
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidatively induced exposure of active surface area during microwave assisted formation of Pt3Co nanoparticles for oxygen reduction reaction†
Robin Sandström ,
Joakim Ekspong,
Eduardo Gracia-Espino and
Thomas Wågberg*
,
Joakim Ekspong,
Eduardo Gracia-Espino and
Thomas Wågberg*
Department of Physics, Umeå University, Umeå 90187, Sweden. E-mail: thomas.wagberg@umu.se
First published on 7th June 2019
Abstract
The oxygen reduction reaction (ORR), the rate-limiting reaction in proton exchange membrane fuel cells, can efficiently be facilitated by properly manufactured platinum catalysts alloyed with late 3d transition metals. Herein we synthesize a platinum![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cobalt nanoparticulate catalyst with a 3:1 atomic ratio by reduction of a dry metalorganic precursor blend within a commercial household microwave oven. The formed nanoparticles are simultaneously anchored to a carbon black support that enables large Pt surface area. Two separate microwave treatment steps were employed, where step one constitutes a fast oxidative treatment for revealing active surface area while a reductive secondary annealing treatment promotes a Pt rich surface. The resulting Pt3Co/C catalyst (∼3.4 nm) demonstrates an enhanced ORR activity directly attributed to incorporated Co with a specific and mass activity of 704 μA cmPt−2 and 352 A gPt−1 corresponding to an increase by 279% and 66% respectively compared to a commercial Pt/C (∼1.8 nm) catalyst measured under identical conditions. The method's simplicity, scalability and novelty is expected to further assist in Pt–Co development and bring the catalyst one step closer toward commercialization and utility in fuel cells.
:cobalt nanoparticulate catalyst with a 3:1 atomic ratio by reduction of a dry metalorganic precursor blend within a commercial household microwave oven. The formed nanoparticles are simultaneously anchored to a carbon black support that enables large Pt surface area. Two separate microwave treatment steps were employed, where step one constitutes a fast oxidative treatment for revealing active surface area while a reductive secondary annealing treatment promotes a Pt rich surface. The resulting Pt3Co/C catalyst (∼3.4 nm) demonstrates an enhanced ORR activity directly attributed to incorporated Co with a specific and mass activity of 704 μA cmPt−2 and 352 A gPt−1 corresponding to an increase by 279% and 66% respectively compared to a commercial Pt/C (∼1.8 nm) catalyst measured under identical conditions. The method's simplicity, scalability and novelty is expected to further assist in Pt–Co development and bring the catalyst one step closer toward commercialization and utility in fuel cells.
1. Introduction
Proton exchange membrane fuel cells (PEMFCs), an important device for efficient conversion of hydrogen into electricity, greatly benefit from new compounds capable of catalyzing the sluggish oxygen reduction reaction (ORR). The ORR however still plagues the PEMFC cathode and depends on a high loading of platinum-based catalysts to facilitate long term ORR operation with minimal degradation after several thousands of hours in PEMFCs,1–4 posing significant challenges in catalyst development. Moreover, it must maintain a full four electron ORR process for highest efficiency and to avoid hydrogen peroxide intermediates that over time will degrade the ionomer membrane.5,6 Ideally, the catalyst manufacturing process should also be industrially and environmentally benign, capable of large scale production to suit the needs of primarily world-wide vehicular applications.Although relatively efficient, platinum alone is not optimal. In fact, theoretical and practical studies have shown that modification of the electron configuration within the Pt lattice can dramatically alter the surface adsorption of oxygen in a favorable direction such that the ORR results in a lowered overpotential.7,8 This can be achieved by alloying Pt into binary or ternary configurations with 3d type transition metals such as Fe,9–11 Ni12–14 or Co,15–17 which may enhance the ORR properties by mechanisms involving the creation of more close-packed surface facets of the face centered cubic (fcc) Pt lattice and/or inducing ligand effects. It is clear that this approach is highly convenient as it also dilutes the Pt within the nanoparticle core and may thus further contribute to an enhanced Pt mass activity. The main challenges with such materials however is to minimize de-alloying at ORR potentials18–20 and to fabricate membrane electrode assemblies with similar activities as the impressive ex situ measured mass activities on glassy carbon electrodes.21–23
The alloy between platinum and cobalt at a 3:1 atomic ratio is an established catalyst that has shown remarkable mass and specific activities for ORR in acidic media for various nanoparticle (NP) size configurations. The choice of Co is also often preferred owing to promising reported stability data and is thereby generally considered more stable than other late transition metal Pt alloy alternatives.24,25 Furthermore, Pt–Co particles have found use-case in catalyzing the methanol oxidation reaction (MOR) with greater activity than pure Pt attributed to the shift in d-band center of the Pt surface facet directly caused by the incorporated Co.26,27 The NPs even demonstrated enhanced stability and higher tolerance toward CO poisoning.
Owing to the great promise Pt–Co based materials has shown, a large variety of reported synthesis methodologies has emerged over the past two decades. The various methods include solvothermal synthesis,15,28 wet impregnation,29 polyol processes,30,31 electro-deposition32 and magnetron sputtering.33 Microwave (MW) based synthesis on the other hand, an intriguing concept often with advantages such as rapid heating and high heating efficiency, in particular for carbonaceous materials,34–37 has however been scarcely exploited for Pt–Co production. Thus, high heating selectivity as well as rapid green-synthesis are some of the main advantages of properly designed microwave reactors, while also using samples with suitable microwave absorption characteristics. Komarneni et al.38 investigated a microwave based polyol route toward Pt and Ag NP production wherein a greatly enhanced reaction rate was observed. Later, more solvothermal microwave processes were explored for the production of Pt–Co nanowires39 and nanoparticles40,41 as active catalyst materials. Most of the methods reported do however involve multiple, sometimes complex, steps that result in relatively complicated procedures and ultimately impair the commercial feasibility.
In this work, we investigate further the use of microwaves for the preparation of Pt–Co NPs and evaluate their ORR properties in acidic media. The method is capable of producing Pt3Co NPs simultaneously decorated on carbon black support by using a microwave treatment directly on a dry metalorganic precursor mixture in a modified household microwave oven. The rapid and simple procedure is carried out with only two MW treatment steps for an improved ORR activity. First, the Pt surface area could be exposed, without a blocking carbonaceous layer, by a fast oxidative treatment followed by a longer reductive routine to enrich the NP surfaces with Pt. Signs of successful Co incorporation were confirmed by both physical and electrochemical characterization techniques. The ORR performance was comprehensively evaluated and the enhancement of the Pt3Co catalysts can be associated directly with a Pt rich skin configuration on top of a Pt–Co core.
