
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing the local conformational flexibility in receptor recognition: mechanistic insight from an atomic-scale investigation†
Fei Ding ab and
Wei Peng*cd
ab and
Wei Peng*cd
aSchool of Environmental Science and Engineering, Chang'an University, Xi'an 710064, China
bKey Laboratory of Subsurface Hydrology and Ecological Effect in Arid Region of Ministry of Education, Chang'an University, No. 126 Yanta Road, Yanta District, Xi'an 710064, China
cCollege of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China. E-mail: crystalw.peng@outlook.com; Fax: +86-29-87092367; Tel: +86-29-87092367
dDepartment of Chemistry, China Agricultural University, Beijing 100193, China
First published on 7th May 2019
Abstract
Inherent protein conformational flexibility is important for biomolecular recognition, but this critical property is often neglected in several studies. This event can lead to large deviations in the research results. In the current contribution, we disclose the effects of the local conformational flexibility on receptor recognition by using an atomic-scale computational method. The results indicated that both static and dynamic reaction modes have noticeable differences, and these originated from the structural features of the protein molecules. Dynamic interaction results displayed that the structural stability and conformational flexibility of the proteins had a significant influence on the recognition processes. This point related closely to the characteristics of the flexible loop regions where bixin located within the protein structures. The energy decomposition analyses and circular dichroism results validated the rationality of the recognition studies. More importantly, the conformational and energy changes of some residues around the bixin binding domain were found to be vital to biological reactions. These microscopic findings clarified the nature of the phenomenon that the local conformational flexibility could intervene in receptor recognition. Obviously, this report may provide biophysical evidence for the exploration of the structure–function relationships of the biological receptors in the human body.
Introduction
In general, molecular recognition between the biological receptors and ligands plays a key role in nearly all biochemical processes. A variety of physiological effects can be achieved through biomolecular recognition of active substances with specific receptors such as enzymes, nucleic acids and proteins.1 These events, e.g. cellular signaling, enzyme catalysis, protein crowding, reactant transport, and receptor–ligand association, are considered to be the essential preconditions for maintaining life activities.2 The investigation of these crucial reaction processes, particularly the structure–function relationships of receptors, could help us to understand the different biological responses, for example, pathological changes, pharmacological and toxicological actions, and physiological regulations in the body.3–6 For this reason, receptor recognition has emerged as one of the most interesting subjects in the areas of chemical biology, medicinal chemistry and toxicology.Biological receptors are frequently regarded as the main participant in almost all physiological activities. They have become the primary objects in the study of molecular recognition due to their intricate microenvironment, special spatial structure, and unique function.7 It is discovered so far that most of the functional receptors belong to protein molecule, and these macromolecules can recognize and selectively bind to active ligands. These events would induce conformational alterations in proteins and activate a series of biological responses, and finally represent as different biochemical effects.8 Usually, protein folds into a tightly flexible conformation. However, such conformation is not fully rigid, and its chief characters and total form are decided by the protein's amino acid sequence. This means that the biopolymer is highly dynamic, and its motions are often crucial to its function.9 It is popularly accepted that the role of a compact protein is greatly dependent on its conformation and its capacity to deform.10 The ability to perform biological functions such as catalysts, mechanical effectors, sensors, signaling molecules and transporters depends on the conformational changes of the cohesive protein and on the dynamics of these deformations.11 As a result, a complete understanding of protein function requires an exploration of both the dynamic behaviors of a protein and its static conformational features.
Typically, protein molecule contains a number of loop components. This structural characteristic endows macromolecular conformation with the great flexibility property.12 For example, the conformations of ligand-gated ion channels and albumin from human serum (HSA) possesses a large degree of flexibility, and they can recognize numerous agonists and antagonists with different molecular characters and volumes.13,14 Likewise, α1-acid glycoprotein from human plasma (AGP) holds a flexible ligand recognition patch. The sophisticated geometry of the domain, together with the structural flexibility of loop 1 (β-strands A/B) at its gate, interprets a broad variety of chemicals that could be recognized by this lipocalin.15,16 Therefore we should take conformational flexibility into consideration so as to accurately study the interaction between a receptor and an active ligand. Recently, the accumulating evidences suggested that conformational flexibility coupled to ligand binding played a major role in receptor recognition process, and the conformational changes of the residues in the reaction region and the large-scale loop reorganization are observed upon ligand binding to receptor.17,18 Unfortunately, the physiological significance of the structural feature and the biological role of flexibility of these loops in receptors are still largely unclear as yet.
Given the above-mentioned research background, the present effort was to elaborate the effects of the local conformational flexibility on receptors recognition by employing an atomic-scale computational technique from a biophysical point of view. The concrete content may be divided into the following parts: (I) analysis of the static and dynamic recognition modes of the receptors–bixin conjugates; (II) elucidation of the influences of the local conformational flexibility on the recognition processes; and (III) clarification of the energy bases during receptors recognition. We expect that this attempt can shed physicochemical light on the investigation of the structure–function relationships of the central receptors in the body.
Experimental
Materials
Bixin (05989, ≥90%, CAS number 6983-79-5), albumin from human serum (A8763, lyophilized powder, essentially globulin free, ≥99%, CAS number 70024-90-7) and α1-acid glycoprotein from human plasma (G9885, ≥99%, CAS number 66455-27-4) were received from Sigma-Aldrich (St. Louis, MO) and utilized without further purification, and deionized water was generated by a Milli-Q Ultrapure Water Purification Systems from Millipore (Billerica, MA). Tris (0.2 M)–HCl (0.1 M) buffer of pH = 7.4, with an ionic strength 0.1 in the presence of NaCl, and the pH was determined with an Orion Star A211 pH Benchtop Meter (Thermo Scientific, Waltham, MA). Dilutions of the protein stock solution (20 μM) in Tris–HCl buffer were prepared immediately before use, and the concentration of proteins was measured by the method of Lowry et al.19 All other reagents used were of analytical grade and purchased from Sigma-Aldrich.In silico docking
Molecular docking of the protein–bixin bioconjugates was executed on SGI Fuel Visual Workstation (Silicon Graphics International Corporation, Milpitas, CA). The crystal structures of HSA (entry codes 1E7I) and AGP (entry codes 3APU),20,21 determined at a resolution 2.7 Å and 2.1 Å, were respectively retrieved from the Brookhaven Protein Data Bank (http://www.rcsb.org/pdb). After being imported in the program Sybyl version 7.3 (http://www.certara.com), protein structures were carefully checked for atom and bond type correctness assignment. Hydrogen atoms were computationally added using the Sybyl Biopolymer and Build/Edit menus. To avoid negative acid/acid interactions and repulsive steric clashes, added hydrogen atoms were energy minimized with the Powell algorithm with 0.5 kcal mol−1 energy gradient convergence criteria for 1000 cycles.22 This procedure does not change positions to heavy atoms, and the potential of the three-dimensional structures of proteins were assigned according to the AMBER force field with Kollman all-atom charges.23The two-dimensional structure of bixin was downloaded from PubChem (http://pubchem.ncbi.nlm.nih.gov), and the initial structure of the molecule was generated by Sybyl 7.3. The geometry of bixin was subsequently optimized to minimal energy (tolerance of 0.5 kcal mol−1) utilizing the Tripos force field with Gasteiger–Hückel charges,24 and the lowest energy conformer was applied for the docking analysis. The Surflex-Dock program which uses an automatic flexible docking algorithm was utilized to analyze the possible conformation of the ligand that binds to proteins, and the program PyMOL (http://www.schrodinger.com) was finally employed for visualization of the ligand docking results. Furthermore, we selected the CABS-dock web server to perform the full flexible docking so as to validate the rationality of the above semi-flexible docking.25–31 This process can be used to check whether there are similar reaction modes in the output results. As an online software, it could use the crystal structures of HSA and AGP as the protein parts, respectively, and the long-chain structure of bixin may be considered as a peptide. The three-dimensional structure was imported and then the top 10 scored models of the HSA–bixin and AGP–bixin adducts will automatically be exported from the software. These models can be used to verify the semi-flexible docking results derived from Sybyl 7.3.
Molecular dynamics simulation
Molecular dynamics (MD) simulation of the HSA–bixin complex was performed using Gromacs program, version 2019, with the Gromos96 ffG43a1 force field.32,33 Simulation processes were run under physiological conditions (pH = 7.4), and the amino acid residues possessed acidity and basicity were adjusted to the protonation states at neutrality condition. Initial conformations of HSA and bixin were, respectively, taken from the original X-ray diffraction crystal structure that was solved at 2.7 Å resolution (entry codes 1E7I) and the optimal structure originated from molecular docking. The topology of protein was yielded by Gromacs package directly, whereas bixin by PRODRG2.5 Server.34 The simulation system was solvated with a periodic cubic box (the volume is 8.771 × 5.061 × 8.664 nm3) filled with TIP3P water molecules and an approximate number (14) of sodium counterion to neutralize the charge.35 Totally, there are 43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 648 crystallographic solvent molecules, and the shortest distance between the complex and the edge of the box is set to 10 Å. Simulations were operated utilizing the isothermal–isobaric (NPT) ensemble with an isotropic pressure of 1 bar,36 and the temperature of the ligand, protein and solvent (water and counterion) was separately coupled to an external bath held at 300 K, using the Berendsen thermostat with 0.2 ps relaxation time.37 The LINCS algorithm was employed to constrain bond lengths,38 and the long-range electrostatic interactions beyond 10 Å were modeled exploiting the Particle Mesh Ewald (PME) method with a grid point density of 0.1 nm and an interpolation order of 4.39 A cutoff of 14 Å was used for van der Waals' interactions. The MD integration time step was 2.0 fs and covalent bonds were not constrained, and the system configurations were saved every 2.0 ps. To decrease the atomic collisions with each other, both gradient descent and conjugate gradient algorithm were used to optimize the whole system.40,41 First the solvated starting structure was preceded by a 1000-step gradient descent and then by conjugate gradient energy minimization. Subsequently, 100 ps equilibration with position restraints uses to remove possible unfavorable interactions between solute and solvent, and after thorough equilibration, MD simulations were conducted for 100 ns. Moreover, the top two ligand structures in the reaction system were selected to execute the parallel MD simulations, in order to confirm the rationality of the optimal energy conformation for MD simulation. The pure protein was also selected to carry out a time period (50 ns) MD simulation so as to compare with the optimal ligand docking adduct. The results of MD simulations were ultimately illustrated by Visual Molecular Dynamics 1.9.4,42 and the program Discovery Studio Visualizer 4.0 (BIOVIA, San Diego, CA) was used to exhibit the patterns of the MD simulations.
