
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly efficient hydrogen peroxide direct synthesis over a hierarchical TS-1 encapsulated subnano Pd/PdO hybrid†
Jinghui Lyu a,
Jun Weia,
Lei Niua,
Chunshan Lu*a,
Yiwei Hua,
Yizhi Xiangb,
Guofu Zhanga,
Qunfeng Zhanga,
Chengrong Ding*a and
Xiaonian Li*a
a,
Jun Weia,
Lei Niua,
Chunshan Lu*a,
Yiwei Hua,
Yizhi Xiangb,
Guofu Zhanga,
Qunfeng Zhanga,
Chengrong Ding*a and
Xiaonian Li*a
aCollege of Chemical Engineering, Zhejiang University of Technology, Hangzhou, 310032, People's Republic of China. E-mail: lcszjcn@zjut.edu.cn; dingcr@zjut.edu.cn; xnli@zjut.edu.cn
bDave C. Swalm School of Chemical Engineering, Mississippi State University, Mississippi 39762, USA
First published on 30th April 2019
Abstract
We report a hierarchical TS-1 encapsulated subnano Pd/PdO hybrid catalyst that shows unprecedented activity in H2O2 direct synthesis from H2 and O2. The macro reaction rate in 30 min is up to 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 010 mmol gPd−1 h−1 at ambient temperature. Such high catalytic activity is achieved due to the hierarchical porous structure of TS-1 and the formation of the encapsulated subnano Pd/PdO hybrid after oxidation/reduction/oxidation treatment.
010 mmol gPd−1 h−1 at ambient temperature. Such high catalytic activity is achieved due to the hierarchical porous structure of TS-1 and the formation of the encapsulated subnano Pd/PdO hybrid after oxidation/reduction/oxidation treatment.
Hydrogen peroxide as a clean and strong oxidant is one of the commonly used chemicals in various fields of chemical industry, such as the pulp and paper industry, the textile industry, wastewater treatment, green chemical synthesis metallurgy, electronics manufacture, propulsion and the food industry.1 Compared to the traditional anthraquinone process (sequential hydrogenation and oxidation of alkyl anthraquinone), the direct synthesis of hydrogen peroxide (DSHP) from hydrogen and oxygen was recognized as an efficient and environmental alternative process owing to its remarkable adherence to green chemistry perspectives, such as low energy consumption, minimized toxicity and infrastructure investment.2–5
Pd supported catalysts were the most extensively and earliest studied catalysts for the DSHP since 1914.6 Both DFT and experimental results indicated that subnano Pd particles were most effective for the selective oxygen hydrogenation to hydrogen peroxide,7 and the activity and selectivity are also highly dependent upon the oxidation state of the Pd particles.8 However, there were limitations in applying Pd nanoparticles catalyst to the reaction due to the thermal vulnerability in a calcination and reduction activation process.9 To solve this problem, many preparation methods have been adopted to stabilise Pd nanoparticles and control the particle size and morphology, such as yolk–shell structure,10 core–shell structure11 and other encapsulation structure supports. But there were still problems that the size of metal particles is larger than 2.5 nm. Encapsulation of Pd species by mercaptosilane-assisted dry gel conversion (DGC) synthesis method can provide a precise control over the nanoparticle size as well as limitating the aggregation under high temperature during activation.12 However, active sites deep inside the encapsulated nanoparticles were often hardly accessible since the internal configuration diffusion limitations of reactants and products in micropores, leading to low H2 conversion and decomposition of the long residence time of synthetic H2O2.13 So, the role of the porous structured catalyst was essential for encapsulated metal nanoparticles.
Titanium silicalite-1 (TS-1) has already been used as an excellent catalyst for a variety of selective oxidation reactions employing hydrogen peroxide as oxidant.14,15 Moreover, in situ H2O2 generation coupled with these selective oxidation reactions leading to the desired products such as propylene,16,17 benzyl alcohol,18 cyclohexene19 was a desirable, green and lower cost route. More importantly, the Ti–OOH species formed on the TS-1 during selective oxidation might improve the stability of OOH, which is a key reaction intermediate during the DSHP.20 Hutchings et al. reported that hierarchical titanium silicalite supported Au–Pd catalysts showed high peroxide production rate and benzaldehyde production rate for oxidation of benzyl alcohol by in situ generated H2O2.21 In this report, the encapsulation of subnano-sized Pd metal particles within conventional (Pd@TS-1) and hierarchical titanium silicalite-1 (Pd@HTS-1) has been achieved (see Scheme 1). The Pd@HTS-1 catalyst after oxidation–reduction–oxidation pre-treatment showed unprecedented activity in direct synthesis of hydrogen peroxide from hydrogen and oxygen under ambient temperature without any promoter.
