DOI:
10.1039/C9RA02002K
(Paper)
RSC Adv., 2019,
9, 12928-12935
Gram-scale carbasugar synthesis via intramolecular seleno-Michael/aldol reaction†
Received
15th March 2019
, Accepted 17th April 2019
First published on 26th April 2019
Abstract
Carbasugars represent an important category of natural products possessing a broad spectrum of biological activities. Lots of effort has been done to develop gram scale synthesis. We are presenting a new approach to gram scale synthesis of the carbasugar skeleton via intramolecular seleno-Michael/aldol reaction. The proposed strategy gave gram amounts of 6-hydroxy shikimic ester in a tandem process in 36% overall yield starting from D-lyxose. We have attempted to demonstrate the synthetic utility of 6-hydroxyshikimic acid derivatives by covering the important synthetic modifications and related applications, namely synthesis of protected (−)-gabosine E, (−)-MK7606, (−)-valienamine and finally unprotected methyl (−)-shikimate.
Introduction
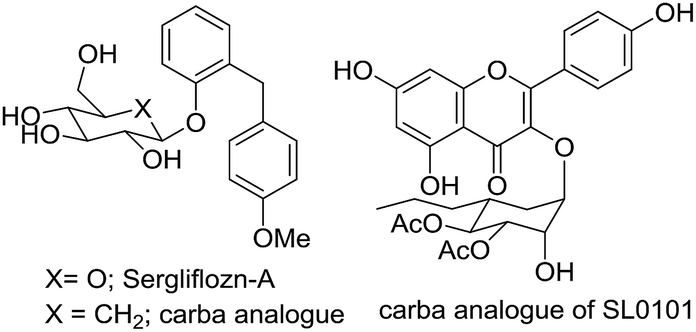
Carbasugars are analogues of sugars that have attracted the interest of medicinal chemists due to their potential application as active pharmaceutical ingredients. These compounds are structurally related to carbohydrates where the ring oxygen is replaced with a methylene group.1,2 Carbasugar fragments have been found in various natural products.3,4 The structural similarity of carbasugar fragments to conventional monosaccharides suggests that they are likely to bind and inhibit the same protein targets. The advantage of carbasugar containing compounds is the lack of a glycosidic linkage resulting in the increased stability toward enzymatic degradation.5,6 For example, synthetic nucleotide analogues containing thiosugar or carbasugars have been reported to be glycosyltransferase inhibitors.7–9 The mechanism of glycosidase inhibition has been studied and several carba-analogues, such as Sergliflozin-A10 or SL0101,11 have been introduced (Fig. 1). Growing interest of carbasugar use is limited due to their low availability from natural sources.
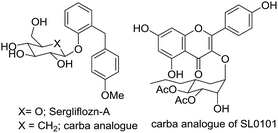 |
| | Fig. 1 Active carbasugars containing compounds. | |
General strategies to obtain carbapyranoses can be broadly classified into two groups: (i) synthetic methods that employ non-carbohydrates as starting materials and (ii) protocols that utilize carbohydrates as precursors. Multi gram synthesis of carbasugar fragments is still challenging and extremely important.
Carbohydrates, especially monosaccharides, are excellent starting material for total synthesis of various natural and valuable synthetic compounds. Their availability is usually very high, prices are low and chemistry of carbohydrates is well known. An application of monosaccharides to carbohydrate mimetic synthesis seems to be a natural choice.12 General synthesis of carbasugars moieties is employing Grubbs cross metathesis reaction,13–16 aldol-type cyclization,17,18 Corey–Fuchs reaction19 and others.3 Total synthesis of gabosines has been recently reviewed by Mac and co-workers.20
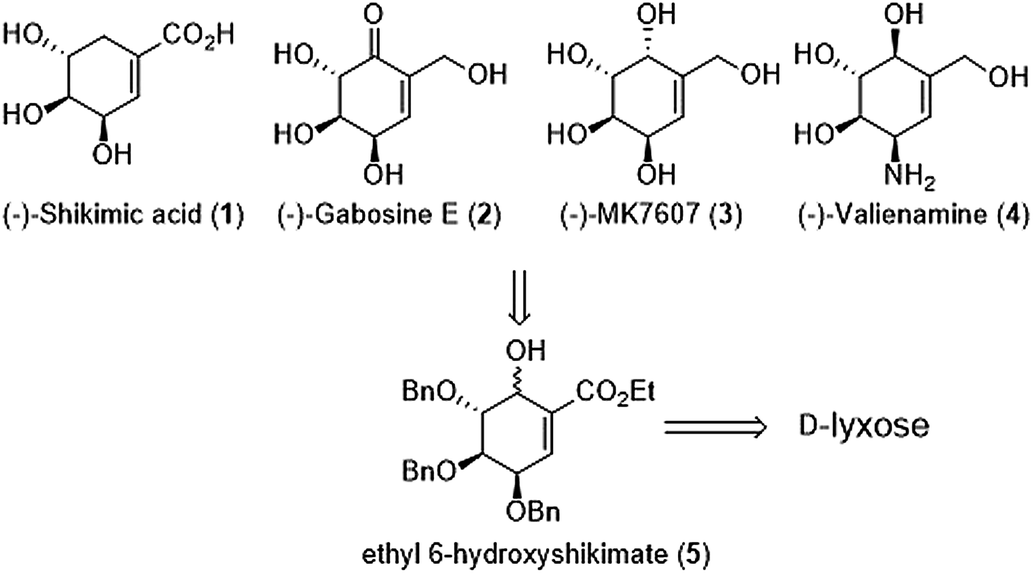
Our proposal of shikimic-type esters synthesis and their reduced analogues is based on modification of 6-hydroxyshikimic ester21,22 obtained via intramolecular seleno-Michael/aldol reaction from D-lyxose (Scheme 1).