648 crystallographic solvent molecules, and the shortest distance between the complex and the edge of the box is set to 10 Å. Simulations were operated utilizing the isothermal–isobaric (NPT) ensemble with an isotropic pressure of 1 bar,36 and the temperature of the ligand, protein and solvent (water and counterion) was separately coupled to an external bath held at 300 K, using the Berendsen thermostat with 0.2 ps relaxation time.37 The LINCS algorithm was employed to constrain bond lengths,38 and the long-range electrostatic interactions beyond 10 Å were modeled exploiting the Particle Mesh Ewald (PME) method with a grid point density of 0.1 nm and an interpolation order of 4.39 A cutoff of 14 Å was used for van der Waals' interactions. The MD integration time step was 2.0 fs and covalent bonds were not constrained, and the system configurations were saved every 2.0 ps. To decrease the atomic collisions with each other, both gradient descent and conjugate gradient algorithm were used to optimize the whole system.40,41 First the solvated starting structure was preceded by a 1000-step gradient descent and then by conjugate gradient energy minimization. Subsequently, 100 ps equilibration with position restraints uses to remove possible unfavorable interactions between solute and solvent, and after thorough equilibration, MD simulations were conducted for 100 ns. Moreover, the top two ligand structures in the reaction system were selected to execute the parallel MD simulations, in order to confirm the rationality of the optimal energy conformation for MD simulation. The pure protein was also selected to carry out a time period (50 ns) MD simulation so as to compare with the optimal ligand docking adduct. The results of MD simulations were ultimately illustrated by Visual Molecular Dynamics 1.9.4,42 and the program Discovery Studio Visualizer 4.0 (BIOVIA, San Diego, CA) was used to exhibit the patterns of the MD simulations.
Calculation of free energies
Binding free energies of the proteins–bixin reactions were computed by using the Amber Molecular Dynamics Package (University of California, San Francisco, CA) based on the method of Molecular Mechanics/Generalized Born Surface Area (MM/GBSA), and the relevant relationships for the MM/GBSA enumerations are given by43,44| ΔGbind = Gcomplex − (Gprotein + Gligand) | (1) |
| EMM = EvdW + Eele | (2) |
| G = 〈EMM〉 = 〈Gnonpol,sol〉 + 〈Gpol,sol〉 − T〈S〉 | (3) |
| Gnonpol,sol = γ × SASA + b | (4) |
In these equations the binding free energy, ΔGbind, is constituted of the classical Eproducts − Ereactants (the endpoints), where Eproducts = ΔGcomplex and Ereactants is composed of Gprotein and Gligand. The molecular mechanics energy (EMM) is made up of the van der Waals energy (including the internal energy) (EvdW) and the electrostatic energy (Eele). The polar solvation ingredient (Gpol,sol) is estimated utilizing the generalized Born method. The nonpolar solvation element (Gnonpol,sol) is reckoned using solvent accessible area with the γ parameter set to 0.00542 kcal (mol Å2)−1, and the b parameter set to 0.92 kcal mol−1. The Solvent Accessible Surface Area (SASA) is measured employing the linear combination of pairwise overlaps (LCPO) model.45
Circular dichroism
Circular dichroism (CD) spectra were conducted with a J-810 Circular Dichroism Spectropolarimeter (Jasco, Japan) equipped with a microcomputer, the apparatus was sufficiently purged with 99.9% dry nitrogen gas before starting the instrument and then it was calibrate with d-10-camphorsulfonic acid. All the CD spectra were gathered at 298 K with a PFD-425S Peltier temperature controller attached to a water bath with an accuracy of ±0.1 °C. Each spectrum was recorded with use of a precision quartz cuvette of 10 mm path length and taken at wavelengths between 200 and 260 nm range that provides a signal extremely sensitive to small secondary conformational disturbances. Every determination was the average of five continual scans encoded with 0.1 nm step resolution and got at a speed of 50 nm min−1 and response time of 1.0 s. All observed CD data were baseline subtracted for buffer and the evaluation of the secondary structure components was gained by employing Jasco Spectra Manager II, which calculates the different designations of secondary structures by comparison with CD spectra, determined from diverse proteins for which high-quality X-ray diffraction data are available.46Results and discussion
Static recognition profiles of the receptors–bixin complexes
To study the static reaction modes between the proteins and bixin, a ligand docking method has been employed to check the molecular biosystems based on the three-dimensional structures of both HSA and AGP.47,48 The optimal reaction modes of the two molecular systems received from the CABS-dock web server were found to be very similar to the optimal reaction patterns obtained by Sybyl software, showing that the results of molecular docking are reasonable. Meanwhile, the top two conformations in the docking conformations were selected and superimposed on the optimal conformations, and the results are displayed in Fig. S2.† The superposition results indicated clearly that the top-ranking ligand conformations can well be superposed with the best-scoring conformations in the two conjugated reactions, and the conformational differences are very small. This phenomenon suggested that the use of the conformation with the highest scoring as the initial conformation for MD simulations is fully reasonable. Fig. 1 is the optimal recognition pattern of the two biological reactions. Apparently, the binding region of bixin is situated within domain III on HSA (residues 404–558), and the binding conformation of bixin favors the middle and later part of the domain III, that is subdomain IIIB (Fig. 1(A)). The Gibbs free energy (ΔG°) of the HSA–bixin is −34.78 kJ mol−1, which coincides with the result of wet experiments reported by Zsila et al.49 We may observe that such molecular recognition is proceeded by hydrogen bonds and hydrophobic effects. Specifically, the two oxygen atoms of the carboxyl group in bixin can make hydrogen bonds with the hydrogen atom of the amino group in Ser-419, and the hydrogen atoms of the hydroxyl groups in Thr-422 and Thr-467 residues (Fig. 1(B)), and the bond lengths are 2.05 Å, 2.39 Å, and 3.28 Å, respectively. Meantime, considering the property of domain III and the hydrophobic feature around the reaction patch, it could be seen that the bixin lie at the location with strong hydrophobicity within domain III, and the surrounding residues are Val-415, Val-418, Leu-423, Leu-430, Leu-457, Val-473, Phe-488, and Val-530. Further, the middle part (polyene chain) of the bixin is a hydrophobic module except for the hydrophilic groups in both ends of long-chain structure. This molecular trait would help bixin to bind to domain III, and thus promoting the recognition strength between the protein and bixin.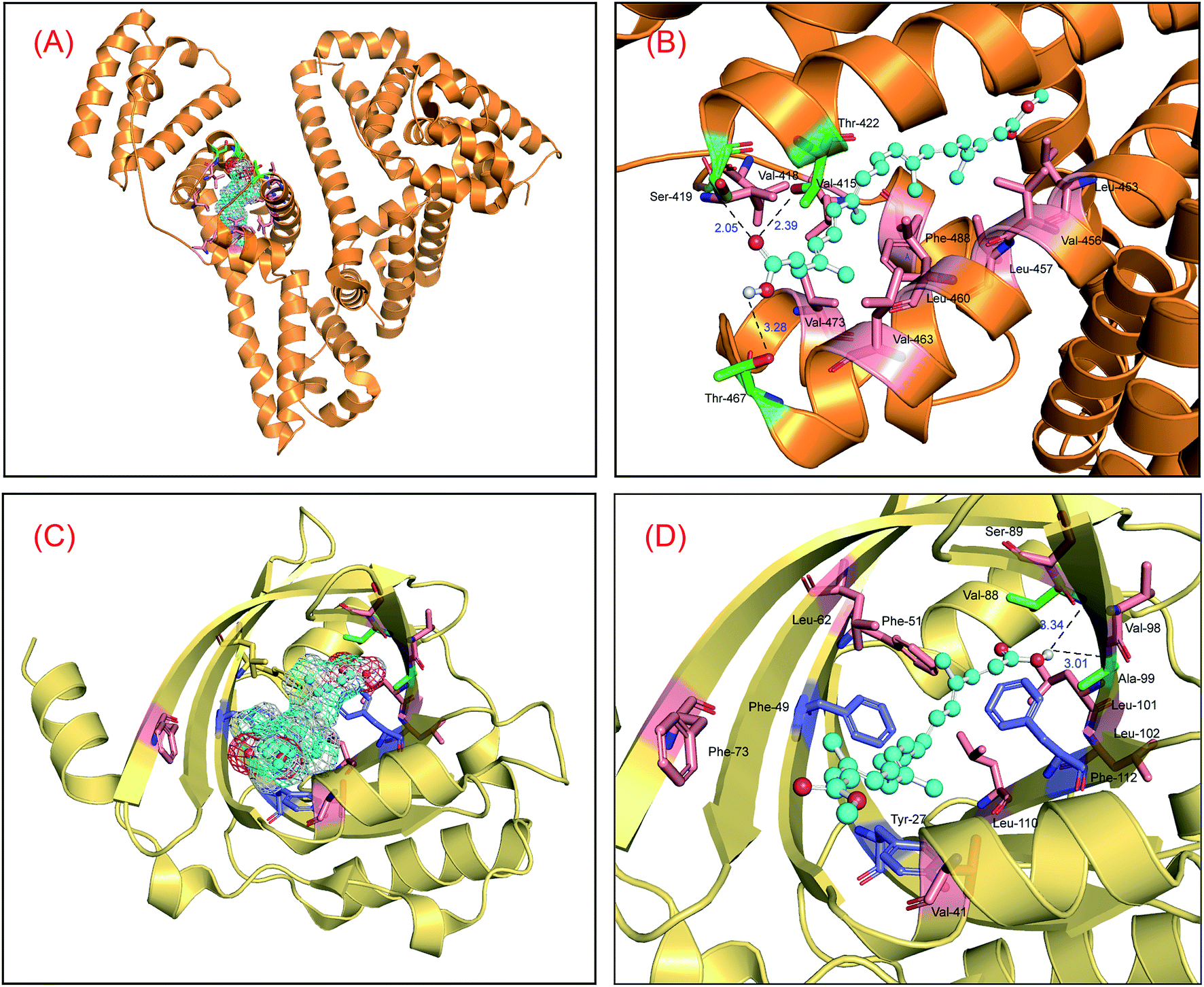 | ||