2. Experimental methods
2.1. Two-step microwave synthesis of Pt3Co nanoparticles
Pt3Co/C nanoparticles decorated on Vulcan XC 72R (Cabot corp.) were produced through a one-pot dry-state microwave reaction. The full scheme of the synthesis procedure is illustrated in Fig. 1. First, 46 mg of platinum(II) acetylacetonate (Pt(acac)2, Aldrich) and 10 mg of cobalt(II) acetate tetrahydrate ((CH3COO)2Co·4H2O, Aldrich), were dispersed in 2 ml tetrahydrofuran (THF), thus adjusted such that an atomic ratio of 3:1 (Pt:Co) was achieved. In a separate vial, 35 mg of ammonium nitrate (NH4NO3, Aldrich) was dissolved in 2 ml methanol. The contents of the two vials were then combined into a single solution by brief sonication (∼30 s) until a clear solution was achieved. The solution was thereafter poured into an agate mortar containing 100 mg of Vulcan XC 72R (target loading: 20 wt%) and ground for a few minutes until a completely dry powder had formed. Important to note is that the ratio of NH4NO3 to Vulcan XC 72R was not exceeded due to safety concerns as the risk of deflagration in the reaction chamber was deemed too high at higher fractions of NH4NO3. The powder was loaded into a quartz crucible and placed in a dead-ended quartz tube connected to a custom modified household microwave oven (800 W), capable of supplying gases into the reaction vessel. After 1 h purge in a flow of Ar (100 ml min−1), the microwave oven was set to full power for 5 min, although it was noted that the actual continuous irradiation time was 4 min & 45 s due to a maximum limit set by the manufacturer of the microwave oven. The sample acquired already after this step is henceforth denoted Pt3Co/C* (note the asterisk). Afterwards, the chamber was purged with 5% H2 in Ar for at least 30 min after which the microwave oven was set to 50% power for 1 h and the resulting sample is denoted Pt3Co/C. It is crucial to note that conventional microwave ovens do not lower the power directly. Rather, they tend to operate under pulse-width modulation (PWM) such that a desired reduced output is reached. In particular, the oven used in this study sent alternating pulses of 14 s pulse widths to achieve 50% power.
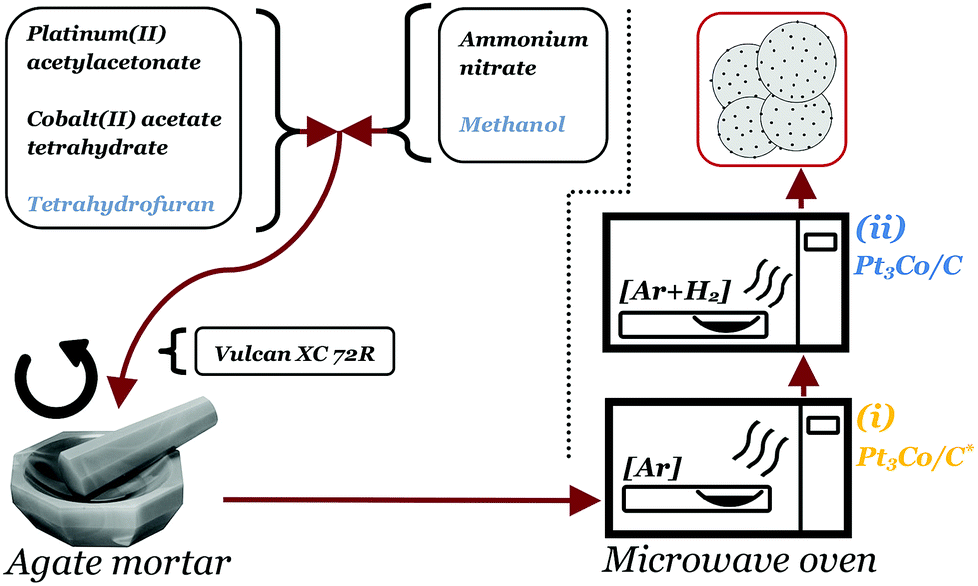 | ||
| Fig. 1 Scheme depicting the novel microwave assisted synthesis route of Pt3Co/C. The metalorganic precursor solution is first combined with the ammonium nitrate solution, ground and dried with the carbon support followed by a 5 min treatment at 800 W in Ar(I) and 400 W for 1 h in H2 + Ar(II). | ||
2.2. Characterization
The metal loading was determined by thermogravimetric analysis (TGA) carried out with a METTLER Toledo TGA/DSC 1 instrument. The flow rate was kept to 30 ml min−1 (air) with a heating rate of 10 K min−1 from 30–900 °C. X-ray diffractograms were acquired with a Panalytical X'Pert3 Powder diffractometer using a CuKα (λ = 1.5406 Å) source. An interval between 20° ≤ 2θ ≤ 75° was recorded and the full width at half maximum (FWHM) at the Pt(220) reflection was selected for calculation of NP average sizes using the Scherrer equation.42 X-ray photoelectron spectra (XPS) were acquired with a Kratos Axis Ultra DLD spectrometer using a monochromated Al Kα source operated at 120 W. Analyzer pass energy of 160 and 20 eV was used for acquisition of wide spectra and individual photoelectron lines respectively. Transmission electron microscopy (TEM) was performed with a JEOL-1230 microscope with an accelerating voltage of 80 kV.Glassy carbon (GC) electrodes (0.196 cm2 geometric surface area) were prepared by rigorous polishing in 300 and 50 nm alumina powder aqueous slurries (Electron Microscopy Sciences inc.) respectively. The GC electrodes were then sonicated for one minute each in acetone, distilled water (H2ODI) and ethanol respectively to remove any residue remaining from the polishing steps. Inks were prepared by dispersing the catalyst powders in a H2ODI:2-propanol mixture (6:1 volumetric ratio) at a concentration of 2.5 mg ml−1 with 12 wt% of dry Nafion resin (from solution; DE520, EW1000, 5 wt%, Ion-power inc.). Portions of 8 μl droplets were applied by a micropipette and dried under a constant rotation of 700 rpm (ca. 15 min).43 The total platinum loadings on the fabricated electrodes were calculated by also taking the TGA results and atomic ratio into account. A commercial Pt/C catalyst (20 wt% on Vulcan XC 72R, Premetek) was used for reference purposes.