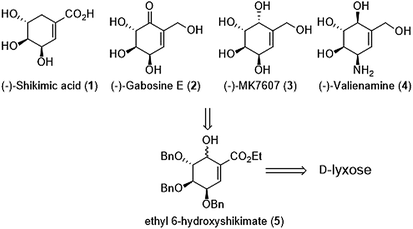 |
| | Scheme 1 General strategy of cyclitols synthesis. | |
Results and discussion
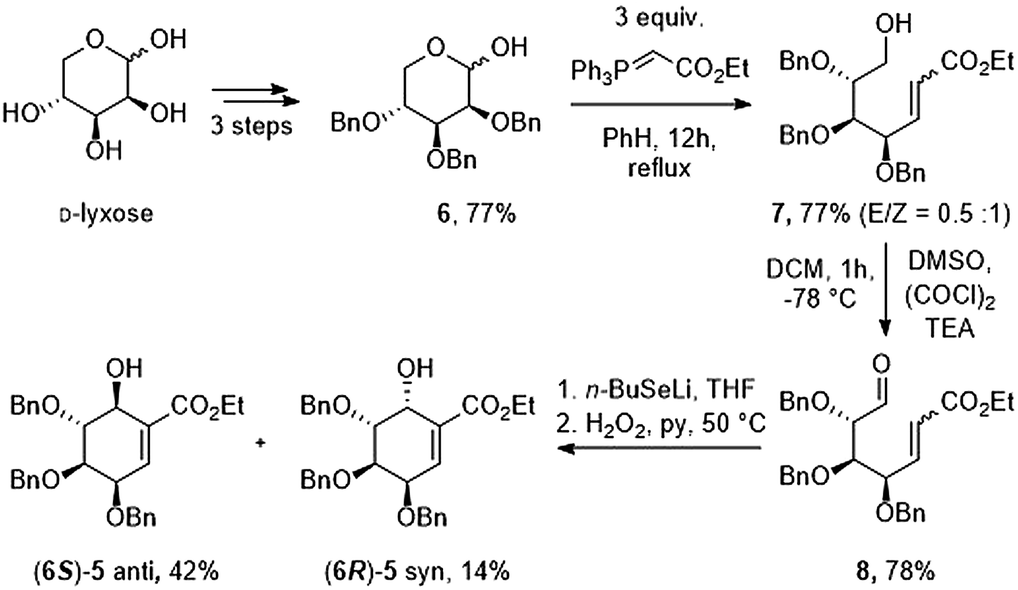
First, we prepared oxo-α,β-unsaturated ester (8) according to the literature method in multi-gram scale.23 Mixture of 2,3,4-tri-O-benzyl-D-lyxopyranoses (6) have been refluxed with excess of ylide over 12 h yielding mixture of E/Z isomers of 7. Alcohol 7 has been oxidized to corresponding aldehyde 8 using Swern oxidation in 78% yield. Compound 8 has been subjected to intramolecular seleno-Michael/aldol reaction with consecutive oxidation-elimination process to afford a mixture of 6-hydroxyshikimic esters (5) in 56% yield for 7 h (Scheme 2).24
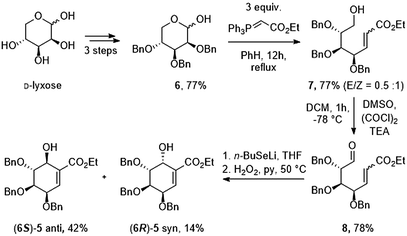 |
| | Scheme 2 Synthesis of key intermediate. | |
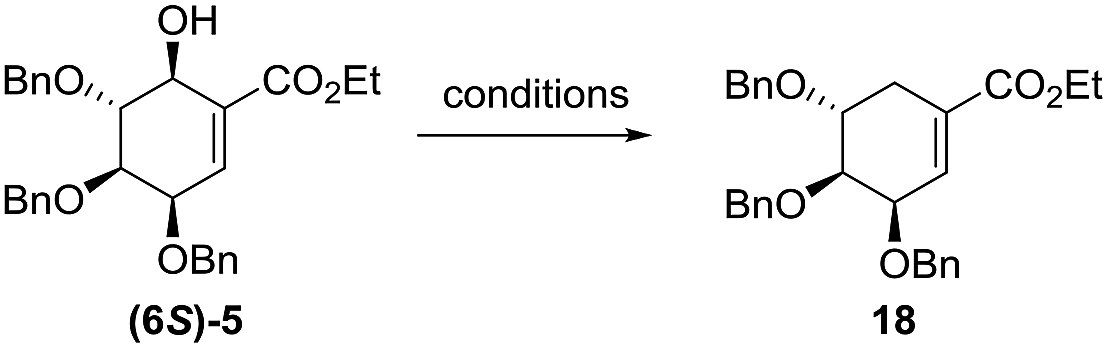
After short optimization, multistep tandem reaction gave the carbocyclic products (6S)-5 and (6R)-5 in 76% yield over 3 steps as an 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.33 mixture of axial and equatorial alcohol (Table 1). The 6S/R-isomers of 5 have been separated easily by column chromatography (Experimental section). Having established the optimized conditions for the cyclic core synthesis, further functionalization has been achieved.
:0.33 mixture of axial and equatorial alcohol (Table 1). The 6S/R-isomers of 5 have been separated easily by column chromatography (Experimental section). Having established the optimized conditions for the cyclic core synthesis, further functionalization has been achieved.
Table 1 Scale optimizationa
| Entry |
Scale |
Conditions |
Yield |
Anti/syn |
| Reaction conditions: THF, 1.2 equiv. n-BuSeLi to RT (30 min), 10 equiv. H2O2, 5 equiv. py, 50 °C (1 h). |
| 1 |
174 mg (0.35 mmol) |
−20 °C (6 h) |
56% |
10.32 |
| 2 |
1080 mg (2.21 mmol) |
−78 °C (30 min) |
76% |
10.32 |
| 3 |
5600 mg (11.45 mmol) |
−78 °C (30 min) |
76% |
10.33 |
Synthesis of protected (−)-valienamine
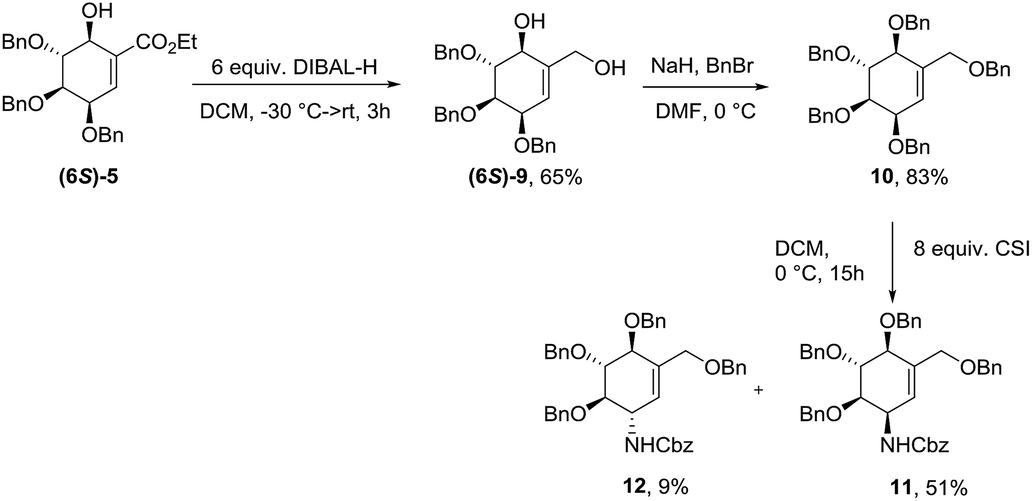
The 6S-hydroxyshikimic ester (6S)-5 has been transformed to non-natural valienamine derivative (11) with known protocol in 27% yield over 3 steps.16,24 Exposure of 10 to chlorosulfonyl isocyanate in DCM afforded the desired product (11) in 51% yield. Minor product has been assigned by NMR analysis as C3-epimer of (−)-valienamine (12) (Scheme 3).
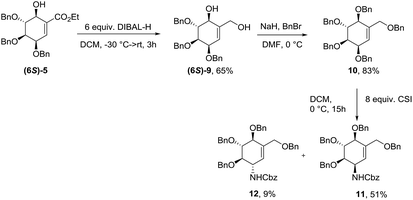 |
| | Scheme 3 Synthesis of protected (−)-valienamine. | |
Synthesis of protected (−)-MK7607
Syn-isomer of compound 5 was transformed to partially protect unnatural (−)-MK7607. Simple reduction with DIBAL-H gave compound (6R)-9 in 33% yield which is surprising to the result obtained for 6S-isomer (65%) at the same conditions (Scheme 4).
 |
| | Scheme 4 Synthesis of unnatural (−)-MK7607. | |
Synthesis of protected (−)-gabosine E
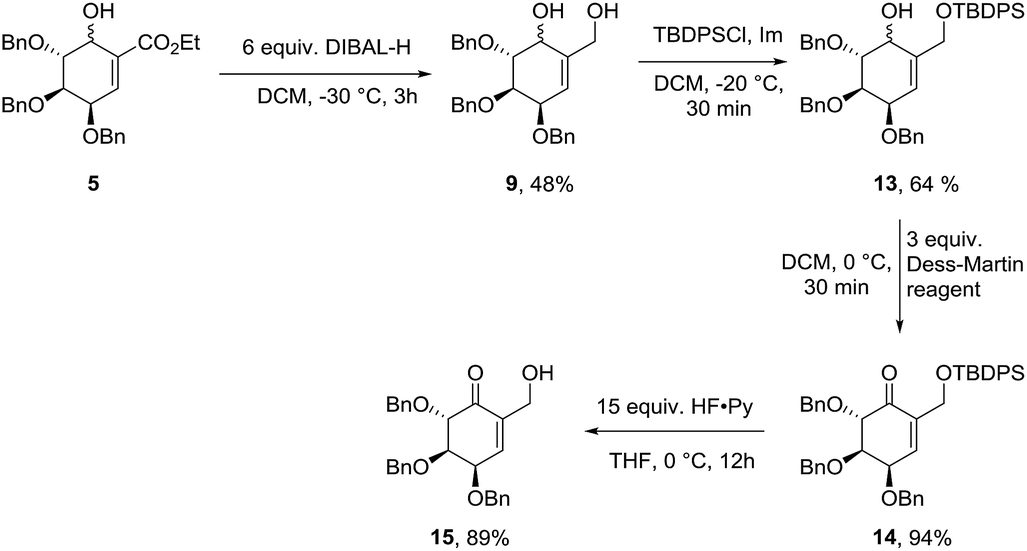
Starting from diastereomeric mixture of 5, ester fragment has been reduced in the presence of DIBAL-H to give diol 9 in 48% yield. Selective protection of primary hydroxyl group gave mixture of isomers 13 in 64% yield. Oxidation with Dess–Martin reagent in DCM to unsaturated ketone (14) and consecutive silyl ether deprotection gave 2,3,4-tri-O-benzylated (−)-gabosine E (15) in 84% yield in two steps (Scheme 5).