| Fig. 1 Molecular docking of bixin docked to HSA (panel (A) and panel (B)) and AGP (panel (C) and panel (D)). HSA and AGP showed in surface colored in orange and yellow, respectively, and the ball-and-stick model displays bixin, colored as per the atoms and possess meshy surface of electron spin density. The key amino acid residues around bixin have been described in stick model, green and salmon stick model indicates hydrogen bonds and hydrophobic effects between the proteins and bixin, respectively; and blue stick model explains π–π conjugated effects between the Tyr-27, Phe-49, Phe-112 residues and bixin (panel (D)). (For clarification of the references to color in this figure legend, the reader is referred to the web version of the article). | ||
Likewise, the reaction center of bixin on AGP is noted to be located at the apolar ligand patch (Fig. 1(C)), residues 27–127, and the Gibbs free energy (ΔG°) of the AGP–bixin is −24.65 kJ mol−1. Obviously, the reaction intensity of the HSA–bixin adduct outweighs the AGP–bixin conjugate to some extent. It may be observed from the recognition mode in Fig. 1(D) that the hydrogen atoms of the carboxyl group in bixin might yield two hydrogen bonds with the oxygen atoms of the carbonyl groups in Val-88 and Ala-99 residues, and the bond lengths are 3.34 Å and 3.01 Å, respectively. Furthermore, the conjugated double bonds of long-chain structure in bixin can generate the π–π stacking with the conjugated ring structures in some aromatic residues, e.g. Tyr-27, Phe-49, and Phe-112. Meanwhile, we could discover that several hydrophobic residues such as Val-41, Phe-49, Phe-51, Leu-79, Val-88, Leu-101, and Leu-110 close to the binding domain, illuminating that the hydrophobic effects existed between the AGP and bixin. These characteristics may facilitate the occurrence of the lipocalin–bixin interaction. Such experimental findings also agrees well with the previous speculation, namely the contemplated binding place was abundant in aromatic residues, and hydrophobic effects seemed to be contained in the receptor recognition.50
Nonetheless, further examination of the hydrophobicity of the domains on two proteins, we will find that bixin insert deeply into the interior of ligand binding areas (Fig. 2), which related greatly to the structural feature of bixin. This exploration perceived that hydrophobic effects provided a prerequisite for the recognition of bixin by proteins besides hydrogen bonds and conjugated effects, since this noncovalent interaction benefits the molecular reactions between the receptors and bixin. In other words, it may enable bixin to situate stably within the domain, and then the chemical can be shipped to target cells in the form of the protein–bixin complexes. This biochemical event plays a significant role in the aimed cargo of active substances to effector cells involved in biological responses such as immunological and inflammatory, because some macromolecules (e.g. AGP) have been observed on the surface of different mammalian cells.51 Based on the static results of in silico docking, we may conclude that the strength of noncovalent interactions in the AGP–bixin adduct is smaller than the HSA–bixin bioconjugate, and the recognition intensity of the AGP–bixin complex is relatively weak as compared with the HSA–bixin reaction. This fact can be ascribed to the structural polymorphism of AGP, and the existing results have revealed that the initial β-sheet-plentiful structure component of AGP turns into an α-helix-bountiful structure element after the lipocalin interact with phospholipid biomembranes, with an accompanying reduction of ligand binding capacity.52
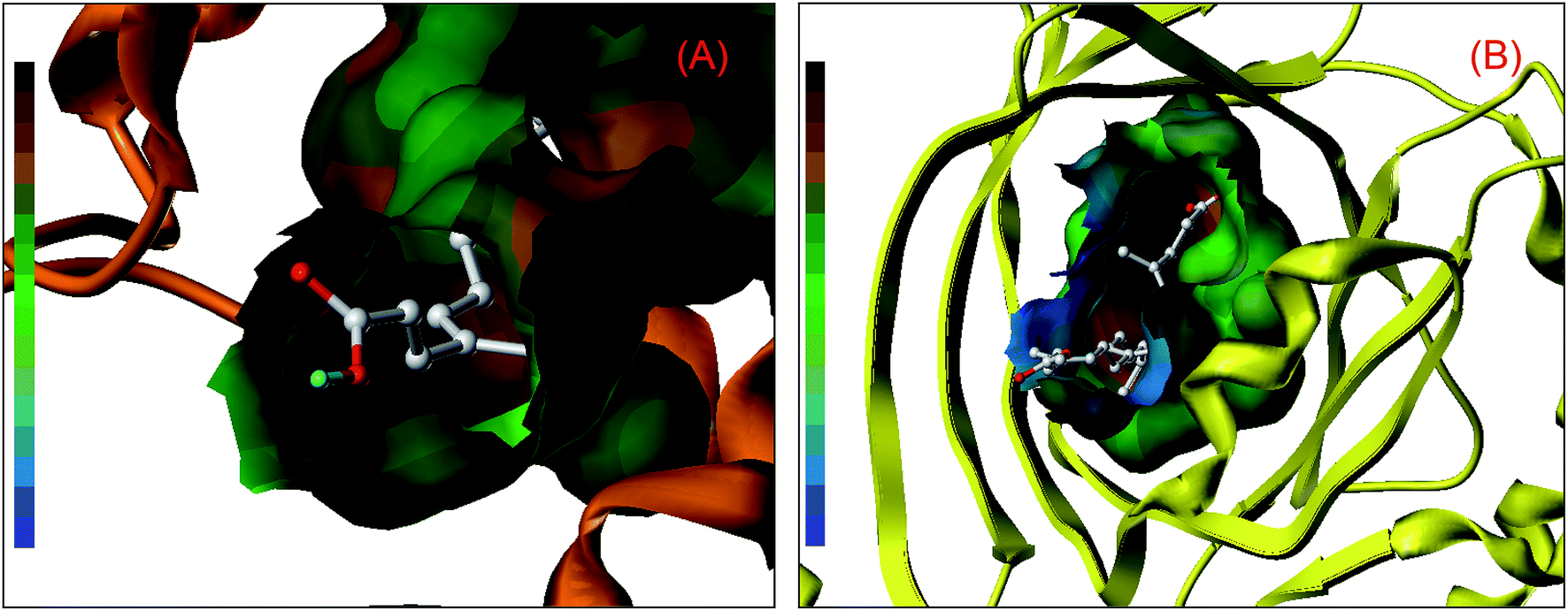 | ||
| Fig. 2 Hydrophobic effects between the amino acid residues comprising the binding domains on HSA (panel (A)) and AGP (panel (B)) and the bixin. The polypeptide chain of HSA and AGP implied in surface colored in orange and yellow, respectively, and the ball-and-stick model suggests bixin, colored as per the atoms. The multi-colored ribbon on the left-hand side of the graph hints the strength of hydrophobicity and the brown signifies strong hydrophobicity while the blue symbolizes forceful hydrophilicity. (For illumination of the references to color in this figure legend, the reader is referred to the web version of the article). | ||
Dynamic reaction patterns of the receptors–bixin conjugates
To investigate the biological recognition of bixin with the proteins from a dynamic viewpoint and clarify the influences of protein conformational flexibility on the processes, an atomic-scale MD simulation technique will be used to probe the proteins–bixin reactions, and the results are exhibited in Fig. 3. Meantime, the top two conformations of the two reaction systems have been selected from the docking experiments and then performed the parallel MD simulations with a timescale of 100 ns, in order to verify the rationality of the optimal conformations used for MD simulations. The Root-Mean-Square Deviation (RMSD) is displayed in Fig. S3.† It can be seen from Fig. S3† that these RMSD fluctuations have similar changing trends, signifying the study of MD simulations regarding the HSA–bixin and AGP–bixin complexes is reasonable. Notably, all dynamic biosystems changed from a relatively unstable state to a gradually stable state after the bioconjugates formation. For instance, the pure HSA (Fig. 3(A)) began to enter the plateau from the time point 10000 ps till the end of the MD simulation, 50000 ps. And in the HSA–bixin system (Fig. 3(B)), the RMSD of protein fluctuates within the time range of 0–40000 ps and the bound protein came to reach the equilibrium state at the starting time 60000 ps. Whereas the RMSD of bixin fluctuates stably after 20000 ps, and a slight increase can be observed and then fluctuated at ∼0.22 nm until the MD simulation finished over a 100000 ps period.