Electrochemical measurements were performed with the electrodes attached to a rotating disk electrode (RDE) setup (Pine Instruments) connected to a potentiostat (Autolab PGSTAT302N). The electrolyte used was 0.1 M HClO4 (99.999% purity, Aldrich) prepared in H2ODI (18.2 Mohm) and the potential was measured vs. an RHE (Gaskatel, Hydroflex®). Prior to any measurements, a conditioning procedure consisting of 100 cycles between 0 and 1.2 V vs. RHE was performed at a scan rate of 100 mV s−1 in Ar saturated electrolyte. Linear sweep voltammetry (LSV) was performed in a still O2 saturated electrolyte with anodic sweeps between 0.05 to 1.05 V vs. RHE at 20 mV s–1 under rotation (400, 600, 900, 1200, 1600 and 2000 rpm). The electron transfer number (n) was estimated by the Koutecky–Levich equation (see ESI eqn (S1)† for details), and specific and mass activities were calculated with the rotation rate 1600 rpm. In order to estimate stability, LSVs were also recorded before and after 6000 cycles between 0.6 and 1.0 V vs. RHE with a scan rate of 400 mV s−1 at continuous 20 ml min−1 O2 bubbling. All LSVs were background corrected and iR compensated (post-measurement) assuming a solution resistance of 20 Ω as confirmed by current-interrupt measurements. Membrane electrode assemblies (MEA) for H2/O2 fuel cell testing were also produced as described in the ESI.†
Electrochemically active surface areas (ECSA) were measured both by hydrogen underpotential deposition (HUPD) and CO-stripping measurements before (separate electrodes) and after the stability measurements. HUPD cyclic voltammograms were acquired in Ar saturated electrolyte by cycling the potential between 0.05 and 1.0 V vs. RHE at a scan rate of 50 mV s−1 three times to ensure stable area and repeatable CV curves. CO-stripping voltammograms were measured by first saturating the electrolyte in Ar, followed by bubbling of the electrolyte in 100% CO gas (100 ml min−1) for 20 min, while keeping the potential fixed at 0.1 V vs. RHE. Then the gas was turned off and switched to Ar for an additional 30 min in order to remove any traces of CO from the electrolyte. The potential was then swept between 0.05 and 1.2 V vs. RHE (initially anodic) at 20 mV s−1 three times to ensure that all CO was stripped in the initial cycle and to establish a reliable baseline.
3. Results and discussion
Nanoparticles consisting of Pt and Co at a 3:1 ratio decorated on high surface area carbon support (Vulcan XC 72R) were synthesized in a “one-pot” manner using a modified conventional household microwave oven. As illustrated in the scheme in Fig. 1, a dry mixture of the carbon support with inter-mixed catalyst precursors plus an oxidative agent (NH4NO3) were first directly exposed to continuous microwaves for 4 min & 45 s. In this step, a rapid release of gaseous products could be observed typically within the first 20 s, followed by a mild continuous orange glow for the remaining duration of the reaction. Previous similar experiments using solely an organic Pt precursor (Pt(acac)2) resulted in Pt particles coated in a carbonaceous layer causing low ECSA.44 Similarly, in the present work it was also evident that samples synthesized by organic precursors alone, with the absence of NH4NO3, produced catalysts with low ECSA (see Fig. S1†). In contrast, the presence of NH4NO3 significantly exposed the platinum surface area, attributed to controlled oxidative removal of carbonaceous species in the very initial stages of the NP formation. The exact exothermal decomposition mechanism of NH4NO3 is known to be complex and highly dependent of a large variety of environmental and heating factors.45–47 Thus, the precise intermediates responsible for the oxidative treatment cannot be easily determined. We note however that NH4NO3 and other nitrate salts have been employed as an additive in a number of combustion synthesis methods,48 even as a combustion aid in solution-phase microwave synthesis.49 In our case, it is therefore reasonable to assume that NH4NO3 release oxidants (intermediates or products; N2O, NO, O2, HNO3), where the acetate and acetylacetonate ligands (organic functional groups) functions as a “fuel” causing initial combustion-like synthesis conditions. The thermogravimetric analysis of the samples (Fig. S2†) are consistent with literature reports as it demonstrates that (i) NH4NO3 leaves no traces and (ii) even the carbon support within the sample was likely etched by oxidative species signified by a slightly higher metal loading of 22.5 wt% contrary to the sample synthesized without NH4NO3 (20.8 wt%). In fact, higher concentrations of NH4NO3 results in visible deflagration phenomena in the reaction chamber.
The subsequent step, consisting of a longer treatment in reductive atmosphere, took place in 5% H2 in Ar with the microwave ovens power output set to 50% and thus preventing any significant heat build-up, which was evident by the absence of any visible “glow” throughout the whole process. The X-ray diffraction patterns in Fig. 2 show no notable peak shifts between these two samples and only a slight change in FWHM in the Pt fcc reflections, that can be ascribed to increments in NP size. Using the Scherrer relation, the samples show an average crystallite size of 3.2 and 3.4 nm for Pt3Co/C* and Pt3Co/C respectively. To not compromise further on the particle sizes, a higher power setting of the secondary MW treatment was not pursued. In addition, the XRD results show absence of reflections that stem from pure cobalt or cobalt oxides, which support that the synthesized NPs consist exclusively of Pt–Co fcc lattices with no preferential ordering. The slight upshift in all fcc peaks as compared to the commercial Pt/C catalyst (Fig. S3†) may also suggest that the Co is successfully incorporated into the lattice causing a more close-packed contracted Pt framework,50 although shifts due to the relatively small Pt/C average NP sizes cannot be excluded as a partial explanation to the observed discrepancy.
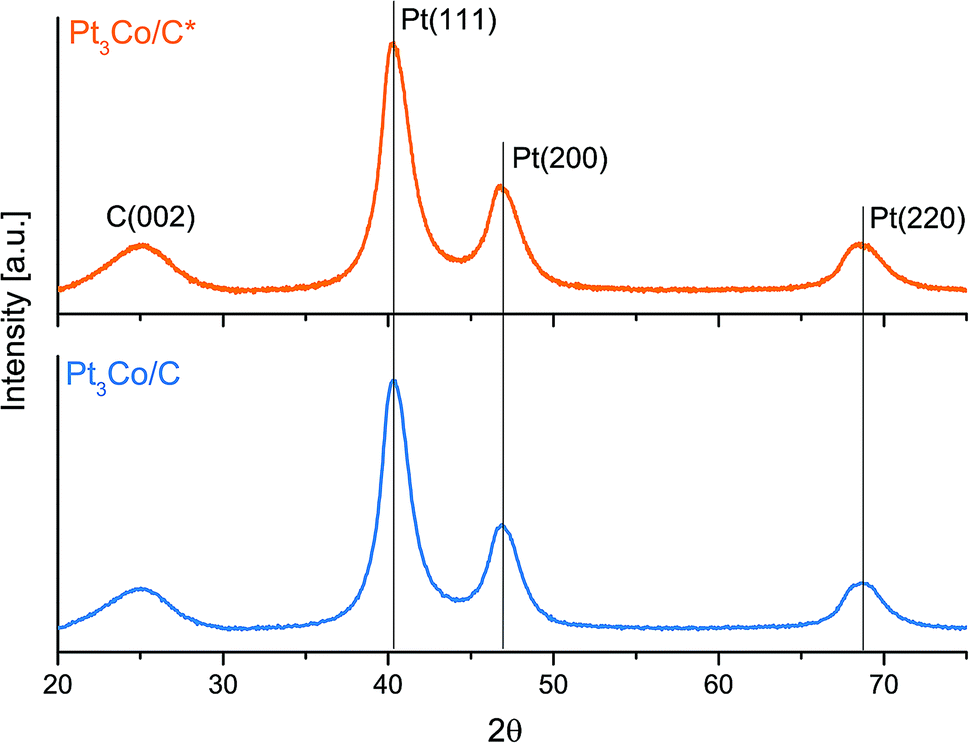 | ||
| Fig. 2 X-ray diffraction patterns of as-produced Pt3Co/C* (oxidative treatment) and Pt3Co/C (reductive treatment). The carbon (Vulcan XC 72R) peak and the Pt fcc reflections are highlighted respectively. | ||
While heating a dry carbonaceous powder with microwaves, some degree of temperature gradient ought to be expected, where the hottest region should be facing the magnetron. Although precautions were taken such as limiting the support amount to 100 mg per batch, this phenomenon may still likely cause multiple NP morphologies to form. Moreover, the unconventional synthesis methodology using consumer-grade apparatus as a MW source may further contribute to local hotspots in the powder mixture. The TEM images in Fig. 3 show representative regions found during screening of the prepared TEM grid. No notable differences between the two samples could be detected, but it was clear that the NPs varied in sizes from a few nm to sometimes well over 10 nm. However, regions with small-diameter NPs such as in Fig. 3a and d constituted the majority, which is in line with the XRD results.
