
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeptaketides from the endophytic fungus Pleosporales sp. F46 and their antifungal and cytotoxic activities†
Gang Li a,
Ke Xub,
Wen-Qi Chena,
Zhi-Hao Guoa,
Yu-Tong Liua,
Ya-Nan Qiaob,
Yong Sunb,
Gang Suna,
Xiao-Ping Penga and
Hong-Xiang Lou*ab
a,
Ke Xub,
Wen-Qi Chena,
Zhi-Hao Guoa,
Yu-Tong Liua,
Ya-Nan Qiaob,
Yong Sunb,
Gang Suna,
Xiao-Ping Penga and
Hong-Xiang Lou*ab
aDepartment of Natural Medicinal Chemistry and Pharmacognosy, School of Pharmacy, Qingdao University, Qingdao 266021, People's Republic of China. E-mail: louhongxiang@sdu.edu.cn; Tel: +86-531-8838-2012
bDepartment of Natural Product Chemistry, Key Laboratory of Chemical Biology of Ministry of Education, School of Pharmaceutical Sciences, Shandong University, Jinan 250012, People's Republic of China
First published on 26th April 2019
Abstract
Six new heptaketides, pleosporalins A–F (1–5, and 7), and a new heptaketide derivative, pleosporalin G (9), together with four biosynthetically related known compounds (6, 8, 10, and 11), were isolated from an endophytic fungus, Pleosporales sp. F46, found in the medicinal plant Mahonia fortunei. The structures and stereochemistry of these compounds were established by extensive spectroscopic analyses including LC-HRMS, NMR spectroscopy, optical rotations, ECD calculations, and single-crystal X-ray diffraction. The antifungal activities of isolated compounds 1–11 were investigated against Candida albicans, and their cytotoxic activities were evaluated against A549, SMMC-721, and MDA-MB-231 cancer cell lines. Compound 1 was active against C. albicans with an MIC80 of 128 μg mL−1, and compound 7 showed moderate cytotoxicity against MDA-MB-231 with an IC50 of 22.4 ± 1.1 μM. By comparing compounds 1 and 7 with structurally related metabolites, it was revealed that alterations to their C-1 or C-2 substitutions could significantly influence their antifungal or cytotoxic efficacies.
Introduction
Endophytes, which mainly include fungi and bacteria, colonize the living tissues of healthy plants and are associated with specific ecological niches.1–3 They have been recognized as a valuable source of skeletally diverse and biologically active secondary metabolites and are attracting considerable attention from chemists and biologists.4–6 As part of our ongoing search for fungal endophytes capable of producing biologically active compounds,7 we recently isolated an endophytic fungus, Pleosporales sp. F46, from the medicinal plant Mahonia fortunei collected from Qingdao, People's Republic of China.7 The OSMAC (One Strain Many Compounds) approach with the aid of LC-ESI-HRMS was employed to explore the chemical diversity of this endophyte.8 When cultured in rice medium, Pleosporales sp. F46 biosynthesized secondary metabolites with UV and MS features similar to those of antifungal heptaketides we reported previously (Experimental section).9Herein we report the isolation and characterization of six new heptaketides, pleosporalins A–F (1–5, and 7), a new biosynthetically related polyketide derivative, pleosporalin G (9), as well as four structurally related known compounds, (+)-pleorubrin B (6),10 biatriosporin A (8),9 (−)-balticol A (10),11 and (−)-balticol B (11)11 from endophytic Pleosporales sp. F46. Following our previous work on the antifungal activities of heptaketides,9 we reused the wild-type strain Candida albicans to evaluate the antifungal activities of the isolated compounds. Considering the increasing focus on the cytotoxic efficacies of this kind of polyketide,12 the cytotoxic activities of the isolated polyketides were also evaluated against the cancer cell lines A549, SMMC-721, and MDA-MB-231.