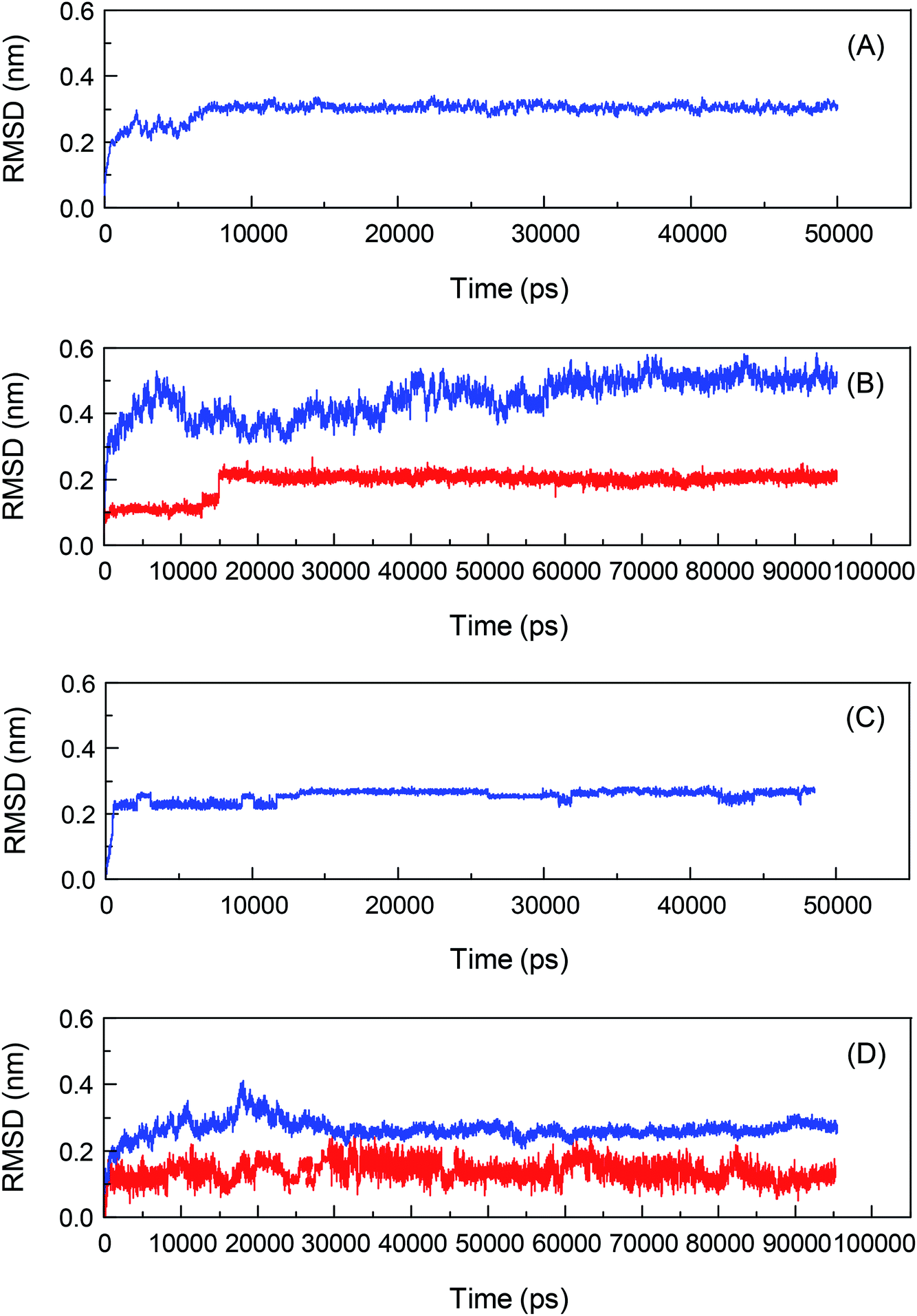 | ||
| Fig. 3 Calculated Root-Mean-Square Deviation (RMSD) for the initial X-ray crystal structures for the backbone Cα atoms of HSA (panel (A)) and AGP (panel (C)), and the bixin and the backbone Cα atoms of HSA (panel (B)) and AGP (panel (D)) from MD simulation at a temperature of 300 K with respect to their docking results as a function of the simulation time. The blue and red trajectories illustrate RMSD data for the backbone Cα atoms of proteins and the bixin, respectively. | ||
In order to disclose the changes of noncovalent interactions of the HSA–bixin adduct between the original conformation and the average conformation, the conformations of protein and bixin between 60000 ps and 100000 ps after equilibration were selected and superimposed on the optimal conformation from ligand docking, and the superposition results are displayed in Fig. 4. As can be seen from Fig. 4(A) and (B), the conformational change of HSA is relatively significant. Concretely, the original conformation of protein is observed to be more loose as compared with the average conformation, that is, protein structure has a tendency to converge at the heart of macromolecule during the dynamic HSA–bixin recognition, and thereby making the helical structure of protein more compact. We believe that such conformational alteration would promote the interactions of the protein with bixin. In contrast to the protein, the molecular conformational change of bixin is insignificant, mainly the translation of the conformation. This phenomenon may be explained by the fluctuation difference of RMSD, in particular, the rise of RMSD of bixin at ∼15000 ps primarily represent the spatial translation of the conformation rather than its conformational torsion of bixin. Further examination of the dynamic character of bixin in the binding region (Fig. 4(C)), we can see that the initial conformation of bixin overlaps the average conformation at equilibrium. However, it is worthwhile to note that the terminal carbonyl group in bixin has the spatial torsion to some degree, but hydrogen bonds could also be observed between the bixin and the residues Ser-419, Thr-422, and Thr-467, and the bond lengths are 2.16 Å, 2.27 Å, and 3.33 Å, respectively. Moreover, no dramatic alteration of the critical noncovalent interactions could be detected in dynamic recognition, and the hydrophobic areas constituted by nonpolar residues are still occurred at both ends of the average conformation in equilibrium state. These events should contribute bixin to stably locate within the domain III on HSA.
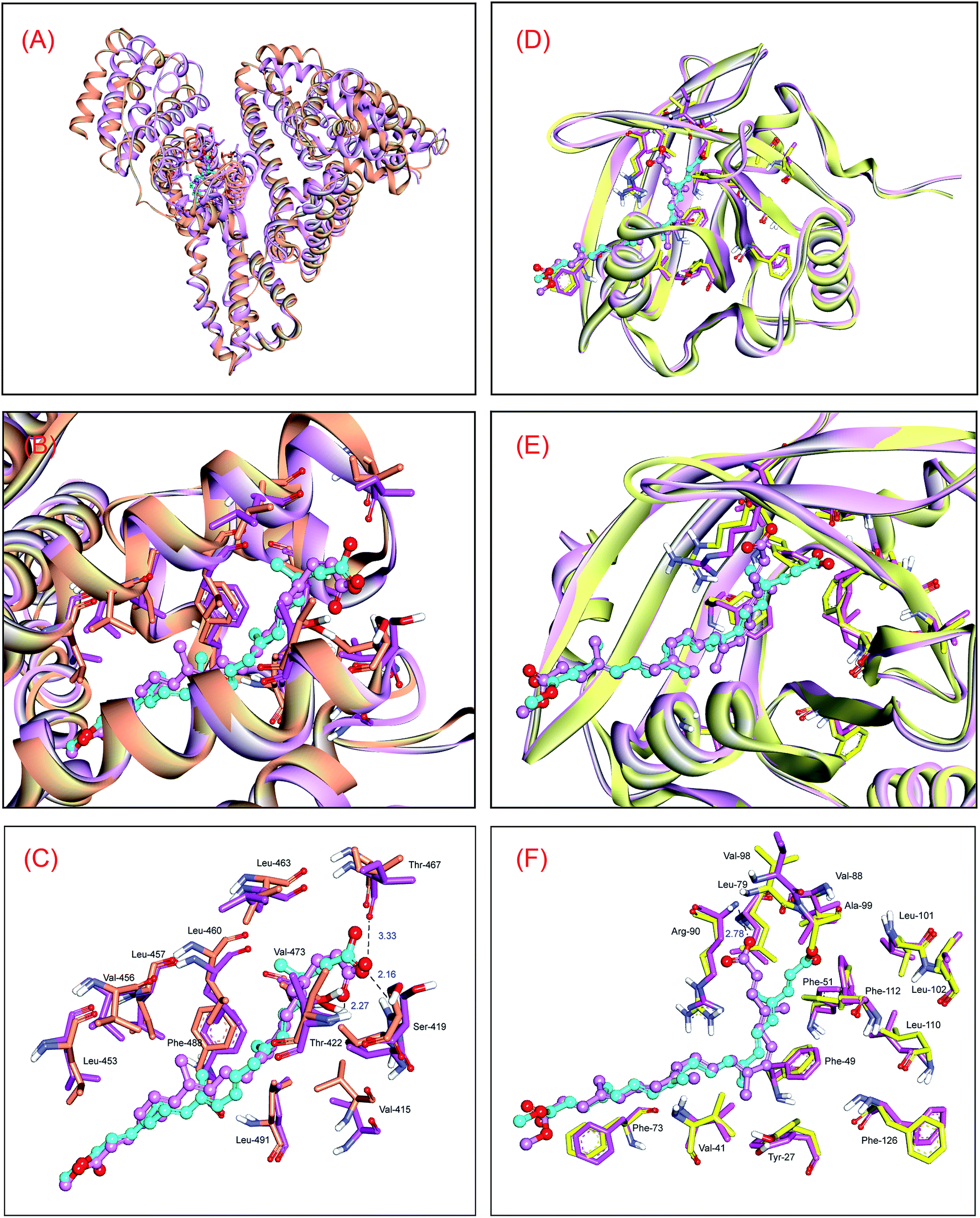 | ||
| Fig. 4 Superimposition of the average conformations of MD simulation on the original conformations of molecular docking resulting from the HSA–bixin (panel (A), panel (B) and panel (C)) and the AGP–bixin (panel (D), panel (E) and panel (F)) adducts. HSA and AGP revealed in surface colored in orange and yellow (initial) and both pink (average), respectively, and the original and average conformations of bixin divulged in cyan and pink ball-and-stick model, respectively. The critical amino acid residues around bixin have been portrayed in stick model, orange and yellow stick model depicts initial conformations of the residues in HSA and AGP, respectively, whereas dark pink stick model represents average conformations of the residues in both proteins. (For explication of the references to color in this figure legend, the reader is referred to the web version of the article). | ||
Similarly, the changing trend of the RMSD of AGP is indicated in Fig. 3(C). It is obviously that the RMSD of the backbone Cα atoms of the lone AGP started going equilibrious state in 4000 ps until the MD simulation finished over a 50000 ps, whereas in the AGP–bixin biosystem (Fig. 3(D)), the RMSD of protein is almost in equilibrium at time node 35000 ps. Meantime, the fluctuation of the RMSD of bixin in the AGP–bixin adduct is remarkable as compared to the HSA–bixin complex, but the RMSD began to fluctuate stably at the time point ∼40000 ps, and subsequently, a slight increase might be noticed and finally the RMSD returned to the equilibrium state till the 100000 ps time terminal. This phenomenon evidenced that the formation of the AGP–bixin conjugate could reinforce the molecular instability of bixin in the dynamic process. Hence, the average conformations of both AGP and bixin in the time frame from 60000 ps to 100000 ps have been selected and overlapped the initial conformations, and the superimposed results are listed in Fig. 4.
