
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-covalent loading of ionic liquid-functionalized nanoparticles for bovine serum albumin: experiments and theoretical analysis†
Xingang Jiaab,
Xiaoling Hu *a,
Wenzhen Wangb and
Chunbao Dub
*a,
Wenzhen Wangb and
Chunbao Dub
aSchool of Natural and Applied Science, Northwestern Polytechnical University, Xi'an 710072, P. R China. E-mail: huxl054@163.com
bCollege of Chemistry and Chemical Engineering, Xi'an Shiyou University, Xi'an 710065, P. R. China
First published on 18th June 2019
Abstract
Biomacromolecule-based nanomaterials have attracted much attention due to their excellent function in sensing, catalysis, medicine, biology and recognition. In this work, a silane-coupling ionic liquid, 1-(3-trimethoxysilylpropyl)-3-methylimidazolium chloride ([TMIM]Cl), was synthesized and applied to prepare ionic liquid-functionalized nanoparticles (SiO2@IL) using surface grafting technology. By employing multiple non-covalent interactions, including electrostatic interactions, hydrogen bonding and π–π stacking, the obtained functional nanoparticles were able to bind bovine serum albumin (BSA) with strong binding affinity, which has been illustrated through experiments and theoretical calculations. Moreover, the stability of SiO2@IL further demonstrated that it is promising in applications for biomacromolecule immobilization.
Introduction
Biomacromolecules, a promising molecular recognition vehicle, have received extensive attention owing to their precise sites, exceptional functionality and easily degradable properties.1–4 Due to their superior biocompatible properties, different types of biomacromolecules, such as DNA, peptides, proteins, enzymes, viruses, etc., have been integrated with nano-based materials, and the fabricated nanomaterial-biomacromolecules have been used in extensive applications in materials science, nanotechnology and biomedicine.5–10 Proteins are an important building block in organisms, for genetic information storage and transfer, and this has been utilized in combination with nano-based materials to create many functional nanomaterials.11–14 Silica (SiO2) nanoparticles are one of the most commonly used nanomaterials in the biological field due to their favorable biocompatibility, and they have been applied to many areas of protein research.15–17 However, at present, there are still many challenges in constructing functional SiO2 for the immobilization of protein; for example, monotonous groups or chains grafted onto the surface of SiO2 can result in defects of unstable immobilization. Moreover, because of the complex structure and multiple types of binding sites of proteins, monotonous binding sites of groups or chains might not satisfy the demands of the protein. Therefore, there are great opportunities to design and exploit multi-functional SiO2 to immobilize protein for biomedical applications.Recently, ionic liquids (ILs) have been regarded as promising “green materials” in various fields, such as electrochemistry, separation chemistry, catalytic chemistry and biological chemistry, due to their excellent properties and wide applications.18,19 Because of their abilities in functional design, ILs have been used in many aspects of biomolecules. Our group have utilized an imidazole-based amphiphilic IL, 1-dodecyl-3-methylimidazolium chloride as a surfactant to immobilize bovine serum albumin (BSA) in the molecular imprinting process, which exhibited a good stabilization effect on BSA.20 Some studies of the application of IL-functionalized materials for biological application are those of Song et al., who prepared an acidic IL-modified SiO2 gel for selective separation of BSA and bovine hemoglobin (BHb).21 Subsequently, Qian et al. immobilized BSA on the surface of ionic liquid-functionalized magnetic Fe3O4 nanoparticles for the development of a surface imprinting strategy.22 Therefore, ILs not only could be used directly, but also could be grafted on the surface of nanomaterials for various biological applications. With respect to practicality and facility, the design and construction of IL-functionalized, modified or grafted nanoparticles is very promising.
In this work, a silane-coupling ionic liquid, 1-(3-trimethoxysilylpropyl)-3-methylimidazolium chloride ([TMIM]Cl) was first synthesized and grafted onto SiO2 nanoparticles to construct functional SiO2 nanoparticles (SiO2@IL). Thereafter, the SiO2@IL nanoparticles obtained were applied to the immobilization of BSA by using the multiple binding sites via multiple non-covalent bonds (shown in Scheme 1). To reveal the binding mechanism, theoretical calculation of density functional theory (DFT) was employed to investigate the multifunctionality of the imidazolium group on the surface of SiO2@IL. The nanoparticles were well characterized to monitor every step of processing. Subsequently, the adsorption equilibrium, kinetics and stability of SiO2@IL for BSA immobilization were investigated.