Results and discussion
Compound 1 (Fig. 1) was isolated as yellow crystals with the molecular formula C15H12O5 (ten double-bond equivalents) as established by ESI-HRMS. The 1H NMR spectrum of 1 (Fig. S1†) showed the presence of two singlet methyl signals, three singlet aromatic protons, and three protons associated with hydroxyl or aldehyde groups. The 1D NMR data in combination with the 2D NMR spectra (Table 1, and Fig. S1–S3†) identified two methyl groups (one oxygenated), twelve aromatic/olefinic carbons (three methine carbons and five oxygenated carbons), and one carbonyl carbon. These data accounted for all 1H and 13C NMR resonances of 1 and indicated an aromatic skeleton. The planar structure of compound 1 was further elucidated by detailed analysis of its HMBC spectrum (Fig. S4,† and 2). A naphthalene ring substructure containing a meta-substituted phenyl ring was tentatively deduced from the presence of two singlet aromatic protons H-5 (δH 6.92) and H-7 (δH 6.47), together with their HMBC correlations from H-5 to C-4, C-7 and C-9, and from H-7 to C-1, C-5, and C-9 (Fig. 2). The HMBC correlations of H3-13/C-11 and C-12, and H-11/C-3 and C-4 constructed the C-4–C-3–C-11–C-12–C-13 fragment (Fig. 2). An aldehyde unit (C-14, δC 190.5) and a methoxy group (δC 55.8) were located at C-2 (δC 129.1) and C-6 (δC 164.2), respectively, and these assignments were consistent with their chemical shifts and the HMBC correlations from 6-OMe to C-6 and from H-14 to C-3 (Fig. 2). The shifts of the oxygenated aromatic carbons (C-1 at δC 160.7 and C-8 at δC 164.5) indicated that the two hydroxyl groups were attached to C-1 and C-8. The remaining one degree of unsaturation and the chemical shifts of C-4 (δC 143.6) and C-12 (δC 156.7) suggested that C-4 and C-12 were both attached to the same oxygen atom, forming a furan ring fused to the naphthalene ring. Finally, single-crystal diffraction analysis with Cu Kα radiation confirmed the structure of 1 (Fig. 4). Compound 1 was named pleosporalin A, and its carbon skeleton is similar to that of biatriosporins D–F, which we reported previously.9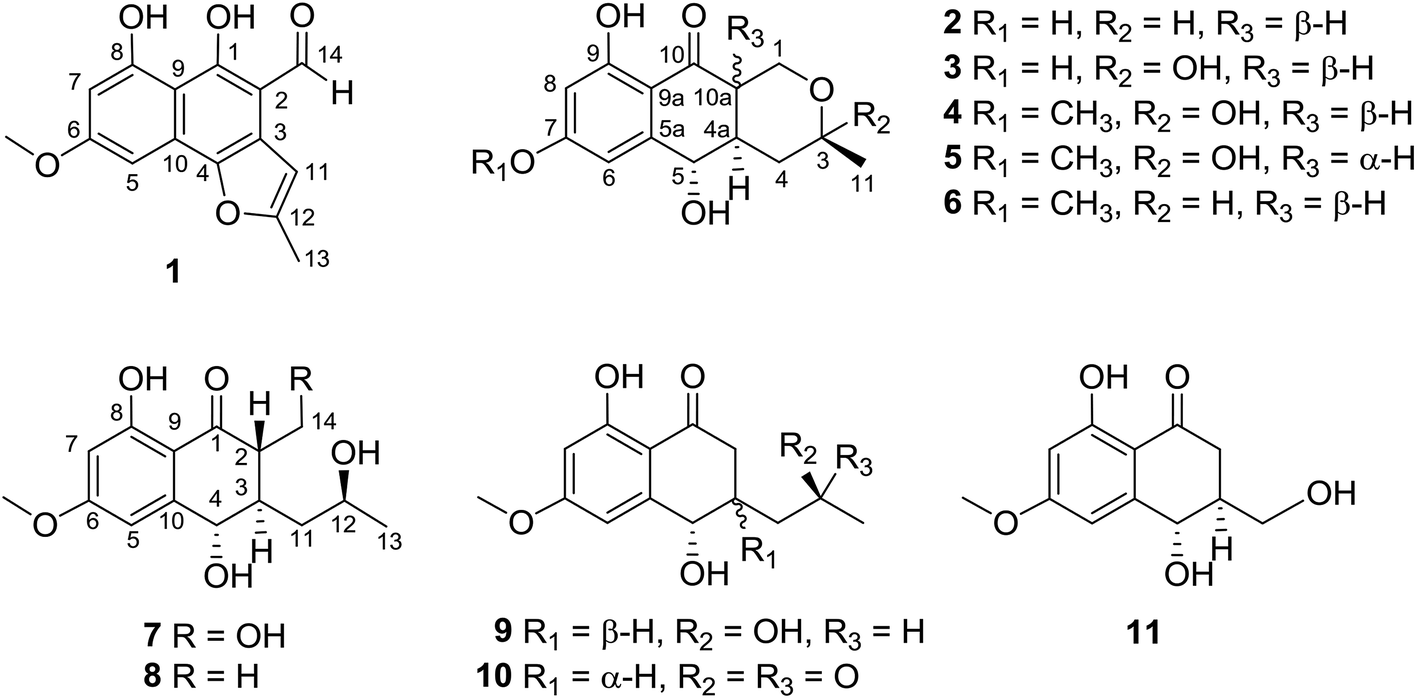 | ||
| Fig. 1 Chemical structures of compounds 1–11. | ||
| Position | δC, mult.a | δH mult.b (J in Hz) |
|---|---|---|
| a Recorded in CDCl3 at 100 MHz; 13C multiplicities were determined by HSQC experiment.b Recorded in CDCl3 at 400 MHz.c Maybe interchanged.d Maybe interchanged. | ||
| 1 | 160.7, Cq | |
| 2 | 129.1, Cq | |
| 3 | 122.4, Cq | |
| 4 | 143.6, Cq | |
| 5 | 93.0, CH | 6.92, s |
| 6 | 164.2,c Cq | |
| 7 | 101.0, CH | 6.47, s |
| 8 | 164.5,c Cq | |
| 9 | 105.5,d Cq | |
| 10 | 105.7,d Cq | |
| 11 | 100.4, CH | 6.59, s |
| 12 | 156.7, Cq | |
| 13 | 14.4, CH3 | 2.53, s |
| 14 | 190.5, CH | 9.91, s |
| 6-OMe | 55.8, CH3 | 3.95, s |
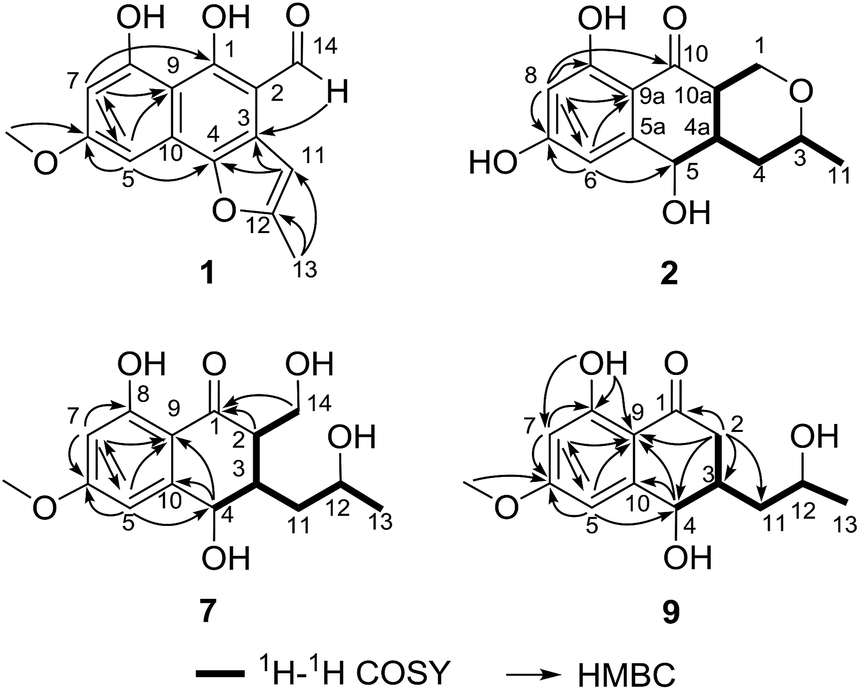 | ||
| Fig. 2 Key 1H–1H COSY and HMBC correlations of compounds 1, 2, 7, and 9. | ||