It is obvious that the original conformation of AGP superposes nicely its average conformation, and no significant changes can be detected in biomolecular structure. This means that the AGP–bixin reaction did not cause considerable alterations in the conformational stability of protein (Fig. 4(D) and (E)). Conversely, bixin has relatively unstable conformational stability during the dynamic recognition, and such point may be detailed interpreted by the RMSF data. Furthermore, we might notice that the average conformation of bixin mainly form hydrogen bond with the residue Arg-90 (Fig. 4(F)), which has notable difference with the hydrogen bonds in the initial conformation. We hold that the principal reason for such disparity could be explained as follows: in static recognition, the strength of hydrogen bonds between the bixin and the residues (Val-88 and Ala-99) is comparatively weak. This will induce a direct result that the original configuration of the carbonyl group in bixin can not exist quite stable in the dynamic reaction, but rather incline to generate large torsion so as to seek a more stable recognition mode. Such issue has also yielded some changes in the conjugated effects during the dynamic reaction. For example, the conjugated effects between the conjugated double bonds in bixin and the benzene rings in the Tyr-27 and Phe-112 residues have a downward tendency, while the conjugated effects between the bixin and the aromatic Phe-49 residue are tending upwards. In addition, because the conformational changes of AGP are faint, several hydrophobic residues could still be surrounded the bixin and produced hydrophobic effects with the active ligand, guaranteeing that bixin may situate stably in the binding domain on the lipocalin.
Impacts of the local conformational flexibility on the recognition processes
To greatly explore the influences of inherent local conformational flexibility on the receptors recognition processes, the Root-Mean-Square Fluctuation (RMSF) has been utilized to comparatively analyze the conformational discrepancies and the stability changes of the identical residue in pure proteins and the proteins–bixin adducts after the equilibrium state. Fig. 5(A) shows the alterations of RMSF for pure HSA and the HSA–bixin bioconjugate. Evidently, the whole instability of the residues in pure HSA is slightly stronger than the HSA–bixin system, which in particular reflect in the domain III (residues 404–558). The main cause for this phenomenon is that the binding of bixin at the domain III would decrease the instability of the residues in this domain upon the HSA–bixin reaction. Meantime, examination of the RMSF of the key residues (i.e. Ser-419, Thr-422, and Thr-467) involved in the formation of hydrogen bonds, one can find that the RMSF data of the three residues are 0.153, 0.179, and 0.245 in pure HSA, respectively, whereas in the HSA–bixin complex, the RMSF values are 0.124, 0.105, and 0.164, respectively. It is very clear that the RMSF of the residues in the HSA–bixin biosystem is subordinate to pure protein, suggesting that there is an upward trend in the stability of the domain III during the bioconjugation of HSA with bixin.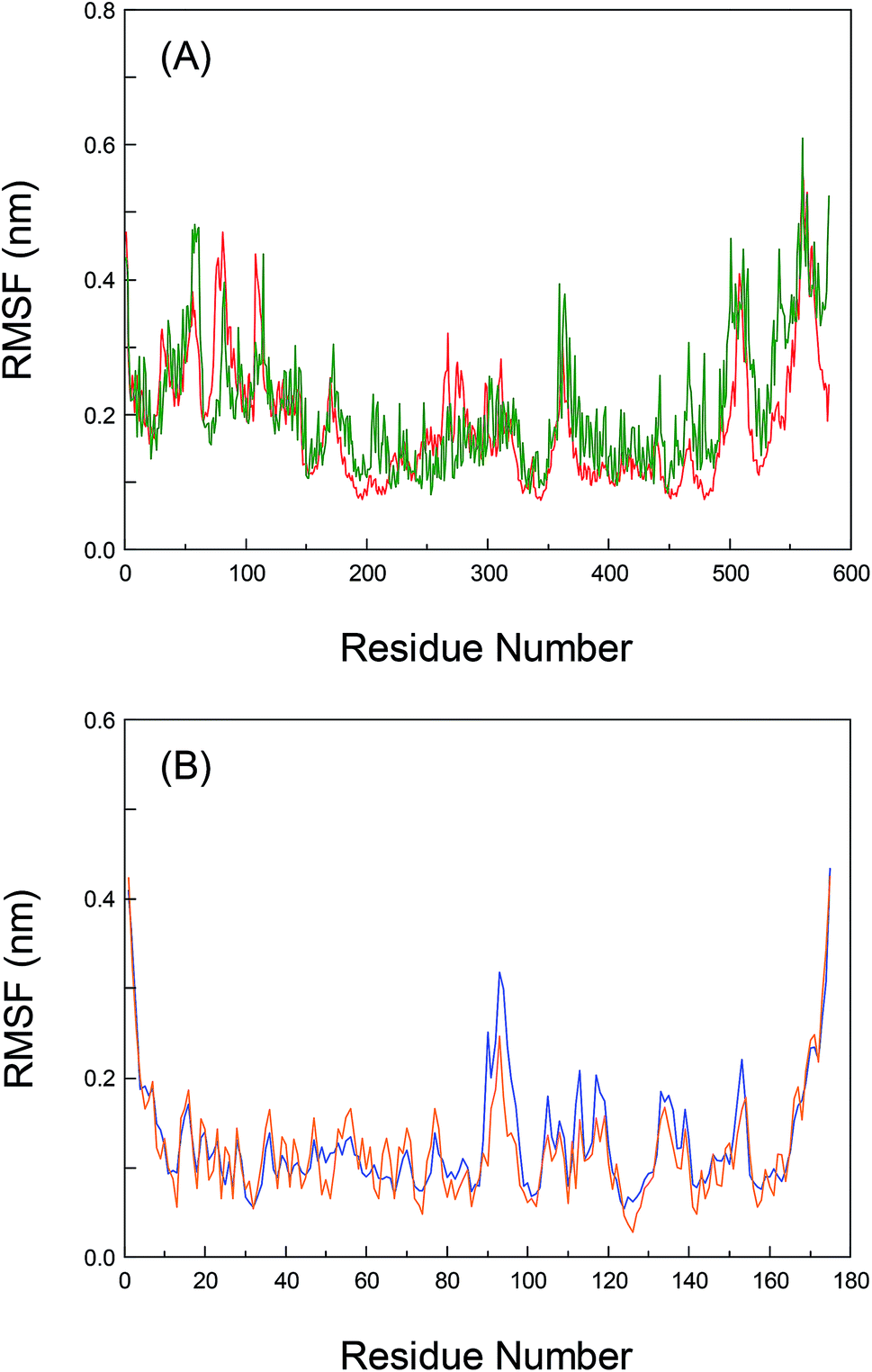 | ||
| Fig. 5 Root-Mean-Square Fluctuation (RMSF) of the backbone of each residue atomic positions for the unbound (olive (panel (A)) and orange (panel (B))) and bound (red (panel (A)) and blue (panel (B))) HSA (panel (A)) and AGP (panel (B)) as a function of the atom location along the polypeptide chain, respectively. | ||
As noted earlier, although the whole protein reveals a tendency to gather in the center of protein throughout the dynamic process, this event has little effect on the domain III, instead it may increase structural stability in the functional patch. Furthermore, as for the residues of subdomain IIA (Lys-199∼Glu-292), the RMSF values of pure HSA are observed to be less than the HSA–bixin conjugate, which should chiefly originate from the structural features of HSA, that is, subdomain IIA is nearing the domain III (especially subdomain IIIA) in a manner of “face-to-face”. Thus the instability of the two crucial domains will show a status during the dynamic recognition: the exhibition of a relatively stable state by one of the domains could easily trigger the reduction of the stability in other neighboring domains and vice versa. This finding can also help to better understand the correlations between the recognition reactions and biomolecular self-assembly, namely ligand binding could effectively fix the amino acid residues near the reaction region through multiple noncovalent interactions such as hydrogen bond networks, van der Waals' interactions and hydrophobic effects. This fact may decrease the flexibility of binding patch, and it can be used to confirm the reverse relationships between self-assembly and conformational flexibility.25,53 Of course, whether this characteristic could be compatible with other receptor proteins which contain many adjacent ligand binding regions, we will detailedly investigate this issue in later research contents.
