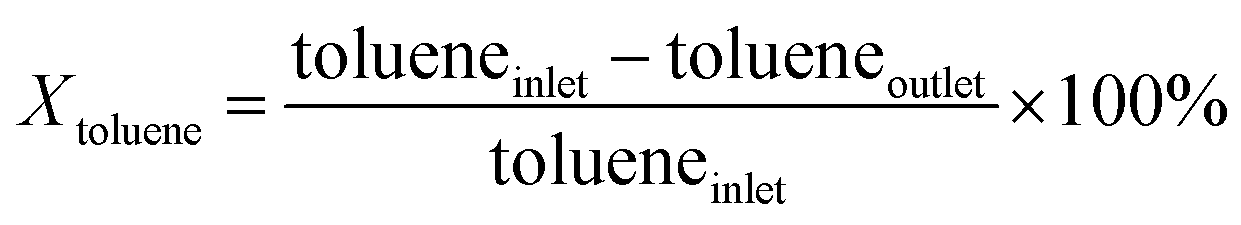
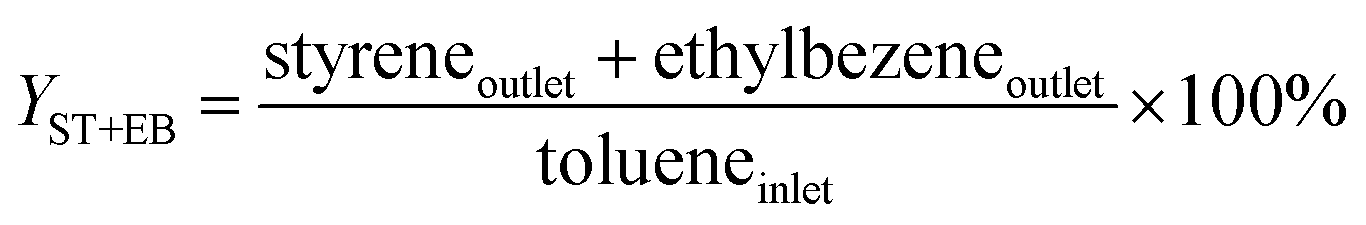
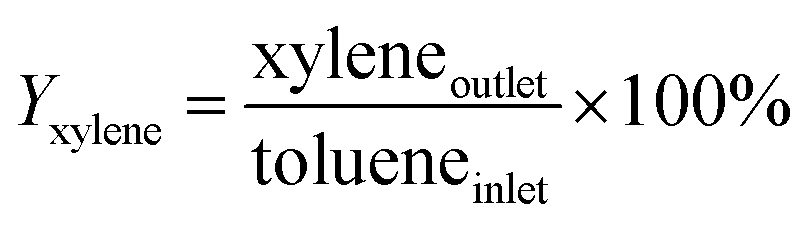
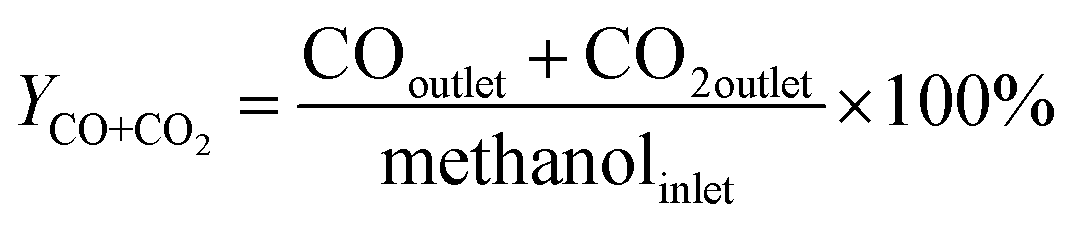
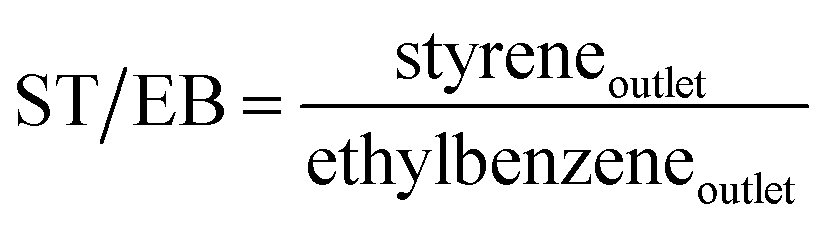
All data were obtained at the reaction time of 50 min.
