
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly active and robust Ni–MoS2 supported on mesoporous carbon: a nanocatalyst for hydrodeoxygenation reactions†
Swathi Mukundan ab,
Md A. Wahabc,
Luqman Atandad,
Muxina Konarovaa and
Jorge Beltramini*de
ab,
Md A. Wahabc,
Luqman Atandad,
Muxina Konarovaa and
Jorge Beltramini*de
aNanomaterials Center-AIBN, School of Chemical Engineering, The University of Queensland, Brisbane, QLD-4072, Australia
bDepartment of Applied Chemistry, Cochin University of Science and Technology, Kochi, Kerala-682022, India
cSchool of Chemistry, Physics and Mechanical Engineering, Faculty of Engineering, Queensland University of Technology, Brisbane, QLD-4000, Australia
dCentre for Tropical Crops and Biocommodities, Queensland University of Technology, Brisbane, QLD-4000, Australia. E-mail: jorge.beltramini@qut.edu.au
eIROAST, Department of Chemistry, Faculty of Advanced Science and Technology, Kumamoto University, Kumamoto 860-8555, Japan
First published on 3rd June 2019
Abstract
NiMoS2 nanoparticles supported on carbon, synthesized by a microemulsion method were used as a nanocatalyst for hydrodeoxygenation (HDO) of a lignin model compound – guaiacol. Two types of carbon supports – mesoporous carbon (CMK-3) and activated carbon (AC) with a predominantly microporous structure, were studied to investigate the role of porosity and nature of the porous structure in catalyst activity. The activity of NiMoS2/AC resulted in the complete guaiacol conversion at 13 h of reaction time to produce phenol (31.5 mol%) and cyclohexane (35.7 mol%) as the two main products. Contrastingly, NiMoS2/CMK-3 needed a much lesser reaction time (6 h) to attain a similar conversion of guaiacol but gave different selectivities of phenol (25 mol%) and cyclohexane (55.5 mol%). Increased cyclohexane production with NiMoS2/CMK-3 implied better deoxygenation of MoS2 and enhanced hydrogenation capacity of Ni since phenol is a partially deoxygenated product of guaiacol while cyclohexane is a completely deoxygenated and hydrogenated product. The superior catalytic activity and deoxygenating behavior of NiMoS2/CMK-3 catalysts could be attributed to the organized mesoporosity of the CMK-3 support in relation to the improved active phase distribution and access to active sites that facilitate the conversion of the reaction's product. Recyclability study implied NiMoS2/CMK-3 was more stable without significant changes in the catalytic activity even after three reaction cycles.
Introduction
Lignin is the most abundant renewable aromatic resource in nature, however, it has been underutilized as a feedstock for biofuel production.1,2 Meanwhile, lignin is currently used mostly as plasticizers or low-grade fuel by burning.3,4 With the current depletion of non-renewable fossil fuels and the associated environmental issues of pollution, production of chemicals and fuels from lignin can be expected to gain more interest in the foreseeable future. Owing to the vast availability of lignin naturally in biomass or in the waste stream of pulp and paper industry, its transformation to useful products i.e. liquid fuel or chemical feedstock via catalytic biorefining technologies will serve as a renewable energy production.5,6 Among the various studies that have received increasing attention in recent years is the catalytic upgrade of lignin via hydrodeoxygenation (HDO), sometimes referred to as catalytic hydrotreatment.7 Catalytic hydrodeoxygenation (HDO) of lignin using molecular hydrogen is considered to be more advantageous than other conversion processes since it produces oil with a small amount of oxygen that can be easily upgraded to hydrocarbon fuels. Alternatively, HDO can be employed in the downstream processing of intermediate bio-oil obtained by pyrolysis or liquefaction of lignin in order to improve the oil energy density by removal of the oxygenated groups. During the HDO process of lignin, liquid oxygenates such as guaiacol, phenol, catechol, etc. are generated.8 Product selectivity can be tailored based on the choice of catalyst, hence crucial to improving yields of targeted valuable chemicals is catalyst design.A remarkable effort has been dedicated to exploring the ideal hydrotreating catalysts for the HDO process in the quest of producing high-quality fuels. Among the studied catalysts, carbon is considered an excellent support for hydroprocessing reactions due to its high surface area, amphoteric nature and less tendency to generate coke.9–12 For example, Ru–MnOx supported on carbon-black selectively catalyzed the conversion of guaiacol to cyclohexanol and methanol.13 The nature of support is also critical to the reaction pathway of the HDO reaction. A study of guaiacol conversion using CoMo catalyst supported on carbon and γ-Al2O3 showed that the demethoxylation reaction pathway that produces higher phenol/catechol ratio and less coke formation was favoured on CoMo/C as compared to CoMo/γ-Al2O3,14 and found to be consistent with the report of Centeno et al.15 who observed that the rate of phenol formation is faster on CoMo supported on carbon.15 Among the widely used carbon support is activated carbon, nonetheless, its characteristic micropores can be easily blocked when using large reactant molecules which often leads to catalyst deactivation as was observed in our previous study of guaiacol conversion using MoS2/C catalysts.16 In this context, the influence of surface porosity of carbon support on the activity of the catalyst for HDO reaction has yet to be explored. Mesoporous carbon is proven to be superior catalytic support because of its organised and controllable pore geometry that enables excellent distribution of active species, thus enhancing catalytic activity and stability.17 Moreover, the wide pores of mesoporous carbon are beneficial for the diffusion of reactants easily to reach the active sites of the catalysts.18 For example, improved cycling performance of MoS2/CMK-3 nanocomposites as an electrocatalytic material was attributed to the large pores of CMK-3.19
Among the several methods that have been employed for the synthesis of metal nanoparticles, incipient wetness impregnation synthesis route is a widely used technique for preparing supported catalysts. This synthesis method adopts the concept of distribution of metal precursor over the support, followed by calcination. With this technique, nanosized metal particles could be obtained but difficult to achieve both the homogeneous and narrow particle size distribution of metal species in the carbon supports. Moreover, the aggregation of metal particles at the pore entrance and the loosely bound metal particles can leach out during the catalyst preparation step such as washing with a solvent. One of the synthesis technique to overcome this problem is microemulsion (ME), which has been utilized as a good strategy to synthesize nanoscale metal particles with narrow particle size distribution.20 High dispersion of metals over the support, less synthesis time and in situ reduction of metals during synthesis are some other benefits associated with the microemulsion technique.21 Therefore, in this article, we utilise the ME method for the synthesis of NiMoS2 nanocatalyst supported on two carbon supports with different surface porosity and pore structures. The catalytic activity of these nanomaterials was tested for the conversion of guaiacol, a lignin model compound. The selectivity of liquid products and the reaction mechanism were influenced by the type of carbon support used.