 | ||
| Scheme 1 Schematic diagram of preparation of SiO2@IL and BSA bound onto the surface of SiO2@IL. | ||
Experimental
Materials
N-Methylimidazole (MI, Shanghai Dibo Chemical Technology Co., Ltd), γ-chloropropyl(trimethoxy)-silane (CPTMO, Aladdin), and benzoquinone (BQ, Aladdin) were used without further purification. Bovine serum albumin (BSA, Mw 66.4 kDa, Sigma-Aldrich) was used in immobilization studies. Ethyl acetate, tetraethoxysilane (TEOS), ammonium hydroxide, and methylbenzene were purchased from Sinopharm Chemical Reagent Co., Ltd, China.Characterization
The morphology, chemical surface characterization, and surface potential of the nanoparticles were characterized by scanning electron microscopy (SEM, FEI Quanta 400 FEG), X–ray photoelectron spectroscopy (XPS, Shimadzu) and zeta (ζ)-potential measurement (ZEN3690 Zetasizer Nano detector, Malvern), respectively. The concentration of BSA in the adsorption experiments was detected using a UV-2550 (Shimadzu) spectrophotometer at 277 nm.Synthesis of [TMIM]Cl
[TMIM]Cl was synthesized by a one-step alkylation reaction.23,24 Typically, CPTMO (10.06 mL), MI (5.00 mL) and BQ (52 mg) were dispersed together and the mixture was treated ultrasonically to form the homogeneous solution. After exposure to nitrogen purge for 30 min, the solution was placed in a water bath for 72 h at 80 °C. After that, the crude fluid was washed with ethyl acetate four times and dried under vacuum at 55 °C for 48 h to obtain the pure product with a dark brown color.Preparation and functionalization of SiO2 nanoparticles
SiO2 nanoparticles were prepared using the method reported by Stöber.25 Typically, ammonium hydroxide (27 mL), ethyl alcohol (48.75 mL) and distilled water (74.25 mL) were mixed together. Then TEOS (13.5 mL) and ethyl alcohol (136.5 mL) were added into the mixture rapidly under vigorous stirring for 2 h at 25 °C. After the reaction, the white product was washed with ethyl alcohol and distilled water by centrifugal separation, respectively. Then the obtained SiO2 nanoparticles were dried under 45 °C until the weight remained constant.The functionalization of SiO2 was conducted as follows. [TMIM]Cl (0.7 g), ethyl alcohol (10 mL) and methylbenzene (40 mL) were mixed together to form the homogeneous solution. Then the obtained SiO2 nanoparticles (0.6 g) were added to the above solution and kept at 50 °C for 8 h under nitrogen. After the reaction, the obtained SiO2@IL was washed with ethyl alcohol by centrifugation at 6500 rpm for 6 min and dried under vacuum at 40 °C for 24 h.
Adsorption and elution experiments
SiO2@IL nanoparticles (10 mg) were first washed once using phosphate buffer saline (PBS, 0.01 M) and BSA solution (PBS, 10 mL, pH = 7.0) added to certain concentrations. The mixture was stirred at 200 rpm for 24 h at 25 °C to facilitate adsorption of non-covalent self-assembly BSA with SiO2@IL. Then, the nanoparticles were washed with deionized water until no BSA was detected using the UV-2550 spectrophotometer at 277.0 nm detection wavelength.The immobilization capacity (Qe) of the nanoparticles for BSA was calculated using the following equation:
 | (1) |
The nanoparticles were washed for a few cycles with a solution containing acetic acid (10%, v/v) and purified water. The complete removal of BSA was confirmed using the UV-2550 spectrophotometer.
Theoretical calculation
To investigate the multiple binding mechanisms of imidazolium groups for biomolecules, DFT at B3LYP/6-311+G (d, p) level using Gaussian 16 was used to calculate the binding energies of imidazolium and the special groups of BSA.26,27 Here, the value of BSSE was ignored because of the large basis sets used in this work.28 Therefore, the binding strength could be described by the binding energy ΔE, and ΔE for each compound can be obtained via eqn (2) as follows:| ΔE = ΔE(imidazolium group of BSA) − [ΔE(imidazolium) + ΔE(group of BSA)] | (2) |
Results and discussion
Synthesis of [TMIM]Cl
The successful synthesis of [TMIM]Cl (C10SiO3N2ClH21) was characterized by using Fourier-transform-infrared (FTIR) and 1H NMR spectroscopy. As shown in Fig. S1,† typical characteristic peaks were found at 1548 and 1454 cm−1, which were assigned to the stretching vibration of the imidazolium groups. Moreover, the typical peaks of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, Si–O and Si–C stretching vibrations could be seen at 1654, 810/1055 and 1240/1410 cm−1, respectively. In order to confirm the synthesis of [TMIM]Cl accurately, the 1H NMR spectrum was acquired. As shown in Fig. S2,† the chemical shifts were at 8.86 (1H, s), 7.67 (1H, s), 6.82 (1H, s), 4.39 (2H, t), 3.89 (3H, s), 3.16 (9H, m), 1.59 (2H, m) and 0.67 (2H, m), respectively. Due to the fact that the reactants could be washed with the solvent ethyl acetate, and that ethyl acetate was easily removed under vacuum, the obtained product was relatively pure. For the IL-functionalization of SiO2 in this work, the obtained product could meet requirements because some subsequent washing procedures were conducted repeatedly, which undoubtedly eluted the reactants.