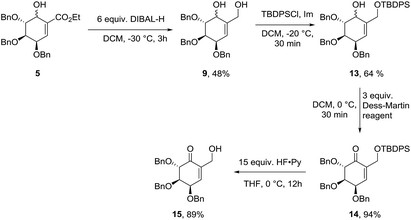 |
| | Scheme 5 Synthesis of (−)-gabosine E derivative. | |
Synthesis of methyl (−)-shikimate
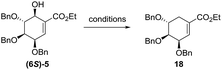
(−)-Shikimic acid is an important metabolite in plants and microorganisms. This compound can be isolated from the Japanese star anise or synthetized starting from chiral and achiral substrates. Recently, Candeias et al.,25 summarized known methods of (−)-shikimic acid synthesis. Starting from diastereomeric mixture of alcohols 5 several deoxygenation methods have been tested (Table 2).
Table 2 Deoxygenation of 6-hydroxyshikimic acid
|
| Entry |
Conditions |
Yield |
| Xanthate not formed. |
| 1 |
Et3SiH, BF3·Et2O, DCM, rt, 1 h |
nr |
| 2 |
EtO3SiH, TFA, DCM, rt, up to 24 h |
nr |
| 3 |
Tf2O, DCM −40 °C, 30 min, 0 °C, 30 min then NaBH4, EtOH |
nr |
| 4 |
NaH, Im, 0 °C, 30 min, THF then CS2, 30 min, rt, then MeI, 30 min, rt, 50 °C, 1 h |
nra |
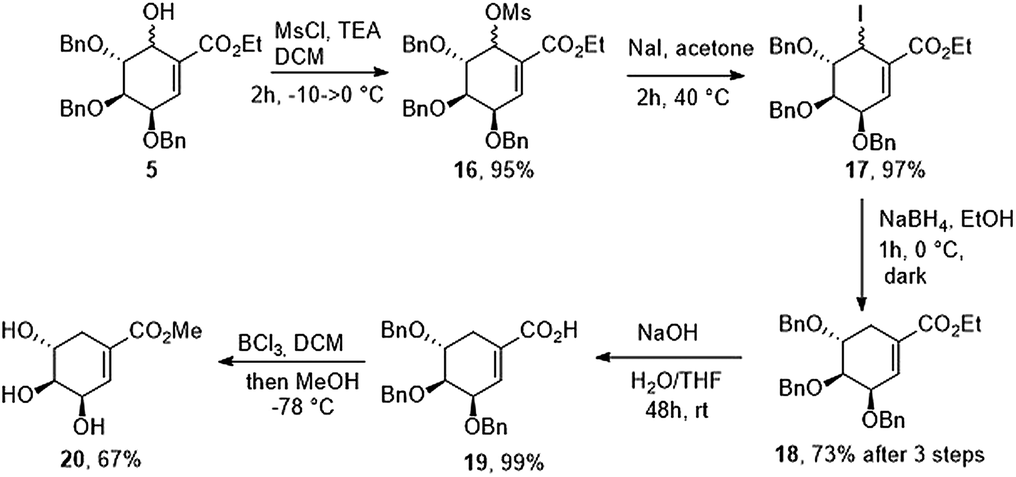
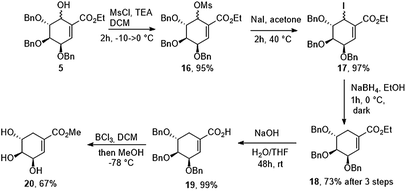
Finally, alcohol 5 was converted to mesylate 16. Unfortunately, all of our attempts to displace the mesylate group with hydride reagents resulted only in the 1,4-displacement. We decided to change mesylate group to iodide (17) and we found that halogenated compound (17) is unstable in the presence of light. In the end, reduction of iodide crude mixture carried out in the dark gave ethyl (−)-tri-O-benzyl-shikimate (18) in 2 h in 73% yield over 3 steps. Ethyl ester has been removed under basic conditions to give (−)-3,4,5-tri-O-benzylshikimic acid in quantitative yield. Exposure of 19 to BCl3 gave methyl (−)-shikimate (20) in 67% yield (Scheme 6).
 |
| | Scheme 6 Synthesis of methyl (−)-shikimate. | |
Conclusions
In summary, we have developed simple methods that provide a rapid entry into the synthesis of a series of shikimate ester and shikimate analogues, including (−)-gabosine E and (−)-MK7606. Our synthesis of 6-hydroxy shikimates from D-lyxose has been efficiently achieved in six steps with a 35% overall yield. The strategies described take place through short, high-yield reaction sequences. Partial protection of D-lyxose derivative enable access to selective modification of hydroxyl groups. Deoxygenation of 6-hydroxyshikimic ester gave methyl (−)-shikimate in 49% yield total.
Experimental
General experimental procedures
All starting materials and reagents were purchased from commercial sources and used without purification. Reactions were controlled using TLC on silica [alu-plates (0.2 mm)]. Plates were visualized with UV light (254 nm) and by treatment with: aqueous cerium(IV) sulfate solution with molybdic and sulfuric acid followed by heating. All organic solutions were dried over anhydrous magnesium sulfate. Reaction products were purified by column chromatography using silica gel 60 (240–400 mesh). Optical rotations were measured at room temperature with a digital polarimeter. CDCl3, D2O, CD3OD were used as NMR solvents. 1H spectra were recorded with 600 and 300 MHz, and referenced relative to: CDCl3 – solvent residual peak (δ = 7.26 ppm); D2O – solvent residual peak (δ = 4.79 ppm); CD3OD – solvent residual peak (δ = 3.31 ppm). Data are reported as follows: chemical shift in parts per million (ppm), multiplicity (s = singlet, d = doublet, t = triplet, dd = doublet of doublets, ddd = doublet of doublet of doublets, dddd = doublet of doublet of doublet of doublets, m = multiplet), coupling constants (in hertz) and integration. 13C NMR spectra were measured at 150 and 75 MHz with complete proton decoupling. Chemical shifts were reported in ppm from the residual solvent as an internal standard: CDCl3 (δ = 77.16 ppm); CD3OD (δ = 49.00 ppm). High-resolution mass spectra were acquired using ESI-TOF method.