3. Results and discussion
3.1. Structural and physicochemical characteristics of the catalysts
The structural and physicochemical characteristics of the CsX-a, CsX-b and CsX-c (exchange degrees were 21.8%, 32.3% and 45.9%, respectively) have been displayed and analyzed in our previous work.4 The XRD patterns and composition of all the CsX modified with alkali oxides are presented in Fig. 1 and Table S2.† As shown in Fig. 1, all the modified CsX samples clearly showed typical peaks of zeolite X structure, and characteristic diffraction patterns of zeolite-β were obtained over pure silica zeolite β before and after loading cesium oxide, indicating the zeolite structures were retained after modifying with alkali oxides. However, the relative peak intensities of all the modified zeolites were reduced with the introduction of alkali oxides. Romero et al. found that there was no resonance shift of the 29Si MAS NMR signals before and after the modification of zeolite X with cesium oxide and no extra-framework aluminum was detected by 27Al MAS NMR spectrum after the modification, although the XRD pattern of the zeolite X reduced sharply after the modification.16 Thereof, the reduction of the XRD relative peak intensities of the modified CsX might be mainly attributed to the change of X-ray absorption behavior with the introduction of alkali oxide rather than the collapse of the zeolite framework.8,26,27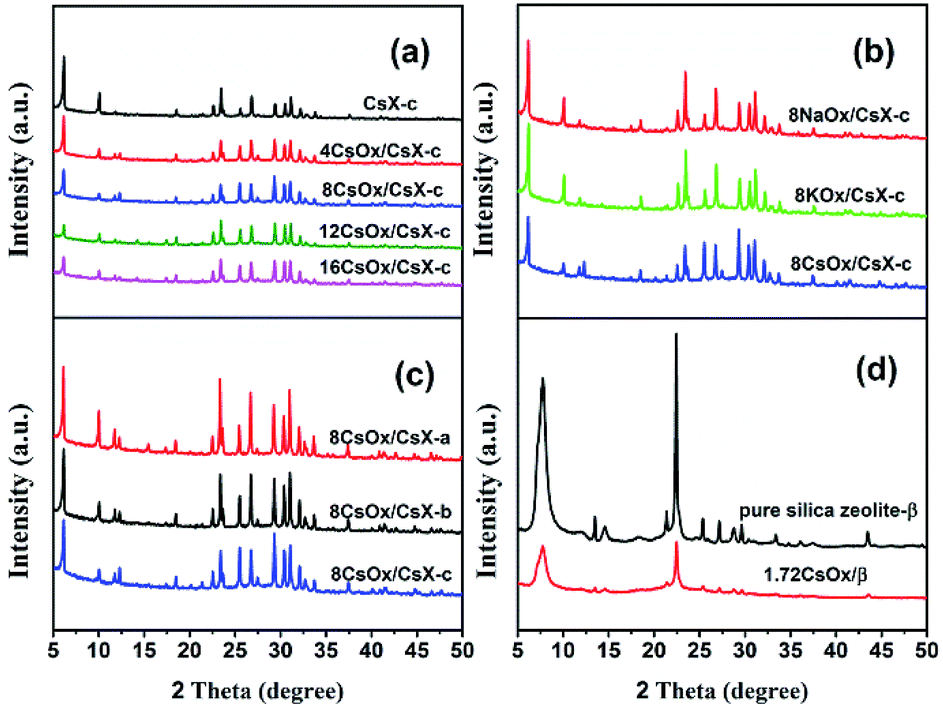 | ||
| Fig. 1 XRD patterns of the indicated catalysts (a) CsX-c loaded with different cesium oxide amount; (b) CsX-c loaded with different kinds of alkali oxides; (c) CsX with different cesium ion exchange degree loaded with 8CsOx per unit cell; (d) pure silica zeolite-β before and after loading cesium oxide. | ||
The textural properties of CsX-c with different cesium oxide amounts are displayed in Table S1† and the BET surface area (SBET) and micropore volume (Vmicro) of CsX-c decreased after introduction of the cesium oxide. According to current knowledge, the cesium oxide is located primarily in the supercages of CsX when the loaded cesium atoms content is less than 24 cesium atoms per unit cell.26,28 The maximum loading amount of CsOx in present work is 16 cesium atoms per unit cell, suggesting the cesium oxide is mainly in the pore of CsX. Therefore, the decrease of BET surface area and micropore volume might be attributed to the occupation of the available void volume by the loaded cesium oxide. What is more, the decrease of the micropore volume as a function of the loaded cesium oxide amount is almost a straight line (Fig. S1†), indicating that there is almost no obvious collapse of the zeolite structure after modifying with cesium oxide.29
3.2. Characterization and analysis of base properties of the alkali oxides modified CsX
CO2-TPD curves for all the catalysts are shown in Fig. 2, from which it can be seen that all the CO2 desorption peaks appeared below 400 °C. And both the CO2 desorption temperature and area increased when the CsX was modified with alkali oxides, indicating that stronger base sites were introduced. And the CO2 desorption temperature of the alkali oxides modified CsX increased with the increase of the alkali oxides loaded amount and the increase of the atomic number of the alkali metal in the loaded alkali oxides as well as the initial base strength of the parent zeolite. Moreover, it should be noticed that a gradual uptrend is observed at temperature higher than 500 °C over alkali oxides modified zeolite, especially for samples with high loading amount.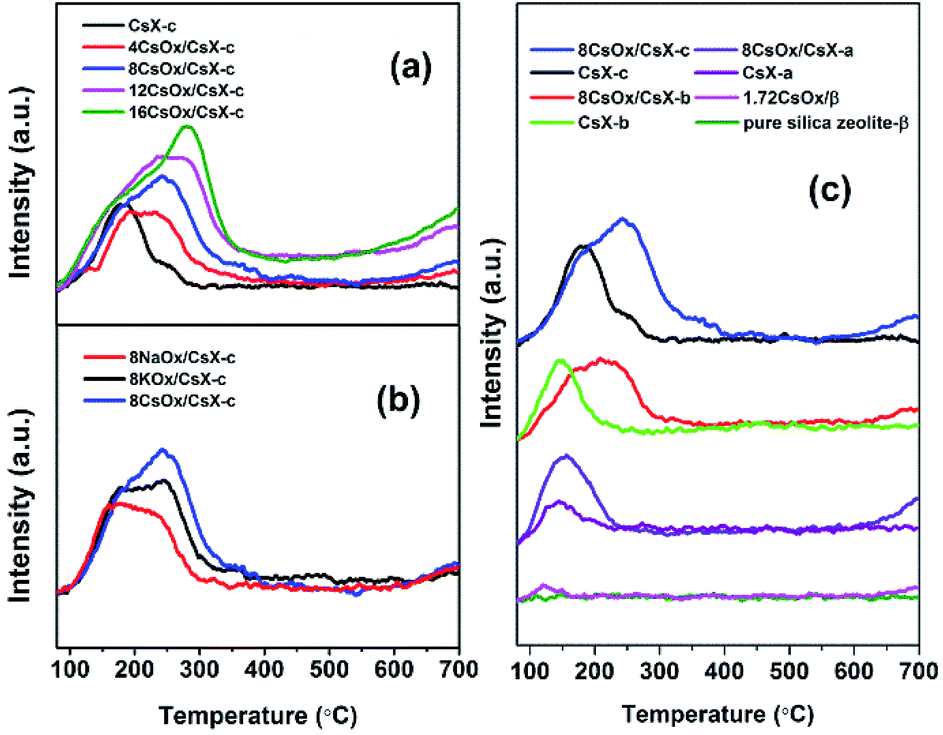 | ||
| Fig. 2 CO2-TPD curves of the indicated catalysts (a) CsX-c loaded with different cesium oxide amount; (b) CsX-c loaded with different kinds of alkali oxides; (c) different zeolite parents before and after loading cesium oxide. | ||
Two kinds of base sites could be introduced when base zeolite was modified with cesium oxide: the strong base sites created on cesium oxide and the base strength enhanced oxygen atoms with negative charge (Oδ−) in the zeolite framework.24 As shown in Fig. 2(c), the CO2 desorption temperature of the cesium oxide modified CsX modified with same cesium oxide amount increased with the base strength of their parent CsX strengthened, which finds difficulty to understand when it comes to explain these introduced base sites created on the cesium oxides. That is to say if these introduced strong base sites is created by the cesium oxide itself, a similar CO2 desorption peak at high temperature should be detected for all the modified zeolite with same cesium oxide amount in Fig. 2(c) instead of relating closely with the desorption temperature of their parent CsX. Thus, it is suggested that the introduced base sites which correspond to the CO2 desorption below 400 °C may be caused by the base strength enhanced Oδ− in the CsX framework.