Experimental section
Materials
Nickel(II) nitrate hexahydrate (Ni(NO3)2·6H2O) (>98.5% purity), ammonium molybdate tetrahydrate ((NH4)6Mo7O24·4H2O) (81.0–83.0% purity MoO3 basis), Brij 30, ammonium sulphide solution (20.0% in H2O), tetraethyl orthosilicate (TEOS) (98.0% purity), sucrose (99.5% purity), sulphuric acid (99.9% purity), dodecane (99.0% purity), guaiacol (99.0% purity), catechol (99.0% purity), anisole (99.7% purity), cyclohexanol (99.0% purity), cyclohexane (99.5% purity), cyclohexene (analytical grade), veratrole (99.0% purity), cresol (99.0% purity), pluronic P-123, activated carbon (norit SX ultra) were all purchased from Sigma-Aldrich.Catalyst preparation
The highly ordered mesoporous carbon (CMK-3) support with a high surface area of 1258 m2 g−1 and uniform pore size of 3.85 nm was synthesised by using mesoporous silica (SBA-15) as the hard templating agent and sucrose as a carbon source.17,22 At first, SBA-15 was prepared by dissolving 2 g of pluronic P-123 surfactant in 60 mL of 2 M HCl at 38 °C under stirring. Then, 4.2 g of the silica source, TEOS, was added dropwise to the above solution under vigorous stirring. After stirring for 6–8 min, the mixture was kept at 38 °C for 24 h without stirring. Then, the solution was transferred into an autoclave and heated for 48 h at 100 °C, followed by cooling, filtration, washing with excess water and the collected sample was dried at 100 °C overnight. Finally, the dried sample was calcined at 550 °C for 6 h in a muffle furnace to obtain a highly organized mesoporous SBA-15 that possesses a high surface area of 719 m2 g−1 and pore size of 9.15 nm.CMK-3 was prepared by impregnating the pore of SBA-15, which now serves as the silica template, with 1.25 g of sucrose dissolved in H2SO4 solution (0.14 g of H2SO4 in 5 mL deionized H2O). Then the mixture was stirred for 6 h, followed by heating at 100 °C for 6 h and 160 °C for 6 h. This impregnation procedure was repeated with 0.8 g sucrose in 0.09 g H2SO4 in 5 mL deionized H2O. The same heating protocol was used as mentioned before. After heating the sample, the carbon–silica nanocomposite sample was carbonised at 850 °C for 5 h under a flow of N2 gas. Finally, the SBA-15 silica template from as-prepared carbon–silica nanocomposite was removed by washing with 5 wt% hydrofluoric acid (HF) solution at room temperature, leaving behind mesoporous carbon material (CMK-3) with a high surface area of 1258 m2 g−1 and a pore size of 3.85 nm. The CMK-3 was dried at 200 °C overnight and used thereafter as a catalyst support.
Supported NiMoS2 catalysts were prepared by W/O (water in oil) microemulsion synthesis method as reported previously.16,23 Typically, 5.0 g of non-ionic brij-30 surfactant was dissolved in 100 mL of cyclohexane to form an oil phase. Then 5 mL of ammonium sulphide solution was added to the oil phase and stirred for 1 h. Again, 2 mL aqueous solution of ammonium molybdate hexahydrate was slowly added to the above solution, followed by 1 mL aqueous solution of nickel nitrate tetrahydrate and allowed to stir for 4 h to form a black microemulsion. The NiMoS2 nanoparticles thus formed were supported over carbon supports (AC or CMK-3) by adding to the above microemulsion an already dispersed carbon material in 50 mL of deionized water, then stirred for an hour. Cyclohexane was subjected to slow evaporation at room temperature followed by heat treatment at 550 °C for 4 h under a flow of N2. The prepared catalysts are labelled as NiMoS2/AC and NiMoS2/CMK-3. The concentrations of molybdenum and nickel were calculated to be 12 wt% and 3 wt%, respectively. MoS2/AC and MoS2/CMK-3 were also synthesised by the same procedure as mentioned above without the addition of the nickel precursor.
Catalyst characterization
X-ray diffraction (XRD) patterns of the used samples for this study were recorded using Rigaku Miniflex with monochromatic Co Kα radiation (30 kV, 15 mA) for the range of 10° ≤ 2θ ≤ 90° (step size of 0.1°) whereas Bruker radiation D8 advanced diffractometer with Cu Kα radiation was used for the range of 1° ≤ 2θ ≤ 5°. Nitrogen adsorption–desorption isotherms were obtained at −196 °C using a Micrometrics Tristar II 3020 system. The catalysts were degassed at 200 °C overnight on a vacuum line, prior to analysis. The specific surface area was calculated by Brunauer–Emmett–Teller (BET) method and pore size distribution curve using the BJH model on the desorption isotherm curve. The chemical structure of the catalysts was analysed by Raman spectroscopy using Renishaw inVia Raman Microscope equipped with a Leica DM LM Microscope using a 514 nm HPNIR diode laser as an excitation source and 0.1 mW laser power. The Raman emission was collected by 50× objective lens in a backscattering geometry in the region of 100–2000 cm−1 at 4 cm−1 resolution and acquisition time of 1.0 s. Transmission Electron Microscopy (TEM) JOEL 2100 microscope operated at 200 kV, fitted with energy dispersive X-ray (EDS) detector was used for morphology characterisation. Before TEM imaging, the samples were prepared by ultrasonically dispersing the particles in ethanol. The solution was settled on carbon-coated Cu grids to prepare the TEM samples. Inductively Coupled Plasma (ICP) analysis in a Varian Vista Pro ICPOES instrument was used for bulk elemental analysis. The samples were digested using a milestone Ethos 1 microwave digester. CHON–S analyser (FLASH EA 1112 series, Thermo Electron Corporation) was used to quantify sulphur. X-ray photoelectron spectra (XPS) was acquired using a Kratos Axis ULTRA X-ray photoelectron spectrometer equipped with a 165 mm hemispherical electron energy analyser using a monochromatic Al Kα (1486.6 eV) X-ray source at 150 W (15 kV, 10 mA). A survey wide scan was collected at analyser pass energy of 160 eV and multiplex (narrow) high resolution scans at 20 eV. Base chamber pressure in the analysis chamber was 1.0 × 10−9 torr and it increased to 1.0 × 10−8 torr during sample analysis. The binding energies were referenced to the C 1s peak of adventitious carbon at 284.6 eV to account for the charging effects. Data analysis was done using Casa-XPS (v 2.3.12) employing a Shirley-background subtraction prior to fitting the spectra using Gaussian–Lorentzian curves.Guaiacol conversion tests
Guaiacol conversion tests were carried out in a 300 mL Parr batch reactor. Before the reaction, 10 wt% of the catalyst was reduced under the flow of H2 at 450 °C for 3 h. Then the reduced catalyst was transferred into the reactor vessel containing guaiacol and dodecane (solvent). The vessel was sealed, flushed with argon, pressurised to 50 bars with H2 gas and the temperature ramped to 300 °C and 450 rpm (H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :guaiacol initial mole ratio = 11:1). The reactions were conducted until the complete conversion of guaiacol. The liquid and gas products were collected every hour and analysed. At the end of the reaction, the catalyst was retrieved by filtration, washed with ethanol and dried overnight at 50 °C. For the recyclability tests, the catalysts were reduced before the next reaction cycle. Each experimental run for the guaiacol HDO reaction was conducted in triplicate and the results agreed within ±4%.
:guaiacol initial mole ratio = 11:1). The reactions were conducted until the complete conversion of guaiacol. The liquid and gas products were collected every hour and analysed. At the end of the reaction, the catalyst was retrieved by filtration, washed with ethanol and dried overnight at 50 °C. For the recyclability tests, the catalysts were reduced before the next reaction cycle. Each experimental run for the guaiacol HDO reaction was conducted in triplicate and the results agreed within ±4%.