2,3,4-Tri-O-benzyl-D-lyxopyranose (6)
To methanolic solution of 1% HCl (50 mL) (prepared by dissolving 1 mL SOCl2 in anhydrous methanol) D-lyxose (5.0 g, 33.3 mmol) was added in one portion and resulting mixture was refluxed for 5 h under argon (reaction was monitored by TLC CH2Cl2/MeOH 2:1). The reaction mixture was allowed to cool to room temperature, neutralized by addition of solid NaHCO3 and concentrated under reduced pressure. The residue was dissolved in EtOH (40 mL) and concentrated to the half of the original volume, toluene (25 mL) was added and the mixture was concentrated to dryness. The residue crude mixture oil was used without further purification in the next step. The viscous oil from the previous step was dissolved in anhydrous DMF (50 mL) and anhydrous THF (50 mL) and in 5 portion was added to 60% NaH suspension in mineral oil (6.8 g, 170 mmol) cooled to 0 °C. After hydrogen evolution was complete the mixture was treated with BnBr (14.8 mL, 122 mmol) and stirred for 30 min in 0 °C and then 12 h in room temperature. The reaction mixture was carefully quenched with cold, 10% aqueous solution of NH4Cl (30 mL) followed by addition of water (75 mL). The aqueous layer was extracted with EtOAc (3 × 50 mL) and combined organic layers were washed with water (2 × 60 mL) and brine (50 mL), then dried over anhydrous MgSO4 and concentrated under reduced pressure. Crude mixture of D-lyxosides from previous step was heated under reflux for 15 h with 1 M H2SO4 (42 mL), AcOH (48 mL) and 1,4-dioxane (44 mL). Mixture was cooled to rt and extracted with EtOAc (2 × 100 mL). Organic phase was washed twice with water (2 × 50 mL) and brine (50 mL) and dried over anhydrous MgSO4. After solvent evaporation residue was purified by column chromatography (Hex/EtOAc 2:1 to 3:2) to afford mixture of anomers of 2,3,4-tri-O-benzyl-D-lyxopyranose as a colorless oil (10.97 g, 26.1 mmol, yield = 78%). 1H and 13C spectra were compared with lit.23
Ethyl (4R,5R,6R)-4,5,6-tri-O-benzyl-7-hydroxyhept-2-enoate (7)
To a solution of 6 (10.0 g, 23.8 mmol) in anhydrous benzene (200 mL), ethyl (triphenylphosphoranylidene)acetate (16.5 g, 47.6 mmol) was added in one portion and mixture was heated to reflux for 8 h. After that time next portion of ethyl (triphenylphosphoranylidene)acetate (8.3 g, 23.8 mmol) was added and heating to reflux was continued for next 4 h. Reaction mixture was cooled to room temperature and benzene was evaporated under reduced pressure. Most of triphenylphosphine oxide was precipitated from the mixture of Et2O/Hex 7:3 and filtered by suction. Solvent was removed under reduced pressure and crude product was purified by column chromatography (Hex/EtOAc 3:1 to 7:3) to afford product as a colorless syrup (8.95 g, 18.2 mmol, 77%, Z/E = 1:0.6). 1H NMR (600 MHz, CDCl3) δ 7.36–7.26 (m, 24H), 7.06 (dd, J = 15.9, 6.6 Hz, 1H), 6.40 (dd, J = 11.8, 9.1 Hz, 0.6H), 6.14 (dd, J = 15.8, 0.9 Hz, 0.6H), 5.97 (dd, J = 11.8, 0.8 Hz, 1H), 5.48–5.44 (m, 1H), 4.80 (d, J = 11.5 Hz, 1H), 4.68 (d, J = 11.8 Hz, 1.6H), 4.63 (d, J = 11.6 Hz, 1H), 4.60 (d, J = 5.1 Hz, 1H), 4.59–4.57 (m, Hz, 2.4H), 4.51 (d, J = 11.6 Hz, 1H), 4.47 (d, J = 11.7 Hz, 1H), 4.30 (d, J = 11.5 Hz, 0.6H), 4.28–4.25 (m, 0.6H), 4.22 (q, J = 7.1 Hz, 1.2H), 4.17 (qd, J = 7.1, 0.9 Hz, 2H), 3.89 (dd, J = 6.0, 3.7 Hz, 1H), 3.80–3.68 (m, 3.8H), 3.66–3.61 (m, 1.6H), 1.31 (t, J = 7.1 Hz, 1.8H), 1.27 (t, J = 7.1 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 166.0, 146.5, 145.4, 138.6, 138.5, 138.3, 138.2, 137.8, 137.7, 128.6, 128.6, 128.5, 128.5, 128.4, 128.4, 128.2, 128.1, 128.0, 127.9, 127.9, 127.8, 127.8, 127.8, 127.8, 127.7, 124.0, 122.5, 81.4, 81.1, 80.2, 79.2, 78.6, 74.8, 74.6, 73.7, 73.1, 73.1, 71.6, 71.5, 61.9, 61.9, 60.7, 60.5, 14.4, 14.3; HRMS (ESI): calcd for C30H34O6Na [M + Na]+ 513.2248, found 513.2258.
Ethyl (4R,5R,6R)-4,5,6-tri-O-benzyl-7-oxohept-2-enoate (8)
A solution of oxalyl chloride (3.91 mL, 45.7 mmol) in anhydrous methylene chloride (200 mL) was cooled to −78 °C, after that DMSO (6.49 mL, 91.4 mmol) was added dropwise and stirred for 30 min. The 7 (7.47 g, 15.2 mmol) was dissolved in anhydrous methylene chloride (100 mL), slowly added to oxidative mixture and kept in −78 °C for 30 min. The triethylamine (19.26 mL, 137.0 mmol) was slowly added and stirring was continued for next 30 min, after that reaction mixture was allowed to slowly warm to 0 °C. The reaction was quenched with addition of sat. NH4Cl (100 mL), followed by addition of water (100 mL). The aqueous layer was extracted with methylene chloride (3 × 100 mL), and combined organic layers were washed with water (2 × 100 mL), and brine (100 mL). The organic phase was dried over anhydrous MgSO4 and concentrated under reduced pressure (below 35 °C). Crude product was purified by column chromatography Hex/EtOAc (1:0) to (85:15) to give the title compound as a mixture of diastereoisomers (Z/E 1:0.5) as almost colorless oil (5.75 g, 11.8 mmol, 77%). 1H NMR (600 MHz, CDCl3) δ 9.73 (s, 1H), 9.61 (d, J = 1.6 Hz, 0.5H), 7.34–7.26 (m, 19.5H), 7.25–7.20 (m, 3H), 6.99 (dd, J = 15.8, 6.7 Hz, 0.5H), 6.28 (dd, J = 11.7, 8.7 Hz, 1H), 6.13 (dd, J = 15.8, 1.0 Hz, 0.5H), 5.94 (dd, J = 11.7, 1.2 Hz, 1H), 5.49 (dd, J = 8.7, 3.5 Hz, 1H), 4.69–4.45 (m, 7.5H), 4.41 (d, J = 11.8 Hz, 1H), 4.28–4.20 (m, 1.5H), 4.19–4.15 (m, 2.5H), 4.07 (dd, J = 3.5, 1.6 Hz, 0.5H), 4.00–3.97 (m, 2H), 3.87 (dd, J = 7.6, 3.5 Hz, 0.5H), 1.31 (t, J = 7.1 Hz, 1.5H), 1.27 (t, J = 7.1 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 202.7, 201.3, 165.9, 165.9, 147.2, 145.2, 138.0, 137.9, 137.3, 137.1, 137.0, 128.7, 128.6, 128.5, 128.5, 128.4, 128.4, 128.1, 128.1, 128.0, 128.0, 127.8, 127.7, 124.6, 122.5, 83.7, 83.3, 81.2, 74.6, 74.3, 73.6, 73.3, 73.2, 71.7, 71.3, 60.7, 60.5, 14.3, 14.3; HRMS (ESI): calcd for C30H22O6Na [M + Na]+ 511.2091, found 511.2091.