N, Si–O and Si–C stretching vibrations could be seen at 1654, 810/1055 and 1240/1410 cm−1, respectively. In order to confirm the synthesis of [TMIM]Cl accurately, the 1H NMR spectrum was acquired. As shown in Fig. S2,† the chemical shifts were at 8.86 (1H, s), 7.67 (1H, s), 6.82 (1H, s), 4.39 (2H, t), 3.89 (3H, s), 3.16 (9H, m), 1.59 (2H, m) and 0.67 (2H, m), respectively. Due to the fact that the reactants could be washed with the solvent ethyl acetate, and that ethyl acetate was easily removed under vacuum, the obtained product was relatively pure. For the IL-functionalization of SiO2 in this work, the obtained product could meet requirements because some subsequent washing procedures were conducted repeatedly, which undoubtedly eluted the reactants.
Preparation of SiO2 nanoparticles
The morphological features of the SiO2 nanoparticles were first observed using SEM. Fig. 1a and b show that the SiO2 nanoparticles are spherical in shape and exhibit a relatively uniform morphology. Moreover, the average size of the SiO2 nanoparticles was obtained by dynamic light scattering (Fig. 1c), and the results established that the average size of the SiO2 nanoparticles was about 350 nm, with a narrow size distribution. In addition, XPS was performed to characterize the surface chemical composition of SiO2 nanoparticles using Gaussian and Lorentzian curve fitting.29 Fig. 1d demonstrates that the SiO2 nanoparticles consisted of C, O and Si, with the atomic compositions of C, O and Si calculated as 63.2, 27.5 and 9.3 atom%, respectively.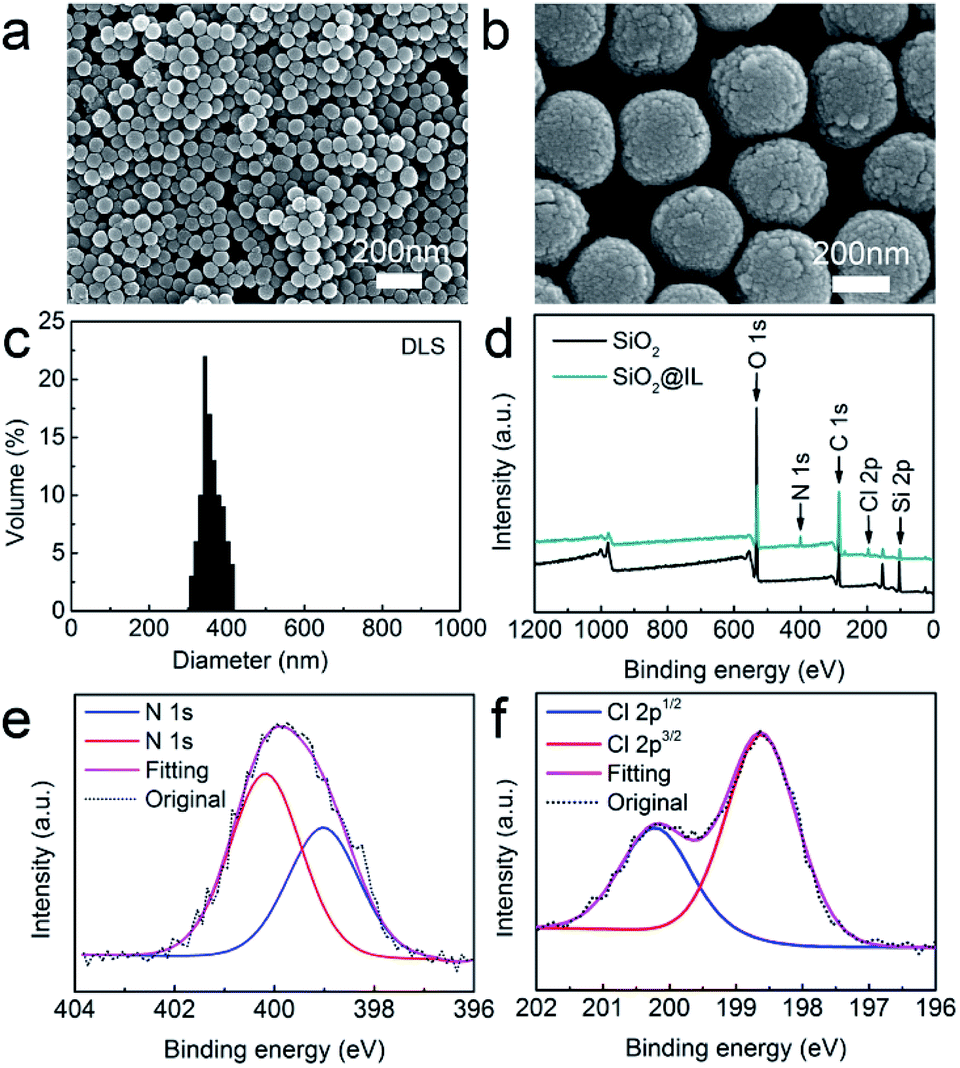 | ||
| Fig. 1 (a) SEM images of SiO2 nanoparticles. (b) SEM images of SiO2 nanoparticles at high resolution. (c) DLS size distribution of SiO2 nanoparticles. (d) XPS survey spectra of SiO2 nanoparticles and SiO2@IL. (e) N 1s XPS spectra of SiO2@IL. (f) Cl 2p XPS spectra of SiO2@IL. | ||
Ionic liquid functionalization of SiO2 nanoparticles
In order to determine the surface functionalization of SiO2 nanoparticles, XPS was also employed to investigate the surface elements and chemical composition. As shown in Fig. 1d, SiO2@IL consisted of C, O, Si, N and Cl, which were calculated with compositions of 61.7, 25.4, 8.2, 3.9 and 0.8 atom%, respectively. Fig. 1e shows that there were two peaks at 400.6 and 398.5 eV in N 1s, which was assigned as the N of the imidazolium groups. In addition, Fig. 1f shows that only one component of Cl 2p (Cl 2p3/2 and Cl 2p1/2) existed after deconvolution. This binding energy of Cl 2p3/2 showed that it was solely associated with Cl− which was in good agreement with the reference.30 According to analysis of the XPS results, it was confirmed that the ionic liquid functionalization of SiO2@IL had been carried out successfully.Batching isotherm