 | ||
| Fig. 3 Transmission electron microscopy images showcasing representative diverse NP types found on the TEM grid of Pt3Co/C* (a–c) and Pt3Co/C (d–f). The majority of regions showed that NP sizes were typically of a few nm, but also significant amounts of NPs up to (and above) 10 nm could be found. | ||
X-ray photoelectron spectroscopy was employed to gain insight into the elemental composition and respective oxidation states. Fig. 4a shows the XPS survey spectra where the primary elements are highlighted. Despite using no sample washing procedure, only C, O, Pt and Co could exclusively be detected above the noise level, once more implying a full decomposition of NH4NO3 without remaining N-traces. This result emphasizes an important advantage over solvothermal methods where removal of solvent impurities can be problematic.15,28 The Pt:Co atomic ratio derived from the XPS was 3.0:1.0 and 4.2:1.0 for Pt3Co/C* and Pt3Co/C respectively. Since XPS is a highly surface sensitive technique probing only the top few nanometers of the sample, it is likely that the higher Pt loading in Pt3Co/C could be explained by surface migration of Pt during the reductive treatment, similar to previously observed phenomena of Pt–Co NPs under annealing conditions.51 The similar metal loading provided by the TGA experiments (Fig. S2†) supports that there are no significant losses of Co after the second MW step. Also taking into account that at no point the reductive treatment reach temperatures sufficient for Co evaporation (temperatures visibly below that of step one), it is then safe to conclude that the actual bulk composition is very close to the target 3:1 (Pt:Co) for both samples.
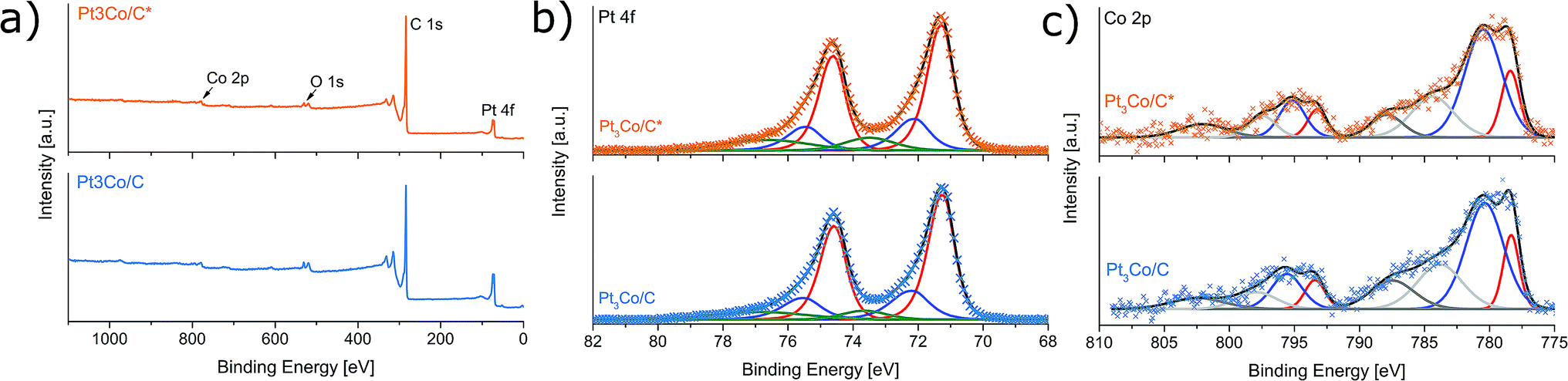 | ||
| Fig. 4 X-ray photoelectron spectra of Pt3Co/C* and Pt3Co/C showing a survey spectra (a) and high resolution spectra scanned over the core-level regions of Pt 4f (b) and Co 2p (c). Both core-level spectra are background subtracted and the satellite peaks in (c) are highlighted in gray. No surface N-species (NH4NO3 induced doping) could be found in any samples. | ||
The Pt 4f and the Co 2p XPS spectra at the core-level regions are shown in Fig. 4b and c respectively. The Pt 4f region indicate three predominant doublets with Pt 4f7/2 binding energies of 71.3, 72.1 and 73.5 eV corresponding to metallic Pt, Pt–C and Pt–O respectively with the majority of Pt being in a metallic phase. Excluding the emerging satellite peaks, there are two Co 2p3/2 peaks centered on binding energies of 778.4 eV (metallic Co) and 780.5 eV (CoO).
Thin homogeneous films of the respective catalysts were fabricated from a dispersion mixture and drop-cast onto well-polished GC electrode surfaces. It is worth noting that in order to avoid a severe “coffee-ring” or other inhomogeneities, it was found that the synthesized samples fared significantly better when the Nafion content was as low as 12 wt%. As a result, our measured activity of the Pt/C reference was also moderately enhanced compared to earlier observations of Pt/C from the same manufacturer but with higher Nafion content,44 consistent with evaluations in literature where Nafion concentration are shown to have a significant influence.52–56 In this fashion, the ability of the synthesized NPs to facilitate the oxygen reduction reaction was evaluated by LSV in O2 saturated 0.1 M HClO4 electrolyte, as shown in Fig. 5. ORR screening tests were also performed with the use of two other organic cobalt precursors shown in Fig. S4† (Co(II) and Co(III) acetylacetonates respectively) and Pt3Co/C* with a Co-nitrate precursor similar to ref. 44 (Fig. S5†), but were not further pursued due to inferior activity. The trend in kinetic current at 0.9 V vs. RHE was determined to Pt3Co/C > Pt3Co/C* > Pt/C (see Table S1.†). MEAs were also produced and tested in H2/O2 conditions (see ESI† for more details). In contrast to the LSV characterization (see Fig. S7†), Pt/C here outperforms Pt3Co/C owing to both a higher ohmic drop and overpotential in Pt3Co/C. As the sole electrolyte in PEMFCs is Nafion, this issue may be related to the Nafion sensitivity of the Pt3Co catalysts discussed above. In fact, it has previously been noted that surface support modification can alter catalysts compatibility with Nafion such as surface graphitization,54,57 posing a challenge in MEA fabrication or additional support functionalization for materials synthesized in this fashion.