Product analysis
The liquid products retrieved on an hourly basis during the reaction were collected in 2 mL Eppendorf tube and centrifuged at 13000 rpm for 3 minutes. 0.2 μL of the supernatant samples were injected manually into a Shimadzu GC-17A gas chromatography unit. The GC was equipped with a flame ionisation detector (FID). CP-Sil 5 CB capillary column (30 m × 0.25 mm × 0.39 m) was employed to analyse the reaction products. The standards for guaiacol and other products were prepared in ethyl acetate. The injector and detector temperatures were set at 200 °C and 230 °C, respectively. The column temperature was held at 50 °C for 5 min, followed by ramping at 15 °C min−1 to 250 °C. Then the temperature was ramped at 5 °C min−1 to 280 °C. A volume of 0.25 μL liquid product was injected in a split mode. A mass spectrometer (Shimadzu GCMS-QP2010 Ultra instrument with Rxi-5ms 30 m, 0.25 × 1 μm column) was used to determine unknown liquid products. Conversion (% C) of guaiacol and product yield (% Y) were calculated in mol% as follows: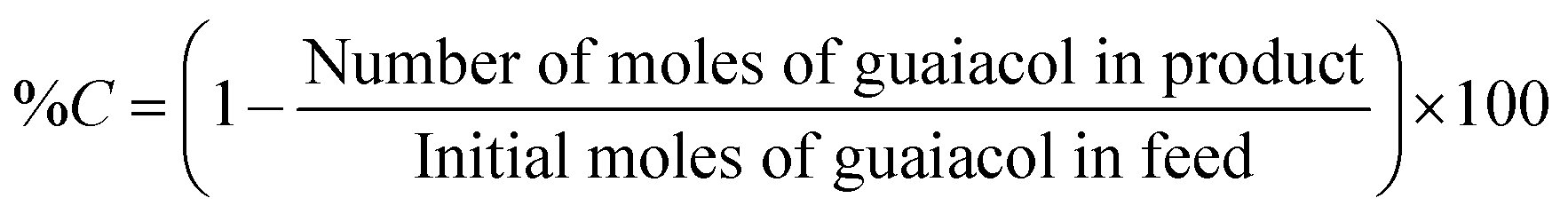
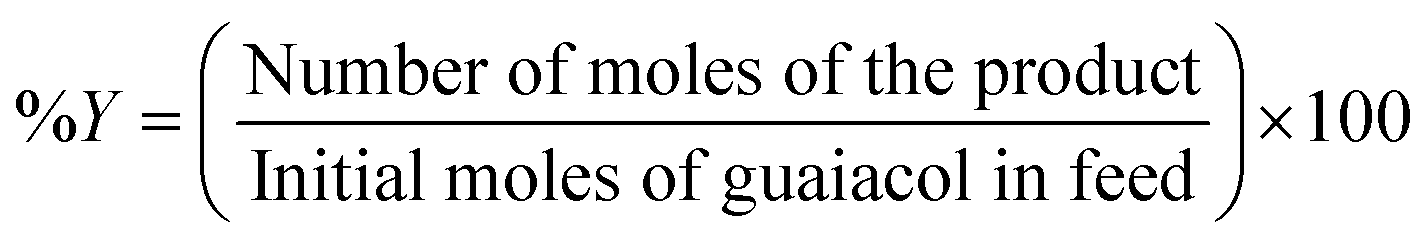
Results and discussion
Hydrodeoxygenation of guaiacol with NiMoS2/AC and NiMoS2/CMK-3 catalysts
High interest in utilizing carbon materials as catalyst support has tremendously increased due to its amphoteric nature, remarkable flexibility and stability. In order to be used in this field, carbon supports should not only have the high surface area but also a highly organized pore structure, adequate pore volume and uniform pore size that can easily accommodate the active species within the pores of carbon supports during the incorporation or functionalization process. Therefore, this study has considered two types of carbon supports: (i) microporous carbon – activated carbon (AC) which was used as received without modification (Fig. S1a and c†) and (ii) mesoporous carbon – CMK-3 that was prepared via the mesoporous SBA-15 silica hard templating method. The detailed characterisation of the activated carbon (AC) and mesoporous carbon (CMK-3) supports along with the supported metal catalysts are discussed in ESI.†17,24–41The catalytic activity at the complete conversion of guaiacol with MoS2 supported on AC and CMK-3 is depicted in Fig. 1. Phenol was the major product of the reaction and was an intermediate for the formation of hydrogenated products such as cyclohexanol, cyclohexene and cyclohexane. The other products evolved during the reaction were anisole, catechol, veratrole, benzene, cresol and methanol. Almost all the products were partially/completely deoxygenated, except catechol and veratrole. However, these products are observed in minor quantities. H2, CO and CH4 were among the gaseous products observed. It is known that guaiacol produces phenol by two pathways as shown in Scheme 1; namely (i) demethoxylation (DMO) – direct production of phenol from guaiacol by cleaving the C–OCH3 bond, producing methanol as a by-product7,42,43 and (ii) demethylation (DME) – where the guaiacol is first converted to catechol by cleaving O–CH3 bond. Thus, the formation of catechol was also accompanied by the cogeneration of CH4. The formation of catechol increases the coke formation by condensation reactions.44 The acidity of the catalyst support was reported to be the key reason for catechol formation.45 Bui et al.46 reported both catechol and methyl catechol to be the major products with CoMoS/γ-Al2O3 catalyst. However, interestingly with CoMoS/ZrO2 catalyst, demethoxylation was the major pathway producing phenol directly. Irrespective of the slightly acidic nature of ZrO2 support, the promoter effect of Co on ZrO2 created a unique active site which promotes direct demethoxylation of guaiacol.46 Hence, it is already concluded that acidity is not the only reason that favours catechol formation. Similarly, Ruiz et al.47 examined the activity of MoS2 supported on four different activated carbons with different textural and chemical properties. Regardless of the neutral character of the carbon support,7 the authors still observed the formation of catechol. Considering the phenol/catechol ratio, the value was constant (ca. 0.3) with all the supported catalysts irrespective of textural and chemical properties differences between the four supports.47 Similarly, with our catalysts, DME and DMO of guaiacol to phenol, both were observed on MoS2/AC. However, DME did not take place when MoS2/CMK-3 was used but DMO was the only route for phenol formation. The conversion of guaiacol using MoS2/AC catalyst as function of time, as well as the reaction mechanism was discussed in our previous work.16 Apart from the non-formation of catechol with MoS2/CMK-3 catalyst, we did not observe considerable difference in the selectivity of products at complete conversion as can be seen from Fig. 1.