Compound 2 (Fig. 1), isolated as white crystals, has the molecular formula C14H16O5 as determined by ESI-HRMS. Its 1D NMR spectra and HSQC data (Table 2 and Fig. S9–S11†) showed the presence of one secondary methyl group, two methylenes (one oxygenated), four methines (two oxygenated), one carbonyl group, and six aromatic/olefinic carbons including two methine carbons. Analysis of the 1H–1H COSY NMR spectrum (Fig. S13,† and 2) identified correlations between C-3 and C-5, between C-1 and C-10a, between C-3 and C-11, and between C-4a and C-10a (Fig. 2). The strong HMBC correlations of H-6/C-8 and C-9a, and H-8/C-6 and C-9a also suggested a meta-substituted phenyl ring, as found in compound 1 (Fig. 2). Two hydroxyl groups were attached to C-7 (δC 166.8) and C-9 (δC 166.6) of the phenyl ring, based on analysis of their chemical shifts. The key HMBC correlations of H-6/C-5 and H-8/C-10 established the connection of C-5a to C-5, and of C-9a to C-10 (Fig. 2). A linkage between C-10 (δC 201.8) and C-10a (δC 48.3) was proposed based on their chemical shifts. The remaining one degree of unsaturation and the chemical shifts of C-1 (δC 67.7) and C-3 (δC 74.5) indicated that C-1 and C-3 were both attached to the same oxygen atom, forming a tetrahydrofuran ring, which was also consistent with the molecular formula. Accordingly, the planar structure of 2 was determined as depicted. The relative configuration of 2 was determined from its NOESY spectrum (Fig. S14,† and 3). The key NOESY correlations of H-5/H-10a, H-5/H-4β, H-4a/H-3, and H-4a/H-1α suggested that H-4β, H-5 and H-10a had the same orientation, whereas H-1α, H-3, and H-4a possessed the opposite orientation (Fig. 3). The relative configuration of 2 was further confirmed by single crystal X-ray diffraction analysis with Cu Kα radiation (Fig. 4). However, the Flack parameter [0.1(6)] was large and can't assign the absolute configuration. Compound 2 was named pleosporalin B and has structural features similar to those of biatriosporin G.9 Compound 2 and biatriosporin G9 have the similar positive optical rotation value, tentatively suggesting the same absolute configuration.
| Position | 2 | 3 | 4 | 5 | 6 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| δC, mult.a | δH mult.b (J in Hz) | δC, mult.c | δH mult.d (J in Hz) | δC, mult.c | δH mult.b (J in Hz) | δC, mult.a | δH mult.b (J in Hz) | δC, mult.e | δH mult.f (J in Hz) | |
| a Recorded in CD3OD at 100 MHz.b Recorded in CD3OD at 400 MHz.c Recorded in CD3OD at 150 MHz.d Recorded in CD3OD at 600 MHz.e Recorded in DMSO-d6 at 100 MHz.f Recorded in DMSO-d6 at 400 MHz.g Signals overlapped. | ||||||||||
| 1 | 67.7, CH2 | α, 3.44, t (11.2) | 60.3, CH2 | α, 3.92, t (11.4) | 60.2, CH2 | α, 3.93, t (11.6) | 59.5, CH2 | α, 4.04, dd (2.8, 11.2) | 65.6, CH2 | α, 3.37, t (10.8) |
| β, 4.43, dd (4.8, 11.2) | β, 4.12, dd (4.8, 11.4) | β, 4.12, dd (4.8, 11.6) | β, 4.51, br d (11.2) | β, 4.27, dd (4.8, 11.6) | ||||||
| 2 | ||||||||||
| 3 | 74.5, CH | 3.46, m | 96.0, Cq | 96.0, Cq | 95.6, Cq | 72.3, CH | 3.41, m | |||
| 4 | 37.7, CH2 | β, 1.19, m | 39.7, CH2 | β, 1.45, mg | 39.7, CH2 | β, 1.46, t (13.2) | 37.1, CH2 | β, 1.11, t (12.8) | 36.3, CH2 | β, 1.10, t (7.2) |
| α, 2.17, m | α, 2.29, mg | α, 2.29, dd (3.6, 13.2) | α, 1.65, dd (3.6, 12.8) | α, 2.09, d (12.0) | ||||||
| 4a | 46.6, CH | 1.96, m | 42.2, CH | 2.31, mg | 42.2, CH | 2.33, mg | 37.5, CH | 2.96, m | 44.6, CH | 1.96, m |
| 5 | 72.7, CH | 4.39, d (11.6) | 72.8, CH | 4.36, d (10.2) | 72.8, CH | 4.39, d (10.8) | 72.2, CH | 4.42, d (1.2) | 70.4, CH | 4.39, d (10.4) |
| 5a | 151.9, Cq | 152.1, Cq | 151.8, Cq | 146.0, Cq | 151.5, Cq | |||||
| 6 | 106.3, CH | 6.67, s | 106.4, CH | 6.68, dd (1.2, 2.4) | 105.3, CH | 6.78, d (2.4) | 110.2, CH | 6.48, d (2.0) | 104.4, CH | 6.73, s |
| 7 | 166.8, Cq | 167.1, Cq | 168.0, Cq | 168.2, Cq | 166.0, Cq | |||||
| 8 | 102.2, CH | 6.14, s | 102.2, CH | 6.15, d (2.4) | 100.5, CH | 6.33, d (2.4) | 101.6, CH | 6.41, d (2.0) | 99.2, CH | 6.36, s |