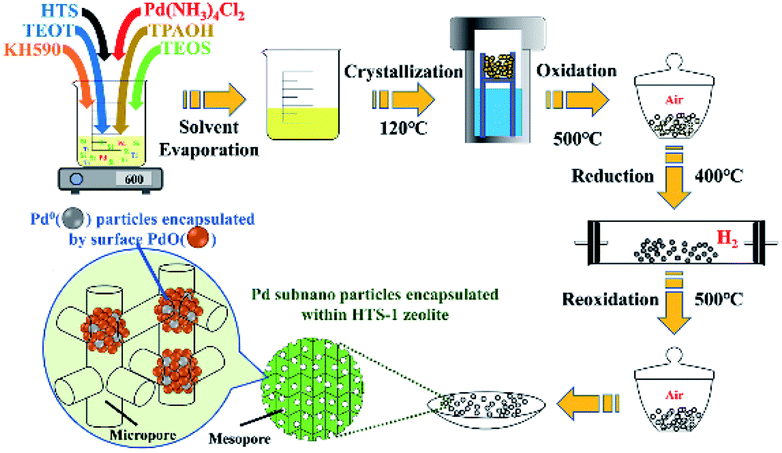 | ||
| Scheme 1 Schematic diagram of the preparation method for Pd@HTS-1. | ||
The TS-1 and HTS-1 encapsulated Pd sub-nanoparticles were first synthesized via solvent evaporation-assisted dry gel conversion method, where the Pd was encapsulated in situ through hydrothermal crystallization in assistance of 3-mercaptopropyl-trimethoxysilane (Scheme 1). The results of ICP analysis confirmed that total Pd contents in Pd@TS-1 and Pd@HTS-1 were 0.094 and 0.106 wt%, respectively. The characteristic diffraction “finger peak” on the X-ray diffraction in Fig. S1† proved that the TS-1, Pd@TS-1 and Pd@HTS-1 had a well-crystallized MFI structure,22 which was further confirmed by the asymmetric stretching of Si–O–Ti in the spectra of Fourier Transform Infrared Spectroscopy (FT-IR, see Fig. S2†). For all of the samples, the diffraction peak at 2θ of 25.4° was not observed. Meanwhile, the diffraction peak of crystalline Pd was also not detected for Pd@HTS-1 and Pd@TS-1, indicating that the Pd particles were well dispersed in the zeolite.7 Besides, the diffuse reflectance UV-vis spectra of the TS-1, Pd@TS-1 and Pd@HTS-1 were shown in Fig. S3.† The band at 210 nm in three samples confirmed the tetrahedral structural geometry of Ti in these silicates, and the weak band at 280 nm was assigned to small amounts of penta/hexacoordinated Ti species.23 Moreover, the absorption band around 300 nm indicated that the three samples contain anatase TiO2.24
The textural properties of the synthesized Pd@TS-1 and Pd@HTS-1 were characterized by N2 adsorption/desorption and the results were shown in Fig. 1 and Table S1.† Notably, typical irreversible type IV adsorption isotherms with an H1 hysteresis loop were observed over the Pd@HTS-1 sample (Fig. 1b), indicating the presence of a mesoporous structure. The mesopore size of Pd@HTS-1, obtained through the BJH method, and the obtained graph peaked at about 7.0 nm. Volume of the micropores was around 0.14 cm3 g−1 for both Pd@TS-1 and Pd@HTS-1, but the surface area of Pd@HTS-1 (509.9 m2 g−1) was 48.9 m2 g−1 larger than that of Pd@TS-1 (461.0 m2 g−1) due to its mesoporous structure, which is beneficial for the diffusion of reactants and products through the catalysts.25
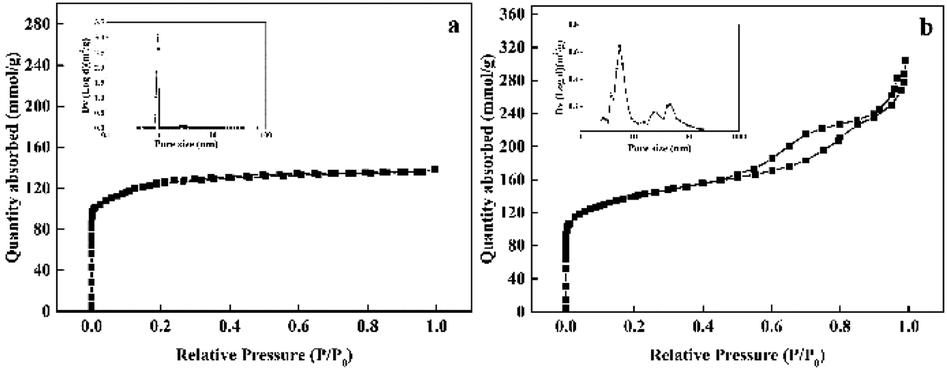 | ||
| Fig. 1 Nitrogen adsorption–desorption isotherms of the synthesized TS-1: (a) Pd@TS-1 and (b) Pd@HTS-1. | ||