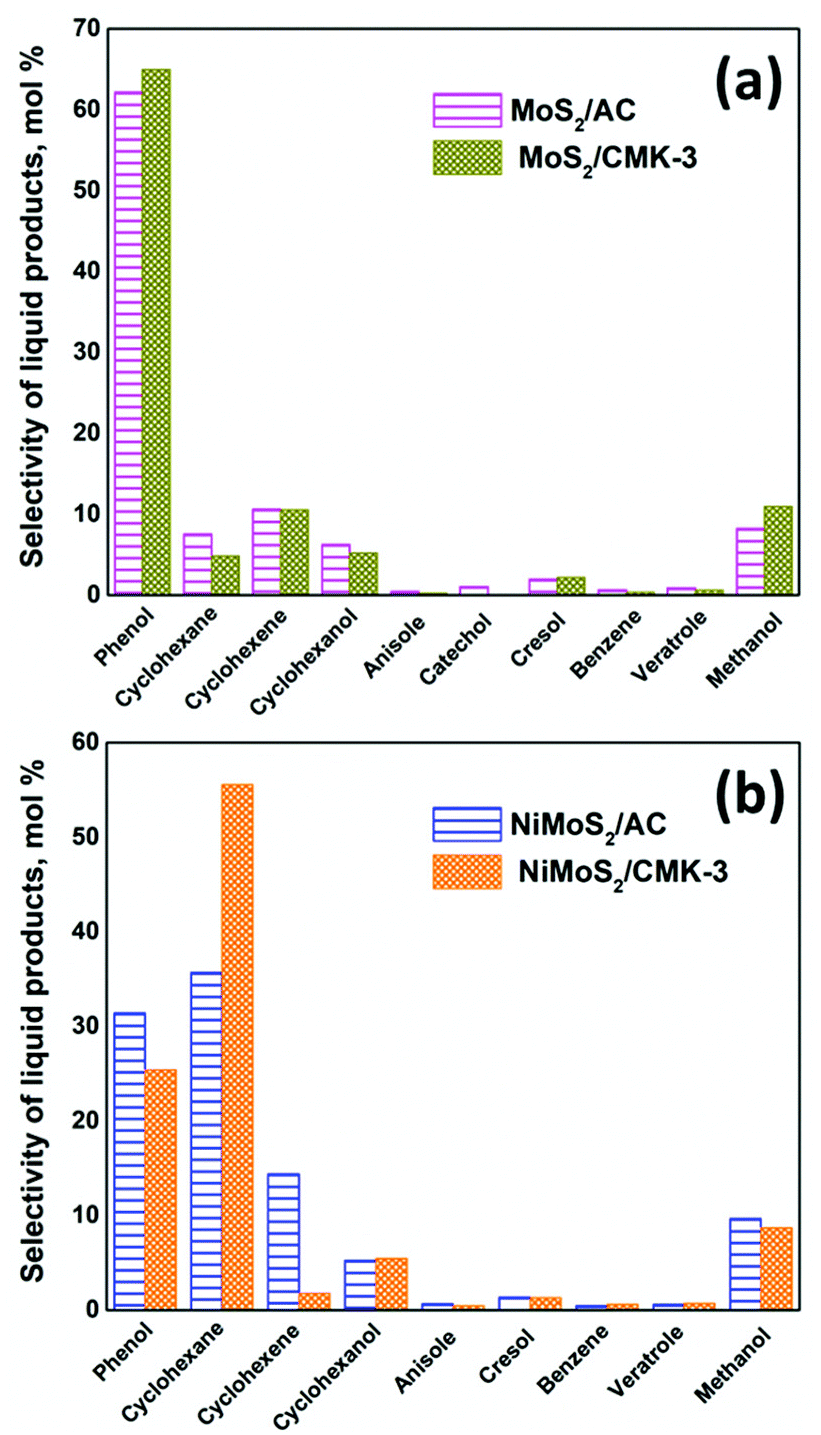 | ||
| Fig. 1 Comparison of the selectivities of liquid products (mol%) by (a) MoS2/AC and MoS2/CMK-3 catalysts (b) NiMoS2/AC and NiMoS2/CMK-3 catalysts for the conversion of guaiacol after complete conversion. | ||
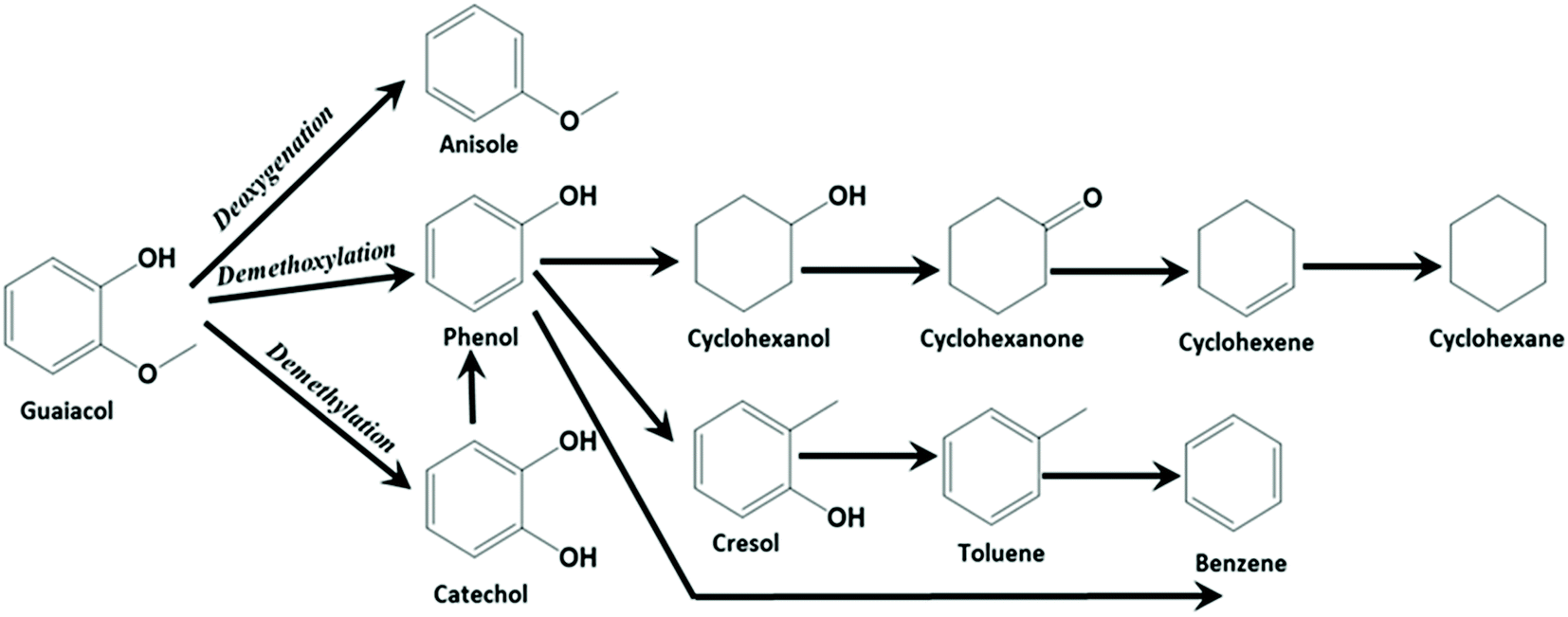 | ||
| Scheme 1 Reaction network of hydrodeoxygenation of guaiacol by carbon supported NiMoS2 catalyst. | ||
In order to increase the deoxygenation tendency of the catalyst, we introduced Ni as a promoter for the active MoS2 species in this study. Among metal catalysts, nickel is an inexpensive and available transition metal which has been employed traditionally for hydrogenation reactions.48 Application of Ni-based catalysts for HDO reactions primarily produce hydrogenated products where the order of deoxygenation is low.49 For instance, with La1−xCexNiO3 (x = 0 to 0.9) catalyst, hydrogenation of guaiacol ring was primarily the observed pathway that produced cyclohexanol and methoxy cyclohexanol.50 Similarly, cyclohexane, cyclohexanone and 1-methylcyclohexane-1,2-diol were the major products observed by Bykova et al.51 with CeO2, ZrO2, Al2O3 and SiO2 supported Ni catalysts for HDO of guaiacol. The formation of the hydrogenated products was also accompanied by the consumption of a significant amount of hydrogen, which is expensive. However, when Ni was used as a promoter for MoS2 catalyst, Ni–Mo–S species was formed which created a synergistic effect between Ni and Mo that promoted the deoxygenating tendency of the catalyst.52
Hence in this work, NiMoS2/CMK-3 catalyst was tested for the conversion of guaiacol, and the activity was compared with that of AC supported catalyst. Complete conversion of guaiacol was achieved after 6 h reaction time with NiMoS2/CMK-3, in contrast, 13 h was needed for the case of using NiMoS2/AC (Fig. 2). The fast conversion of guaiacol with CMK-3 supported catalysts can be ascribed to the presence of more surface area available for the reaction when compared to NiMoS2/AC (as observed from BET analysis result in Table S1†).