| 9 | 166.6, Cq | 166.7, Cq | 166.6, Cq | 166.7, Cq | 164.5, Cq | |||||
| 9a | 109.9, Cq | 109.9, Cq | 110.6, Cq | 110.6, Cq | 109.2, Cq | |||||
| 10 | 201.8, Cq | 201.9, Cq | 202.4, Cq | 202.7, Cq | 201.5, Cq | |||||
| 10a | 48.3, CH | 2.52, dt (4.4, 11.2) | 47.7, CH | 2.54, ddd (4.8, 10.8, 12.0) | 47.8, CH | 2.57, dt (4.8, 11.6) | 41.8, CH | 2.93, m | 46.5, CH | 2.66, dt (4.4, 12.0) |
| 11 | 22.0, CH3 | 1.23, d (6.0) | 29.8, CH3 | 1.43, s | 29.8, CH3 | 1.42, s | 29.7, CH3 | 1.24, s | 21.7, CH3 | 1.14, d (6.0) |
| 7-OMe | 56.1, CH3 | 3.85, s | 56.3, CH3 | 3.86, s | 55.7, CH3 | |||||
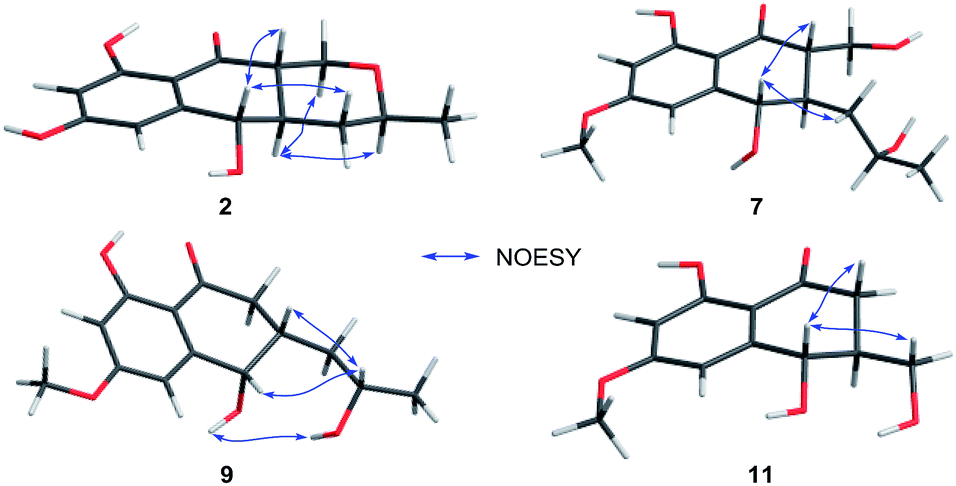 | ||
| Fig. 3 Key NOESY correlations of compounds 2, 7, 9, and 11. | ||
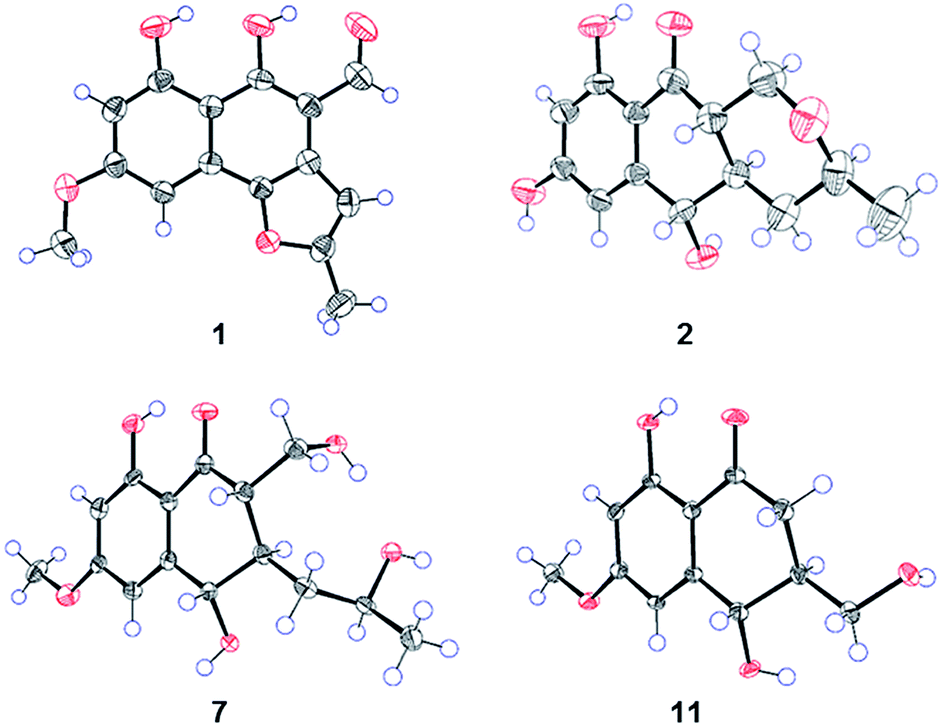 | ||
| Fig. 4 ORTEP drawing of compounds 1, 2, 7, and 11. | ||
The spectroscopic data (IR, UV, 1H NMR and 13C NMR; Fig. S17–S48†) of pleosporalins C–E (3–5, Fig. 1) and (+)-pleorubrin B (6, Fig. 1) established that they contained the basic carbon skeleton and ring system seen in compound 2. Of these, compound 3 was a white powder and was assigned the molecular formula C14H16O6, corresponding to one oxygen atom more than the formula of compound 2. The 1H and 13C NMR data of 3 (Table 2, and Fig. S17 and S18†) were similar to those of 2, except for the significantly downfield shift of C-3 from 74.5 in 2 to 96.0 in 3. Further analysis of the HMBC correlations of H2-1/C-3, H3-11/C-3, and H3-11/C-4 in conjunction with the 1H–1H COSY data confirmed a tetrahydropyran ring with a hydroxyl group, making C-3 a hemiacetal carbon in 3 (Fig. S20 and S21†). The overall structural assignment was supported by the 2D NMR data and was in accordance with the molecular formula. Compound 4 possessed a molecular formula of C15H18O6 as assigned from its quasimolecular ion at m/z 295.1180 [M + H]+. Careful comparison of its 1H and 13C NMR data (Table 2) with those of compound 3 indicated that compound 4 has one more oxygenated methyl group than is present in 3. This additional methoxy group in 4 instead of the hydroxyl group in 3, was located at C-7 based on the HMBC correlation from OCH3-7 to C-7 (Fig. S28†). For metabolite 5, its ESI-HRMS data showed that it had the same molecular formula, C15H18O6, as that of 4. Further comparison of the 1D and 2D NMR spectroscopic data of 5 (Table 2, and Fig. S33–S37†) with those of 4 confirmed that they shared the same planar structure. However, a major difference in 5 was the small coupling constant between H-4a and H-10a (Fig. S33,† and Table 2), indicating these protons were in the cis-configuration. Furthermore, the presence of correlations of H-4a/H-1α and H-5/H2-4 and the absence of a correlation of H-5/H-10a in the NOESY spectrum of 5 confirmed that H-4a and H-10a were in the same orientation (Fig. S38†). Compound 6 was assigned the molecular formula C15H18O5, corresponding to one CH2 group more than is seen in 2. On the basis of its 1D and 2D NMR data (Table 2 and Fig. S41–S46†), the structure of 6 was similar to that of 2, and the hydroxyl group at C-7 in 2 was replaced by a methoxy group in 6. Further analysis of its NMR data established that compound 6 possessed the planar and relative structure of pleorubrin B with an unclear absolute configuration.10 From a biogenetic standpoint, compounds 3–6, which have positive optical rotations, should have the same absolute configuration as 2 ([α]20D +39.3, MeOH). This is the first report of the absolute configuration of 6.