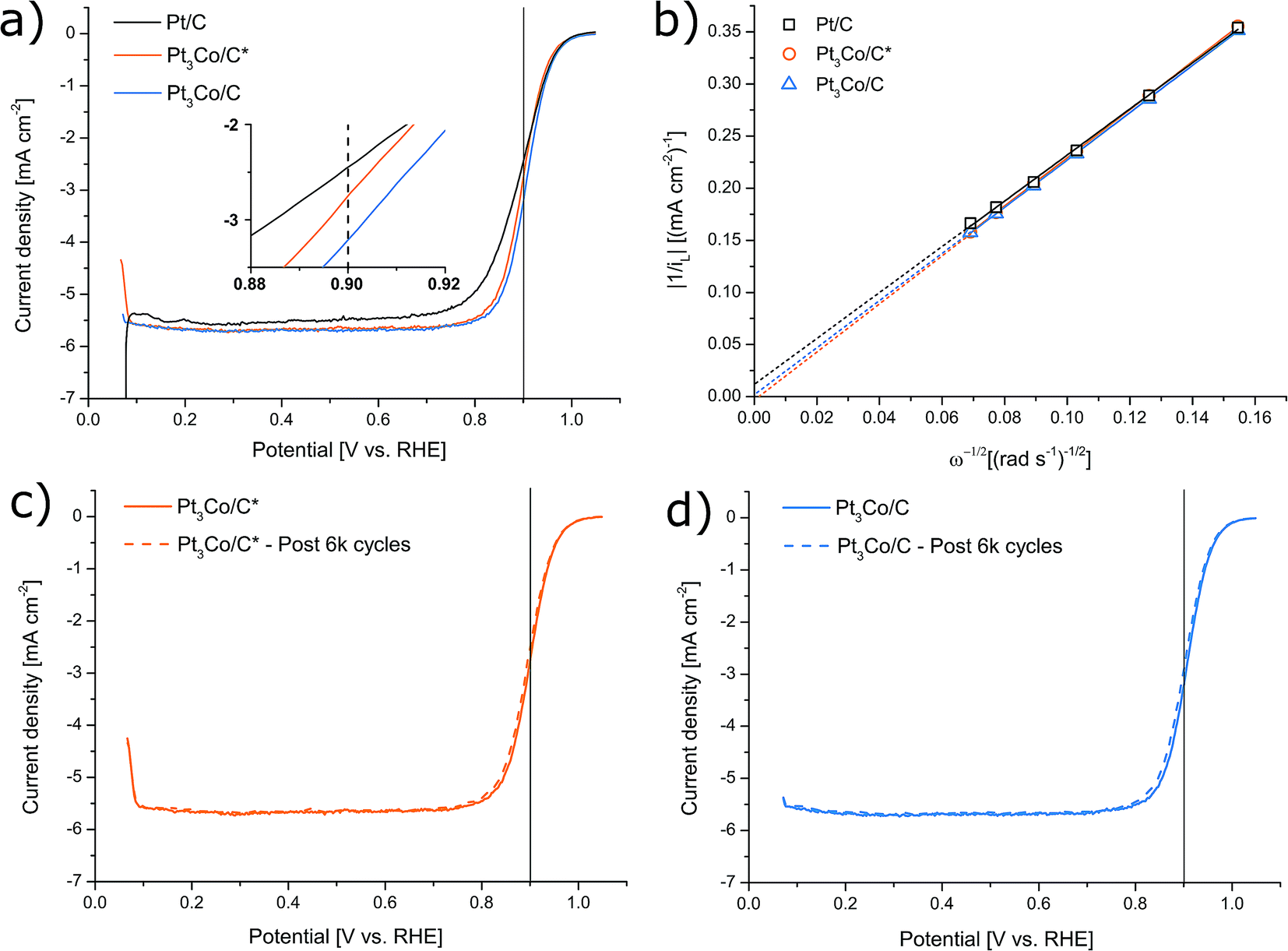 | ||
| Fig. 5 Linear sweep voltammetry curves of respective samples (a) for oxygen reduction performance evaluation recorded in 0.1 M HClO4 at 1600 rpm. Corresponding Koutecky–Levich analysis (b) was deduced from rotational speeds of 400, 600, 900, 1200, 1600 and 2000 rpm, where iL is the measured current density limited by diffusion and ω is the angular rotation rate. Accelerated degradation tests of Pt3Co/C* and Pt3Co/C are shown in (c) and (d) respectively. Here, LSV curves were recorded before and after 6000 cycles between 1.0 and 0.6 V vs. RHE in O2 saturated electrolyte. | ||
After an experiment consisting of 6000 consecutive cycles between 0.6 and 1.0 V vs. RHE, serving as an accelerated degradation test, the samples displayed a loss of their initial ik by 22%, 19% and 18% corresponding to Pt3Co/C, Pt3Co/C* and Pt/C respectively (see Fig. S6† for Pt/C LSVs). Both before and after the degradation, the electron transfer number was measured to 3.7 (Pt3Co/C), 3.6 (Pt3Co/C*) and 3.8 (Pt/C), demonstrating that the measured ORR activities stem from a near full four-electron reaction pathway.
The ECSA was determined before and after the accelerated degradation experiments by both CO-stripping and HUPD with the CV-scans plotted in Fig. 6a and b respectively. The data presented in the full electrochemical evaluation in Table 1, demonstrate that the ECSA determined by CO-stripping is higher than for HUPD, where the discrepancy is even more pronounced in the Pt3Co samples. This is consistent with earlier observations where ECSA measured by HUPD can greatly underestimate the surface area of Pt-alloys.58,59 We thus follow the literature recommendations and use the ECSA derived from CO-stripping for calculations of all specific activity values. Initially, the mass and specific activities for Pt3Co/C were 66% and 279% higher respectively than Pt/C and were lowered to 58% and 218% after degradation respectively. Interestingly, while no loss of ECSA after the degradation cycles could be measured for the synthesized Pt3Co samples, Pt/C lost 17% of its initial ECSA. As a consequence, the specific activity for Pt/C remained almost unchanged after degradation while the loss of ik in the Pt3Co catalysts influenced both specific and mass activity by nearly the same degree. This observation suggests that the degradation mechanism of Pt and Pt3Co is not entirely the same. In fact, it is well known that Pt-transition metal alloys primarily degrade through de-alloying processes, justifying the decline in SA. Meanwhile, the main reasons for Pt/C degradation may in our case be attributed to other factors causing a decline in ECSA, such as Pt dissolution or Ostwald ripening phenomenon, where the latter is more likely.