Ethyl 3,4,5-tri-O-benzyl-6-hydroxyshikimate (5)
A suspension of elemental selenium (1.09 g, 13.75 mmol) in anhydrous THF (250 mL) was cooled in an ice bath and n-BuLi (1.6 M in hexanes, 8.6 mL, 13.75 mmol) was added dropwise (a clear solution was produced) and the mixture was stirred for 15 min in 0 °C. After that reaction mixture was cooled to −78 °C, solution of 8 (5.60 g, 11.46 mmol in 100 mL of anhydrous THF) was added dropwise and stirring was continued through the next 30 min and then cryo-bath was removed and mixture was allowed to warm to room temperature and stirring was continued for the next 1 h. After that time, reaction was cooled to 0 °C and hydrogen peroxide (35% v/v, 10.1 mL, 114.6 mmol) and pyridine (4.6 mL, 57.3 mmol) were added. Ice bath was removed and mixture was heated to 50 °C for 1 h (solution became clear and colorless). After solvent evaporation under reduced pressure oily residue was dissolved in ethyl acetate (200 mL), washed twice with water (2 × 80 mL) and brine (100 mL) and dried over anhydrous MgSO4. Solvent was evaporated under reduced pressure and crude product was purified by column chromatography (Hex/EtOAc 4:1 to 3:1) to give title product as colorless syrup (4.22 g, 8.65 mmol, 76%, 6S(anti)/6R(syn) 1:0.33). 1 g sample of 5 was separated by column chromatography (Hex/EtOAc 5:1 to 3:1) to give (6S)-5 (0.75 g) and (6R)-5 (0.24 g). Ethyl (6S)-3,4,5-tri-O-benzyl-6-hydroxyshikimate (6S-5): [α]20D = −73.1 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.39–7.27 (m, 13H), 7.24–7.21 (m, 2H), 6.96 (dd, J = 2.5, 0.9 Hz, 1H), 4.77 (d, J = 11.9 Hz, 1H), 4.67 (dd, J = 11.9, 6.3 Hz, 3H), 4.63 (d, J = 11.9 Hz, 1H), 4.56 (d, J = 11.8 Hz, 1H), 4.50 (dd, J = 9.4, 2.8 Hz, 1H), 4.38 (t, J = 2.8 Hz, 1H), 4.29–4.22 (m, 2H), 4.05 (dd, J = 5.5, 2.9 Hz, 1H), 3.94–3.91 (m, 1H), 3.30 (d, J = 9.3 Hz, 1H), 1.31 (t, J = 7.1 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 166.0, 138.0, 137.9, 137.7, 137.4, 132.1, 128.6, 128.2, 128.1, 128.0, 127.8, 127.8, 77.4, 74.3, 73.7, 73.7, 72.8, 71.9, 66.2, 61.0, 14.3; HRMS (ESI): calcd for C30H32O6Na [M + Na]+ 511.2091, found 511.2091; Ethyl (6R)-3,4,5-tri-O-benzyl-6-hydroxyshikimate (6R-5): [α]20D = −82.9 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.38–7.27 (m, 15H), 6.88 (d, J = 3.7 Hz, 1H), 4.80 (t, J = 3.2 Hz, 1H), 4.76 (d, J = 11.7 Hz, 1H), 4.73–4.66 (m, 4H), 4.62 (d, J = 11.9 Hz, 1H), 4.30 (t, J = 3.3 Hz, 1H), 4.23 (qd, J = 7.1, 1.0 Hz, 2H), 4.04 (dd, J = 7.9, 4.1 Hz, 1H), 3.94 (dd, J = 7.9, 3.7 Hz, 1H), 3.19 (d, J = 3.3 Hz, 1H), 1.29 (t, J = 7.1 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 166.3, 138.4, 138.2, 137.9, 132.1, 128.6, 128.6, 128.5, 128.1, 128.0, 128.0, 127.8, 76.3, 75.5, 73.9, 73.4, 72.5 (2C), 65.4, 61.2, 14.3; HRMS (ESI): calcd for C30H32O6Na [M + Na]+ 511.2091, found 511.2091.
3,4,5-Tri-O-benzyl-(−)-MK7607 ((6R)-9)
A solution of compound (6R)-5 (0.10 g, 0.2 mmol) in anhydrous methylene chloride (5 mL) was cooled to −30 °C and solution of DIBAL-H (1 M in methylene chloride, 1.2 mL, 1.2 mmol) was added dropwise. When the addition was complete, the mixture was stirred at −10 °C for 3 h. After that time, methanol (1 mL) was added to quench the reaction, and mixture was stirred at −10 °C for 30 min. After water addition (5 mL) and few drops of 1 M aqueous HCl, water phase was extracted with methylene chloride (3 × 5 mL) and combined organic phases were washed with water (2 × 5 mL) and brine (10 mL) and dried over anhydrous MgSO4. Solvent was evaporated under reduced pressure. Crude oil was purified by column chromatography (Hex/EtOAc 2:1 to 1:1) to give title compound as a colorless syrup (0.03 g, 0.067 mmol, 33%). [α]20D = −78.5 (c 1.1, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.36–7.27 (m, 15H), 5.83 (d, J = 3.9 Hz, 1H), 4.75–4.68 (m, 4H), 4.65 (d, J = 11.5 Hz, 1H), 4.62 (d, J = 12.1 Hz, 1H), 4.43 (s, 1H), 4.24 (d, J = 13.4 Hz, 1H), 4.18–4.13 (m, 2H), 4.02 (dd, J = 8.2, 4.3 Hz, 1H), 3.92 (dd, J = 8.2, 3.6 Hz, 1H), 2.83 (d, J = 4.0 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 139.7, 138.5, 137.8, 128.5, 128.4, 128.0, 127.8, 127.8, 127.7, 123.8, 76.7, 75.6, 73.6, 73.2, 72.2, 72.0, 67.3, 64.9; HRMS (ESI): calcd for C28H30O5Na [M + Na]+ 469.1985, found 469.1987.
6-Epi-3,4,5-tri-O-benzyl-(−)-MK7607 ((6S)-9)
A solution of compound (6S)-5 (0.22 g, 0.45 mmol) in anhydrous methylene chloride (10 mL) was cooled to −30 °C and solution of DIBAL-H (1 M in methylene chloride, 2.7 mL, 2.7 mmol) was added dropwise. When the addition was complete, the mixture was stirred at −10 °C for 3 h. After that time, methanol (2 mL) was added to quench the reaction, and mixture was stirred at −10 °C for 30 min. After water addition (10 mL) and few drops (0.5 mL) of 1 M aqueous HCl, water phase was extracted with methylene chloride (3 × 10 mL) and combined organic phases were washed with water (2 × 10 mL) and brine. (20 mL) and dried over anhydrous MgSO4. Solvent was evaporated under reduced pressure. Crude oil was purified by column chromatography (Hex/EtOAc 2:1 to 1:1) to give title compound as a white solid (0.13 g, 0.29 mmol, 65%). Mp. 82–83 °C; [α]20D = −48.5 (c 0.65, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.38–7.27 (m, 15H), 5.81 (d, J = 1.7 Hz, 1H), 4.76 (d, J = 11.9 Hz, 2H), 4.69–4.65 (m, 4H), 4.22 (s, 3H), 4.11 (dd, J = 7.5, 4.8 Hz, 1H), 3.98 (dd, J = 7.2, 4.7 Hz, 1H), 3.79 (dd, J = 7.1, 3.6 Hz, 1H), 2.87 (d, J = 8.0 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 140.0, 138.4, 138.2, 137.9, 128.5, 128.4, 128.4, 127.9, 127.9, 127.9, 127.8, 127.7, 127.7, 122.4, 78.9, 76.5, 73.5, 73.1, 72.7, 71.7, 70.1, 64.5; HRMS (ESI): calcd for C28H30O5Na [M + Na]+ 469.1985, found 469.1981.