Compounds 7 and 8 (Fig. 1) should share the same carbon skeleton based on their 1D NMR data (Table 3 and Fig. S49, S50, S57, and S58†), and compound 8, isolated as a white powder, was identified as a known compound, biatriosporin A, based on a comparison of its NMR data with those reported in the literature.9 Compound 7 was obtained as white crystals from MeOH, and the molecular formula C15H20O6 was assigned to 7 based on its ESI-HRMS data. Analysis of the 1H NMR, 13C NMR, and HSQC spectra of 7 (Fig. S49–S51†) indicated that it also has a 2,3-substituted-6-methoxy-4,8-dihydroxy-3,4-dihydronaphthalen-1(2H)-one carbon skeleton and is therefore related to 8. The side chain located at C-3 was determined from the 1H–1H COSY data (Fig. 2) and was the same as that of 8. The 1H–1H COSY correlation of H2-14/H-2 and the downfield shift of C-14 (δC 59.0) in the 13C NMR spectrum of 7 suggested that a hydroxyl group was attached to C-14, which was also consistent with its molecular formula. The key NOESY correlation of H-2/H-4 indicated that H-2 and H-4 are on the same side and are β oriented (Fig. 3). Finally, a single crystal X-ray diffraction experiment with Cu Kα radiation [Flack parameter: 0.12(14)] confirmed the relative and absolute structures of 7, and compound 7 was named pleosporalin F (Fig. 4).
| Position | 7 | 9 | 10 | 11 | ||||
|---|---|---|---|---|---|---|---|---|
| δC, mult.a | δH mult.b (J in Hz) | δC, mult.a | δH mult.b (J in Hz) | δC, mult.c | δH mult.d (J in Hz) | δC, mult.e | δH mult.d (J in Hz) | |
| a Recorded in DMSO-d6 at 100 MHz.b Recorded in DMSO-d6 at 400 MHz.c Recorded in CD3OD at 150 MHz.d Recorded in CD3OD at 400 MHz.e Recorded in CD3OD at 100 MHz.f Signals overlapped. | ||||||||
| 1 | 203.1, Cq | 203.3, Cq | 202.8, Cq | 203.5 | ||||
| 2 | 52.8, CH | 2.53, m | 39.5, CH2 | β, 2.47, dd (3.6, 17.6) | 42.3, CH2 | β, 2.48 dd (16.8, 7.2) | 39.8, CH2 | β, 2.61, dd (11.2, 17.6) |
| α, 2.73, dd (10.4, 17.6) | α, 2.80, dd (16.8, 2.8) | α, 2.77, dd (4.4, 17.6) | ||||||
| 3 | 39.3, CH | 2.24, m | 36.6, CH | 2.29, m | 39.6, CH | 2.56, m | 46.0, CH | 2.20, m |
| 4 | 69.9, CH | 4.55, d (7.2) | 67.8, CH | 4.64, t (4.8) | 72.1, CH | 4.48, d (8.8) | 69.7, CH | 4.66, d (9.6) |
| 5 | 105.5, CH | 6.65, d (2.4) | 106.9, Cq | 6.52, d (2.4) | 106.3, Cq | 6.71, brs | 106.2, CH | 6.74, d (2.4) |
| 6 | 166.0, Cq | 165.8, Cq | 168.2, Cq | 168.2 | ||||
| 7 | 99.3, CH | 6.38, d (2.4) | 99.7, Cq | 6.39, d (2.4) | 100.6, Cq | 6.35, brs | 100.5, CH | 6.34, d (2.4) |
| 8 | 164.4, Cq | 164.4, Cq | 166.6, Cq | 166.4 | ||||
| 9 | 109.7, Cq | 109.3, Cq | 110.8, Cq | 110.8 | ||||
| 10 | 149.6, Cq | 148.8, Cq | 150.7, Cq | 151.1 | ||||
| 11 | 40.0, CH2 | 1.49, m | 39.4, CH2 | 1.58, ddd (4.4, 7.6, 13.6) | 46.4, CH2 | 2.55, m | 63.2, CH2 | 3.76, m |
| 1.61, m | ||||||||
| 1.23, dt (13.6, 6.8) | 2.97, m | |||||||
| 12 | 65.1, CH | 3.83, mf | 63.8, CH | 3.77, m | 210.2, Cq | |||
| 13 | 24.2, CH3 | 1.08, d (6.4) | 24.3, CH3 | 1.06, d (6.0) | 30.4, CH3 | 2.18, s | ||
| 14 | 59.0, CH2 | 3.68, m, 4.17, m | ||||||
| 6-OMe | 55.7, CH3 | 3.83, s | 55.8, CH3 | 3.82, s | 56.2, CH3 | 3.85, s | 56.1, CH3 | 3.86, s |
| 4-OH | 5.81, d (6.4) | 5.43, d (4.8) | ||||||
| 8-OH | 13.00, s | 12.82, s | ||||||
| 12-OH | 4.79, d (4.4) | 4.46, d (4.8) | ||||||
The molecular formula, C14H18O5, of compound 9 (Fig. 1) was determined by ESI-HRMS. Analysis of its 1D and 2D NMR data (Table 3, and Fig. S65–S70†) suggested its structural features were similar to those of compounds 7 and 8. The main difference observed in compound 9 was the connectivity around C-2 (CH2, δH 2.47, 2.73; δC 39.5). A detailed analysis of the 1H–1H COSY spectrum indicated the connection of C-2 to C-4 (Fig. 2). The HMBC correlations from H2-2 to C-1 and C-9 established the C-2–C-1–C-9 fragment (Fig. 2). The above data of 9 showed that a methylene group was present at C-2 instead of the methyl (or hydroxymethyl)-substituted methine in compounds 7 or 8. The large coupling constant between Hα-2 and H-3 (J = 10.4) and the small coupling constants between H-2β and H-3 (J = 3.6) and between H-3 and H-4 (J = 4.8), established the relative configuration of 9 as shown (Fig. 3). Compound 10 (Fig. 1) had the molecular formula, C14H16O5, as determined by ESI-HRMS. Its 1D NMR spectral data (Table 3, and Fig. S73 and S74†) confirmed that it has the same planar and relative configuration as that of balticol A with an unclear absolute configuration.11 Compound 11 (Fig. 1) had the molecular formula, C12H14O5, based on ESI-HRMS. Analysis of its 1D and 2D NMR data (Table 3, and Fig. S77–S79†) indicated that the planar structure of 11 was identical to those of balticol B and iso-balticol B.11,12 Specifically, the relative configuration of 11 should be the same as that of balticol B based on the NOESY correlations of H-4/H-2β and H-4/H2-11 (Fig. 3) in conjunction with the large coupling constants between H-3 and H-4 and between H-3 and H-2β (Table 3). However, the absolute configuration of 11 is unknown.11 Using single-crystal X-ray diffraction analysis with Cu Kα radiation [Flack parameter: 0.18(15)] (Fig. 4), its absolute configuration was unambiguously determined for the first time as depicted.
