
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot detection of telomerase activity with high sensitivity and specificity via RNA FRET probes and RNase H-assisted signal cycling amplification†
Honghong Wang ,
Hui Wang*,
Yuting Jia,
Mai Zhang and
Zhengping Li*
,
Hui Wang*,
Yuting Jia,
Mai Zhang and
Zhengping Li*
School of Chemistry and Biology Engineering, University of Science and Technology Beijing, Beijing, 100083, P. R. China. E-mail: winscavin@ustb.edu.cn; lzpbd@ustb.edu.cn
First published on 14th May 2019
Abstract
Human telomerase is a universal cancer biomarker and a promising anticancer therapeutic target. Sensitive and specific detection of telomerase activity is of great significance for cancer diagnosis and treatment. Up to now, many methods have been established to detect the activity of telomerase, but most of these methods require complex probe design and tedious experimental steps generally including telomere extension reaction, amplification of the extended products and signal detection. Herein, we propose a one-pot method to detect the telomerase activity via RNA FRET probes and RNase H-assisted signal cycling amplification, and the proposed assay can integrate the telomere extension reaction, signal amplification and readout in one step without requirement of amplification of the extended products, which greatly simplifies the experimental design and operation steps. Additionally, the proposed one-pot method has high sensitivity and can unequivocally detect the telomerase activity in as few as 5 cancer cells, which holds great potential in telomerase-related fundamental and clinical studies.
1 Introduction
Telomerase is a unique ribonucleoprotein enzyme complex with reverse transcriptase activity that can add species-dependent telomeric repeat sequences (such as “TTAGGG” in humans) onto the 3′ end of linear chromosomes to protect the end of the chromosome from fusion with neighboring chromosomes and undesired degradation.1,2 Due to the lack of telomerase activity in normal cells, telomeres gradually shorten during each cycle of cell division, and activate DNA damage signals to trigger cellular senescence and apoptosis. On the contrary, telomerase activity is reactivated in most (∼85% to 90%) human cancer cells, maintaining the telomere length and leading to cellular immortalization.3–5 Telomerase has been deemed as a universal diagnostic and prognostic biomarker for cancers, as well as a promising anti-cancer therapeutic target.6–8 Therefore, the sensitive and specific detection of telomerase activity is of great significance for cancer diagnosis and therapy.Since the first identification of telomerase in 1985,9 a variety of methods have been reported for the detection of telomerase activity. The PCR-based telomeric repeat amplification protocol (TRAP)10 is the most widely applied method for telomerase activity assay, in which radioactive label primer and multiple time-consuming steps are required, and the results are not easily quantitated. Subsequently, many modified TRAP strategies have been developed to overcome these limits by redesigning the primers, simplifying post-processing steps and adopting non-radioactive detection technologies.11–15 However, the TRAP assays require thermal cycles and the false positive or negative signals resulting from PCR-derived artifacts are difficult to be avoided. Recently, many PCR-free methods have also been designed for detection of telomerase activity, such as colorimetric,16,17 fluorescent,18–21 chemiluminescent,22,23 electrochemical,24,25 electrochemiluminescent26 methods. Although some of the strategies have been used for monitoring intracellular telomerase activity,18,19 most of these methods also challenge to satisfactory sensitivity owing to the lack of effective signal amplification. More recently, Weizmann,27 Zhang23 and their coworkers reported the sensitive methods for the telomerase activity assay at single cell level based on telomerase-triggered isothermal exponential amplification, but these approaches need the synergistic reactions catalyzed by nicking enzyme and DNA polymerase, which increase the cost and inevitably produce nonspecific amplification signals.28 Moreover, strict experimental conditions such as reaction buffer, temperature and divalent metal ions should be carefully optimized to ensure the activity of different enzymes. In 2016, we developed a stem-loop primer-mediated exponential amplification (SPEA) strategy for accurately and specifically detecting the telomerase-elongated telomere repeats with near zero nonspecific signal.29 The SPEA-based assay can detect telomerase activity in a single cell with one-type of DNA polymerase under isothermal conditions. Although the sensitivity has been pushed to extremely low detection limits, this method requires DNA amplification with a complex design, using two probes and two primers. Nevertheless, all of the above-mentioned methods require complex design and multiple operating steps to complete telomere extension reaction, amplification of the extended products and the signal detection. In this regard, the analysis of telomerase activity with simple operation process and satisfactory clinical research sensitivity is still a great demand.