The adsorption isotherm experiments of SiO2@IL were conducted with BSA concentrations in the range 0.20–1.60 mg mL−1 (pH 7.0, PBS) to confirm the superiority of the properties of the ionic liquid. As shown in Fig. 2a, the immobilization capacity of SiO2@IL for BSA increased with the initial concentration of BSA in the range 0.20–0.80 mg mL−1. In addition, a stable immobilization capacity of 23.1 mg g−1 was obtained with the increasing of the concentration of BSA from 0.80 mg mL−1. As for SiO2, the immobilization capacity was always at a low value, indicating that the functional groups on the surface of SiO2@IL could facilitate the binding of SiO2@IL for BSA. The maximum immobilization capacity of SiO2@IL for BSA was improved more than tenfold. In PBS with pH 7.0, the functional groups and imidazolium groups could be ionized and form multiple binding sites with BSA. Considering BSA with a pI of 4.7, it was easier for imidazolium groups to bind BSA. For unmodified SiO2, only a very small amount of hydroxyl groups existed, so it was very difficult for SiO2 to bind BSA. | ||
| Fig. 2 (a) Immobilization capacity of SiO2@IL and SiO2 for BSA. (b) Adsorption isotherm curves of SiO2@IL for BSA. (c) Adsorption kinetic curves of SiO2@IL and SiO2. (d) Effect of pH on immobilization capacity of SiO2@IL and SiO2 for BSA. | ||
In addition, Langmuir and Freundlich isotherm models were performed to investigate the adsorption mechanism of SiO2@IL for BSA as follows:31
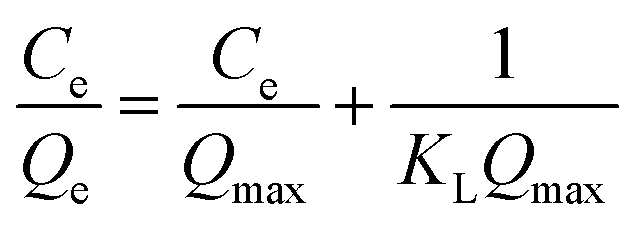 | (3) |
| Qe = KFCe1/n | (4) |
Fig. 2b shows the Langmuir and Freundlich adsorption equilibrium tendency of SiO2@IL for BSA. According to the correlation coefficients (R2), the Langmuir isotherm model (R2 = 0.9822) was found to be more suitable than the Freundlich model (R2 = 0.9182) for revealing the binding mechanism of SiO2@IL for BSA, indicating that the binding was uniform with a maximal immobilization capacity Qm of 37.9 mg g−1, that is, there was only one kind of binding site on the surface of SiO2@IL. It should be noted that one kind of binding site does not mean one type of interaction. Multiple interactions behaving synergistically could be regarded as one kind of binding site, which will be revealed clearly in the following quantum calculation. From another point of view, the ionic liquid functionalization on the surface of SiO2 nanoparticles was homogeneous, leading to the uniform properties of the surface.