6-Epi-3,4,5,6,7-penta-O-benzyl-(−)-MK7607 (10)
To a stirred suspension of NaH (0.027 g, 0.67 mmol, 60% in mineral oil) in anhydrous DMF (5 mL) was added a solution of 9 (0.130 g, 0.29 mmol) and BnBr (0.08 mL, 0.67 mmol) at 0 °C under argon. After 30 min of stirring, ice bath was removed and stirring was continued for 12 h in room temperature. The reaction mixture was carefully quenched with a cold, 10% aqueous solution of NH4Cl (5 mL). The aqueous layer was extracted with ethyl acetate (3 × 5 mL). The organic layer was washed with H2O (5 mL) and brine (5 mL), dried over anhydrous MgSO4, and concentrated under reduced pressure. The residue was purified by column chromatography (Hex/EtOAc 9:1 to 7:1) to give title compound as a colorless syrup (0.117 g, 0.186 mmol, 83%). [α]24D = −150.3 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.41–7.27 (m, 25H), 5.92 (d, J = 5.3 Hz, 1H), 5.05 (d, J = 11.0 Hz, 1H), 4.85–4.66 (m, 7H), 4.49 (d, J = 11.8 Hz, 1H), 4.45 (d, J = 11.8 Hz, 1H), 4.32 (dd, J = 10.0, 7.1 Hz, 1H), 4.24–4.17 (m, 2H), 4.12–4.08 (m, 1H), 3.96 (d, J = 12.5 Hz, 1H), 3.60 (dd, J = 10.1, 3.5 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 140.2, 139.0, 138.9, 138.8, 138.6, 138.2, 128.5, 128.4, 128.4, 128.1, 128.0, 127.9, 127.8, 127.8, 127.7, 127.7, 127.6, 127.6, 123.5, 80.6, 80.0, 79.8, 75.0, 73.8, 72.7, 72.6, 71.8, 71.0, 70.4; HRMS (ESI): calcd for C42H42O5Na [M + Na]+ 649.2924, found 649.2927.
4,5,6-Tri-O-benzyl-(−)-valienamine-N-benzyl carbamate (11) and 4,5,6-tri-O-benzyl-(+)-β-valienamine-N-benzyl carbamate (12)
To a stirred solution of 10 (0.69 g, 0.11 mmol) in anhydrous toluene (0.74 mL) was added Na2CO3 (0.132 g, 1.25 mmol) and chlorosulfonyl isocyanate (0.07 mL, 0.83 mmol) at 0 °C under argon. The reaction mixture was stirred for 15 h at 0 °C and quenched carefully with addition H2O (1 mL). The aqueous layer was extracted with ethyl acetate (2 × 2 mL). The organic layer was added to a solution of aqueous 25% Na2SO3 (2 mL). The reaction mixture was stirred for 24 h at room temperature. The organic layer was washed with H2O (∼3 mL) and brine (∼3 mL), dried over anhydrous MgSO4 and concentrated under reduced pressure. The solid residue was purified by column chromatography (Hex/EtOAc 7:1 to 4:1) to give title compounds 11 (0.038 g, 0.055 mmol, 51%), 12 (0.006 g, 0.01 mmol, 9%) and substrate 10 (0.013 g, 0.021 mmol, 19%) was recovered. 4,5,6-Tri-O-benzyl-(−)-valienamine-N-benzyl carbamate (11): [α]20D = −28.9 (c 1.0, CHCl3). 1H NMR and 13C NMR spectra are identical as reported in lit. for enantiomer;16 1H NMR (600 MHz, CDCl3) δ 7.38–7.27 (m, 24H), 7.25 (s, 1H), 5.82 (s, 1H), 5.15–5.05 (m, 3H), 4.75 (d, J = 11.4 Hz, 1H), 4.72–4.55 (m, 6H), 4.48 (d, J = 11.8 Hz, 1H), 4.40 (d, J = 11.8 Hz, 1H), 4.24 (d, J = 12.1 Hz, 1H), 4.06 (d, J = 3.4 Hz, 1H), 3.91 (d, J = 12.1 Hz, 1H), 3.81 (dd, J = 7.4, 4.8 Hz, 1H), 3.77–3.71 (m, 1H); 13C NMR (150 MHz, CDCl3) δ 156.2, 138.3, 138.2, 137.9, 137.2, 136.6, 128.6, 128.5, 128.5, 128.4, 128.2, 128.1, 128.0, 127.9, 127.7, 125.2, 76.3, 75.9, 74.4, 73.8, 72.2, 72.1, 70.6, 66.9, 47.2; HRMS (ESI): calcd for C43H43NO6Na [M + Na]+ 692.2983, found 692.2982. 4,5,6-Tri-O-benzyl-(+)-β-valienamine-N-benzyl carbamate (12): 1H NMR and 13C NMR spectra are identical as we previously reported;24 1H NMR (600 MHz, CDCl3) δ 7.35–7.26 (m, 25H), 5.65 (s, 1H), 5.13–5.06 (m, 2H), 4.83–4.62 (m, 7H), 4.47 (d, J = 11.8 Hz, 1H), 4.43 (d, J = 11.8 Hz, 1H), 4.21 (d, J = 8.0 Hz, 2H), 3.88 (d, J = 12.3 Hz, 2H), 3.53 (t, J = 7.7 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 155.9, 138.4, 138.3, 138.2, 136.6, 136.4, 128.6, 128.6, 128.5, 128.3, 128.2, 128.1, 128.0, 127.9, 127.9, 127.8, 127.8, 126.2, 82.3, 79.7, 77.9, 74.7, 74.3, 74.2, 72.6, 70.4, 66.8, 52.1.
3,4,5-Tri-O-benzyl-(−)-MK7607 and 6-epi-3,4,5-tri-O-benzyl-(−)-MK7607 – mixture of diastereoisomers (9)
A solution of compound 5 (0.30 g, 0.61 mmol) in anhydrous methylene chloride (10 mL) was cooled to −30 °C and solution of DIBAL-H (1 M in methylene chloride, 3.7 mL, 3.7 mmol) was added dropwise. When the addition was complete, the mixture was stirred at −10 °C for 3 h. After that time, methanol (2 mL) was added to quench the reaction, and mixture was stirred at −10 °C for 30 min. After water addition (10 mL) and few drops (<1 mL) of 1 M aqueous HCl, water phase was extracted with methylene chloride (3 × 15 mL) and combined organic phases were washed with water (2 × 15 mL) and brine (20 mL) and dried over anhydrous MgSO4. Solvent was evaporated under reduced pressure. Crude oil was purified by column chromatography (Hex/EtOAc 2:1 to 1:1) to give title mixture of diastereoisomers as semi-solid residue (0.131 g, 0.29 mmol, 48%). Products were characterized as pure diastereoisomers (6S)-9 and (6R)-9.
(4R,5R,6R/S)-4,5,6-Tri-O-benzyl-2-(((tert-butyldiphenylsilyl)oxy)methyl)cyclohex-2-en-1-ol (13)
A solution of mixture of compounds 9 (0.120 g, 0.27 mmol) in anhydrous methylene chloride (3 mL) was cooled to −20 °C and TBDPSCl (0.089 g, 0.32 mmol) was added in one portion followed by dropwise addition of solution of imidazole (0.045 g, 0.65 mmol in 1 mL anhydrous DMF) and stirring was continued in −20 °C for 30 min. Ice bath was removed and mixture was allowed to warm to room temperature. After addition of water (5 mL), product was extracted with EtOAc (3 × 10 mL) and combined organic layers were washed with water (2 × 10 mL), brine (15 mL) and dried over anhydrous MgSO4. After solvent evaporation, oily residue was purified by column chromatography (Hex/EtOAc 6:1 to 5:1) to give pure compound 13 (mixture of diastereoisomers) as a clear syrup (0.118 g, 0.17 mmol, 64%). 1H NMR (600 MHz, CDCl3) δ 7.70–7.63 (m, 4.84H), 7.44–7.27 (m, 25.41H), 5.96 (d, J = 4.5 Hz, 0.21H), 5.87 (d, J = 2.3 Hz, 1H), 4.82–4.63 (m, 7.26H), 4.35–4.27 (m, 2.42H), 4.20 (d, J = 20.1 Hz, 1.21H), 4.16 (t, J = 3.9 Hz, 0.21H), 4.07 (s, 1H), 4.04 (dd, J = 9.0, 4.2 Hz, 1H), 4.00 (dd, J = 7.2, 4.7 Hz, 1H), 3.94 (dd, J = 9.0, 3.8 Hz, 0.21H), 3.77 (dd, J = 7.2, 3.7 Hz, 1H), 2.69 (s, 0.21H), 2.61 (d, J = 5.6 Hz, 1H), 1.08–1.05 (m, 10.89H); 13C NMR (150 MHz, CDCl3) δ 140.4, 140.0, 138.9, 138.9, 138.7, 138.6, 138.4, 138.3, 135.7, 135.7, 134.9, 133.5, 133.5, 133.4, 129.8, 128.6, 128.5, 128.5, 128.0, 127.9, 127.9, 127.8, 127.8, 127.7, 127.7, 127.6, 121.8, 120.8, 79.4, 77.0, 76.3, 73.6, 73.2, 73.1, 72.6, 72.1, 72.0, 71.5, 69.6, 66.6, 64.8, 64.4, 27.0, 19.4; HRMS (ESI): calcd for C44H48O5SiNa [M + Na]+ 707.3163, found 707.3165.