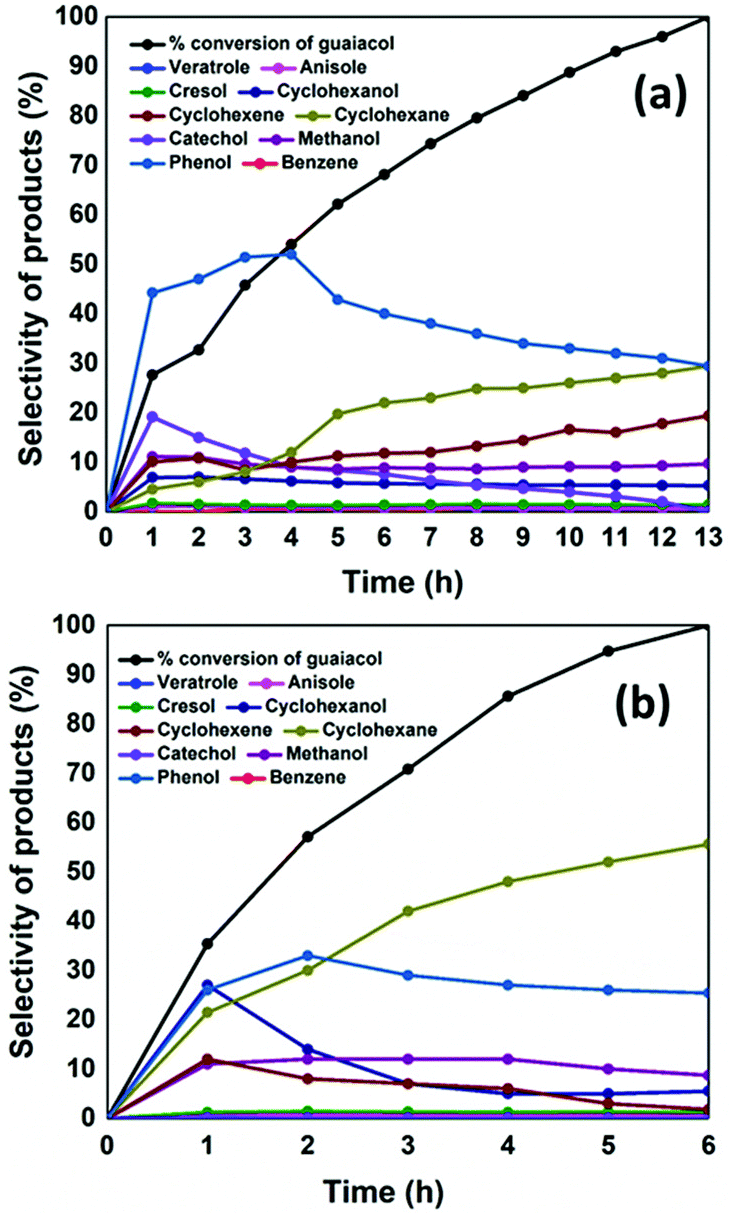 | ||
| Fig. 2 Conversion (mol%) of guaiacol and selectivities of liquid products produced (mol%) with respect to increase in reaction time with (a) NiMoS2/AC and (b) NiMoS2/CMK-3 catalysts. | ||
The conversion of guaiacol with time and the selectivity of liquid products are given in Fig. 2. At 1 h reaction time, 27% of guaiacol was converted with NiMoS2/AC, where the liquid product selectivity was 44% of phenol and 19% of catechol. There is also a co-generation of 11% methanol formed as result of the direct deoxygenation of guaiacol to phenol. Methane which is coproduced during the formation of catechol was also observed in the gaseous product. The hydrogenation of phenol to cyclohexanol was observed from the start of the reaction (7%). The formation of cyclohexene from cyclohexanol occurs through the production of cyclohexanone which is an intermediate compound. As cyclohexanone was not detected in the reaction product, indicates a rapid conversion of cyclohexanone to cyclohexene (10.1%). The further hydrogenation of cyclohexene to cyclohexane also occurred (4.6%) within 1 h reaction time. On the other hand, 35% guaiacol was converted at 1 h with NiMoS2/CMK-3 catalyst. Contrastingly, the main products produced were phenol (25%), cyclohexanol (26%) and 20% cyclohexane respectively. On the other hand, it was also observed that 10% cyclohexene and 10% methanol were produced within 1 h reaction time. There was no trace of catechol detected by GC-MS. On NiMoS2/C catalyst, increasing reaction time, the selectivity towards phenol constantly increased up to 4 h where it reached a 52% maximum, followed by a constant drop till the end of the reaction. The decrease in phenol selectivity was accompanied by an increase in cyclohexane and cyclohexene selectivity, which concludes that phenol was converted to cyclohexane. The catechol selectivity decreased sharply after 1 h, and the final product collected at the complete conversion of guaiacol showed no trace of catechol. The other products such as cresol, anisole, veratrole, benzene were produced at the onset of reaction but there was no increase in selectivity after 1 h of reaction. We can also infer from this reaction pattern that during the first hour, the kinetics of the reaction was fast for all deoxygenation, hydrogenation and methylation reactions that were taking place. After 1 h, the methylation reaction was suppressed. Looking into the reaction mechanism with NiMoS2/CMK-3, there was no trace of catechol from the onset of reaction. However, methane was still detected in the gas product analysis, assuming that might be produced through decomposition of methanol.
The organized mesopore structure of CMK-3 can facilitate the better distribution of active species (Ni and MoS2) that enhances the contact time of guaiacol with the catalyst active phases. On the other hand, on NiMoS2/AC, larger metal species are deposited on the micropores of activated carbon reducing the active surface area of the catalyst that can be noticed from Table S1.† Hence, we may conclude that the active metal species are deposited on the surface of the AC support, wherein interaction of the reactants with active species occurs mostly at the surface level. The channel-like pores of the CMK-3 act as a nanoreactor containing the active catalyst species. Thus, guaiacol has easy accessibility to get in contact with the active metal sites, as illustrated in Fig. 3. Ferrari et al.34 concluded that the dispersion of catalytic metal species and the accessibility of active sites are the crucial steps in guaiacol conversion. This observation was strongly supported by Ghampson et al.53 who have used four different activated carbons as support for Mo2N catalyst.