From a biogenetic perspective with a focus on the chiral center at C-5 in compounds 2–6 or C-4 in compounds 7–11, the identical absolute configurations of compounds 2–11 were suggested, which was supported by single-crystal X-ray diffraction studies (Fig. 4). To further confirm their absolute configurations, ECD calculations were performed at the mPW1PW91/6-311G(d)//B3PW91/TZVP level of theory. The calculated ECD spectra of selected compounds 2, 10, and 11, are in good agreement with the experimental curves (Fig. 5,S82 and S83†).
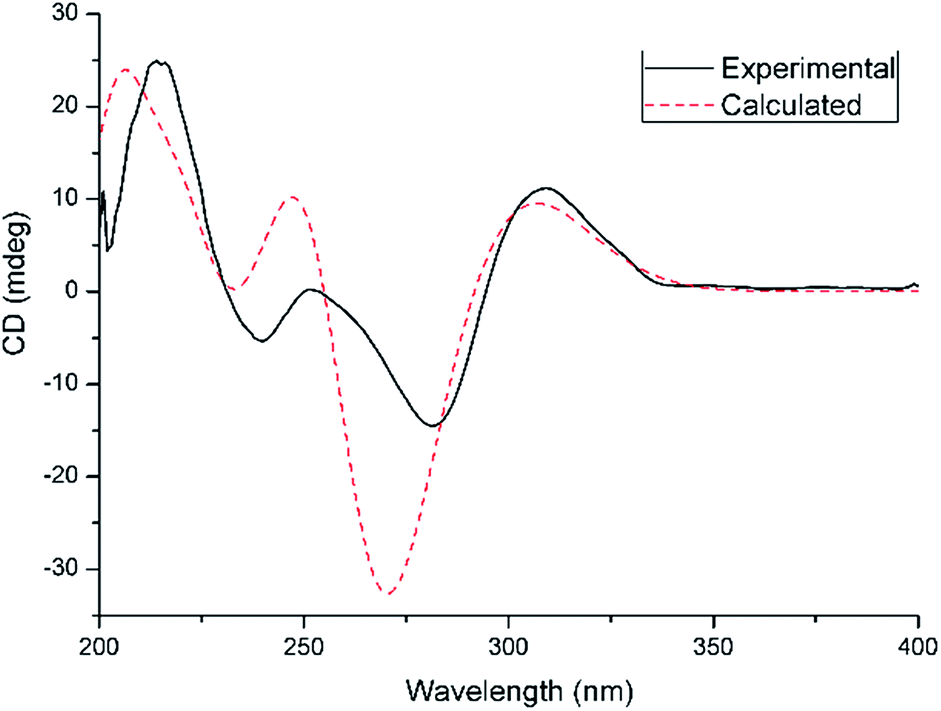 | ||
| Fig. 5 Experimental (black, solid) and calculated (red, dash) ECD curves of compound 2. | ||
Based on our previous results showing that several heptaketides displayed potent inhibitory activities against C. albicans,9 the antifungal activities of the isolated metabolites (1–11) were investigated in the present study to further test this hypothesis. Only compound 1, which has a core structure similar to that of our previously reported compound biatriosporin D with high antifungal activity, exhibited weak antifungal activity against C. albicans with an MIC80 of 128 μg mL−1. The above results indicated that for this type of polyketide, the substituents at C-1 and C-2 play an important role in increasing the antifungal activity against the selected fungus. Considering the increasing attention on the cytotoxic efficacies of these kinds of secondary metabolites and their derivatives,12 the cytotoxicities of all the isolated compounds were also evaluated against the cancer cell lines A549, SMMC-721, and MDA-MB-231. Only compound 7 showed moderate cytotoxicity against MDA-MB-231 cells with an IC50 of 22.4 ± 1.1 μM. By comparing compound 7 with 8–11, it was revealed that the substituent at C-2 could increase the cytotoxicity of compound 7. These results indicated the potential of compounds 1 and 7 as bioactive compounds for the further development of new derivatives with increased activity.