To further confirm this speculation, the base properties of cesium oxide modified pure silica zeolite β were investigated considering that both of zeolite β and zeolite X have three dimensional 12-membered ring pore structure. As shown in Fig. 2(c), there was no CO2 desorption peak on pure silica zeolite β and only a small peak can be detected at 118 °C on cesium oxide modified zeolite β, which may be attributed to the effects on defective sites of the zeolite by cesium oxide. For the CsX, the base sites are the oxygen atoms with negative charge (Oδ−) in the zeolite framework and the base properties could be enhanced with increase of the cesium ion exchange degree.4,30 This base sites cannot be generated over pure silica zeolite due to lack of Oδ− in the framework as Oδ− only present in the framework of zeolite possessing the Si–O–Al species.17 This result also suggested that the introduced base sites which correspond to the CO2 desorption below 400 °C should be attributed to the increased base strength of Oδ− in zeolite framework of CsX, and the CO2 adsorbed on strong base sites created by alkali oxides was desorbed over 500 °C.
In order to investigate the existence state of the adsorbed CO2 in the zeolite, the FT-IR spectra of the adsorbed CO2 on CsX-c, 8CsOx/CsX-c and 16CsOx/CsX-c were measured, and the results are shown in Fig. 3. A pair of strong bands at 1648 cm−1 and 1385 cm−1 was detected on CsX-c after adsorbing CO2, which can be ascribed to asymmetric stretching bands and symmetric stretching bands of bidentate carbonate, respectively.5,13 As is shown in Fig. 3, the samples of 8CsOx/CsX-c, 16CsOx/CsX-c and CsX-c revealed similar CO2 absorption bands. And after sweeping with N2 at 300 °C for CsX-c and 400 °C for 8CsOx/CsX-c and 16CsOx/CsX-c, all the absorption bands disappeared. Combined the results of the CO2-TPD in Fig. 2, it can be concluded that those absorption bands are corresponding to the CO2 desorption peaks in the CO2-TPD in Fig. 2 below 400 °C. It can be found that the bands for bidentate carbonate decreased with the sweeping temperature increased, and its stability on the cesium oxide modified CsX-c increased with the increasing loading amount, which is consistent with the increase of CO2 desorption temperature in CO2-TPD after alkali oxides modification.
 | ||
| Fig. 3 FT-IR spectra of adsorbed CO2 on CsX-c, 8CsOx/CsX-c and 16CsOx/CsX-c. The backgrounds were recorded after the samples were pretreated at 500 °C in vacuum for 2 h, after adsorbing CO2 at 80 °C, the samples were further swept with N2 at the indicated temperature for 30 min. All the spectra were recorded at 80 °C and the sweeping N2 flow was 40 mL min−1 (STP). | ||
Additionally, no absorption band was detected on the CsX-c after pretreated at 500 °C, while three peaks appeared in the region of 1300–1500 cm−1 on 8CsOx/CsX-c, which should be responsible for the uptrend after 500 °C in the CO2-TPD shown in Fig. 2. Absorption bands was also found in this region in the work of Alabi et al., which was ascribed to the formation of ionic carbonate and unidentate carbonate on cesium oxide by adsorbing CO2.5 Unidentate carbonate is generally formed on strong base sites by adsorbing CO2, and it is inherent before the introduction of CO2 during the experimental process, suggesting that the adsorbed CO2 on the strong base sites of cesium oxide itself was from the air and could not be removed by pretreatment at 500 °C.