Herein, we wish to present a one-pot method for the detection of telomerase activity with greatly simplified principle design and operation process. The proposed method does not require complex DNA amplification and can put the telomerase reverse transcriptional extension reaction and signal amplification reaction all in one tube. The two reactions were carried out in coordination at 37 °C. For signal amplification reaction, an RNA probe (18 nucleotides) based on fluorescence resonance energy transfer (FRET) (RNA FRET probe) is rationally designed, which can specifically identify and hybridize with the telomerase-elongated telomere repeat sequences. The RNA FRET probes hybridized with telomerase extension products were hydrolyzed specifically to produce fluorescence signal in the presence of RNase H, resulting in cycling signal amplification, which makes the detection sensitivity of the telomerase activity greatly improved. Taking only one-step reaction, and at a constant temperature, the telomerase activity can be unequivocally detected in as low as 5 HeLa cells, which can generally satisfy the needs for detection of telomerase activity for biological studies and clinical diagnostics.
2 Results and discussion
2.1. Principle of the telomerase activity assay
In this study, we report a simple and sensitive fluorescence assay that only needs one-step to complete the detection of telomerase activity based on RNA probe and RNase H-assisted signal recycling amplification. The mechanism of the assay for detecting telomerase activity is schematically illustrated in Fig. 1. The assay system composed of telomerase substrate (TS) primer, RNase H, RNA probe and telomerase extracted from crude cancer cells. TS primer (18 deoxyribonucleotides) contains a non-telomeric repeat sequence and can be specifically identified by telomerase as a substrate. Once the telomerase extracted from the cancer cells is added into the solution, the TS will be elongated with catalysis of telomerase by adding tandem repeats (5′-TTAGGG-3′)n at its 3′ terminus to form a longer single-stranded DNA molecule in the presence of the dNTP mixture. We design an RNA probe (14 nucleotides) containing a complementary sequence to the telomerase extension products is modified with 6-carboxyfluorescein (FAM) as a fluorophore at its 5′ terminus and Black Hole Quencher® (BHQ1) as a quencher at its 3′ terminus. The RNA probe gives a very weak fluorescence signal from FAM due to fluorescence resonance energy transfer (FRET) from FAM to BHQ1. The extended TS telomeric repeat strand can hybridize with multiple RNA probes to form RNA–DNA hybrids. RNase H is a ribonuclease endonuclease that specifically hydrolyzes the phosphodiester bonds of RNA hybridized to DNA strand. The enzyme cannot digest single strand or double strand DNA. These RNA–DNA hybrids can be recognized by RNase H, leading to the digestion of the RNA probe incorporated in the hybrids. | ||
| Fig. 1 Schematic illustration of RNA probe and RNase H-assisted signal recycling amplification for one-step detection of the telomerase activity. | ||
More importantly, this digestion process not only makes the fluorophore–quencher pair of the RNA probe separated from each other, but also releases the telomerase extended products, which then hybridized with fresh RNA probe to continually initiate the recycling digestion process leading to significantly enhanced fluorescence. The digestion process can be performed in coordinately with telomerase extension reaction at 37 °C. Therefore, the fluorescence signal of the telomerase extension reaction can be real-time monitored, that is to say, only one step is needed to complete the detection of telomerase activity, which greatly simplifies the operation process of the telomere activity analysis.