Comparison between the experimentally obtained results from ammonia temperature-programmed desorption (NH3-TPD) analysis (Fig. S4†) and the previously reported data showed that the peaks observed were corresponding to weak acid sites, medium acid sites, and strong acid sites of the catalysts.26 Furthermore, pyridine adsorption peak on the FT-IR spectra of these samples (Fig. S5†) revealed that titanium silicate (TS-1) was an acidic support with a large number of Lewis acid segments and few Brønsted acid segments. As shown in scanning electron microscopy (SEM) image (Fig. 2), Pd@TS-1 particles were crystallites with a morphology close to cuboids and a mean particle size of about 3–5 μm, while the Pd@HTS-1 has spherical morphology with a particle size of about 1.3 μm.
 | ||
| Fig. 2 SEM images of the synthesized Pd-modified TS-1: (a) Pd@TS-1 and (b) Pd@HTS-1. | ||
The synthesized TS-1 and HTS-1 encapsulated Pd sub-nanoparticles were then subjected to oxidation/reduction/oxidation treatment to adjust the valence states of Pd.27 Such heat treatment cycle can switch off the sequential hydrogenation and decomposition reactions in the DSHP. However, Ostwald ripening, thus the migration and coalescence of metal clusters, will occur at a higher temperature. Therefore, high temperature treatments was used to emulate the conditions used in the literature mentioned before,28,29 and the thermal stability of the encapsulated Pd@TS-1 catalysts before and after the treatments were also evaluated and compared to investigate the effect of high temperature and the thermal treatments on the catalysts. The Pd@TS-1 and Pd@HTS-1 samples after an air/H2/air thermal treatments at 500/400/500 °C for 4/2/6 h were denoted as Pd@TS-1-O, Pd@TS-1-OR, Pd@TS-1-ORO, Pd@HTS-1-O, Pd@HTS-1-OR, Pd@HTS-1-ORO respectively with O denoting oxidation and R denoting reduction. The Pd particle size distribution after such treatments was first released by the high-resolution transmission electron microscopy (HRTEM) image in Fig. 3 and S6.