XPS studies were carried out to investigate the influence of the loaded alkali oxides on the framework of CsX, and the results are shown in Fig. 4. It has been widely accepted that Sint (Sanderson intermediate electronegativity) of the zeolite is related to the acid–base properties of zeolite, and the lower Sint of the base zeolite, the higher the base strength.31–35 For alkali cation exchanged zeolite, the lower binding energy (BE) of Si 2p and O 1s means lower Sint.36,37 It can be seen that the BE of Si 2p and O 1s decreases after loading alkali oxides, indicating that the base strength of the Oδ− in the zeolite framework was promoted by the loaded alkali oxides. This result clearly demonstrates the chemical interaction imposed zeolite framework by the loaded alkali oxides.38
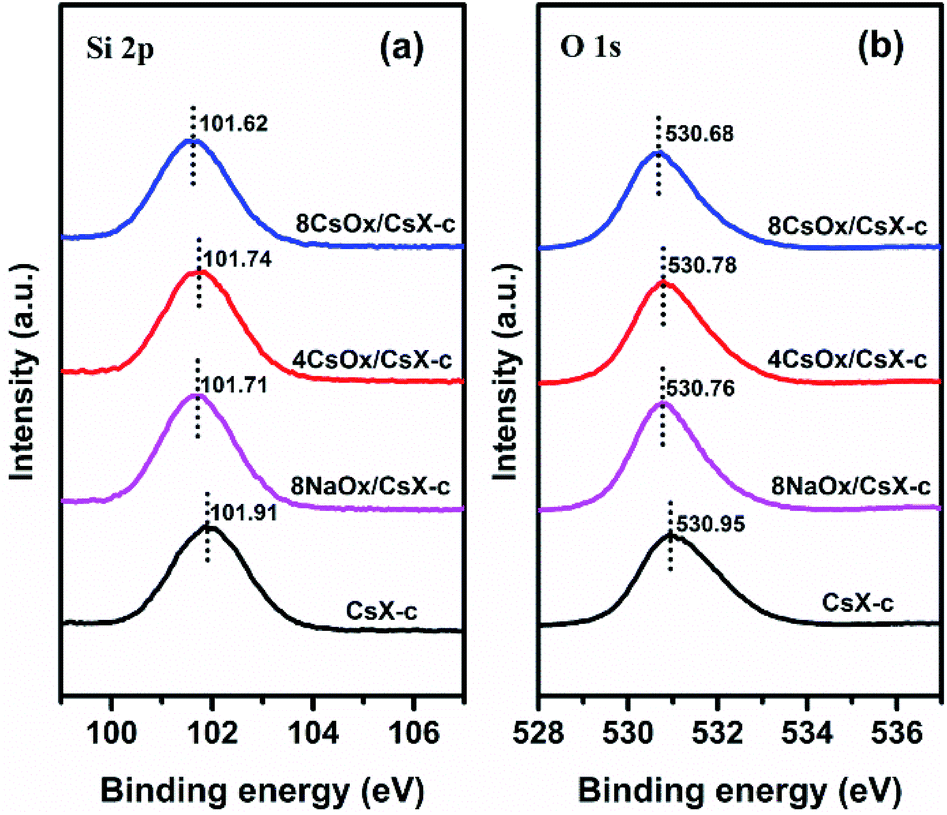 | ||
| Fig. 4 XPS spectra of the indicated catalysts: (a) spectra of Si 2p; (b) spectra of O 1s. | ||
According to the principle of Sanderson electronegativity equalization, different atoms will be adjusted to the same electronegativity within the compound when they are combined chemically, and Sint is the geometric mean value of electronegativity of the compound.31 Assuming the abundant existing state of alkali oxides in the CsX is in formula of M2O (where M is Na, K and Cs), when alkali oxides insert into the CsX by chemical combination, the electronegativity of M2O and CsX will reach a new equilibrium value according to the principle of Sanderson electronegativity equalization, and the negative charge will transfer to the framework of CsX from alkali oxides as the Sint for CsX is higher than that of M2O (Table S2†).39 Hence, the electron donating function of the loaded alkali oxides for the zeolite framework should be the intrinsic reason for the promotion of base strength of the Oδ− in the zeolite framework. What is more, the BE of Si 2p and O 1s decreases in the order of CsX-c > 4CsOx/CsX-c > 8NaOx/CsX-c > 8CsOx/CsX-c, indicating that the electron donating function for the framework of CsX increases with increasing alkali oxides loading amount and atomic number of alkali metal in the alkali oxides. The increase of atomic number of the alkali metal means the decrease of its metallicity which will lead to the increase of the electron donating ability of its oxide. Meanwhile, it can be found that the increasing trend of the CO2 desorption temperature for CsX modified with different cesium loading amount and different kinds of alkali oxides according well to the above electron donating regularities (Fig. 2(a) and (b)).
Moreover, it should be noticed that the area of desorption peak in CO2-TPD increases when CsX is modified by alkali oxides. The CO2 desorption amount and the amount of Oδ− in the zeolite framework are listed in Table S2.† The amount of Oδ− in the zeolite framework is far more than that of the desorbed CO2, suggesting that only part of the Oδ− sites with strong enough base strength in the zeolite framework could adsorb CO2. When the CsX was modified with alkali oxide, the base strength of Oδ− would be enhanced so that more Oδ− sites would have enough base strength to adsorb CO2.