2.2. Feasibility of the proposed telomerase assay
To investigate feasibility of the RNA probe and RNase H-assisted signal recycling amplification assay, we first detected synthetic telomerase extension product (TEP) with eight TTAGGG repeats. As shown in Fig. 2A, the RNA probe showed a very weak fluorescence signal, which indicated the fluorescence of FAM was quenched effectively by BHQ1 (curve a). After incubating with the TS primer and RNase H at 37 °C for 120 min, no significant fluorescence change of RNA probe could be observed as well (curve b), demonstrating that RNase H cannot digest the RNA probe due to its high specificity to hydrolyze the phosphodiester bonds of RNA hybridized to DNA strand. In contrast, a strong fluorescence signal was obtained (curve d) when 1 nM TEP is mixed with 1 μM RNA probe, suggesting that the RNA probe was digested by RNase H only after being hybridized with the synthetic TTAGGG repeats, resulting in the separation of fluorescence and quenching groups and the enhanced of fluorescence signal. Interestingly, when the added RNA probe concentration (1 nM) was equal to the concentration of the TEP (1 nM) (curve c), the fluorescence signal showed a much smaller increase. This means that, without the presence of an excess of RNA probes, TEP cannot repeatedly hybridize with the excess RNA probes, causing the target recycling amplification reaction to fail to occur. Therefore, we conclude that our telomerase activity detection scheme is specific to TTAGGG repeats, and a great of signal amplification is obtained. Next, we used this scheme to test the telomerase extract. The telomerase extract, TS, RNA probe and RNase H were incubated together at 37 °C for 2 h. That is, along with the telomerase reverse transcriptional extension reaction and RNase H hydrolysis reaction were also conducted. As shown in Fig. 2B, in the presence of telomerase extracts from 800 HeLa cells, obviously enhanced fluorescence was observed (curve c). No noticeable fluorescence signal was obtained using telomeres extract-free lysis buffer (curve a). This is due to the high selectivity of RNase H for hydrolyzing RNA probes hybridized with the extended products of telomerase. A negative control that telomerase extracts from 800 HeLa cells were preheated at 95 °C for 5 min to inactivate telomerase activity is simultaneously detected and no observed fluorescence signal was produced (curve b), confirming that enhanced fluorescence is specifically derived from telomerase activity. Such phenomenon is further validated by polyacrylamide gel electrophoresis (inset of Fig. 2B). TS primer shows a defined band corresponding to 18 bp (lane 1). After incubation of TS with HeLa extracts, as depicted in lane 2, the TS primers with 18 bases were dispersed on the polyacrylamide gels because telomerase added different numbers of repeats (TTAGGG)n to the TS. Meanwhile, the negative control (lane 3), in which the HeLa cell extract is heat-inactivated, has no telomerase reverse transcriptional extension reaction. However, there are some light-colored stripes in lane 2 and lane 3, and we also performed electrophoresis with the telomerase extracts (lane 4) and heat-inactivated telomerase extracts (lane 5). Lane 4 and lane 5 also have light-colored stripes as same as lane 2 and lane 3, which confirmed that these stripes are not reaction products of elongate TS by telomerase and are from the cell extract containing large amounts of proteins and nucleic acids. These results demonstrated that the proposed telomerase activity scheme was feasible. | ||
| Fig. 2 (A) Fluorescence spectra of (a) 1 μM RNA probe, (b) reaction products with 1 μM RNA probe and 1 μM TS primer, (c) reaction products with 1 nM RNA probe and 1 nM TEP, and (d) reaction products with 1 μM RNA probe and 1 nM TEP. (B) Fluorescence spectra of reaction products with (a) lysis buffer, (b) heat-inactivated telomerase extracts, (c) telomerase extracts equivalent to 800 HeLa cells. Inset: non-denaturing PAGE analysis of the telomerase activity. Lane M: double strand DNA ladder, lane 1: TS primer (100 nM), lane 2: reaction products with telomerase extracts, lane 3: reaction products with heat-inactivated telomerase extracts, lane 4: telomerase extracts, lane 5: heat-inactivated telomerase extracts equivalent to 800 HeLa cells. | ||