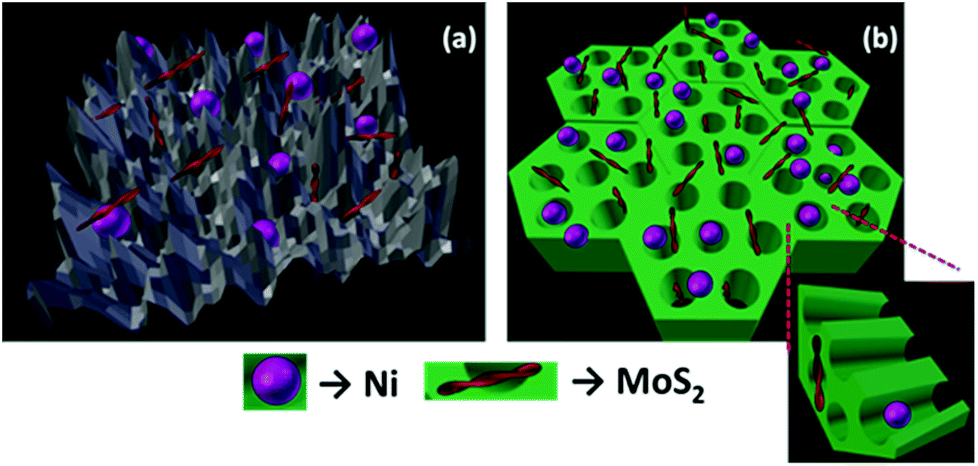 | ||
| Fig. 3 Figure illustrating the Ni and MoS2 active phase distribution over (a) activated carbon (b) CMK-3 supports. | ||
While testing these catalysts for the conversion of guaiacol, they noticed that the support with high mesopore content possessed the highly exposed active sites for reaction. Lee et al.54 proposed that CoMoS/nanoporous carbon exhibit higher activity for HDS reaction when compared to alumina and activated carbon supported catalyst. The high surface area and the organized pore geometry are the main reasons for the enhanced activity. They noted similar observation while evaluating the activity of Pt/mesoporous carbon for methanol oxidation. The superior activity is attributed to the high dispersion of Pt species in the pores, the orientation of nanochannels and easy accessibility of the pores by the reactant.55
Catalyst recyclability – NiMoS2/CMK-3
The catalyst after being used for the reaction was recovered by filtration, washed with ethanol and dried overnight at 50 °C. Afterwards, was reduced before each recyclability test. The reactions were conducted for 13 h and 6 h with NiMoS2/AC and NiMoS2/CMK-3 catalysts, respectively. These are the times that the catalysts attained complete conversion at the first catalytic run. The selectivity of liquid products (mol%) and the conversion of guaiacol by NiMoS2/AC and NiMoS2/CMK-3 catalysts for three cycles is depicted in Fig. 4.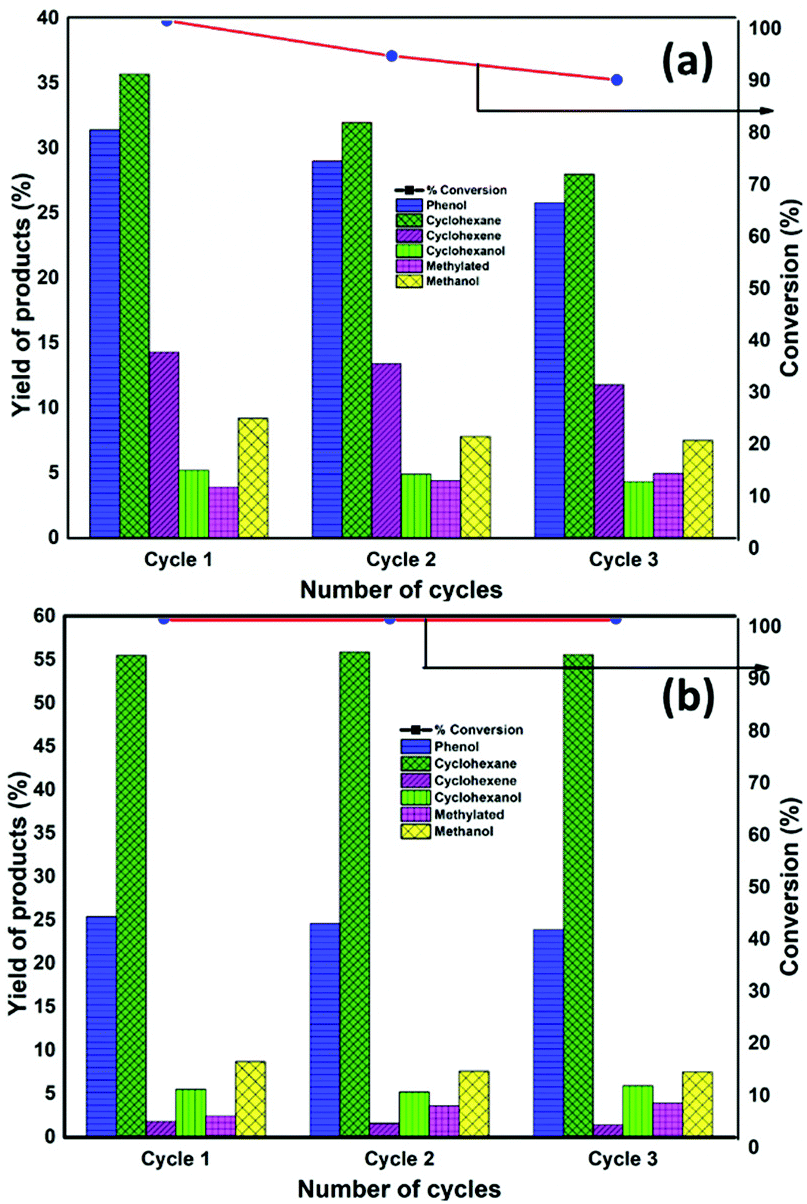 | ||
| Fig. 4 The selectivities of liquid products (mol%) and the conversion of guaiacol by NiMoS2/AC and NiMoS2/CMK-3 catalysts for three cycles. | ||
During the recyclability experiment, there was a drastic drop in the catalytic activity with NiMoS2/AC catalyst. 93.2% guaiacol was converted during the second cycle which was further reduced to 88.6% during the third cycle in 13 h of reaction time. Further discussing on the selectivity of products, the selectivity towards phenol decreased from 31.4% to 25.8% respectively after the third cycle which is an overall 18% decrease in selectivity. Similarly, the cyclohexane selectivity decreased from 35.7% to 28%. On the other hand, the methylated product increased from 3.9% to 4.9% by third cycle. We have already reported that the increase in methylated products was mainly due to the formation of MoOx species which was also noticed by MoS2/AC catalyst recyclability test.16 It can also be supported by the decrease in the S:Mo ratio in bulk (2.05:1 to 1.95:1) and surface (1.92:1 to 1.79:1) as estimated by ICP and XPS analyses (Table 1). There was also a decrease in catalyst surface area from 153 m2 g−1 to 102 m2 g−1 after third cycle accounting for a 33% loss in active surface area. From the current result, we can infer that one of the main reason for the catalyst deactivation could be the loss of catalyst surface area available for catalytic reaction. Zanuttini et al. also described rapid deactivation of Pt/zeolite, for the conversion of m-cresol due to the micropore blockage of the support.56 The coke formation in the catalyst was also calculated using CHNSO analysis and the amount of coke formed after 3 cycles was found to be 1.1%. Generally, carbon supported catalyst were reported to produce less coke when compared to acidic catalysts such as alumina, ZrO2 etc. which was attributed to the neutral/less acidic nature of carbon.57,58
:Mo ratio, BET surface area and pore volume of spent NiMoS2/C catalyst after cycle 1, 2 and 3
| Catalyst | Amount of sulphur (at%) | S/Mo (at. ratio) | BET surface area (m2 g−1) | Pore volume (cm3 g−1) | Coke depositionb (%) | ||
|---|---|---|---|---|---|---|---|
| Surfacea | Bulkb | Surfacea | Bulkb | ||||
| a Analyzed by XPS.b Analyzed by CHNS elemental analyzer. | |||||||
| NiMoS2/AC – fresh | 9.8 | 3.2 | 1.92:1 |
2.05:1 |
153 | 0.21 | 0 |
| NiMoS2/AC – cycle-1 | 9.9 | 3.0 | 1.88:1 |
2.02:1 |
135 | 0.19 | 0.6 |
| NiMoS2/AC – cycle-2 | 9.7 | 2.9 | 1.82:1 |
1.99:1 |
119 | 0.17 | 1.0 |
| NiMoS2/AC – cycle-3 | 8.7 | 2.7 | 1.79:1 |
1.95:1 |
102 | 0.17 | 1.1 |
| NiMoS2/CMK-3 – fresh | 9.1 | 3.3 | 1.92:1 |
1.98:1 |
453 | 0.33 | 0 |
| NiMoS2/CMK-3 – cycle-1 | 8.9 | 3.2 | 1.90:1 |
1.97:1 |
448 | 0.27 | 0.3 |
| NiMoS2/CMK-3 – cycle-2 | 8.9 | 3.0 | 1.87:1 |
1.97:1 |
441 | 0.24 | 0.6 |
| NiMoS2/CMK-3 – cycle-3 | 8.7 | 2.8 | 1.85:1 |
1.92:1 |
437 | 0.21 | 0.7 |
Regarding the activity of NiMoS2/CMK-3, the complete guaiacol conversion was achieved within 6 h even after 3 reaction cycles. Considering the selectivity, the selectivity towards phenol decreases from 25.4% after first cycle to 23.9% after third cycle, which is a 5.9% selectivity difference. It is noteworthy to mention that the selectivity towards cyclohexane was not affected even after the third cycle.