Rebinding kinetics study
Here, the adsorption dynamics of SiO2@IL and SiO2 for BSA were investigated. As shown in Fig. 2c, there was a rapid increase of adsorption rate on SiO2@IL for BSA in the first 30 min and then the adsorption rate slowed down from 30 min to 2 h. After about 1 h, adsorption equilibrium was achieved, which indicated that BSA could reach the binding sites of SiO2@IL easily and quickly. The large number of multiple binding sites provided by the imidazolium groups of SiO2@IL could attract the functional groups of BSA, which induced the rapid binding of BSA. Moreover, the binding sites of SiO2@IL for BSA were all on the surface of the SiO2@IL, which could also endow lower mass-transfer resistance. Compared with the low affinity of SiO2 for BSA, the results confirmed that the functionalized SiO2 with the properties of the ionic liquid in this work could exhibit high affinity and rapid adsorption rate because of the driving force of multiple binding sites on the surface.Effect of pH on immobilization capacity
Since the pI of BSA is 4.7, the net charge of BSA in PBS would vary with different pH, because the functional groups, the imidazolium groups, which possess the ionization property, and the immobilization capacity of SiO2@IL for BSA can be affected by pH. Therefore, the immobilization capacity of SiO2@IL for BSA was investigated by changing the pH of PBS.As shown in Fig. 2d, the immobilization capacity of SiO2@IL for BSA increased at first and then decreased, with increase in pH. The maximum immobilization capacity (Qe = 23.1 mg g−1) was obtained with pH 7.0. Under pH 4.0, the net charge of BSA was positive. As a result, it might not be easy for SiO2@IL with positive imidazolium groups to bind BSA due to electronic repulsion. On the other hand, when the pH of PBS was higher than 7.0, it should be helpful for SiO2@IL to bind BSA, and the immobilization capacity should be higher than that in PBS with a pH of 7.0. However, the immobilization capacity decreased, which might be owing to the following reasons. The decreased immobilization capacities of SiO2@IL for BSA at pH of 8 and 9 were most likely due to the change in binding sites, which were not as strong as those at pH 7. When the pH is set at 10, the unfolding of BSA might be responsible for the decrease of immobilization capacity of SiO2@IL for BSA for the above reason.32 That is to say, the redistributed binding sites were unfavorable for binding on the surface of SiO2@IL, which would induce the result of a low immobilization capacity of SiO2@IL for BSA. It should be noted that the other non-covalent bonds would also induce binding of SiO2@IL for BSA. As for SiO2, because of the simple binding sites with hydroxyl groups, it was easy to come to the conclusion that the weak binding affinity of SiO2 would not be affected significantly by pH.
Surface charge properties of SiO2@IL for binding BSA
ζ-Potential is a very effective measurement to study the surface charge properties of nanoparticles in solution. In order to reveal the effect of net charge on the surface of SiO2@IL for BSA, ζ-potential measurements were conducted by choosing a pH of 4.0, 5.0, 6.0, 7.0 and 8.0. As shown in Table 1, with increase in pH, the ζ-potential value of the surface of SiO2 and SiO2@IL in PBS is reduced, indicating that the surface properties of SiO2 and SiO2@IL could be controlled by the proton concentration. In other words, the adsorption properties of SiO2 and SiO2@IL for BSA were affected by pH. Some typical results have shown that electrostatic interaction was the important driving force for the adsorption, especially for the SiO2-based nanoparticles.33,34 Due to the differences in the pH environment and the change in the secondary structure of BSA at different pH (pH < 4 and pH > 9),32 the binding sites of SiO2 and SiO2@IL for BSA would also be different. However, compared with SiO2, SiO2@IL exhibited stronger affinity for BSA under the same pH environment (Fig. 1d). In other words, it is clear that electrostatic interactions are a very important driving force for biomolecules to be bound to the surface of nanoparticles.35–37 Therefore, it is acceptable to construct the nanoparticles with special properties on their surface for binding or immobilizing biomacromolecules.| Sample | pH | ζ-Potential (mV) |
|---|---|---|
| SiO2@IL | 4.0 | 35.2 ± 1.1 |
| 5.0 | 32.8 ± 0.9 | |
| 6.0 | 23.6 ± 1.8 | |
| 7.0 | 25.7 ± 0.3 | |
| 8.0 | 8.4 ± 0.7 | |
| SiO2 | 4.0 | 0.6 ± 0.6 |
| 5.0 | −4.1 ± 0.5 | |
| 6.0 | −29.7 ± 0.3 | |
| 7.0 | −42.2 ± 0.4 | |
| 8.0 | −61.6 ± 0.7 |
Binding mechanism of SiO2@IL for BSA
Currently, computer simulation/calculation is often combined with experimental studies to explore detailed interaction mechanisms. In research in molecular simulations, molecular dynamics simulation and quantum mechanics calculations have been applied to different research systems.38,39 For systems with more than 500 atoms, it is not easy to calculate their properties by using quantum mechanics simulation. To uncover their role in adsorption on the surface of SiO2@IL, we have simplified the models and calculated the binding energies of different functional groups of BSA with the functional groups on the surface of SiO2@IL, namely imidazolium groups. For this, various molecules, including CH3–X (X= –OH, –SH, –NH2, –COOH, –SSCH3 and –C6H5), were chosen as the models of the functional groups of BSA.40 Then, to illustrate the functionality of ionic liquid-based properties on the surface of SiO2@IL, first-principles calculations based on DFT were performed to evaluate the physical adsorption of BSA on the surface of SiO2@IL.Fig. 3 describes the stable conformations of the functional groups of BSA with 1-propyl-3-ethylimidazolium (PEIM), which clearly illustrate the non-covalent bindings of the electron bond donors and electron bond acceptors. It was found that the typically polar atoms S, O and N with lone-pair electrons in Fig. 3a–e were more likely to form non-covalent bonding with the H of the imidazolium group. As shown in Fig. 3f, an edge-to-face π–π stacking interaction formed between the H of the imidazolium group and the benzene ring. In the previous discussion, due to the fact that the immobilization capacity of SiO2@IL for BSA changed significantly with changing pH, it was quite clear that there were electrostatic interactions between the imidazolium groups and BSA. What is more, Table 2 reveals that the binding energies (ΔE) of these compounds demonstrated some differences, and the π–π stacking interaction was the weakest non-covalent interaction.