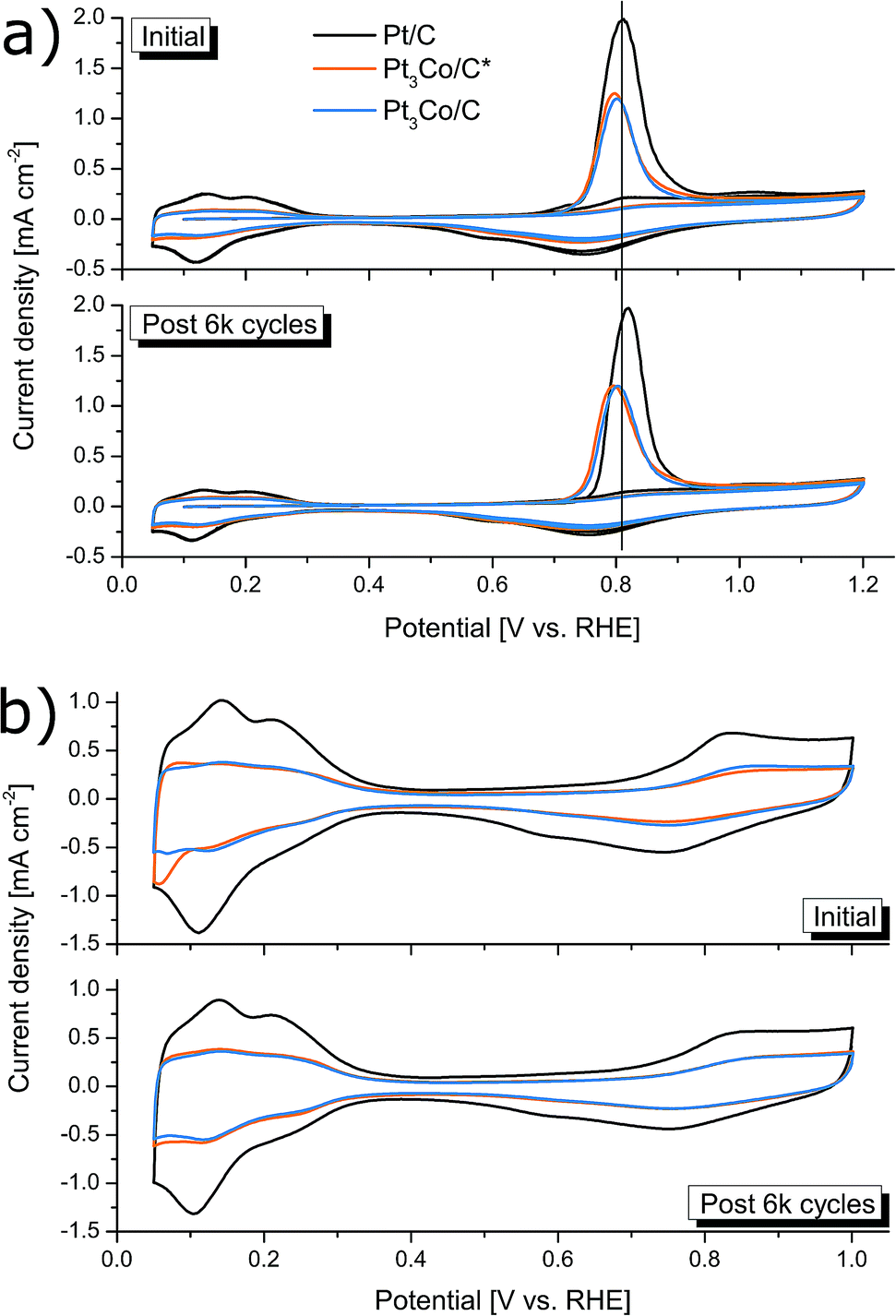 | ||
| Fig. 6 Electrochemically active surface area measurements including CO-stripping (a) and HUPD measurements (b). The ECSA measurements were repeated once the electrodes had been degraded by 6000 ORR cycles in O2 saturated electrolyte for evaluating the degradation mechanism. | ||
| Pt/Ca/(post degr.) | Pt3Co/C*/(post degr.) | Pt3Co/C/(post degr.) | |
|---|---|---|---|
| a Commercial sample. | |||
| Metal loading [wt%] | 20.0 | 22.5 | 23.0 |
| Pt:Co atomic ratio (XPS) |
— | 3.0:1.0 |
4.2:1.0 |
| NP size (XRD) [nm] | 1.8 | 3.2 | 3.4 |
| e− transfer number (n) | 3.8/3.8 | 3.6/3.6 | 3.7/3.7 |
| ECSA (HUPD) [m2 gPt−1] | 79.6/75.0 | 37.3/38.1 | 36.6/35.5 |
| ECSA (CO-stripping) [m2 gPt−1] | 84.2/70.0 | 55.7/56.2 | 50.0/50.5 |
| MA [A gPt−1] | 212/174 | 257/208 | 352/275 |
| SA [μA cmPt−2] | 252/250 | 461/374 | 704/544 |
Interesting to note is that signs of Pt-surface facet types may be hinted at by the CV profiles for ECSA determination, owing to the structurally sensitive nature of H-adsorption/desorption and CO-oxidation.17 In Fig. 6a, no significant changes in the CO-stripping voltammetry profiles can be observed, apart from a slight upshift in the Pt/C peak that may indicate an increased ratio of NPs with large fractions of steps, kinks and edges typically found in smaller NPs.60,61 However, the feature seen in the HUPD curves in Fig. 6b at 0.06–0.07 V vs. RHE can sometimes emerge in Pt NPs with incorporated transition metals, often seen in provided HUPD curves as a peak (or bump) below 0.1 V vs. RHE.14,26,62 Assuming that this is the reason for the peak, being more pronounced in Pt3Co/C*, it infer that near-surface transition metal species is less common in Pt3Co/C than Pt3Co/C*, supporting the above discussion on the occurrence of surface Pt migration during the secondary MW treatment. In addition, the disappearance of this feature after degradation can likely be directly attributed to de-alloying and re-installment of a more “Pt-like” surface.