2.3. Detection of telomerase activity in HeLa cells
To verify the effectiveness of the proposed strategy for detecting the activity of telomerase extracted from cancer cells, the HeLa cell was used as a model cancer cell, which is human cervical carcinoma with positive telomerase activity. 6 U RNase H are found to be optimum for the proposed assay (see Fig. S1 in ESI†). In the experiment, all reaction components including the telomerase, TS, dNTPs, RNase H, and RNA probe were added to the reaction solution, the mixture was incubated at 37 °C for 120 min. As depicted in Fig. 3A, the well-defined fluorescence signals in response to the diluted extracts of telomerase equivalent to a series of different number of HeLa cells can be observed. As expected, the fluorescence intensity increased with the number of HeLa cell in the range from 5 to 1000 cells, and the plot of fluorescence intensity with the number of HeLa cells is shown in Fig. 3B. These results clearly indicated that more telomerase could produce more extension products and result in higher fluorescence intensity. Obviously, telomerase extension reaction and RNase H enzyme digestion reaction can be carried out simultaneously, and telomerase activity in the HeLa cell extracts equivalent to 5–1000 cells could be detected by the proposed strategy. When the fluorescence intensity at 520 nm (IF) were plotted against the number of HeLa cells (N) in the range of 5–80 cells, the standard curve showed a linear relationship with the correlation equation of IF = 2.1N + 220.1(inset of Fig. 3B). According to the rule of three times standard deviation over the blank, the corresponding detection limit for telomerase activity assay was estimated to be equivalent to 2 HeLa cells, which is comparable to the TRAP-based method13,15 and better than that of the recently reported the majority of PCR-free detection.17,21,22,24,25 In addition, we have further investigated feasibility of the proposed one-pot strategy for the detection of telomerase activity in the crude lysates of 5, 10 and 20 cells, respectively. It can be seen from Fig. S2† that as low as 5 cells can also be well detected, which coincides with that for detection of the diluted extracts.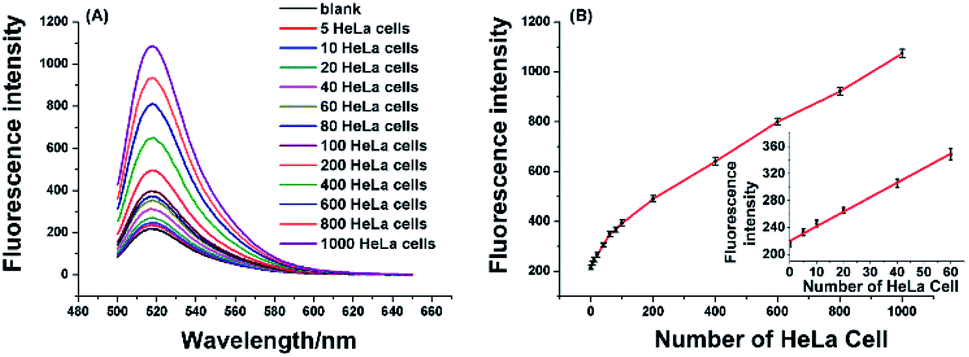 | ||
| Fig. 3 (A) Fluorescence spectra for detecting telomerase activity in diluted extracts equivalent to 5, 10, 20, 40, 60, 80, 100, 200, 400, 600, 800, and 1000 HeLa cells, respectively. The CHAPS lysis buffer was used as the blank. (B) The relationship between the fluorescence intensity at 520 nm and different number of HeLa cells. Inset: the linear plot of the fluorescence intensity versus the number of the HeLa cells (0–80 cells). Error bars represent the standard deviation of five repetitive tests. | ||
2.4 Real-time fluorescence detection of the telomerase activity
As mentioned above, the RNA probe and RNase H-assisted telomerase assay does not require complex DNA amplification reactions. Reverse transcription extension reaction and signal amplification steps by the proposed assay can be completed under uniform constant temperature condition. Telomerase activity detection can be achieved by laboratory conventional fluorescence instrument. By using real-time quantitative PCR instrument, telomerase activity assay can be accomplished in one step. As shown as in Fig. 4, well-defined real-time fluorescence curves can be obtained which are produced by the different concentration of telomerase ranging from 5–1000 HeLa cells. With increasing concentration of telomerase, the corresponding fluorescence intensity is gradually increased, and as low as 5 HeLa cells can be distinctly detected with the proposed telomerase activity assay under isothermal condition. Comparison between the proposed one-pot assay and other reported methods13,15,17,21,22,24,25,29,30 for the detection of telomerase activity is shown in Table S1 in ESI.† The table reveals that present assay is among the sensitive analysis schemes of telomerase. The major advantages of the one-pot method, besides its sensitivity, are, however, reflected by lack for need of sophisticated manipulation and the nucleic acids amplification. | ||
| Fig. 4 Real-time fluorescence signals of telomerase activity assay. From a to m, HeLa cell number was successively 0, 5, 10, 20, 40, 60, 80, 100, 200, 400, 600, 800, 1000. | ||