 | ||
| Fig. 3 Hydrogen bonding and π–π stacking interactions in the complexes of PEIM and models of functional groups of BSA, namely, CH3–SH (a), CH3–OH (b), CH3–SSCH3 (c), CH3–COOH (d), CH3–NH2 (e) and CH3–C6H5 (f). | ||
| Compound | ΔE (kJ mol−1) | Distance (Å) | BCP | ρ | ∇2ρ | |
|---|---|---|---|---|---|---|
| PEIM | CH3–SH | −4.1 | 2.751 | 0 | — | — |
| CH3–OH | −10.7 | 2.049 | 1 | 0.01961 | 0.06287 | |
| CH3–SSCH3 | −24.9 | 2.898 | 0 | — | — | |
| CH3–COOH | −7.5 | 2.136 | 1 | 0.01399 | 0.05031 | |
| CH3–NH2 | −12.6 | 2.129 | 0 | — | — | |
| CH3–C6H5 | −1.0 | — | — | — | — | |
Lipkowski et al. have found that the following criteria could be used to confirm hydrogen bonding and these criteria are universal and essential: existence of bond critical points (BCP), density total (ρ) and Laplacian eigenvalue (∇2ρ) being in the range of approximately 0.002–0.04 a.u. and 0.02–0.15 a.u., respectively.41 The binding energies and distances are listed in Table 2. To further reveal the specific types of hydrogen binding that exist between SiO2@IL and BSA, demonstrating whether the interactions between electron donors of the functional groups of BSA and electron acceptor of PEIM were hydrogen bonding interactions or not, the atoms-in-molecules (AIM) electron density topological analysis (proposed by Bader et al.42–44) for these compounds was conducted. Based on the above theory, the topological properties of BCP for the compounds at MP2/6-311+G(d,p) level was obtained (shown in Table 2). It was found that BCPs existed for CH3–OH (in Fig. 3b) and CH3–COOH (in Fig. 3d), while no BCP could be formed in the other compounds. As shown in Table 2, both ρ and ∇2ρ of the compounds PEIM/CH3–OH and PEIM/CH3–COOH were in the range of 0.002–0.04 a.u. and 0.02–0.15 a.u., respectively, indicating that there were typical hydrogen-bonding interactions in PEIM/CH3–OH and PEIM/CH3–COOH. Kubiak-Ossowska et al. have shown that electrostatics play an important role in the adsorption of charged protein on SiO2 nanoparticles by atomistic molecular dynamics.45 Analogously, electrostatics was also the important driving force for BSA adsorption on the charged SiO2@IL. However, multiple interactions commonly exist due to the diversity of biomacromolecules and carriers, including ion–ion electrostatic interactions (20–80 kcal mol−1), coordination bonding (20–50 kcal mol−1), hydrogen bonding (1–30 kcal mol−1), π–π stacking (0–12 kcal mol−1), and van der Waals interactions.30,46 Combined with the above analysis, it could be confirmed that the adsorption of SiO2@IL was driven by the synergistic interactions of electrostatic interaction, hydrogen bonding and π–π stacking. The differences between these non-covalent interactions were the probability and binding strength, which obey the Boltzmann distribution.47 The non-directional, non-covalent interaction (electrostatic interaction) and directional non-covalent interactions (hydrogen bonding, π–π stacking) synergistically varied the functionalization of biomacromolecule–nanoparticle complexes.