4. Conclusions
In summary, platinum–cobalt nanoparticles with a 3:1 atomic ratio were synthesized and simultaneously decorated onto carbon Vulcan XC 72R in a “one-pot” fashion. The method is entirely microwave based and includes only two steps. Step 1 allowed rapid high temperature oxidative treatment of metalorganic precursors with an exposed Pt surface area, owing to the added NH4NO3 creating initial oxidative conditions for removal of carbon traces. Step 2 consisted of a longer treatment at a 50% power setting in reductive atmosphere, causing a more Pt-rich NP surface with minimal particle size increments. The benefit of both MW steps was evaluated by comprehensive electrochemical testing and in combination, the prepared Pt3Co/C sample demonstrated 66% higher mass activity and 279% higher specific activity than a commercial Pt/C reference measured under identical conditions.
Co incorporation into the Pt fcc lattice was evident by both physical and electrochemical characterization techniques. The developed method is simple, fast and suitable for industrial upscaling. The simplicity of the procedure may also make the method particularly attractive for further research into Pt–Co based NPs by fine-tuning composition, varying support type or even introducing a third metal. The intriguing novel synthesis concept developed here is thus anticipated to assist in further progress toward the full commercialization of Pt–Co catalysts.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
T. W acknowledges support from Vetenskapsrådet (2017-04862) Energimyndigheten (45419-1), and Ångpanneföreningen (15-483). E. G. E. acknowledges support from Vetenskapsrådet (2018-03937) and the Olle Engkvist Foundation (186-0637). The authors acknowledge Umeå Core Facility for Electron Microscopy (UCEM) at Umeå University, Chemical Biological Centre (KBC). Andrey Shchukarev at Umeå University, KBC, is further acknowledged for performing the XPS measurements.References
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- S. Stariha, N. Macauley, B. T. Sneed, D. Langlois, K. L. More, R. Mukundan and R. L. Borup, J. Electrochem. Soc., 2018, 165, F492–F501 CrossRef CAS.
- R. Y. Jiang, S. O. Tung, Z. Tang, L. Li, L. Ding, X. G. Xi, Y. Y. Liu, L. Zhang and J. J. Zhang, Energy Storage Materials, 2018, 12, 260–276 CrossRef.
- J. Stacy, Y. N. Regmi, B. Leonard and M. H. Fan, Renewable Sustainable Energy Rev., 2017, 69, 401–414 CrossRef CAS.
- J. L. Qiao, M. Saito, K. Hayamizu and T. Okada, J. Electrochem. Soc., 2006, 153, A967–A974 CrossRef CAS.
- T. Tsuneda, R. K. Singh, A. Iiyama and K. Miyatake, ACS Omega, 2017, 2, 4053–4064 CrossRef CAS.
- Y. Bing, H. Liu, L. Zhang, D. Ghosh and J. Zhang, Chem. Soc. Rev., 2010, 39, 2184–2202 RSC.
- M. H. Shao, Q. W. Chang, J. P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- S. J. Guo and S. H. Sun, J. Am. Chem. Soc., 2012, 134, 2492–2495 CrossRef CAS PubMed.
- Q. Li, L. H. Wu, G. Wu, D. Su, H. F. Lv, S. Zhang, W. L. Zhu, A. Casimir, H. Y. Zhu, A. Mendoza-Garcia and S. H. Sun, Nano Lett., 2015, 15, 2468–2473 CrossRef CAS PubMed.
- R. Sandström, G. Hu and T. Wågberg, ACS Appl. Energy Mater., 2018, 1, 7106–7115 CrossRef.
- C. Chen, Y. J. Kang, Z. Y. Huo, Z. W. Zhu, W. Y. Huang, H. L. L. Xin, J. D. Snyder, D. G. Li, J. A. Herron, M. Mavrikakis, M. F. Chi, K. L. More, Y. D. Li, N. M. Markovic, G. A. Somorjai, P. D. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- F. Hasche, M. Oezaslan and P. Strasser, J. Electrochem. Soc., 2012, 159, B25–B34 CAS.
- S. P. Chen, Z. Q. Niu, C. L. Xie, M. Y. Gao, M. L. Lai, M. F. Li and P. D. Yang, ACS Nano, 2018, 12, 8697–8705 CrossRef CAS PubMed.
- G. Hu, E. Gracia-Espino, R. Sandstrom, T. Sharifi, S. Cheng, H. Shen, C. Wang, S. Guo, G. Yang and T. Wagberg, Catal. Sci. Technol., 2016, 6, 1393–1401 RSC.
- D. L. Wang, H. L. L. Xin, R. Hovden, H. S. Wang, Y. C. Yu, D. A. Muller, F. J. DiSalvo and H. D. Abruna, Nat. Mater., 2013, 12, 81–87 CrossRef CAS PubMed.
- L. Wang, W. P. Gao, Z. Y. Liu, Z. H. Zeng, Y. F. Liu, M. Giroux, M. F. Chi, G. F. Wang, J. Greeley, X. Q. Pan and C. Wang, ACS Catal., 2018, 8, 35–42 CrossRef CAS.
- F. J. Lai, W. N. Su, L. S. Sarma, D. G. Liu, C. A. Hsieh, J. F. Lee and B. J. Hwang, Chem.–Eur. J., 2010, 16, 4602–4611 CrossRef CAS PubMed.
- W. S. Jung and B. N. Popov, ACS Sustainable Chem. Eng., 2017, 5, 9809–9817 CrossRef CAS.
- D. D. Papadias, R. K. Ahluwalia, N. Kariuki, D. Myers, K. L. More, D. A. Cullen, B. T. Sneed, K. C. Neyerlin, R. Mukundan and R. L. Borup, J. Electrochem. Soc., 2018, 165, F3166–F3177 CrossRef CAS.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- V. Yarlagadda, S. E. McKinney, C. L. Keary, L. Thompson, B. Zulevi and A. Kongkanand, J. Electrochem. Soc., 2017, 164, F845–F849 CrossRef CAS.
- A. Kongkanand and M. F. Mathias, J. Phys. Chem. Lett., 2016, 7, 1127–1137 CrossRef CAS PubMed.
- E. Antolini, J. R. C. Salgado and E. R. Gonzalez, J. Power Sources, 2006, 160, 957–968 CrossRef CAS.
- M. N. Cao, D. S. Wu and R. Cao, ChemCatChem, 2014, 6, 26–45 CrossRef CAS.
- H. Liu, C. Li, D. Chen, P. Cui, F. Ye and J. Yang, Sci. Rep., 2017, 7, 11421 CrossRef.
- H. H. Huang, X. L. Hu, J. B. Zhang, N. Su and J. X. Cheng, Sci. Rep., 2017, 7, 45555 CrossRef CAS PubMed.
- M. K. Carpenter, T. E. Moylan, R. S. Kukreja, M. H. Atwan and M. M. Tessema, J. Am. Chem. Soc., 2012, 134, 8535–8542 CrossRef CAS PubMed.