3.3. Analysis and discussion for side-chain alkylation of toluene with methanol over alkali oxides modified CsX
 | ||
| Fig. 5 The reaction behaviors of side-chain alkylation of toluene with methanol over the indicated catalysts (a) I: CsX-c (20–40 mesh) mechanically mixed with pure silica zeolite β (20–40 mesh), II: CsX-c (20–40 mesh) mechanically mixed with 1.72CsOx/β (20–40 mesh), III: CsX-c mixed with 1.72CsOx/β by grinding in a mortar for 10 min, IV: 4CsOx/CsX-c (20–40 mesh) mechanically mixed with pure silica β (20–40 mesh), the mass ratio of CsX-c or 4CsOx/CsX-c to pure silica zeolite β or 1.72CsOx/β was 2:1; (b) CsX-c loaded with different kinds of alkali oxides; (c) CsX-c loaded with different cesium oxide amount; (d) same cesium oxide amount loaded in CsX with different cesium exchange degree. The calculation of YST+EB was calculated based on the inlet quantities of toluene, and YCO+CO2 was based on the inlet quantities of methanol. | ||
During the reaction process of side-chain alkylation of toluene with methanol, formaldehyde is generated by dehydrogenation of methanol over the base sites of CsX, and it is widely accepted that the generated formaldehyde acts as the actual alkylating reagent for side-chain alkylation of toluene.1,6,40 Meanwhile, a part of the generated formaldehyde that has not reacted with the activated toluene will further decompose to CO over the base sites,14,41 and small part of the formed CO will be further converted into CO2.13 In previous work, we have proved that the dehydrogenation of methanol to formaldehyde, which can be promoted by increasing both the base strength and amount of the base sites, is the crucial step for side-chain alkylation of toluene with methanol over CsX with cesium exchange degree higher than 32.3%.4 Thus, improving the base properties of CsX will promote the conversion of methanol, the formation of styrene and ethylbenzene and the yield of CO and CO2. In addition to the base sites, the acid sites are also required for side-chain alkylation of toluene with methanol, which play a role in adsorbing and stabilizing the benzene ring of toluene.41 In our previous work, it was confirmed that the existence of Brønsted acid sites would lead to the production of xylene, and Lewis acid sites derived from the positive ions in CsX are the essential acid sites involved in side-chain alkylation of toluene.4 As shown in Fig. S2,† no acid site is introduced by the introduction of alkali oxides, and the Lewis acid sites in CsX are little influenced by modifying with alkali oxides. Thus, the major influence for side-chain alkylation of toluene with methanol over the alkali oxides modified CsX in Fig. 5 should be the changes of base properties of the catalysts.
The reaction behaviors of side-chain alkylation of toluene with methanol over CsX-c modified with different kinds of alkali oxides are shown Fig. 5(b). After the CsX-c was modified with alkali oxides, the Xtoluene, Xmethanol, YST+EB and YCO+CO2 increased in the order of 8NaOx/CsX-c < 8KOx/CsX-c < 8CsOx/CsX-c. This is in consistent with the change in the base strength and base sites amount of the three catalysts (Fig. 2(b) and Table S2†), indicating that the introduced base sites in alkali oxides modified CsX promoted the side-chain alkylation of toluene with methanol. The influence for side-chain alkylation of toluene with methanol by with or without CO2 pretreatment for 8CsOx/CsX-c was shown in Table 1. It can be seen that pretreating 8CsOx/CsX-c with CO2 has litter influence on Xtoluene, Xmethanol, YST+EB and YCO+CO2 which is higher than that over CsX-c. Although parts of the strong base sites provided by the alkali oxide itself might be exposed by the pretreatment at 550 °C in this work, it should be occupied again by CO2 formed in situ as CO2 can be formed during the reaction process (Table S3†), which should account for the similar reaction results over 8CsOx/CsX-c with or without CO2 pretreatment. Thus, it indicated that the efficient base sites for side-chain alkylation of toluene with methanol corresponding to the CO2 desorption below 400 °C in Fig. 2. And combined with the analysis of base properties of the catalysts above, it suggested that the promotion for side-chain alkylation of toluene with methanol over the cesium oxide modified CsX was attributed to the enhanced base properties of Oδ− in the zeolite framework.
| Catalysts | Pretreatment conditions | Xtoluene (%) | Xmethanol (%) | YST+EB | YCO+CO2 |