2.5. Evaluation of the generality and selectivity of telomerase assay
To demonstrate the proposed assay is universal for telomerase activity detection, different cancer cell lines, including breast cancer cells (MCF-7), lung adenocarcinoma cells (A549), leukemia cells (CEM), and normal cell line (MRC-5) were tested. These cell lines were purchased from the cell bank of Chinese Academy of Science. As demonstrated in Fig. 5, all the cancer cell lines were positive for telomerase activity, which was consistent with the telomerase overexpression of the well-known tumor cells. The normal cell line did not generate any enhanced fluorescence signal due to the lack of telomerase activity in normal cells. Moreover, after mixing with the same amount of extracts from MRC-5 normal cell line, the telomerase activity of HeLa cancer cell can still be accurately detected. Furthermore, as shown in Fig. S3 (ESI†), the proposed assay have high selectivity towards other proteins including of ProtoScript II reverse transcriptase, bull serum albumin (BSA), horse radish peroxidase (HRP) and glucose oxidase (GOD). All of these results demonstrate the high specificity of the one-pot assay for telomerase activity detection, which implies the present method has great potential to be applied in clinical samples. | ||
| Fig. 5 Fluorescence spectra (A) and histogram (B) in response to telomerase extracts equivalent to 200 cells from different kinds of cell lines including of a normal cell line MRC-5, the fluorescence curves from a to g refer to the blank, MRC-5, CEM, MCF-7, HeLa, the mixture of HeLa and MRC-5, A549, respectively. Error bars represent the standard deviation of three repetitive tests. The RTA of the HeLa cell extract is normalized to be 1.0. | ||
The inhibition assay of telomerase activity was also investigated by adding 3′-azido-3′-deoxythymidine (AZT), a potent inhibitor of telomerase activity, into the reaction mixture of telomerase activity detection. As shown in Fig. S4 in ESI,† fluorescence intensity decreased gradually with increasing concentration of AZT in the range 0.5–30 mM. The result indicated that the RNA probe and RNase H-assisted telomerase assay shows high specificity for detection of telomerase activity and great potential in high-throughput screening for telomerase activity inhibitors and for anticancer drug development.
3 Conclusions
In summary, with the simply designed RNA probe, a sensitive and specific telomerase assay based on RNA probe and RNase H-assisted signal recycling amplification was proposed for the detection of telomerase activity in this study, which can be performed only one-step operation under isothermal conditions. Compared with the conventional multi-step strategies, telomerase reverse transcriptional reaction, the signal amplification and detection of the as-proposed assay can be all accomplished in one-step in a single-tube, which is easy to operate and reduce the possibility of contamination. Additionally, the RNA probe and RNase H-assisted telomerase assay not only has remarkably specificity but also has a high sensitivity, which is feasible for the accurate detection of telomerase activity down to equivalent to 5 HeLa cells. By taking advantage of its various distinct advantages, we believe this one-pot assay but highly sensitive telomerase activity detection strategy holds great potential in telomerase-related fundamental and clinical studies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China [21705008 and 21775012], the China Postdoctoral Science Foundation [2017M620605], and the Fundamental Research Funds for the Central Universities [FRF-TP-17-046A1].Notes and references
- E. H. Blackburn, Nature, 1991, 350, 569–573 CrossRef CAS PubMed.
- E. H. Blackburn, Cell, 2001, 106, 661–673 CrossRef CAS PubMed.
- S. B. Cohen, M. E. Graham, G. O. Lovrecz, N. Bache, P. J. Robinson and R. R. Reddel, Science, 2007, 315, 1850–1853 CrossRef CAS PubMed.
- J. W. Shay and W. E. Wright, Nat. Rev. Drug Discovery, 2006, 5, 577–584 CrossRef CAS PubMed.
- J. W. Shay and S. Bacchetti, Eur. J. Cancer, 1997, 33, 787–791 CrossRef CAS PubMed.
- L. J. Wang, F. Ma, B. Tang and C. Y. Zhang, Chem. Sci., 2017, 8, 2495–2502 RSC.
- X. M. Zhou and D. Xing, Chem. Soc. Rev., 2012, 41, 4643–4656 RSC.