Experimental section
General experimental procedures
Optical rotations were measured with a PerkinElmer 241MC polarimeter. IR data were recorded on a Nicolet iN 10 micro FTIR spectrometer. NMR spectra were measured on Bruker Advance 400 MHz or Bruker Advance 600 MHz spectrometers. ESI-HRMS experiments were performed on an LTQ-Orbitrap spectrometer (Thermo Fisher, USA) equipped with an analytical HPLC system. The semipreparative HPLC system (Tianjin Bonna-Agela Technologies Co., Ltd., China) was equipped with two HP-Q-P050 high-pressure pumps with an HP-Q-UV100S variable UV detector, an ATS-051-H10 automatic sampler, a FL-C100B fraction collector, and an Innoval column (10 × 250 mm, 10 μm). Silica gel (200–300 mesh; Qingdao Haiyang Chemical Co., Ltd., China), Sephadex LH-20 (25–100 μm; Pharmacia, Uppsala, Sweden), and octadecyl carbon chain (C18)-bonded silica (40–60 μm, 100 Å) were employed for column chromatography (CC). Thin-layer chromatography (TLC) separations were conducted with precoated silica gel GF254 glass plates (Qingdao Haiyang Chemical Co., Ltd., China).Fungal material
The endophytic fungus Pleosporales sp. F46 was isolated from the pedicel of M. fortunei collected from Qingdao, People's Republic of China, following a previously established method.13 Briefly, the plant tissues were washed and cut into small fragments, and the surfaces were sterilized by sequential immersion in 70% ethanol for 1 min, sodium hypochlorite (4% available chlorine) for 3 min, 70% ethanol for 30 s, and sterile water for 1 min. These surface-sterilized tissue fragments were then placed on water agar medium in Petri dishes and checked every day to evaluate the growth of the fungi. Fungal hyphal tips that were growing from the plant tissue fragments were subcultured onto potato dextrose agar (PDA) for further biological and chemical investigations. The endophytic fungus F46 was identified by internal transcribed spacer (ITS) sequencing, and its ITS sequence has been deposited at GenBank (accession number MH256138). For large-scale fermentation, the strain was cultured on PDA at 28 ± 2 °C for one week. Agar plugs were cut into small pieces and 40 pieces were used to inoculate 20 flasks (1 L) each containing 80 g of rice, 120 mL of water, and 0.3% peptone. The cultures were maintained in an incubator at 28 ± 2 °C for two months.Extraction and isolation
The fungal cultures were extracted with ethyl acetate (EtOAc) by sonication at room temperature. The organic solvent was evaporated under vacuum to give the crude extract (12.8 g). The extract was further fractionated by CC on silica gel, eluting with a gradient of petroleum ether (60–90 °C)–EtOAc from 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 to 0:100 (v/v). Eight fractions (A–H) were obtained on the basis of TLC analysis. Fraction C (726.6 mg) was subjected to a Sephadex LH-20 column eluting with MeOH and afforded pleosporalin A (1, 135.5 mg). Fraction F (1907.7 mg) was separated on a Sephadex LH-20 column (MeOH) and yielded six subfractions (F1–F6). Subfraction F3 (551.0 mg) was subjected to C18-bonded silica CC eluting with a gradient of methanol (5–100%, 50 min) in water to give nine subfractions (F31–F39). Subfraction F34 (17.4 mg) was further purified by HPLC (MeOH–H2O, 40:60, 2.0 mL min−1) to obtain pleosporalin E (5, 8.2 mg, tR = 33.0 min). Further separation of subfraction F35 (54.6 mg) by HPLC (ZORBAX SB-C18 column, 4.6 × 250 mm, 5 μm; acetonitrile–H2O, 30:70, 1.0 mL min−1) yielded pleosporalin G (9, 11.4 mg, tR = 8.2 min) and (−)-balticol A (10, 1.8 mg, tR = 12.2 min). Following the same procedure as that used for subfraction F35, subfraction F36 (191.2 mg) gave (+)-pleorubrin B (6, 24.3 mg, tR = 15.0 min). Subfraction F37 (72.3 mg) was also purified by HPLC (MeOH–H2O, 70:30, 2.0 mL min−1) and afforded biatriosporin A (8, 8.1 mg, tR = 12.8 min). Separation of subfraction F4 (223.2 mg) by C18-bonded silica CC provided seven subfractions (F41–F47). Subfraction F42 (55.7 mg) was then separated by HPLC (MeOH–H2O, 45:55, 2.0 mL min−1) to yield (−)-balticol B (11, 7.7 mg, tR = 19.1 min). Fraction G (935.0 mg) was subjected to a Sephadex LH-20 column (MeOH) to give three subfractions (G1–G3). Subfraction G1 was separated by filtration and crystallized from MeOH to afford pleosporalin F (7, 7.3 mg). Subfraction G3 (221.6 mg) was subjected to C18-bonded silica CC followed by HPLC (MeOH–H2O, 55:45, 2.0 mL min−1) to afford pleosporalin B (2, 24.8 mg, tR = 16.2 min), pleosporalin C (3, 35.6 mg, tR = 10.8 min), and pleosporalin D (4, 1.0 mg, tR = 18.2 min).
:0 to 0:100 (v/v). Eight fractions (A–H) were obtained on the basis of TLC analysis. Fraction C (726.6 mg) was subjected to a Sephadex LH-20 column eluting with MeOH and afforded pleosporalin A (1, 135.5 mg). Fraction F (1907.7 mg) was separated on a Sephadex LH-20 column (MeOH) and yielded six subfractions (F1–F6). Subfraction F3 (551.0 mg) was subjected to C18-bonded silica CC eluting with a gradient of methanol (5–100%, 50 min) in water to give nine subfractions (F31–F39). Subfraction F34 (17.4 mg) was further purified by HPLC (MeOH–H2O, 40:60, 2.0 mL min−1) to obtain pleosporalin E (5, 8.2 mg, tR = 33.0 min). Further separation of subfraction F35 (54.6 mg) by HPLC (ZORBAX SB-C18 column, 4.6 × 250 mm, 5 μm; acetonitrile–H2O, 30:70, 1.0 mL min−1) yielded pleosporalin G (9, 11.4 mg, tR = 8.2 min) and (−)-balticol A (10, 1.8 mg, tR = 12.2 min). Following the same procedure as that used for subfraction F35, subfraction F36 (191.2 mg) gave (+)-pleorubrin B (6, 24.3 mg, tR = 15.0 min). Subfraction F37 (72.3 mg) was also purified by HPLC (MeOH–H2O, 70:30, 2.0 mL min−1) and afforded biatriosporin A (8, 8.1 mg, tR = 12.8 min). Separation of subfraction F4 (223.2 mg) by C18-bonded silica CC provided seven subfractions (F41–F47). Subfraction F42 (55.7 mg) was then separated by HPLC (MeOH–H2O, 45:55, 2.0 mL min−1) to yield (−)-balticol B (11, 7.7 mg, tR = 19.1 min). Fraction G (935.0 mg) was subjected to a Sephadex LH-20 column (MeOH) to give three subfractions (G1–G3). Subfraction G1 was separated by filtration and crystallized from MeOH to afford pleosporalin F (7, 7.3 mg). Subfraction G3 (221.6 mg) was subjected to C18-bonded silica CC followed by HPLC (MeOH–H2O, 55:45, 2.0 mL min−1) to afford pleosporalin B (2, 24.8 mg, tR = 16.2 min), pleosporalin C (3, 35.6 mg, tR = 10.8 min), and pleosporalin D (4, 1.0 mg, tR = 18.2 min).