3,4,5-Tri-O-benzyl-1-tert-butyldiphenylsilyl-(−)-gabosine E (14)
A solution of compound 13 (0.115 g, 0.17 mmol) in anhydrous methylene chloride (3 mL) was cooled to 0 °C and Dess–Martin periodinane (0.214 g, 0.50 mmol) was added in one portion and stirred in 0 °C for 30 min. After that time ice bath was removed and reaction mixture was stirred for 2 h in room temperature. Reaction was quenched with 0.2 mL of saturated aqueous solution of NaHCO3 and evaporated to dryness with silica gel (<0.5 g). After column chromatography (Hex/EtOAc 7:1 to 6:1) pure product was obtained as a clear syrup (0.108 g, 0.16 mmol, 94%). [α]23D = −187.5 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.62 (ddd, J = 10.4, 8.0, 1.3 Hz, 4H), 7.43–7.27 (m, 21H), 7.04 (dd, J = 4.5, 2.1 Hz, 1H), 4.86 (d, J = 11.4 Hz, 1H), 4.83–4.78 (m, 2H), 4.76–4.69 (m, 2H), 4.65 (d, J = 11.4 Hz, 1H), 4.46–4.37 (m, 3H), 4.35 (d, J = 8.2 Hz, 1H), 3.93 (dd, J = 8.2, 3.3 Hz, 1H), 1.08 (s, 9H); 13C NMR (150 MHz, CDCl3) δ 196.4, 138.8, 138.3, 138.2, 138.0, 135.6, 135.5, 133.2, 133.1, 129.9, 129.9, 128.6, 128.5, 128.4, 128.1, 128.0, 127.9, 127.9, 127.9, 80.4, 78.9, 74.1, 73.3, 72.6, 72.2, 60.5, 27.0, 19.4; HRMS (ESI): calcd for C44H46O5SiNa [M + Na]+ 705.3007, found 705.3007.
3,4,5-Tri-O-benzyl-(−)-gabosine E (15)
A solution of compound 14 (0.038 g, 0.056 mmol) in anhydrous THF (1 mL) was cooled to 0 °C and excess of HF·Py (0.05 mL) complex was added dropwise and ice bath was removed and stirring was continued for next 12 h in room temperature. After that time reaction was quenched with water (2 mL) and product was extracted with EtOAc (3 × 5 mL). Combined organic layers were washed with water (5 mL), brine (10 mL) and dried over anhydrous MgSO4. Solvent was evaporated under reduced pressure. Crude product was purified by column chromatography (Hex/EtOAc 4:1 to 1:1) to afford title compound as a clear syrup (0.022 g, 0.05 mmol, 89%). [α]21D = −120.5 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.39–7.27 (m, 15H), 6.80 (d, J = 4.2 Hz, 1H), 4.86 (d, J = 11.5 Hz, 1H), 4.78 (dd, J = 12.0, 9.2 Hz, 2H), 4.71–4.65 (m, 3H), 4.45 (t, J = 3.7 Hz, 1H), 4.35 (dt, J = 14.3, 1.2 Hz, 1H), 4.32 (d, J = 7.7 Hz, 1H), 4.23 (d, J = 14.3 Hz, 1H), 3.99 (dd, J = 7.7, 3.3 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 197.3, 140.9, 138.1, 138.0, 138.0, 137.7, 128.6, 128.5, 128.5, 128.1, 128.0, 128.0, 128.0, 127.9, 80.0, 78.5, 73.9, 73.4, 72.7, 72.5, 60.9; HRMS (ESI): calcd for C28H28O5Na [M + Na]+ 467.1829, found 467.1829.
Ethyl (−)-3,4,5-tri-O-benzylshikimate (18)
A solution of compound 5 (0.50 g, 1.02 mmol) in anhydrous methylene chloride (15 mL) was cooled to −20 °C and methanesulfonyl chloride (0.238 mL, 3.07 mmol) was added in one portion followed by dropwise addition of triethylamine (0.427 mL, 3.07 mmol). After 30 min cryo-bath was removed and reaction mixture was allowed to warm to room temperature. Methylene chloride was evaporated under reduced pressure (below 30 °C) and crude yellow oil was purified by flash column chromatography (Hex/EtOAc 5:1 to 4:1) to afford mixture of methanesulfonyl derivatives (16) as a clear syrup (0.545 g, 0.96 mmol, 95%). HRMS (ESI): calcd for C31H34O8SNa [M + Na]+ 589.1867, found 589.1867. Compound 16 was dissolved in anhydrous acetone (10 mL) and cooled to 0 °C followed by addition of NaI (1.44 g, 9.61 mmol). Reaction was stirred for 15 min in 0 °C and then heated to 40 °C for 2 h (covered with aluminum foil to avoid light access). Solvent was evaporated under reduced pressure (below 35 °C). Crude semi-solid residue was purified by flash column chromatography (Hex/EtOAc 7:1 to 6:1) to afford iodine intermediate (17) as a clear, colorless syrup (0.560 g, 0.94 mmol, 97%). Crude mixture (17) was dissolved in ethanol (10 mL) and cooled to 0 °C, followed by addition of sodium borohydride (0.071 g, 1.88 mmol) and vigorously stirred in 0 °C for 2 h (conversion of substrate was monitored by TLC). Reaction was quenched with diluted acetic acid (acetic acid/water v/v 1:10, 5 mL). Product was extracted with EtOAc (3 × 5 mL). Combined organic layers were washed with water (2 × 5 mL), brine (10 mL) and dried over anhydrous MgSO4. After solvent evaporation title compound was purified by column chromatography (Hex/EtOAc 7:1 to 6:1) to afford product as clear syrup (0.415 g, 0.88 mmol, 93% including 15% inseparable impurities), total yield after 3 step = 73% of pure 18. Ethyl (−)-3,4,5-tri-O-benzylshikimate (18): 1H NMR (600 MHz, CDCl3) δ 7.39–7.22 (m, 15H), 6.92 (d, J = 1.0 Hz, 1H), 4.75–4.63 (m, 4H), 4.59 (d, J = 11.9 Hz, 1H), 4.51 (d, J = 11.9 Hz, 1H), 4.35 (s, 1H), 4.19 (qd, J = 7.1, 1.4 Hz, 2H), 3.96 (dd, J = 10.3, 4.3 Hz, 1H), 3.83 (dd, J = 6.1, 3.7 Hz, 1H), 2.72 (ddt, J = 18.5, 4.5, 2.2 Hz, 1H), 2.47–2.41 (m, 1H), 1.28 (t, J = 7.1 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 166.6, 138.6, 138.4, 136.1, 129.6, 128.5, 128.4, 127.9, 127.9, 127.8, 127.7, 127.7, 75.2, 73.9, 73.3, 73.1, 71.8, 71.7, 60.7, 27.9, 14.3; HRMS (ESI): calcd for C30H32O5Na [M + Na]+ 495.2142, found 495.2142.