- X. J. Zhang, X. D. Lou and F. Xia, Theranostics, 2017, 7, 1847–1862 CrossRef CAS PubMed.
- C. W. Greider and E. H. Blackburn, Cell, 1985, 43, 405–413 CrossRef CAS PubMed.
- N. W. Kim, M. A. Piatyszek, K. R. Prowse, C. B. Harley, M. D. West, P. L. C. Ho, G. M. Coviello, W. E. Wright, S. L. Weinrich and J. W. Shay, Science, 1994, 266, 2011–2015 CrossRef CAS PubMed.
- N. W. Kim and F. Wu, Nucleic Acids Res., 1997, 25, 2595–2597 CrossRef CAS PubMed.
- A. T. Ludlow, J. D. Robin, M. Sayed, C. M. Litterst, D. N. Shelton, J. W. Shay and W. E. Wright, Nucleic Acids Res., 2014, e104 CrossRef CAS PubMed.
- H. Wege, M. S. Chui, H. T. Le, J. M. Tran and M. A. Zern, Nucleic Acids Res., 2003, 31, e3 CrossRef PubMed.
- B. S. Herbert, A. E. Hochreiter, W. E. Wright and J. W. Shay, Nat. Protoc., 2006, 1, 1583–1590 CrossRef CAS PubMed.
- Y. Xiao, K. Y. Dane, T. Uzawa, A. Csordas, J. Qian, H. T. Soh, P. S. Daugherty, E. T. Lagally, A. J. Heeger and K. W. Plaxco, J. Am. Chem. Soc., 2010, 132, 15299–15307 CrossRef CAS PubMed.
- Y. Xiao, V. Pavlov, T. Niazov, A. Dishon, M. Kotler and I. Willner, J. Am. Chem. Soc., 2004, 126, 7430–7431 CrossRef CAS PubMed.
- E. Sharon, E. Golub, A. Niazov-Elkan, D. Balogh and I. Willner, Anal. Chem., 2014, 86, 3153–3158 CrossRef CAS PubMed.
- R. Qian, L. Ding and H. Ju, J. Am. Chem. Soc., 2013, 135, 13282–13285 CrossRef CAS PubMed.
- R. Qian, L. Ding, L. Yan, M. Lin and H. Ju, J. Am. Chem. Soc., 2014, 136, 8205–8208 CrossRef CAS PubMed.
- R. Qian, L. Ding, L. Yan, M. Lin and H. Ju, Anal. Chem., 2014, 86, 8642–8648 CrossRef CAS PubMed.
- H. B. Wang, S. Wu, X. Chu and R. Q. Yu, Chem. Commun., 2012, 48, 5916–5918 RSC.
- V. Pavlov, Y. Xiao, R. Gill, A. Dishon, M. Kotler and I. Willner, Anal. Chem., 2004, 76, 2152–2156 CrossRef CAS PubMed.
- L. J. Wang, Y. Zhang and C. Y. Zhang, Anal. Chem., 2013, 85, 11509–11517 CrossRef CAS PubMed.
- W. J. Wang, J. J. Li, K. Rui, P. P. Gai, J. R. Zhang and J. J. Zhu, Anal. Chem., 2015, 87, 3019–3026 CrossRef CAS PubMed.
- P. Ling, J. Lei and H. Ju, Anal. Chem., 2016, 88, 10680–10686 CrossRef CAS PubMed.
- X. Zhou, D. Xing, D. Zhu and L. Jia, Anal. Chem., 2009, 81, 255–261 CrossRef CAS PubMed.
- L. Tian and Y. Weizmann, J. Am. Chem. Soc., 2013, 135, 1661–1664 CrossRef CAS PubMed.
- E. Tan, B. Erwin, S. Dames, T. Ferguson, M. Buechel, B. Irvine, K. Voelkerding and A. Niemz, Biochemistry, 2008, 47, 9987–9999 CrossRef CAS PubMed.
- H. Wang, H. Wang, C. Liu, X. Duan and Z. Li, Chem. Sci., 2016, 7, 4945–4950 RSC.
- X. J. Liu, W. Li, T. Hou, S. S. Dong, G. H. Yu and F. Li, Anal. Chem., 2015, 87, 4030–4036 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra01816f |
| This journal is © The Royal Society of Chemistry 2019 |