X-ray crystallographic analysis of compounds 1, 2, 7, and 11
Single crystals for X-ray analysis were obtained by recrystallization from MeOH. The single crystal X-ray diffraction experiments were carried out on a Bruker D8 Venture diffractometer with a graphite monochromator at 296 K with Cu Kα radiation (λ = 1.54178 Å). The crystal structure was solved with the SHELXS structure solution package using direct methods and refined with the SHELXL refinement program by least-squares minimization. Molecular graphics were obtained using Ortep-3 (Version 2014.1).14 Crystallographic data of compounds 1, 2, 7, and 11 were deposited in the Cambridge Crystallographic Data Center (CCDC) with deposition numbers 1874970, 1874969, 1874967, and 1874968, respectively.Computational details
ECD calculations for compounds 2, 10, and 11 were performed as described previously.7 The possible conformers were searched using Frog2 online version.15 Further geometrical optimization and vibrational evaluation with Gaussian using DFT calculations with the B3PW91 functional and the TZVP basis set were carried out. TDDFT at the mPW1PW91/6-311G(d) level of theory was employed to calculate the singlet electronic excitation energies and rotational strengths with the solvation PCM model with methanol. The ECD curve for each conformer was simulated according to eqn (8d) with a half-band of 0.3 eV.16 The individual conformer values were summed to obtain the final ECD spectrum with respect to their Boltzmann distributions.Antifungal assay and cytotoxicity assay
The in vitro antifungal activities and cytotoxicities of compounds 1–11 were evaluated based on our previously described methods (Table S1†).9,17 Fluconazole was employed as the positive control in the antifungal susceptibility testing against C. albicans SC5314, and it showed an MIC80 of 2.0 μg mL−1. Adriamycin was used as the positive control in the cytotoxicity assay against MDA-MB-231 cells, and it exhibited an IC50 of 0.45 ± 0.1 μM. Each test was carried out in triplicate.Conclusions
In summary, we isolated and characterized six new heptaketides, pleosporalins A–F (1–5, and 7), and a new heptaketide derivative, pleosporalin G (9), together with four biosynthetically related known compounds (6, 8, 10, and 11), from an endophytic fungus, Pleosporales sp. F46, found in the medicinal plant Mahonia fortunei. Their structures were established by extensive spectroscopic analyses. The absolute configurations of known compounds 6, 10 and 11 were determined for the first time. Compound 1 was active against C. albicans with an MIC80 of 128 μg mL−1, and compound 7 exhibited moderate cytotoxicity against MDA-MB-231 with an IC50 of 22.4 ± 1.1 μM. By comparing compounds 1 and 7 with structurally related metabolites, it was revealed that alterations to their C-1 or C-2 substitutions could significantly influence their antifungal or cytotoxic efficacies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by Scientific Research Foundation of Qingdao University, Natural Science Foundation of Shandong Province (no. ZR2018BC001), and National Natural Science Foundation of China (no. 40218030034). We gratefully acknowledge Dr Wen-Xuan Wang (School of Pharmaceutical Sciences, South-Central University of Nationalities) for the quantum chemistry calculations and valuable discussions.Notes and references
- A. H. Aly, A. Debbab and P. Proksch, Pharmazie, 2013, 68, 499–505 CAS.
- S. Kusari, C. Hertweck and M. Spiteller, Chem. Biol., 2012, 19, 792–798 CrossRef CAS PubMed.
- H. W. Zhang, Y. C. Song and R. X. Tan, Nat. Prod. Rep., 2006, 23, 753–771 RSC.
- H. Gao, G. Li and H.-X. Lou, Molecules, 2018, 23, 646 CrossRef PubMed.
- G. Li and H.-X. Lou, Med. Res. Rev., 2018, 38, 1255–1294 CrossRef PubMed.
- W. X. Wang, S. Kusari, S. Sezgin, M. Lamshoft, P. Kusari, O. Kayser and M. Spiteller, Appl. Microbiol. Biotechnol., 2015, 99, 7651–7662 CrossRef CAS PubMed.
- Z.-R. Wang, G. Li, L.-X. Ji, H.-H. Wang, H. Gao, X.-P. Peng and H.-X. Lou, Steroids, 2019, 145, 1–4 CrossRef CAS PubMed.
- H. B. Bode, B. Bethe, R. Hofs and A. Zeeck, ChemBioChem, 2002, 3, 619–627 CrossRef CAS PubMed.
- Y. H. Zhou, M. Zhang, R. X. Zhu, J. Z. Zhang, F. Xie, X. B. Li, W. Q. Chang, X. N. Wang, Z. T. Zhao and H.-X. Lou, J. Nat. Prod., 2016, 79, 2149–2157 CrossRef CAS PubMed.
- E. Stodulkova, P. Man, M. Kuzma, J. Cerny, I. Cisarova, A. Kubatova, M. Chudickova, M. Kolarik and M. Flieger, Folia Microbiol., 2015, 60, 259–267 CrossRef CAS PubMed.
- M. A. M. Shushni, R. Mentel, U. Lindequist and R. Jansen, Chem. Biodiversity, 2009, 6, 127–137 CrossRef CAS PubMed.
- C. Padumadasa, Y. M. Xu, E. M. K. Wijeratne, P. Espinosa-Artiles, J. M. U'Ren, A. E. Arnold and A. A. L. Gunatilaka, J. Nat. Prod., 2018, 81, 616–624 CrossRef CAS PubMed.
- S. Kusari, M. Lamshoft, S. Zuhlke and M. Spiteller, J. Nat. Prod., 2008, 71, 159–162 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- M. A. Miteva, F. Guyon and P. Tuffery, Nucleic Acids Res., 2010, 38, W622–W627 CrossRef CAS PubMed.
- P. J. Stephens and N. Harada, Chirality, 2010, 22, 229–233 CAS.
- K. Xu, Y. Gao, Y. L. Li, F. Xie, Z. T. Zhao and H.-X. Lou, J. Nat. Prod., 2018, 81, 2041–2049 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data of compounds 1–11. Experimental and calculated ECD curves of compounds 10 and 11. CCDC 1874967–1874970. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra01956a |
| This journal is © The Royal Society of Chemistry 2019 |