(−)-3,4,5-Tri-O-benzylshikimic acid (19)
18 (0.40 g, 0.85 mmol) was dissolved in THF (5 mL) followed by addition of water (5 mL) and 1 M NaOH (1 mL) and reaction was stirred for 48 h in room temperature. After that time reaction mixture was treated with 1 M HCl (to pH = 2) and product was extracted with Et2O (4 × 5 mL). Combined organic layers were washed with water (10 mL), brine (10 mL) and dried over anhydrous MgSO4. Solvent was evaporated in atmospheric pressure. Product was purified by column chromatography (PhMe/EtOH/EtOAc 5:1:1) to afford title compound as a colorless syrup (0.376 g, 0.846, quant., 88% NMR purity). 1H NMR (600 MHz, CDCl3) δ 7.40–7.20 (m, 15H), 7.05 (s, 1H), 4.76–4.61 (m, 4H), 4.57 (d, J = 11.9 Hz, 1H), 4.49 (d, J = 11.9 Hz, 1H), 4.35 (s, 1H), 3.95 (dd, J = 9.9, 4.3 Hz, 1H), 3.82 (s, 1H), 2.73–2.67 (m, 1H), 2.48–2.40 (m, 1H); 13C NMR (150 MHz, CDCl3) δ 171.4, 138.8, 138.6, 138.3, 138.3, 128.5, 128.4, 127.9, 127.9, 127.8, 127.8, 127.7, 75.0, 73.8, 73.3, 73.1, 71.8, 71.7, 27.6; HRMS (ESI): calcd for C28H28O5Na [M + Na]+ 467.1829, found 467.1829.
(−)-Methyl shikimate (20)
To a solution of 19 (0.210 g, 0.47 mmol) in anhydrous methylene chloride (6 mL) cooled to −78 °C boron trichloride (1 M in methylene chloride, 9.5 mL, 9.50 mmol) was added dropwise and stirring was continued for 12 h. Reaction was quenched by addition of anhydrous methanol (2 mL) and intense stirring was continued in −78 °C for 1 h. After solvent evaporation under reduced pressure (with 2 M NaOH trap) water was added (10 mL), organic impurities were removed by washing water phase with methylene chloride (3 × 5 mL). Water layer was acidified with 1 M HCl (to pH = 5) and solvent was evaporated under reduced pressure. Title compound was crystallized from EtOAc as white solid (0.060 g, 0.186 mmol, 67%). Mp 113–114 °C; [α]21D = −138.0 (c 0.5, MeOH); 1H NMR (600 MHz, CD3OD) δ 6.80 (dq, J = 1.8, 1.4 Hz, 1H), 4.40–4.37 (m, 1H), 4.01 (dt, J = 7.1, 5.2 Hz, 1H), 3.76 (s, 3H), 3.33 (dd, J = 4.0, 2.4 Hz, 1H), 2.71 (ddt, J = 18.2, 4.7, 1.9 Hz, 1H), 2.22 (ddt, J = 18.2, 5.4, 1.7 Hz, 1H); 13C NMR (150 MHz, CD3OD) δ 169.5, 139.9, 131.0, 73.4, 69.2, 68.1, 53.2, 32.3; 1H NMR and 13C NMR are identical as previously reported.26
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support from the Polish National Science Centre (Grant No. 2015/17/D/ST5/01334) is gratefully acknowledged. The research was carried out with the equipment purchased thanks to the financial support of the European Regional Development Fund in the framework of the Polish Innovation Economy Operational Program (contract no. POIG.02.01.00-12-023/08).
Notes and references
- G. E. Mccasland, S. Furuta and L. J. Durham, J. Org. Chem., 1968, 33, 2835–2841 CrossRef CAS PubMed.
- G. E. Mccasland, S. Furuta and L. J. Durham, J. Org. Chem., 1968, 33, 2841–2844 CrossRef CAS.
- O. Arjona, A. M. Gómez, J. C. López and J. Plumet, Chem. Rev., 2007, 107, 1919–2036 CrossRef CAS PubMed.
- S. Roscales and J. Plumet, Int. J. Carbohydr. Chem., 2016, 2016, 1–42 CrossRef.
- W. Ren, R. Pengelly, M. Farren-Dai, S. Shamsi Kazem Abadi, V. Oehler, O. Akintola, J. Draper, M. Meanwell, S. Chakladar, K. Świderek, V. Moliner, R. Britton, T. M. Gloster and A. J. Bennet, Nat. Commun., 2018, 9, 1–12 CrossRef PubMed.
- S. Shamsi Kazem Abadi, M. Tran, A. K. Yadav, P. J. P. Adabala, S. Chakladar and A. J. Bennet, J. Am. Chem. Soc., 2017, 139, 10625–10628 CrossRef CAS PubMed.
- N. Tateda, K. Ajisaka, M. Ishiguro and T. Miyazaki, Bioorg. Med. Chem., 2018, 1–23 Search PubMed.
- H. Yuasa, O. Hindsgaul and M. M. Palcic, J. Am. Chem. Soc., 1992, 114, 5891–5892 CrossRef CAS.
- S. H. Yu and S. K. Chung, Tetrahedron: Asymmetry, 2005, 16, 2729–2747 CrossRef CAS.
- T. K. M. Shing, W. L. Ng, J. Y. W. Chan and C. B. S. Lau, Angew. Chem., Int. Ed., 2013, 52, 8401–8405 CrossRef CAS PubMed.
- M. Li, Y. Li, K. A. Ludwik, Z. M. Sandusky, D. A. Lannigan and G. A. O'Doherty, Org. Lett., 2017, 19, 2410–2413 CrossRef CAS PubMed.
- X. Yang, P. Yuan, F. Shui, Y. Zhou and X. Chen, Org. Biomol. Chem. 10.1039/c9ob00469f.
- D. C. Babu, C. B. Rao, K. Venkatesham, J. J. P. Selvam and Y. Venkateswarlu, Carbohydr. Res., 2014, 388, 130–137 CrossRef CAS PubMed.
- J. P. Rao and B. V. Rao, Tetrahedron: Asymmetry, 2010, 21, 930–935 CrossRef CAS.
- C. Muniraju, J. P. Rao and B. V. Rao, Tetrahedron: Asymmetry, 2012, 23, 86–93 CrossRef CAS.
- Q. R. Li, S. I. Kim, S. J. Park, H. R. Yang, A. R. Baek, I. S. Kim and Y. H. Jung, Tetrahedron, 2013, 26, 10384–10390 CrossRef.
- P. Yuan, X. Liu, X. Yang, Y. Zhang and X. Chen, J. Org. Chem., 2017, 82, 3692–3701 CrossRef CAS PubMed.
- P. Wen and D. Crich, Tetrahedron, 2018, 74, 5183–5191 CrossRef CAS PubMed.
- J. Sardinha, A. P. Rauter and M. Sollogoub, Tetrahedron Lett., 2008, 49, 5548–5550 CrossRef CAS.
- D. H. Mac, S. Chandrasekhar and R. Grée, Eur. J. Inorg. Chem., 2012, 5881–5895 CrossRef CAS.
- G. M. Whited, G. International, K. Way and S. S. Francisco, Tetrahedron: Asymmetry, 1996, 7, 691–698 CrossRef.
- H. A. J. Carless and Y. Dove, Tetrahedron: Asymmetry, 1996, 7, 649–652 CrossRef CAS.
- I. S. Kim, O. P. Zee and Y. H. Jung, Org. Lett., 2006, 8, 4101–4104 CrossRef CAS PubMed.
- P. Banachowicz, J. Mlynarski and S. Buda, J. Org. Chem., 2018, 83, 11269–11277 CrossRef CAS PubMed.
- N. R. Candeias, B. Assoah and S. P. Simeonov, Chem. Rev., 2018, 118, 10458–10550 CrossRef CAS PubMed.
- L. Sánchez-Abella, S. Fernández, N. Armesto, M. Ferrero and V. Gotor, J. Org. Chem., 2006, 71, 5396–5399 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02002k |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Szymon Buda
and
Szymon Buda