|---|---|---|---|---|---|
| a The catalyst was pretreated with 40 mL min−1 CO2 at 80 °C for 30 min, and then swept with helium for 30 min at 400 °C before introducing the reactants. | |||||
| CsX-c | Without CO2 | 6.99 | 22.25 | 6.67 | 17.95 |
| 8CsOx/CsX-c | Without CO2 | 18.85 | 68.38 | 17.20 | 58.18 |
| 8CsOx/CsX-c | With CO2a | 17.46 | 66.06 | 16.56 | 56.71 |
In our previous work,4 we proposed the concept regarding formaldehyde ([HCHO]) that has been generated from methanol during the reaction processes. The [HCHO], virtual yield of [HCHO] (Y[HCHO]) and conversion of [HCHO] to styrene and ethylbenzene (X(HCHO to ST+EB)) were defined as (quantities are molar flow rates):
| [HCHO] = styreneoutlet + ethylbenzeneoutlet + COoutlet + CO2outlet |
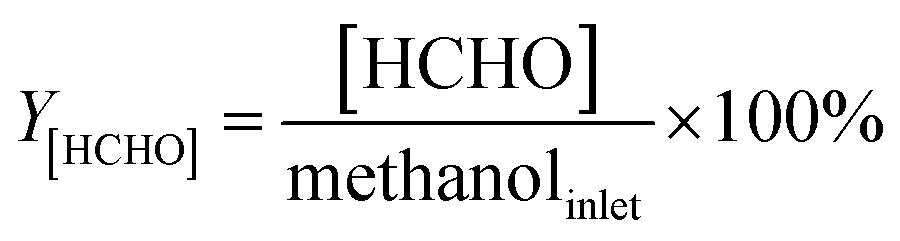
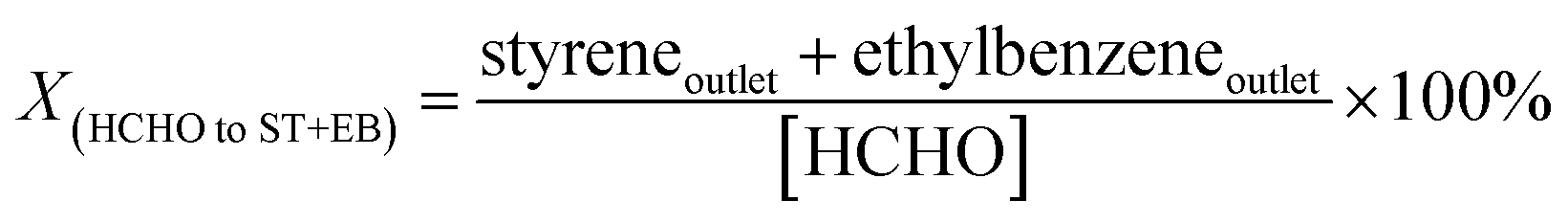
X(HCHO to ST+EB) represents the utilization extent of [HCHO] by side-chain alkylation of toluene. When the cesium ion exchange degree of CsX was higher than 32.3%, the X(HCHO to ST+EB) over CsX remained constant with the exchanged degree further increased.4 However, the X(HCHO to ST+EB) decreased when CsX-c was modified by alkali oxides, and the decrease trend was in the order of 8NaOx/CsX-c > 8KOx/CsX-c > 8CsOx/CsX-c, suggesting that different influences have been imparted on the Oδ− in the zeolite framework by improving cesium ion exchanged degree and modifying with alkali oxides. Reaction results over CsX-6 (cesium exchanged degree is 59.2%), taken from our preceding paper,4 were compared with 8NaOx/CsX-c and 4CsOx/CsX-c (Fig. 6), as the Sint of the three catalysts is similar to each other. The base strength of CsX-6 and 4CsOx/CsX-c was similar to each other, but the base sites amount of CsX-6 was larger than that of 4CsOx/CsX-c (Fig. S3†), however, the Xmethanol over 4CsOx/CsX-c was higher than that over CsX-6, suggesting that the base strength enhanced Oδ− in 4CsOx/CsX-c were more inclined to adsorb and activate methanol instead of toluene, which might be due to the different location of the introduced base sites in 4CsOx/CsX-c with that in CsX-6. Generally, the adsorption of toluene in zeolite X was at SII sites in the supercage of CsX.42 The lower Xtoluene and higher YCO+CO2 over 4CsOx/CsX-c indicated that formaldehyde generated over the strength enhanced Oδ− would preferentially decompose to CO to react with toluene at SII sites, and most of the strength enhanced Oδ− in alkali oxides modified CsX were far away from the SII sites in the supercage. The more easy decomposition of the generated formaldehyde over the introduced base sites was responsible for the decrease of X(HCHO to ST+EB) over the alkali oxides modified CsX.
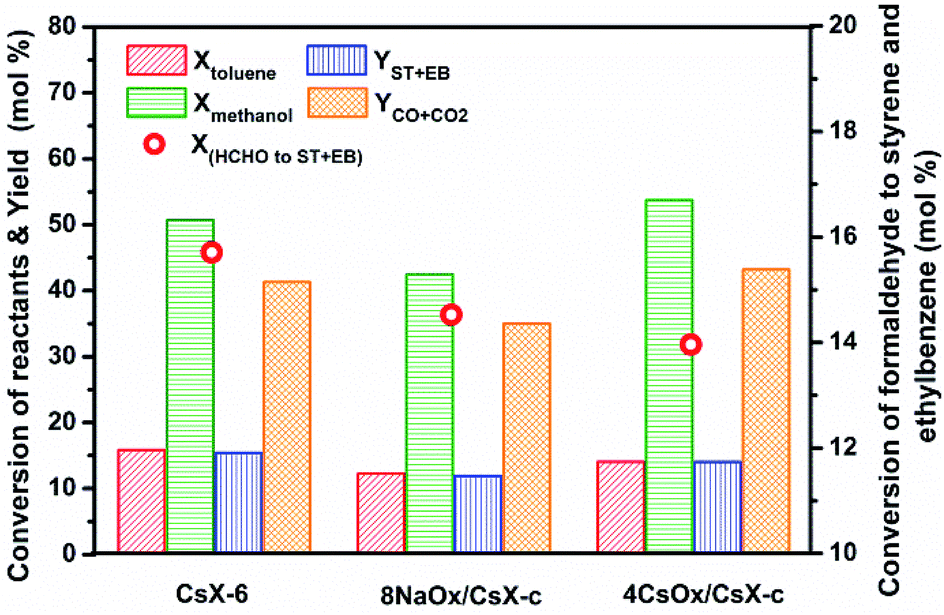 | ||
| Fig. 6 Reaction behaviors of side-chain alkylation of toluene with methanol over catalysts with similar Sint. | ||
As shown in Fig. 5(c) and Table S3,† Xmethanol, YCO+CO2 and Y[HCHO] increased over CsX-c with the increase of cesium oxides amount, which was consistent with the base properties. Nevertheless, Xtoluene and YST+EB increased with increasing cesium oxide amount over CsX-c lower than 8CsOx per unit cell and then decreased. What is more, the X(HCHO to ST+EB) decreased sharply as the loaded cesium oxide increased, indicating that the loading of cesium oxide brought down the utilization of [HCHO], and the lower utilization of [HCHO] at higher cesium oxide loading is the reason for the decrease of Xtoluene and YST+EB.
The cesium ion exchange degree of CsX-a is only 21.8% which is too low to provide enough base strength to activate the side-chain of toluene effectively, thus, the X(HCHO to ST+EB) is lower than that of CsX-b and CsX-c (Fig. 5(d)).4 Meanwhile, there is Brønsted acid sites existed in CsX-a (Fig. S2†) which will result in the formation of xylene,4 and the dehydrogenation of methanol to formaldehyde will also be restricted as the base strength of CsX-a is very low. Thus, the product from reaction between toluene and methanol over CsX-a is mainly xylene instead of styrene and ethylbenzene (Table S3†). When CsX-a is modified with cesium oxide, the base properties of CsX-a is enhanced as well as the Brønsted acid sites was removed (Fig. 2(c) and S2†), which lead to the main hydrocarbon products changed from xylene to styrene and ethylbenzene (Table S2†). Finally, the X(HCHO to ST+EB) increased due to more effective activation of toluene over the base sites of 8CsOx/CsX-a (Fig. 5(d)).