Stability of SiO2@IL
The stability of SiO2@IL is critical for its potential applications. Herein, adsorption–desorption tests for SiO2@IL were conducted for five cycles. As shown in Fig. 4, there was almost no change in immobilization capacity of SiO2@IL for BSA after five cycles at 1.0 mg mL−1 of BSA. These results show that the binding sites on the surface of SiO2@IL were very stable. Moreover, due to the grid structure of SiO2, the core of SiO2@IL was also stable, which ensured the stability of SiO2@IL. Therefore, SiO2@IL possesses the potential for reusability, which lays the foundation for practical applications.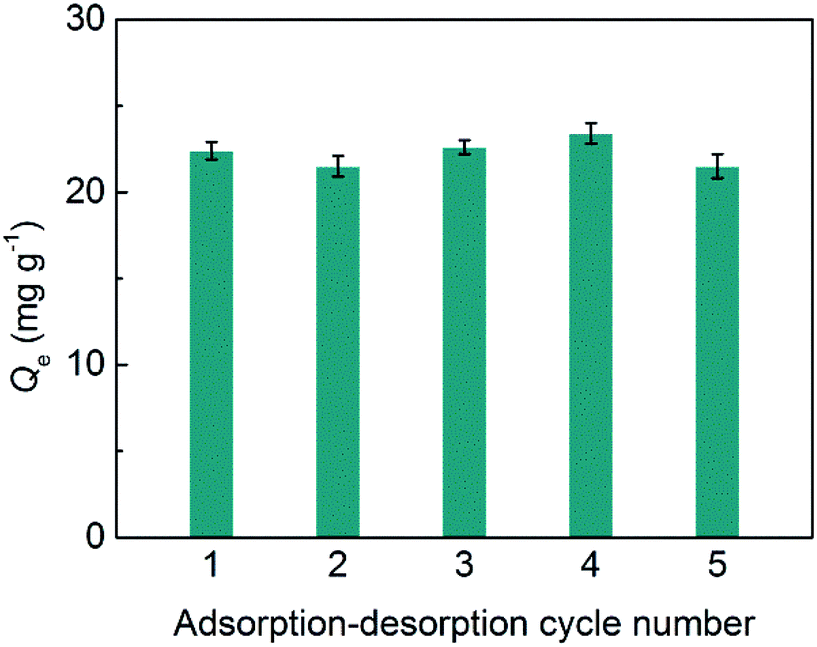 | ||
| Fig. 4 Stability of SiO2@IL for binding BSA. | ||
Conclusions
In this work, a functional nanoparticle with ionic liquid properties was prepared by using a one-step grafting reaction. The obtained SiO2@IL showed good binding ability for BSA and the adsorption experiment and theoretical calculations revealed that the binding of SiO2@IL for BSA was driven by multiple binding interactions, such as electrostatic interactions, hydrogen bonding and π–π stacking. In particular, it was found that electrostatic interactions were the driving force for SiO2@IL to immobilize BSA. The results reported in this work demonstrate the promising potential for exploring designed nanoparticles with ionic liquid properties for biomacromolecule applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
X. Jia and X. Hu are grateful for the financial support provided by the National Natural Science Foundation of China (grant no. 51433008). Moreover, X. Jia, W. Wang and C. Du thank the Modern Analysis and Testing Center of Xi'an Shiyou University.Notes and references
- A. E. Nel, L. Mädler, D. Velegol, T. Xia, E. M. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- M. Mahmoudi, I. Lynch, M. Ejtehadi, M. P. Monopoli, F. Bombelli and S. Laurent, Chem. Rev., 2011, 111, 5610–5637 CrossRef CAS PubMed.
- D. Li, B. Ji, K. Hwang and Y. Huang, J. Phys. Chem. B, 2016, 114, 3060–3069 CrossRef PubMed.
- Y. Cheng, L.-D. Koh, F. Wang, D. Li, B. Ji, J. Yeo, G. Guan, M. Han and Y.-W. Zhang, Nanoscale, 2017, 9, 9181–9189 RSC.
- Z. Li, H. Yin, Z. Zhang, K. L. Liu and J. Li, Biomacromolecules, 2012, 13, 3162–3172 CrossRef CAS PubMed.
- L. Shang and G. U. Nienhaus, Mater. Today, 2013, 16, 58–66 CrossRef CAS.
- Z. Li and X. J. Loh, Chem. Soc. Rev., 2015, 44, 2865–2879 RSC.
- Y. Min, J. M. Caster, M. J. Eblan and A. Z. Wang, Chem. Rev., 2015, 115, 11147–11190 CrossRef CAS PubMed.
- C. Du and B. Han, Acta Phys.-Chim. Sin., 2019 DOI:10.3866/PKU.WHXB201905058.
- C. Du, X. Hu, G. Zhang and Y. Cheng, Acta Phys.-Chim. Sin., 2019 DOI:10.3866/PKU.WHXB201812057.
- N. P. King, W. Sheffler, M. R. Sawaya, B. S. Vollmar, J. P. Sumida, I. André, T. Gonen, T. O. Yeates and D. Baker, Science, 2012, 336, 1171–1174 CrossRef CAS PubMed.
- C. Y. K. Chan, Z. J. Zhao, J. W. Y. Lam, J. Z. Liu, S. M. Chen, P. Lu, F. Mahtab, X. J. Chen, H. H. Y. Sung, H. S. Kwok, Y. G. Ma, I. D. Williams, K. S. Wong and B. Z. Tang, Adv. Funct. Mater., 2012, 22, 3170–3180 CrossRef.
- N. P. King, J. B. Bale, W. Sheffler, D. E. McNamara, S. Gonen, T. Gonen, T. O. Yeates and D. Baker, Nature, 2014, 510, 103–108 CrossRef CAS PubMed.
- G. Guan, J. Xia, S. Liu, Y. Cheng, S. Bai, S. Y. Tee, Y.-W. Zhang and M. Y. Han, Adv. Mater., 2017, 29, 1700326 CrossRef PubMed.
- M. Madliger, M. Sander and R. P. Schwarzenbach, Environ. Sci. Technol., 2010, 44, 8870–8876 CrossRef PubMed.
- Q. Liu, J. Shi, M. Cheng, G. Li, D. Cao and G. Jiang, Chem. Commun., 2012, 48, 1874–1876 RSC.