- J. Zeng and J. Yang Lee, Int. J. Hydrogen Energy, 2007, 32, 4389–4396 CrossRef CAS.
- B. P. Vinayan, R. Nagar, N. Rajalakshmi and S. Ramaprabhu, Adv. Funct. Mater., 2012, 22, 3519–3526 CrossRef CAS.
- C. N. Chinnasamy, B. Jeyadevan, K. Shinoda and K. Tohji, J. Appl. Phys., 2003, 93, 7583–7585 CrossRef CAS.
- R. Ojani, J.-B. Raoof, M. Goli and R. Valiollahi, J. Power Sources, 2014, 264, 76–82 CrossRef CAS.
- R. Fiala, M. Vaclavu, M. Vorokhta, I. Khalakhan, J. Lavkova, V. Potin, I. Matolinova and V. Matolin, J. Power Sources, 2015, 273, 105–109 CrossRef CAS.
- S. Horikoshi and N. Serpone, Catal. Sci. Technol., 2014, 4, 1197–1210 RSC.
- P. Priecel and J. A. Lopez-Sanchez, ACS Sustainable Chem. Eng., 2019, 7, 3–21 CrossRef CAS.
- G. Hu, F. Nitze, E. Gracia-Espino, J. Ma, H. R. Barzegar, T. Sharifi, X. Jia, A. Shchukarev, L. Lu, C. Ma, G. Yang and T. Wågberg, Nat. Commun., 2014, 5, 5253 CrossRef CAS PubMed.
- S. Horikoshi and N. Serpone, Microwaves in Nanoparticle Synthesis: Fundamentals and Applications, Wiley, 2013 Search PubMed.
- S. Komarneni, D. Li, B. Newalkar, H. Katsuki and A. S. Bhalla, Langmuir, 2002, 18, 5959–5962 CrossRef CAS.
- D. C. Higgins, S. Y. Ye, S. Knights and Z. W. Chen, Electrochem. Solid-State Lett., 2012, 15, B83–B85 CrossRef CAS.
- V. Kepeniene, L. Tamasauskaite-Tamasiunaite, J. Jablonskiene, J. Vaiciuniene, R. Kondrotas, R. Juskenas and E. Norkus, J. Electrochem. Soc., 2014, 161, F1354–F1359 CrossRef CAS.
- M. Zhang, Y. Li, Z. Yan, J. Jing, J. Xie and M. Chen, Electrochim. Acta, 2015, 158, 81–88 CrossRef CAS.
- A. L. Patterson, Phys. Rev., 1939, 56, 978–982 CrossRef CAS.
- K. Shinozaki, J. W. Zack, S. Pylypenko, R. M. Richards, B. S. Pivovar and S. S. Kocha, Int. J. Hydrogen Energy, 2015, 40, 16820–16830 CrossRef CAS.
- R. Sandström, E. Gracia-Espino, G. Hu, A. Shchukarev, J. Ma and T. Wågberg, Nano Energy, 2018, 46, 141–149 CrossRef.
- G. Feick and R. M. Hainer, J. Am. Chem. Soc., 1954, 76, 5860–5863 CrossRef CAS.
- S. Chaturvedi and P. N. Dave, J. Energ. Mater., 2013, 31, 1–26 CrossRef CAS.
- Z. Han, S. Sachdeva, M. I. Papadaki and M. S. Mannan, J. Loss Prev. Process Ind., 2015, 35, 307–315 CrossRef CAS.
- A. Varma, A. S. Mukasyan, A. S. Rogachev and K. V. Manukyan, Chem. Rev., 2016, 116, 14493–14586 CrossRef CAS PubMed.
- S. Habibzadeh, Y. Mortazavi and A. A. Khodadadi, J. Nanosci. Nanotechnol., 2010, 10, 6003–6008 CrossRef CAS PubMed.
- J. R. C. Salgado, E. Antolini and E. R. Gonzalez, J. Phys. Chem. B, 2004, 108, 17767–17774 CrossRef CAS.
- M. F. Chi, C. Wang, Y. K. Lei, G. F. Wang, D. G. Li, K. L. More, A. Lupini, L. F. Allard, N. M. Markovic and V. R. Stamenkovic, Nat. Commun., 2015, 6, 8925 CrossRef CAS PubMed.
- S. S. Kocha, J. W. Zack, S. M. Alia, K. C. Neyerlin and B. S. Pivovar, ECS Trans., 2012, 1475 Search PubMed.
- K. Shinozaki, B. S. Pivovar and S. S. Kocha, in Polymer Electrolyte Fuel Cells 13, ed. H. A. Gasteiger, A. Weber, K. Shinohara, H. Uchida, S. Mitsushima, T. J. Schmidt, S. R. Narayanan, V. Ramani, T. Fuller, M. Edmundson, P. Strasser, R. Mantz, J. Fenton, F. N. Buchi, D. C. Hansen, D. L. Jones, C. Coutanceau, K. SwiderLyons and K. A. Perry, Electrochemical Soc Inc, Pennington, 2013, vol. 58, pp. 15–26 Search PubMed.
- J. Chlistunoff and J. M. Sansinena, J. Electroanal. Chem., 2016, 780, 134–146 CrossRef CAS.
- J. Chlistunoff and J. M. Sansinena, J. Phys. Chem. C, 2016, 120, 28038–28048 CrossRef CAS.
- R. K. Singh, R. Devivaraprasad, T. Kar, A. Chakraborty and M. Neergat, J. Electrochem. Soc., 2015, 162, F489–F498 CrossRef CAS.
- J. Chlistunoff and J.-M. Sansiñena, J. Phys. Chem. C, 2016, 120, 28038–28048 CrossRef CAS.
- S. Rudi, C. H. Cui, L. Gan and P. Strasser, Electrocatalysis, 2014, 5, 408–418 CrossRef CAS.
- D. F. van der Vliet, C. Wang, D. G. Li, A. P. Paulikas, J. Greeley, R. B. Rankin, D. Strmcnik, D. Tripkovic, N. M. Markovic and V. R. Stamenkovic, Angew. Chem., Int. Ed., 2012, 51, 3139–3142 CrossRef CAS PubMed.
- Y. Ye, J. Joo, S. Lee and J. Lee, J. Mater. Chem. A, 2014, 2, 19239–19246 RSC.
- R. M. Antoniassi, J. C. M. Silva, T. Lopes, A. O. Neto and E. V. Spinace, Int. J. Hydrogen Energy, 2017, 42, 28786–28796 CrossRef CAS.
- H. X. Nan, D. Dang and X. L. Tian, J. Mater. Chem. A, 2018, 6, 6065–6073 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional electrochemical experimental details, TGA analysis, supporting XRD pattern, LSVs, HUPD and additional electrochemical data. See DOI: 10.1039/c9ra02095k |
| This journal is © The Royal Society of Chemistry 2019 |