| [H] = ([HCHO] + COoutlet + CO2outlet) × 2 |
The ST/EB over CsX-c and all the alkali oxides modified CsX as a function of [H] that has been generated during the side-chain alkylation of toluene with methanol is shown in Fig. 7. CsX-a and CsX-b was not included in Fig. 7 as the Brønsted acid sites can be detected on them which would lead to different ethylbenzene formation path.4 As shown in Fig. 7, the ST/EB decreased with the increase of the generated [H], indicating that the lower ST/EB over the alkali oxides modified CsX was attributed to the high generation of [H]. When CsX was modified with alkali oxides, the increase of both base sites amount and base strength will lead to the promotion of methanol dehydrogenation to formaldehyde, which was responsible for the promoted generation of [H] and reduced ST/EB. On the other hand, compared to CsX, the lower utilization of [HCHO] over the alkali oxide modified CsX which led to more decomposition of [HCHO] was also liable for the low ST/EB over the alkali oxides modified CsX.
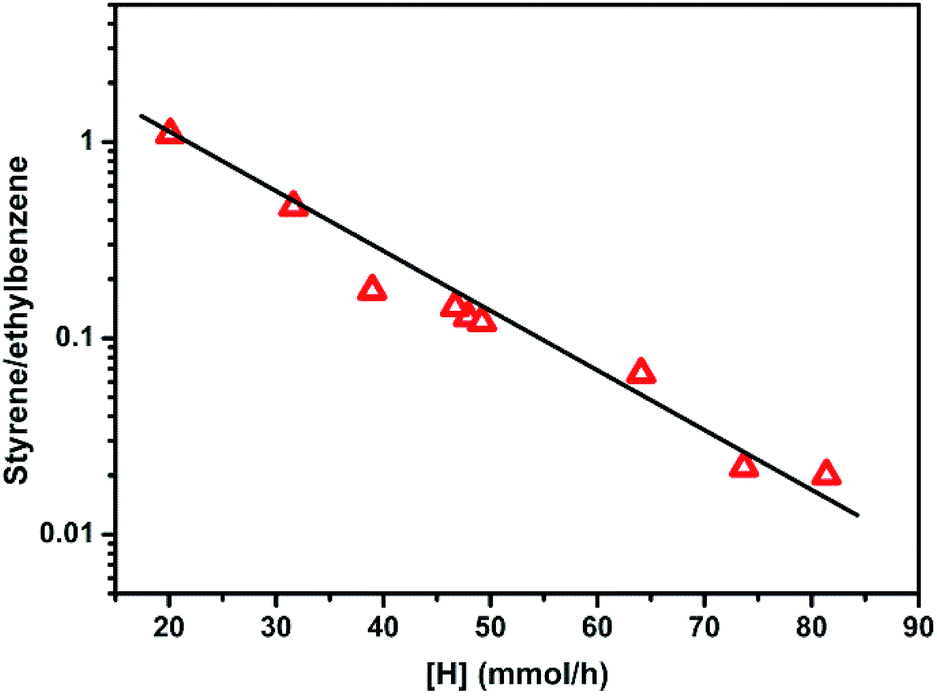 | ||
| Fig. 7 The styrene/ethylbenzene in the products over the alkali oxides modified CsX and CsX-c as a function of [H] that has been generated during the side-chain alkylation of toluene with methanol. | ||
4. Conclusions
In this study, a series of alkali oxides modified CsX with different base properties gradient were prepared together with cesium oxide modified pure silica zeolite β as a comparison sample. Base on the investigation of the base properties of all the samples, it can be concluded that the base properties of Oδ− in the zeolite framework can be promoted by electron donating function of alkali oxides in the supercage of the CsX. The base properties of the Oδ− in the framework of CsX, increased with the increase of loading amount of alkali oxides, electron donating capacity of the alkali oxides and the base strength of the parent zeolite. The relation between the base properties and the reaction performance of side-chain alkylation of toluene with methanol of the indicated catalysts was also analyzed. It is found that the enhanced base strength of Oδ− in the CsX framework could promote dehydrogenation of methanol to formaldehyde which account for the promotion effect on side-chain alkylation of toluene with methanol over the alkali oxides modified CsX. At the same time, the increased base properties of the modified CsX promote the decomposition of the formaldehyde, which results in a decrease in utilization of the generated formaldehyde in side-chain alkylation of toluene. With a loading amount increase of cesium oxide on CsX, the yield of styrene and ethylbenzene rises at first and then goes down. The yield reaches the highest when CsX is loaded with 8CsOx per unit cell. Additionally, the strong base properties of the alkali oxides modified CsX stimulates the formation of active hydrogen atoms, giving rise to a lower ST/EB in the products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by National Natural Science Foundation of China (No. 21576252).References
- Y. N. Sidorenko, P. N. Galich, V. S. Gutyrya, V. G. Ilin and I. E. Neimark, Dokl. Akad. Nauk SSSR, 1967, 173, 132–134 CAS.
- A. J. Martín, S. Mitchell, O. Scholder, R. Verel, R. Hauert, L. Bernard, C. Jensen, M. Schwefer and J. Pérez-Ramírez, ChemPhysChem, 2018, 19, 437–445 CrossRef PubMed.
- H. Chen, X. Li, G. Zhao, H. Gu and Z. Zhu, Chin. J. Catal., 2015, 36, 1726–1732 CrossRef CAS.
- P. Li, Q. Han, X. Zhang, Y. Yuan, Y. Zhang, H. Guo, L. Xu and L. Xu, Catal. Sci. Technol., 2018, 8, 3346–3356 RSC.