- H. Kim, J. Hong, Y. U. Park, K. Jinsoo, H. Insang and K. Kisuk, Adv. Funct. Mater., 2015, 25, 5823–5832 CrossRef.
- X. Gao, X. Hu, P. Guan, C. Du, S. Ding, X. Zhang, B. Li, X. Wei and R. Song, RSC Adv., 2016, 6, 110019–110031 RSC.
- A. J. Holding, A. Parviainen, I. Kilpeläinen, A. Soto, A. W. T. King and H. Rodríguez, RSC Adv., 2017, 7, 17451–17461 RSC.
- N. Zhang, X. Hu, P. Guan, C. Du, J. Li, L. Qian, X. Zhang, S. Ding and B. Li, Chem. Eng. J., 2017, 317, 356–367 CrossRef CAS.
- H. Song, C. Yang, A. Yohannes and S. Yao, RSC Adv., 2016, 6, 107452–107462 RSC.
- L. Qian, J. Sun, C. Hou, J. Yang, Y. Li, D. Lei, M. Yang and S. Zhang, Talanta, 2017, 168, 174–182 CrossRef CAS PubMed.
- J. F. P. Liévanoa and L. A. C. Díaz, Mater. Res., 2016, 19, 534–541 CrossRef.
- T. Ishii, T. Enoki, T. Mizumo, J. Ohshita and Y. Kaneko, RSC Adv., 2015, 5, 15226–15232 RSC.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian, Inc., Wallingford CT, 2016.
- X. Cui, W. Cai and X. Shao, RSC Adv., 2016, 6, 105729–105736 RSC.
- A. Kowalska-Baron, Comput. Theor. Chem., 2015, 1057, 7–14 CrossRef CAS.
- C. Du, X. Hu, P. Guan, X. Gao, R. Song, J. Li, L. Qian, N. Zhang and L. Guo, J. Mater. Chem. B, 2016, 4, 1510–1519 RSC.
- C. Du, X. Hu, P. Guan, L. Guo, L. Qian, R. Song, J. Li and C. Wang, J. Mater. Chem. B, 2015, 3, 3044–3053 RSC.
- J. Li, X. Hu, P. Guan, X. Zhang, L. Qian, R. Song, C. Du and C. Wang, RSC Adv., 2015, 5, 62697–62705 RSC.
- K. Kubiak-Ossowska, K. Tokarczyk, B. Jachimska and P. A. Mulheran, J. Phys. Chem. B, 2017, 121, 3975–3986 CrossRef CAS PubMed.
- B. E. Giverns, N. D. Diklich, J. Fiegel and V. H. Gressian, Biointerphases, 2017, 12, 02D404 CrossRef PubMed.
- B. E. Giverns, E. Wilson and J. Fiegel, Colloids Surf., B, 2019, 179, 374–381 CrossRef PubMed.
- M. Wiśniewska, K. Szewczuk-Karpisz and D. Sternik, J. Therm. Anal. Calorim., 2015, 120, 1355–1364 CrossRef.
- B. E. Givens, Z. Xu, J. Fiegel and V. H. Grassian, J. Colloid Interface Sci., 2017, 493, 334–341 CrossRef CAS PubMed.
- L. Shang and G. U. Nienhaus, Acc. Chem. Res., 2017, 50, 387–395 CrossRef CAS PubMed.
- Y. Cheng, L. Koh, D. Li, B. Ji, Y. Zhang, J. Yeo, G. Guan, M. Y. Han and Y.-W. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 21787–21796 CrossRef CAS PubMed.
- J. Gao and F. Ding, Angew. Chem., Int. Ed., 2014, 53, 14031–14035 CrossRef CAS PubMed.
- G. Guan, S. Zhang, S. Liu, Y. Cai, M. Low, C. P. Teng, I. Y. Phang, Y. Cheng, K. L. Duei, B. M. Srinivasan, Y. Zheng, Y.-W. Zhang and M.-Y. Han, J. Am. Chem. Soc., 2015, 137, 6152–6155 CrossRef CAS PubMed.
- P. Lipkowski, S. J. Grabowski, T. L. Robinson and J. Leszczynski, J. Phys. Chem. A, 2004, 108, 10865–10872 CrossRef CAS.
- R. W. F. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- U. Koch and P. L. A. Popelier, J. Phys. Chem., 1995, 99, 9747–9754 CrossRef CAS.
- P. L. A. Popelier, J. Phys. Chem. A, 1998, 102, 1873–1878 CrossRef CAS.
- K. Kubiak-Ossowska, B. Jachimska and P. A. Mulheran, J. Phys. Chem. B, 2016, 120, 10463–10468 CrossRef CAS PubMed.
- J. E. Lofgreen and G. A. Ozin, Chem. Soc. Rev., 2014, 43, 911–933 RSC.
- C. Du, X. Hu, Y. Cheng, J. Gao, Y.-W. Zhang, K. Su, Z. Li, N. Zhang, N. Chang and K. Zeng, Mater. Sci. Eng., C, 2018, 83, 169–176 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02265a |
| This journal is © The Royal Society of Chemistry 2019 |