The characterisation of spent catalyst in Table 1 shows interesting results. A similar decrease in S:Mo ratio was noticed with NiMoS2/CMK-3 also as previously noticed with NiMoS2/AC. This data supports corelate with an increase in methylated product from 2.4% – cycle 1 to 3.9% – cycle 3. Surprisingly the mesopore surface area decrease after third cycle was only 3.5%. It is to be noted that the pore volume decreased from 0.33 cm3 g−1 to 0.21 cm3 g−1; with an amount of coke deposited of 0.7%. Hence, we can conclude that the coke formed were deposited inside the pores which reduced the pore volume of the catalyst, however coke did not block the mesopore of the catalyst and hence only 3.5% of the surface area was decreased. Hence, the remarkable activity of the NiMoS2/CMK-3 catalyst could then be attributed to the mesoporosity of the CMK-3 support.
Discussion
The influence of the difference in carbon surface porosity was not in an appreciable amount for the conversion of guaiacol with MoS2/C. Notably, when Ni was introduced as a promotor the selectivity of products changed significantly. For example, the use of NiMoS2/AC produced phenol (31.5%) and cyclohexane (35.7%) as the major products compared to the MoS2/AC where phenol (62.2%) was produced to MoS2/AC predominantly followed by cyclohexane (7.6%) (Fig. 1).The main influence of Ni to MoS2 was the increase in deoxygenation and hydrogenation activity. So, the deoxygenation of guaiacol and the hydrogenation of phenol occurred simultaneously which increased the conversion of phenol and selectivity of cyclohexane. Also, comparing the other hydrogenated compounds such as cyclohexene (10.6%) and cyclohexanol (6.3%) were similar for NiMoS2/AC (14.4% – cyclohexene, 5.3% – cyclohexanol). Hence, we can conclude that promotion of MoS2 by Ni, increases the hydrogenation tendency. On the other hand, a similar trend was not observed with CMK-3 supported catalyst. The selectivity of cyclohexene was 10.6% for MoS2/CMK-3 which reduced to 1.8% on NiMoS2/CMK-3 but accompanied by increased cyclohexane (50%) which is a product of intense hydrogenation. Thus, the promotion of Ni to MoS2/CMK-3 increases the hydrogenation as well as the deoxygenation of guaiacol to a greater extent. The kinetic diameter of guaiacol was calculated to be 0.0668 nm.59 Since most of the pores of activated carbon falls in the micropore region, the diffusion of guaiacol into the pores of activated carbon was limited. Hence, the conversion of guaiacol mostly occurred at the surface level. On the other hand, the high surface area of CMK-3 along with its mesoporosity (3.85 nm pore size) facilitates better dispersion of the active sites both within and outside of the pore channels of the support. As a result, there is an improved contact of the reactant with the catalytic active sites, thus increasing the rate of the reaction and hence, the complete conversion of guaiacol was achieved in 6 h with NiMoS2/CMK-3 which was much quicker than the 13 h required for NiMoS2/AC to achieve a similar conversion. This fact is supported by XPS data (Table S2†) that shows that Ni species are predominantly deposited inside the pores of CMK-3 (surfaceNi/Mo = 0.16) whereas, for AC (surfaceNi/Mo = 0.31), Ni was deposited mostly on the catalyst surface.
Conclusions
In summary, the microemulsion technique has been used for the successful synthesis of NiMoS2 nanocatalyst supported on two different carbon materials containing different surface porosity. The catalysts were tested for HDO of guaiacol. Phenol and cyclohexane were produced as the major products. The selectivity of products was influenced by the pore geometry of the carbon supports which is ascribed to the distribution of Ni and MoS2 active species. The Ni–Mo–S phase, along with the easy accessibility to the active phases by guaiacol in the NiMoS2/CMK-3 catalysts exhibited superior deoxygenation and hydrogenation activity than the activated carbon supported catalyst. Mesoporous carbon is a perfect choice to be used as support for hydrodeoxygenation reaction for their mesoporosity and pore geometry. Hence, the use of the high surface area, mesoporous materials as support for HDO reactions are highly recommended.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the Nanomaterials Center (NANOMAC), The University of Queensland. Sincere thanks to Dr Miaoqiang Lyu, Department of chemical engineering, The University of Queensland for the assistance with TEM analysis.References
- P. Azadi, O. R. Inderwildi, R. Farnood and D. A. King, Renewable Sustainable Energy Rev., 2013, 21, 506–523 CrossRef CAS.
- A. Shrotri, H. Kobayashi and A. Fukuoka, in Advances in Catalysis, ed. C. Song, Academic Press, 2017, vol. 60, pp. 59–123 Search PubMed.
- A. Kalliola, T. Vehmas, T. Liitiä and T. Tamminen, Ind. Crops Prod., 2015, 74, 150–157 CrossRef CAS.
- L. Hu, H. Pan, Y. Zhou and M. Zhang, BioResources, 2011, 6, 3515–3525 CAS.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- C. Zhao and J. A. Lercher, ChemCatChem, 2012, 4, 64–68 CrossRef CAS.
- E. Furimsky, Appl. Catal., A, 2000, 199, 147–190 CrossRef CAS.
- B. Joffres, M. T. Nguyen, D. Laurenti, C. Lorentz, V. Souchon, N. Charon, A. Daudin, A. Quignard and C. Geantet, Appl. Catal., B, 2016, 184, 153–162 CrossRef CAS.
- F. Rodríguez-reinoso, Carbon, 1998, 36, 159–175 CrossRef.
- A. Shrotri, H. Kobayashi and A. Fukuoka, Nanoporous Catal. Biomass Convers., 2017, 79–98 Search PubMed.
- J. F. Blandez, S. Navalón, M. Alvaro and H. Garcia, ChemCatChem, 2015, 7, 3020–3026 CrossRef CAS.
- Y. Qin, L. He, J. Duan, P. Chen, H. Lou, X. Zheng and H. Hong, ChemCatChem, 2014, 6, 2698–2705 CrossRef CAS.
- M. Ishikawa, M. Tamura, Y. Nakagawa and K. Tomishige, Appl. Catal., B, 2016, 182, 193–203 CrossRef CAS.
- I. D. Mora, E. Méndez, L. J. Duarte and S. A. Giraldo, Appl. Catal., A, 2014, 474, 59–68 CrossRef CAS.
- A. Centeno, E. Laurent and B. Delmon, J. Catal., 1995, 154, 288–298 CrossRef CAS.
- S. Mukundan, M. Konarova, L. Atanda, Q. Ma and J. Beltramini, Catal. Sci. Technol., 2015, 5, 4422–4432 RSC.