Also, the RMSF of both pure AGP and the AGP–bixin system are illustrated in Fig. 5(B). Unlike the HSA–bixin reaction, the overall stability of the residues in pure AGP is somewhat greater than the AGP–bixin complex, suggesting that biomolecular recognition of bixin by AGP could cause an upward tendency for the conformational flexibility and instability in lipocalin. As we have seen, the disappearance of hydrogen bonds between the bixin and the residues Val-88 and Ala-99 may be detected in the dynamic bioreaction, and meanwhile the bixin has generated a new hydrogen bond with the Arg-90 residue. As a result, there needs to be detailed discussion of the RMSF of the three vital residues. It is evident to see the RMSF data of the residues, i.e. Val-88, Ala-99, and Arg-90, have respectively changed from 0.087, 0.073, and 0.102 in pure AGP to 0.079, 0.079, and 0.251 in the AGP–bixin adduct. Undoubtedly, except the residue Arg-90, the changes of the RMSF of the Val-88 and Ala-99 residues are too small in either pure AGP or the AGP–bixin bioconjugate, which indicated that the two residues failed to yield markedly spatial alterations during the dynamic AGP–bixin biointeraction. On the contrary, the residue Arg-90 has significantly spatial change in the dynamic process. Combining the analyses of the distinctions between the original conformation and average conformation in MD simulation, we know that the AGP–bixin reaction own less ability of mutual restraint as a result of the formation of weak hydrogen bonds between the bixin and the residues Val-88 and Ala-99. Thereby it will highly affect the spatial stability of bixin and lead to the gradually decrease and further obliterate the hydrogen bonds between the bixin and the residues (Val-88 and Ala-99) during the dynamic recognition. As regards the Arg-90 residue, it can yield a strong hydrogen bond with bixin and gradually make the terminal carbonyl group in bixin more stable, as the phenomenon of pendulum motion of the carbonyl group existed in MD simulation.
Nonetheless, evaluation of the RMSF of the residues which may generate conjugated effects with bixin, it would be found that the RMSF data of the aromatic residues, viz. Tyr-27, Phe-49, and Phe-112 in pure AGP are 0.065, 0.068, and 0.076, respectively. However, the RMSF values of the three residues are changed to be 0.105, 0.082, and 0.171 in the AGP–bixin conjugate, respectively. It is quite clear that the RMSF of the Tyr-27 and Phe-112 residues in the AGP–bixin adduct is substantially larger than the RMSF in pure AGP except for the Phe-49 residue. The key reason for this issue is that the conjugated effects are discovered to be maintained between the residues Tyr-27, Phe-49, and Phe-112 and bixin in the dynamic bioreaction, but the impact of the noncovalent interaction on the conformational changes of the Phe-49 residue is relatively small, and the conformational alterations of both the Tyr-27 and Phe-112 residues are largely affected by the conjugated effects. Accordingly, along with the bend of bixin conformation, the conjugated effects between the Tyr-27 and Phe-112 residues and bixin are gradually weakened, and yet there is a trend for increasing the conjugated effects between the residue Phe-49 and bixin. Significantly, these detailed research findings regarding the RMSF are concurred highly with the previous conclusions of superposition analyses based upon the dynamic receptors recognition processes.
Free energies of the receptors–bixin reactions
Investigation of free energy is very useful for disclosing many biophysical characters such as the recognition intensity and the constituents of free energy of the proteins–bixin adducts so as to explore such receptors recognition from an energy of view. According to the RMSD analyses, the decomposition of free energy of the last 40000 ps MD trajectory at dynamic equilibrium was executed via the approach of MM/GBSA (time interval: 2.0 ps), and the specific energy components of each biological response are summarized in Table 1.
| Components | Biosystems | |
|---|---|---|
| HSA–bixin | AGP–bixin | |
| ΔGele | −26.65 ± 0.58 | −18.55 ± 0.61 |
| ΔGvdW | −29.77 ± 1.52 | −33.39 ± 2.11 |
| ΔGMM | −56.42 ± 2.37 | −51.94 ± 2.89 |
| ΔGpol,sol | 47.50 ± 1.81 | 42.04 ± 1.95 |
| ΔGnonpol,sol | −29.37 ± 1.27 | −22.56 ± 1.03 |
| ΔGsol | 18.13 ± 0.79 | 19.48 ± 1.45 |
| ΔGbind | −38.29 ± 2.44 | −32.47 ± 1.76 |
| ΔGbind (docking) | −34.78 | −24.65 |
Evidently, the binding free energy of the HSA–bixin conjugate is slightly greater than the AGP–bixin system, which agrees with the data of the Gibbs free energy. This event further demonstrated that the results of static recognition are logically reasonable. As may be seen from Table 1, the following three terms, i.e. electrostatic contribution (ΔGele), van der Waals' contribution (ΔGvdW), and nonpolar solvation contribution (ΔGnonpol,sol) favors the bioreaction of bixin with the two proteins, whereas the electrostatic solvation free energy (ΔGpol,sol) is observed to be unfavorable to the proteins–bixin complexes. Furthermore, the ΔGvdW occupies a predominant role in the binding free energy of biomolecular recognition, and the energy values are respectively −29.77 kJ mol−1 and −33.39 kJ mol−1 for the HSA–bixin and AGP–bixin reactions, which indicated that the conjugated effects are of considerable significance for the proteins–bixin interactions. Nevertheless, the impact of the conjugated effects on the AGP–bixin biosystem is greater than the HSA–bixin adduct, that is because the AGP–bixin bioconjugate has smaller data of the ΔGvdW as compared with the HSA–bixin complex. Notably, these results strongly support the above discussions with regard to in silico docking and MD simulations.
Meantime, one could note that the proportion of the ΔGele in free energy is somewhat small and the energy values are found to be −26.65 kJ mol−1 and −18.55 kJ mol−1 for the HSA–bixin and AGP–bixin reactions, respectively, and this point reflected particularly in the AGP–bixin biointeraction. Strictly, the formation of hydrogen bond derives in the main from the attractive electrostatic interaction between a hydrogen atom and an electron rich atom,54 and therefore the electrostatic energy can be employed to decode the proteins–bixin conjugations. Combining the strength of hydrogen bonds with the data of electrostatic energies, it is easy to perceive that the action of the electrostatic energy on molecular interaction of the HSA–bixin is more significant. This result has also interpreted the reason that the reactivity of the AGP–bixin complex is comparatively weaker than the HSA–bixin conjugate. Besides, it may be noticed from Table 1 that the influences of the solvation effects on the binding free energy of the two proteins–bixin biosystems are nearly the same under the present circumstances.
Per-residue energy decomposition of the receptors–bixin biointeractions
As has been argued, the conformational alterations of the residues during the dynamic receptors recognition have been explained through the elaboration of the RMSF data. We also deciphered the interaction energies between the residues and bixin in order to deeply understand the proteins–bixin reactions. The contribution of the individual residues in proteins to the whole binding free energy has been calculated by the method of decomposing the free energy of binding into residual contributions to guarantee those residues that should completely be included in the bioreaction with bixin. Fig. 6 displays the interaction energies of various residues in the proteins–bixin systems. Obviously, one can observe that the interaction energies of several residues, e.g. Val-418, Ser-419, Thr-422, Leu-457, Leu-460, Thr-467, Val-473, and Phe-488 in the HSA–bixin adduct (Fig. 6(A)) are −1.535, −7.674, −5.397, −2.755, −2.323, −2.075, −1.932, and −2.075 kJ mol−1, respectively. The absolute values of these energies are relatively large, implying that the above residues should play a key role in the HSA–bixin bioconjugation. This issue could especially be reflected in the residues Ser-419, Thr-422, and Thr-467, which may form hydrogen bonds with bixin, and the interaction energies of the three residues are found to be more powerful than the residues such as Val-418 and Val-473 involved in hydrophobic effects. Furthermore, it needs to be indicated that most of the residues contained in the sequence of 400–550 can show somewhat good recognition capacity with bixin, and such event further validated that the chief binding location of bixin on HSA is explicitly situated within the domain III (residues 404–558).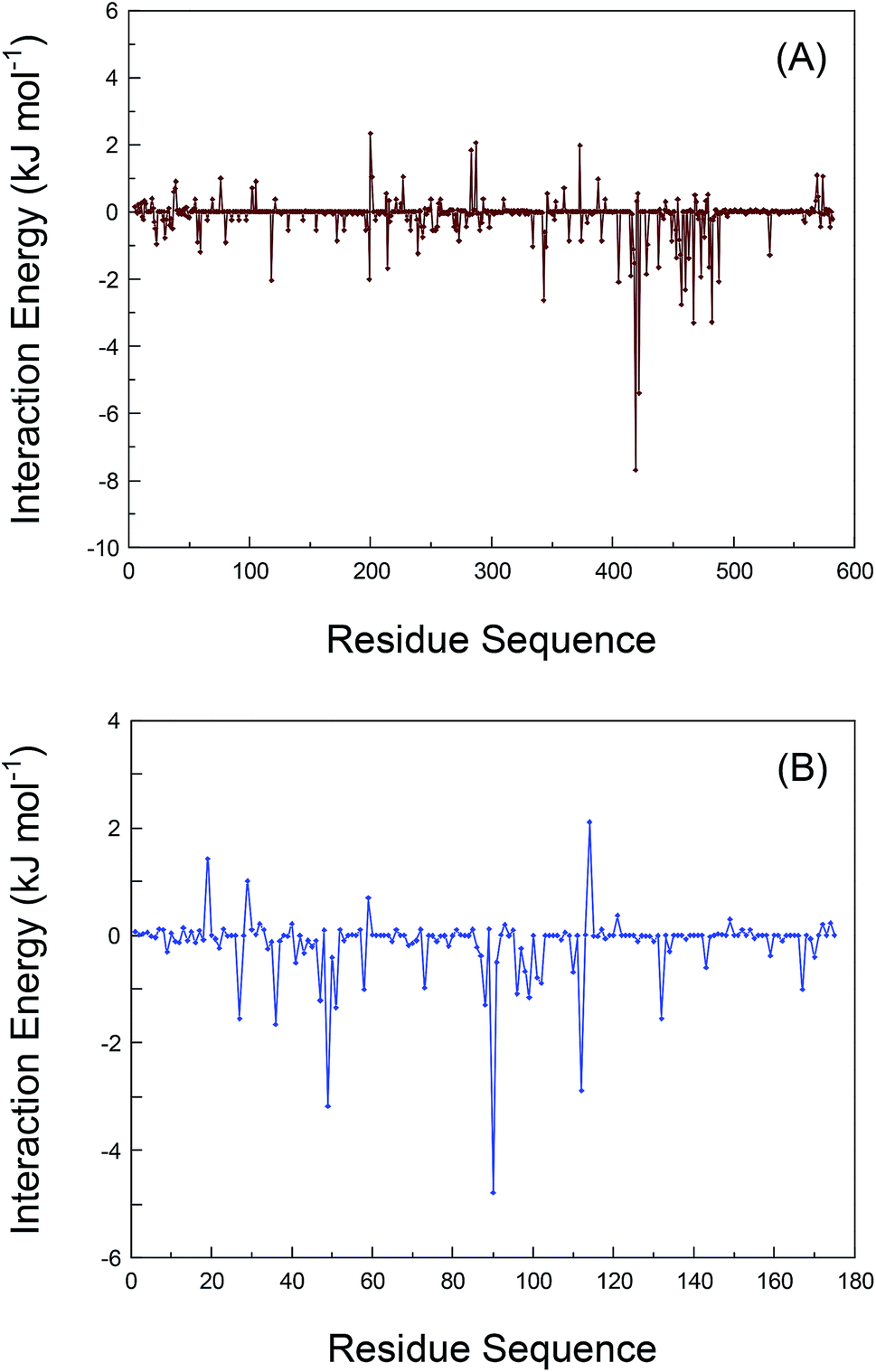 | ||