- W. O. Alabi, B. B. Tope, R. B. Jermy, A. M. Aitani, H. Hattori and S. S. Al-Khattaf, Catal. Today, 2014, 226, 117–123 CrossRef CAS.
- A. E. Palomares, G. Eder-Mirth, M. Rep and J. A. Lercher, J. Catal., 1998, 180, 56–65 CrossRef CAS.
- D. Barthomeuf, Microporous Mesoporous Mater., 2003, 66, 1–14 CrossRef CAS.
- H. Han, M. Liu, X. Nie, F. Ding, Y. Wang, J. Li, X. Guo and C. Song, Microporous Mesoporous Mater., 2016, 234, 61–72 CrossRef CAS.
- X. Li, J. Lu, Y. Li and J. Yu, Chin. J. Chem., 2017, 35, 716–722 CrossRef CAS.
- R. J. Davis, J. Catal., 2003, 216, 396–405 CrossRef CAS.
- E. J. Doskocil and R. J. Davis, J. Catal., 1999, 188, 353–364 CrossRef CAS.
- J. Engelhardt, J. Szanyi and J. Valyon, J. Catal., 1987, 107, 296–306 CrossRef CAS.
- H. Hattori, A. A. Amusa, R. B. Jermy, A. M. Aitani and S. S. Al-Khattaf, J. Mol. Catal. A: Chem., 2016, 424, 98–105 CrossRef CAS.
- P. E. Hathaway and M. E. Davis, J. Catal., 1989, 119, 497–507 CrossRef CAS.
- D. Archier, G. Coudurier and C. Naccache, in Proceedings of the 9th International Zeolite Conference, ed. R. Van Ballucos, et al., Butterworth–Heinemann, 1993, vol. 2, pp. 525–534 Search PubMed.
- M. D. Romero, G. Ovejero, A. Rodríguez and J. M. Gómez, Microporous Mesoporous Mater., 2005, 81, 313–320 CrossRef CAS.
- D. Barthomeuf, Catal. Rev., 1996, 38, 521–612 CrossRef.
- Y. Ono and H. Hattori, Solid Base Catalysis, Tokyo Institute of Technology Press, Tokyo, 2011, pp. 178–184 Search PubMed.
- S. V. Bordawekar and R. J. Davis, J. Catal., 2000, 189, 79–90 CrossRef CAS.
- J. Li and R. J. Davis, Appl. Catal., A, 2003, 239, 59–70 CrossRef CAS.
- H. Tsuji, F. Yagi and H. Hattori, Chem. Lett., 1991, 1881–1884 CrossRef CAS.
- F. Yagi and H. Hattori, Microporous Mater., 1997, 9, 247–251 CrossRef CAS.
- U. Schenk, M. Hunger and J. Weitkamp, Magn. Reson. Chem., 1999, 37, S75–S78 CrossRef CAS.
- M. Hunger, U. Schenk, B. Burger and J. Weitkamp, Angew. Chem., Int. Ed. Engl., 1997, 36, 2504–2506 CrossRef CAS.
- M. A. Camblor, A. Corma and S. Valencia, J. Mater. Chem., 1998, 8, 2137–2145 RSC.
- J. Li and R. J. Davis, J. Phys. Chem. B, 2005, 109, 7141–7148 CrossRef CAS PubMed.
- J. Weitkamp, S. Ernst, M. Hunger, T. Röser, S. Huber, U. A. Schubert, P. Thomasson and H. Knözinger, Stud. Surf. Sci. Catal., 1996, 101, 731–740 CrossRef CAS.
- F. Yagi, N. Kanuka, H. Tsuji, S. Nakata, H. Kita and H. Hattori, Microporous Mater., 1997, 9, 229–235 CrossRef CAS.
- M. Laspéras, H. Cambon, D. Brunel, I. Rodriguez and P. Geneste, Microporous Mater., 1996, 7, 61–72 CrossRef.
- M. Sánchez-Sánchez and T. Blasco, J. Am. Chem. Soc., 2002, 124, 3443–3456 CrossRef.
- W. J. Mortier, J. Catal., 1978, 55, 138–145 CrossRef CAS.
- N. Giordano, L. Pino, S. Cavallaro, P. Vitarelli and B. S. Rao, Zeolites, 1987, 7, 131–134 CrossRef CAS.
- D. Barthomeuf, Stud. Surf. Sci. Catal., 1991, 65, 157–169 CrossRef CAS.
- V. Bosacek, Z. Phys. Chem., 1995, 189, 241–250 CrossRef CAS.
- M. Huang, A. Adnot and S. Kaliaguine, J. Catal., 1992, 137, 322–332 CrossRef CAS.
- M. Huang, A. Adnot and S. Kaliaguine, J. Am. Chem. Soc., 1992, 114, 10005–10010 CrossRef CAS.
- Y. Okamoto, M. Ogawa, A. Maezawa and T. Imanaka, J. Catal., 1988, 112, 427–436 CrossRef CAS.
- T. L. Barr, Zeolites, 1990, 10, 760–765 CrossRef CAS.
- W. J. Mortier, Struct. Bonding, 1987, 66, 125–143 CrossRef CAS.
- S. T. King and J. M. Garces, J. Catal., 1987, 104, 59–70 CrossRef CAS.
- H. Itoh, T. Hattori, K. Suzuki and Y. Murakami, J. Catal., 1983, 79, 21–33 CrossRef CAS.
- J. Zhu, N. Mosey, T. Woo and Y. Huang, J. Phys. Chem. C, 2007, 111, 13427–13436 CrossRef CAS.
- H. Hattori, W. O. Alabi, B. R. Jermy, A. M. Aitani and S. S. Al-Khattaf, Catal. Lett., 2013, 143, 1025–1029 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra01798d |
| This journal is © The Royal Society of Chemistry 2019 |