- B. Kuppan and P. Selvam, Prog. Nat. Sci.: Mater. Int., 2012, 22, 616–623 CrossRef.
- S. H. Joo, S. J. Choi, I. Oh, J. Kwak, Z. Liu, O. Terasaki and R. Ryoo, Nature, 2001, 412, 169–172 CrossRef CAS PubMed.
- X. Zhou, L.-J. Wan and Y.-G. Guo, Nanoscale, 2012, 4, 5868–5871 RSC.
- M. Konarova, F. Tang, J. Chen, G. Wang, V. Rudolph and J. Beltramini, ChemCatChem, 2014, 6, 2394–2402 CrossRef CAS.
- T. Jurkin and M. Gotic, Kem. Ind., 2013, 62, 401–415 CAS.
- M. A. Wahab and J. N. Beltramini, Int. J. Hydrogen Energy, 2014, 39, 18280–18290 CrossRef CAS.
- S. Mukundan, L. Atanda and J. Beltramini, Sustainable Energy Fuels, 2019, 3, 1317–1328 RSC.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, in Handbook of Heterogeneous Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, 2008, DOI:10.1002/9783527610044.hetcat0065.
- L. Rivoira, J. Juárez, H. Falcón, M. G. Costa, O. Anunziata and A. Beltramone, Catal. Today, 2017, 282, 123–132 CrossRef CAS.
- S. Jun, S. H. Joo, R. Ryoo, M. Kruk, M. Jaroniec, Z. Liu, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 2000, 122, 10712–10713 CrossRef CAS.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500 CrossRef CAS PubMed.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- M. A. Wahab, Y. Jia, D. Yang, H. Zhao and X. Yao, J. Mater. Chem. A, 2013, 1, 3471–3478 RSC.
- M. A. Wahab, D. J. Young, A. Karim, S. Fawzia and J. N. Beltramini, Int. J. Hydrogen Energy, 2016, 41, 20573–20582 CrossRef CAS.
- C. Fan, V. Nguyen, Y. Zeng, P. Phadungbut, T. Horikawa, D. Do and D. Nicholson, Microporous Mesoporous Mater., 2015, 209, 79–89 CrossRef CAS.
- C. G. Burgess, D. H. Everett and S. Nuttall, Pure Appl. Chem., 1989, 61, 1845–1852 CAS.
- L. Li, Z. H. Zhu, G. Q. Lu, Z. F. Yan and S. Z. Qiao, Carbon, 2007, 45, 11–20 CrossRef CAS.
- M. Ferrari, B. Delmon and P. Grange, Microporous Mesoporous Mater., 2002, 56, 279–290 CrossRef CAS.
- L. Wang, J. Liu, L. L. Zhang, B. Dai, M. Xu, M. Ji, X. S. Zhao, C. Cao, J. Zhang and H. Zhu, RSC Adv., 2015, 5, 8422–8426 RSC.
- F. Su, J. Zeng, X. Bao, Y. Yu, J. Y. Lee and X. S. Zhao, Chem. Mater., 2005, 17, 3960–3967 CrossRef CAS.
- A. G. Bagnall, W. Y. Liang, E. A. Marseglia and B. Welber, Physica B+C, 1980, 99, 343–346 CrossRef CAS.
- J. L. Pinilla, H. Purón, D. Torres, I. Suelves and M. Millan, Carbon, 2015, 81, 574–586 CrossRef CAS.
- H. Kobayashi, H. Kaiki, A. Shrotri, K. Techikawara and A. Fukuoka, Chem. Sci., 2016, 7, 692–696 RSC.
- D. Lennon, D. Lundie, S. Jackson, G. Kelly and S. Parker, Langmuir, 2002, 18, 4667–4673 CrossRef CAS.
- J. F. Moulder, Handbook of X-ray photoelectron spectroscopy, Physical Electronics, 1995, pp. 230–232 Search PubMed.
- M. Ferrari, R. Maggi, B. Delmon and P. Grange, J. Catal., 2001, 198, 47–55 CrossRef CAS.
- J. Chang, T. Danuthai, S. Dewiyanti, C. Wang and A. Borgna, ChemCatChem, 2013, 5, 3041–3049 CrossRef CAS.
- Y.-C. Lin, C.-L. Li, H.-P. Wan, H.-T. Lee and C.-F. Liu, Energy Fuels, 2011, 25, 890–896 CrossRef CAS.
- V. N. Bui, D. Laurenti, P. Afanasiev and C. Geantet, Appl. Catal., B, 2011, 101, 239–245 CrossRef CAS.
- V. N. Bui, D. Laurenti, P. Delichère and C. Geantet, Appl. Catal., B, 2011, 101, 246–255 CrossRef CAS.
- P. E. Ruiz, B. G. Frederick, W. J. De Sisto, R. N. Austin, L. R. Radovic, K. Leiva, R. García, N. Escalona and M. C. Wheeler, Catal. Commun., 2012, 27, 44–48 CrossRef CAS.
- A. R. Ardiyanti, S. A. Khromova, R. H. Venderbosch, V. A. Yakovlev and H. J. Heeres, Appl. Catal., B, 2012, 117–118, 105–117 CrossRef CAS.
- Y. Yang, C. Ochoa-Hernández, V. A. de la Peña O'Shea, J. M. Coronado and D. P. Serrano, ACS Catal., 2012, 2, 592–598 CrossRef CAS.
- N. Escalona, W. Aranzaez, K. Leiva, N. Martínez and G. Pecchi, Appl. Catal., A, 2014, 481, 1–10 CrossRef CAS.
- M. V. Bykova, O. A. Bulavchenko, D. Y. Ermakov, M. Y. Lebedev, V. A. Yakovlev and V. N. Parmon, Catal. Ind., 2011, 3, 15–22 CrossRef.
- H. Topsøe, B. S. Clausen and F. E. Massoth, in Catalysis, Springer, 1996, pp. 1–269 Search PubMed.
- I. T. Ghampson, C. Sepúlveda, R. Garcia, L. R. Radovic, J. L. G. Fierro, W. J. DeSisto and N. Escalona, Appl. Catal., A, 2012, 439–440, 111–124 CrossRef CAS.
- J. J. Lee, S. Han, H. Kim, J. H. Koh, T. Hyeon and S. H. Moon, Catal. Today, 2003, 86, 141–149 CrossRef CAS.
- Z. Lei, L. An, L. Dang, M. Zhao, J. Shi, S. Bai and Y. Cao, Microporous Mesoporous Mater., 2009, 119, 30–38 CrossRef CAS.
- M. S. Zanuttini, B. O. Dalla Costa, C. A. Querini and M. A. Peralta, Appl. Catal., A, 2014, 482, 352–361 CrossRef CAS.
- A. Popov, E. Kondratieva, J. M. Goupil, L. Mariey, P. Bazin, J.-P. Gilson, A. Travert and F. Maugé, J. Phys. Chem. C, 2010, 114, 15661–15670 CrossRef CAS.
- S. Echeandia, P. L. Arias, V. L. Barrio, B. Pawelec and J. L. G. Fierro, Appl. Catal., B, 2010, 101, 1–12 CrossRef CAS.
- H. Lee, H. Kim, M. J. Yu, C. H. Ko, J.-K. Jeon, J. Jae, S. H. Park, S.-C. Jung and Y.-K. Park, Sci. Rep., 2016, 6, 28765 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The details containing the detailed physical and chemical characterisation of carbon supports and as-synthesised supported metal catalysts. See DOI: 10.1039/c9ra02143d |
| This journal is © The Royal Society of Chemistry 2019 |