| Fig. 6 The decompositions of free energy of recognition per-residue for the proteins–bixin biosystems. The amino acid residues contributing predominantly to the HSA–bixin (panel (A)) and the AGP–bixin (panel (B)) bioconjugations are underlined by wine or blue dash dotted line, respectively. | ||
Still, Fig. 6(B) is the interaction energies of different residues in the polypeptide chain on AGP. It is clear that the interaction energies of the residues formed hydrophobic effects, i.e. Phe-49, Phe-51, Val-88, Ala-99, and Phe-112, are noted to be −3.176, −1.344, −1.289, −1.154, and −2.886 kJ mol−1, which suggested these residues have relatively strong energy contributions to the AGP–bixin bioconjugate, or rather, hydrophobic effects are extremely essential to the AGP–bixin biosystem. Meanwhile, we could see from Fig. 6(B) that the interaction energy of the Arg-90 residue is −4.786 kJ mol−1, revealing the noncovalent interaction between the Arg-90 residue and bixin is more forceful and has the maximal reaction intensity compared to the residues Val-88 and Ala-99. These evidences are enough to explain the utmost importance to the Arg-90 residue in the reaction of AGP with bixin, and also match the results obtained from the dynamic recognition and the RMSF analysis. Moreover, the residues in the sequence 20–130 represent better bioactivity with bixin as a whole, which illustrated the previously experimental conclusions are obviously reasonable, namely the bixin located at the nonpolar patch composed mostly by the residues 27–127 on the lipocalin molecule.
Overall conformational changes of the receptors–bixin interactions
To carefully examine the overall conformational changes of proteins in biological reaction processes, the circular dichroism (CD) spectra of proteins in the absence and presence of different concentrations of bixin were recorded and secondary structure components calculated according to raw CD data were collected in Table 2. Clearly, the CD curves of proteins exhibited two negative peaks in the far-UV CD region at 208 nm and 222 nm, characteristic of the α-helical structure of proteins. The reasonable explanation of this phenomenon is that the negative bands between 208 and 209 nm and 222 and 223 nm are both contributed by π → π* and n → π* transition of amide groups and are simultaneously affected through the geometries of the skeleton of polypeptide chain.55 As displayed in Table 2, free HSA/AGP has 55.4%/24.7% α-helix, 7.9%/38.9% β-sheet, 13.5%/10.5% turn and 23.2%/25.9% random coil, after interacting with bixin, increase of α-helix was seen from 55.4%/24.7% free HSA/AGP to 63.5%/28.5% HSA/AGP–bixin adducts, respectively, while decrease was detected in β-sheet, turn and random coil from 7.9%/38.9%, 13.5%/10.5% and 23.2%/25.9% free HSA/AGP to 6.6%/39.1%, 10.1%/8.9% and 19.8%/23.5% HSA/AGP–bixin complexes at a molar ratio of proteins to bixin of 1:4. The increase of α-helix with a reduction in the β-sheet, turn and random coil demonstrated that bixin yielded markedly noncovalent bonds with some amino acid residues of the polypeptide chain and triggering the conformational changes in proteins, i.e. some degree of proteins stabilization upon bixin conjugation.56,57
| Samples | Secondary structure components (%) | |||
|---|---|---|---|---|
| α-Helix (±2%) | β-Sheet (±1%) | Turn (±1%) | Random (±2%) | |
| Free HSA | 55.4 | 7.9 | 13.5 | 23.2 |
| HSA + bixin (1:2) |
59.1 | 7.3 | 11.9 | 21.7 |
| HSA + bixin (1:4) |
63.5 | 6.6 | 10.1 | 19.8 |
| Free AGP | 24.7 | 38.9 | 10.5 | 25.9 |
| AGP + bixin (1:2) |
26.4 | 38.2 | 9.8 | 25.6 |
| AGP + bixin (1:4) |
28.5 | 39.1 | 8.9 | 23.5 |
Meanwhile, the previously computational results revealed that the conformational change of HSA was relatively obvious and the biomolecular structure becomes compact, while the conformational change of AGP was not significant. It is evident to us that such conclusion has strongly been supported by the results of CD spectra, because the secondary structures (particularly α-helix) of HSA were markedly changed and the hydrophobicity was enhanced, and the secondary structures of AGP were not changed noticeably. Nevertheless, it should be indicated that the overall conformational changes of the proteins induced by bixin binding could not be attributed to the significant alterations of the compact three-dimensional structure in proteins, but just the appropriate adjustment of their own conformations after the reaction of bixin with HSA/AGP. This manner would help to accommodate the ligand more suitable. In other words, by slightly adjusting the regular conformations of proteins, these biomolecules can enable the ligand to locate at the primary binding domain using the lowest energy conformation. And this biological mode will allow the whole protein–ligand system to possess the minimum energy. Undoubtedly, these experimental outcomes obtained by CD spectra have validated the above computational analyses of the proteins–bixin interactions, that is, the research findings of protein conformational alterations based on molecular docking and MD simulations are quite reasonable.
Conclusions
In summary, this study clarified the impacts of the local conformational flexibility on the receptors recognition by utilizing an atomic-scale computational method. Experimental results suggested that the significant distinctions were existed between the static and dynamic interaction patterns, and these biological points were associated closely with the flexible properties of protein conformations. Dynamic reaction researches confirmed that the intrinsic conformational flexibility owned considerable effects on the receptor recognition processes, which reflected mainly in the features of the flexible loop domain structures where bixin situated in protein molecules. The results of energy decomposition and CD spectra further indicated that the conformational and energy alterations in the residues surrounding the ligand binding location played a key role in biological interactions. These issues elucidated the nature of the fact that the local conformational flexibility can intervene greatly in receptor recognition at the microscopic level. Based on the present research findings derived from MD simulations, we could detect and obtain the significant differences in protein conformational changes, particularly the changes in the interested ligand binding regions. This event has largely affected the reaction features between the proteins and ligand. The subsequent investigations will deeply explore the changes of structural flexibility and frequency induced by ligand binding by employing other methods such as elastic networks models and normal mode analysis.58 At the moment the intrinsic protein conformational flexibility has often been ignored in several studies such as biochemistry, chemical biology, medicinal chemistry, and toxicological science,59–62 and this event could cause relatively large deviations in the experimental results. Consequently, this attempt should contribute biophysical evidence to elaborating the structure–function relationships of the important receptors in human beings.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We are extremely indebted to Professor Ulrich Kragh-Hansen of Department of Biomedicine, University of Aarhus, for the precious gift of his doctoral dissertation. We thank Professor Yong-Jun Liu of School of Chemistry and Chemical Engineering, Shandong University, for his warm support during the manuscript processing. Thanks also go to the reviewers of this manuscript for their insightful and valuable suggestions. This work was funded with grant support from the “Chang'an Scholars Construction Project” (No. 201806CT016) and partially supported by the State Key Laboratory of the Discovery and Development of Novel Pesticide (No. 2016NYRD02), the Shandong Provincial Natural Science Foundation (No. ZR2016CQ02 and No. ZR2016CP19), and the Project of Shandong Province Higher Educational Science and Technology Program (No. J16LF05).References
- F. Biedermann and H.-J. Schneider, Chem. Rev., 2016, 116, 5216–5300 CrossRef CAS PubMed.
- R. Baron and J. A. McCammon, Annu. Rev. Phys. Chem., 2013, 64, 151–175 CrossRef CAS PubMed.
- H. Iranfar, O. Rajabi, R. Salari and J. Chamani, J. Phys. Chem. B, 2012, 116, 1951–1964 CrossRef CAS PubMed.
- E. Froehlich, J. S. Mandeville, D. Arnold, L. Kreplak and H. A. Tajmir-Riahi, Biomacromolecules, 2012, 13, 282–287 CrossRef CAS PubMed.
- X. Ma and Y. L. Zhao, Chem. Rev., 2015, 115, 7794–7839 CrossRef CAS PubMed.
- S. L. Zhuang, X. Lv, L. M. Pan, L. P. Lu, Z. W. Ge, J. Y. Wang, J. P. Wang, J. S. Liu, W. P. Liu and C. L. Zhang, Environ. Pollut., 2017, 220, 616–624 CrossRef CAS PubMed.
- U. Ryde and P. Söderhjelm, Chem. Rev., 2016, 116, 5520–5566 CrossRef CAS PubMed.
- J. G. Kim, T. W. Kim, J. Kim and H. Ihee, Acc. Chem. Res., 2015, 48, 2200–2208 CrossRef CAS PubMed.
- B. Gipson, D. Hsu, L. E. Kavraki and J.-C. Latombe, Annu. Rev. Anal. Chem., 2012, 5, 273–291 CrossRef CAS PubMed.
- E. Papaleo, G. Saladino, M. Lambrughi, K. Lindorff-Larsen, F. L. Gervasio and R. Nussinov, Chem. Rev., 2016, 116, 6391–6423 CrossRef CAS PubMed.
- R. O. Dror, R. M. Dirks, J. P. Grossman, H. F. Xu and D. E. Shaw, Annu. Rev. Biophys., 2012, 41, 429–452 CrossRef CAS PubMed.
- M. A. Lill, Biochemistry, 2011, 50, 6157–6169 CrossRef CAS PubMed.
- C. Maffeo, S. Bhattacharya, J. Yoo, D. Wells and A. Aksimentiev, Chem. Rev., 2012, 112, 6250–6284 CrossRef CAS PubMed.
- O. K. Abou-Zied, N. Al-Lawatia, M. Elstner and T. B. Steinbrecher, J. Phys. Chem. B, 2013, 117, 1062–1074 CrossRef CAS PubMed.
- T. Maruyama, M. Otagiri and A. Takadate, Chem. Pharm. Bull., 1990, 38, 1688–1691 CrossRef CAS PubMed.
- S. Schlehuber and A. Skerra, Drug Discovery Today, 2005, 10, 23–33 CrossRef CAS PubMed.
- M. C. Ferrolino, A. Zhuravleva, I. L. Budyak, B. Krishnan and L. M. Gierasch, Biochemistry, 2013, 52, 8843–8854 CrossRef CAS PubMed.
- F. Feixas, S. Lindert, W. Sinko and J. A. McCammon, Biophys. Chem., 2014, 186, 31–45 CrossRef CAS PubMed.
- O. H. Lowry, N. J. Rosebrough, A. L. Farr and R. J. Randall, J. Biol. Chem., 1951, 193, 265–275 CAS.
- A. A. Bhattacharya, T. Grüne and S. Curry, J. Mol. Biol., 2000, 303, 721–732 CrossRef CAS PubMed.
- K. Nishi, T. Ono, T. Nakamura, N. Fukunaga, M. Izumi, H. Watanabe, A. Suenaga, T. Maruyama, Y. Yamagata, S. Curry and M. Otagiri, J. Biol. Chem., 2011, 286, 14427–14434 CrossRef CAS PubMed.
- M. J. D. Powell, Comput. J., 1964, 7, 155–162 CrossRef.
- S. J. Weiner, P. A. Kollman, D. A. Case, U. C. Singh, C. Ghio, G. Alagona, S. Profeta Jr. and P. Weiner, J. Am. Chem. Soc., 1984, 106, 765–784 CrossRef CAS.
- J. Gasteiger, Nat. Chem., 2015, 7, 619–620 CrossRef CAS PubMed.
- M. Kouza, N. T. Co, P. H. Nguyen, A. Kolinski and M. S. Li, J. Chem. Phys., 2015, 142, 145104 CrossRef PubMed.
- M. Kurcinski, M. Jamroz, M. Blaszczyk, A. Kolinski and S. Kmiecik, Nucleic Acids Res., 2015, 43, W419–W424 CrossRef CAS PubMed.
- M. Blaszczyk, M. Kurcinski, M. Kouza, L. Wieteska, A. Debinski, A. Kolinski and S. Kmiecik, Methods, 2016, 93, 72–83 CrossRef CAS PubMed.
- M. P. Ciemny, A. Debinski, M. Paczkowska, A. Kolinski, M. Kurcinski and S. Kmiecik, Sci. Rep., 2016, 6, 37532 CrossRef CAS PubMed.
- M. P. Ciemny, M. Kurcinski, K. J. Kozak, A. Kolinski and S. Kmiecik, in Modeling Peptide-Protein Interactions: Methods and Protocols, ed. O. Schueler-Furman and N. London, Springer Science+Business Media, New York, NY, 2017, vol. 1561, pp. 69–94 Search PubMed.
- M. P. Ciemny, M. Kurcinski, M. Blaszczyk, A. Kolinski and S. Kmiecik, Biomed. Eng. Online, 2017, 16, 71 CrossRef PubMed.
- M. Blaszczyk, M. P. Ciemny, A. Kolinski, M. Kurcinski and S. Kmiecik, Briefings Bioinf., 2018 DOI:10.1093/bib/bby080.
- D. van der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- W. R. P. Scott, P. H. Hünenberger, I. G. Tironi, A. E. Mark, S. R. Billeter, J. Fennen, A. E. Torda, T. Huber, P. Krüger and W. F. van Gunsteren, J. Phys. Chem. A, 1999, 103, 3596–3607 CrossRef CAS.
- A. W. Schüttelkopf and D. M. F. van Aalten, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 1355–1363 CrossRef PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- H. A. Stern, J. Comput. Chem., 2004, 25, 749–761 CrossRef CAS PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- M. R. Hestenes and E. Stiefel, J. Res. Natl. Bur. Stand., 1952, 49, 409–436 CrossRef.
- J. A. Snyman, in Practical Mathematical Optimization: An Introduction to Basic Optimization Theory and Classical and New Gradient-Based Algorithms, Springer Science+Business Media, New York, NY, 2005, vol. 97, pp. 1–231 Search PubMed.
- J. Hsin, A. Arkhipov, Y. Yin, J. E. Stone and K. Schulten, Curr. Protoc. Bioinf., 2008, 24, 5 Search PubMed.
- J. E. Kerrigan, in In Silico Models for Drug Discovery, ed. S. Kortagere, Springer Science+Business Media, New York, NY, 2013, vol. 993, pp. 95–113 Search PubMed.
- S. Genheden and U. Ryde, Expert Opin. Drug Discovery, 2015, 10, 449–461 CrossRef CAS PubMed.
- J. Weiser, P. S. Shenkin and W. C. Still, J. Comput. Chem., 1999, 20, 217–230 CrossRef CAS.
- L. Whitmore and B. A. Wallace, Biopolymers, 2008, 89, 392–400 CrossRef CAS PubMed.
- D. C. Carter, X.-M. He, S. H. Munson, P. D. Twigg, K. M. Gernert, M. B. Broom and T. Y. Miller, Science, 1989, 244, 1195–1198 CrossRef CAS PubMed.
- D. L. Schönfeld, R. B. G. Ravelli, U. Mueller and A. Skerra, J. Mol. Biol., 2008, 384, 393–405 CrossRef PubMed.
- F. Zsila, P. Molnár and J. Deli, Chem. Biodiversity, 2005, 2, 758–772 CrossRef CAS PubMed.
- F. Zsila, P. Molnár, J. Deli and S. F. Lockwood, Bioorg. Chem., 2005, 33, 298–309 CrossRef CAS PubMed.
- Z. H. Israili and P. G. Dayton, Drug Metab. Rev., 2001, 33, 161–235 CrossRef CAS.
- K. Nishi, T. Maruyama, B. Halsall, T. Handa and M. Otagiri, Biochemistry, 2004, 43, 10513–10519 CrossRef CAS PubMed.
- P. H. Nguyen, M. S. Li, G. Stock, J. E. Straub and D. Thirumalai, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 111–116 CrossRef CAS.
- M. Nishio, Y. Umezawa, J. Fantini, M. S. Weiss and P. Chakrabarti, Phys. Chem. Chem. Phys., 2014, 16, 12648–12683 RSC.
- L. A. Abriata, J. Chem. Educ., 2011, 88, 1268–1273 CrossRef CAS.
- E. Froehlich, J. S. Mandeville, C. J. Jennings, R. Sedaghat-Herati and H. A. Tajmir-Riahi, J. Phys. Chem. B, 2009, 113, 6986–6993 CrossRef CAS PubMed.
- S. Dubeau, P. Bourassa, T. J. Thomas and H. A. Tajmir-Riahi, Biomacromolecules, 2010, 11, 1507–1515 CrossRef CAS PubMed.
- K. Sankar, S. K. Mishra and R. L. Jernigan, J. Phys. Chem. B, 2018, 122, 5409–5417 CrossRef CAS PubMed.
- K. Teilum, J. G. Olsen and B. B. Kragelund, Biochim. Biophys. Acta, Proteins Proteomics, 2011, 1814, 969–976 CrossRef CAS PubMed.
- J. Janin and M. J. E. Sternberg, F1000Prime Rep., 2013, 5, 2 Search PubMed.
- R. Buonfiglio, M. Recanatini and M. Masetti, ChemMedChem, 2015, 10, 1141–1148 CrossRef CAS PubMed.
- Z. T. Sun, Q. Liu, G. Qu, Y. Feng and M. T. Reetz, Chem. Rev., 2019, 119, 1626–1665 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: A brief introduction to the ligand bixin, detailed protocol of molecular dynamics simulation of the AGP–bixin reaction, elucidation of the overall energy of the receptors–bixin adducts, molecular structure of bixin, superimposition of the top three bixin conformations, and calculated RMSD for the bixin and the backbone Cα atoms of two proteins. See DOI: 10.1039/c9ra01906e |
| This journal is © The Royal Society of Chemistry 2019 |