† The Pd particles encapsulated within microporous TS-1 zeolites were well dispersed and uniformly distributed throughout the zeolite crystals. The average sizes of Pd particles encapsulated in the TS-1 and HTS-1 were in the range of 1–2 nm, which, however, was bigger than those of the MFI topology channels (0.53 × 0.56 nm) and intersectional channels (∼0.9 nm). Nevertheless, the successful encapsulation of the Pd particles in the TS-1 zeolites was verified by comparing the hydrogenation rates of a mixture of nitrobenzene and 1-nitronaphthalene. As shown in Fig. S7,† the reaction rate for the hydrogenation of nitrobenzene and 1-nitronaphthalene was much higher over the Pd@HTS-1-OR compared to the Pd@TS-1-OR. We anticipated that the slightly larger Pd size than the zeolite channels might reflect the local disruption of the crystal structures near the location of the particles during the in situ synthesis. More detailed size distributions of Pd particles encapsulated in the TS-1 and HTS-1 zeolites after air, Ar/H2 and air treatments were shown in Fig. 3d–f and j–l, respectively. The particle sizes of most of the Pd species still remain below 2 nm on average, which indicated the absence of metal clusters migration and coalescence by Ostwald ripening even after such higher temperature treatments. The high thermal stability of the Pd subnano particles resulted from the embedding confinement.30
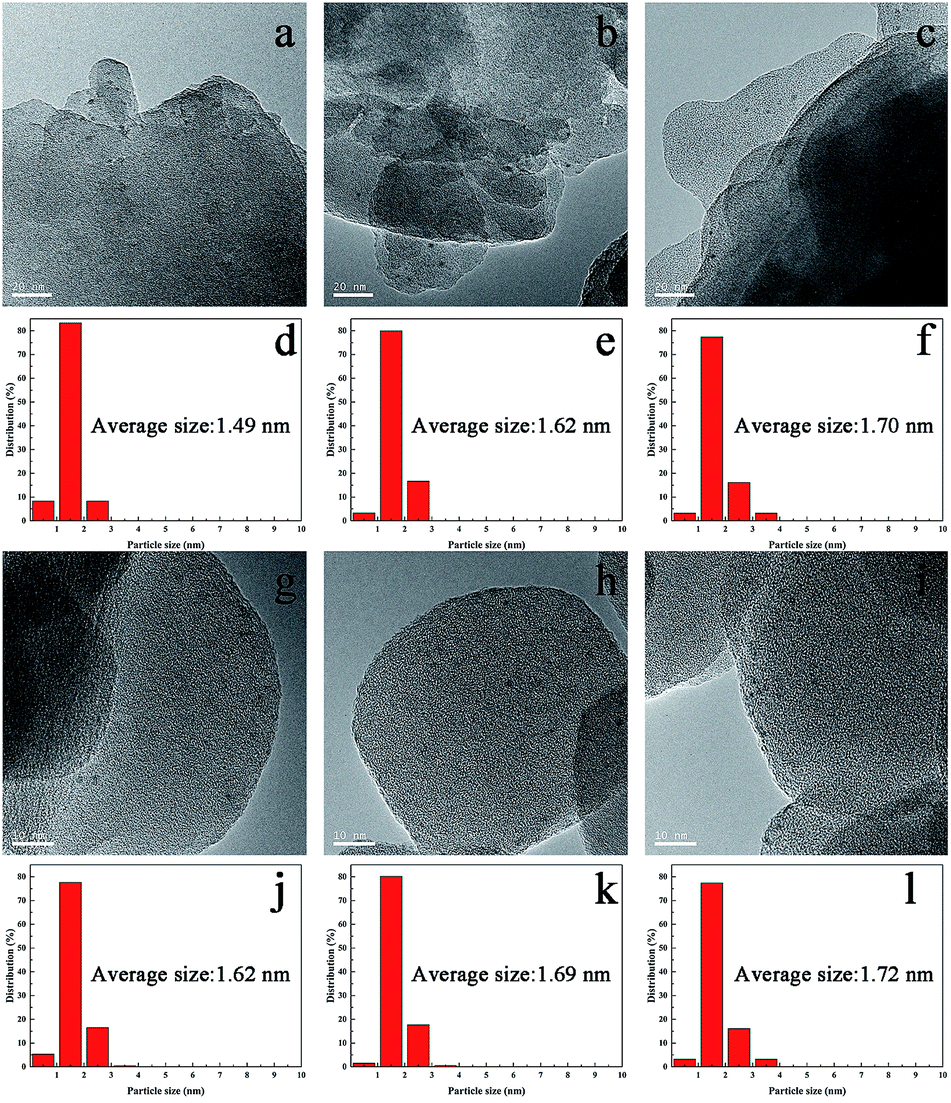 | ||
| Fig. 3 HRTEM images and metal particle size distributions of the Pd@TS-1 and Pd@HTS-1 before and after high-temperature oxidation–reduction–oxidation treatments. (a, d Pd@TS-1-O. b, e Pd@TS-1-OR. c, f Pd@TS-1-ORO. g, j Pd@HTS-1-O. h, k Pd@HTS-1-OR. i, l Pd@HTS-1-ORO.) | ||
The Pd dispersion and average Pd nanoparticle size for Pd@TS-1 and Pd@HTS-1 after the air/H2 treatment were further determined by CO chemisorption measurements (see Table S2†). The dispersions of Pd in Pd@TS-1 and Pd@HTS-1 are 85% and 81%, respectively. The average Pd particle sizes for Pd@TS-1 and Pd@HTS-1 calculated by CO adsorption measurements are 1.06 nm and 1.17 nm, respectively, which was smaller than that estimated from the TEM analysis. This was probably due to the presence of Pd nanocluster or single atoms, which cannot be directly observed by HRTEM.
We now turn to the Pd valence states of the catalysts after the oxidation/reduction/oxidation treatment by the XPS (see Fig. S8†). The Pd3d spectra signals were hardly observed when the concentration of Pd atoms was low, the binding energy peaks for different oxidation states of Pd atoms were collected after peak fitting by prolonging the scanning time.31 The XPS results demonstrated the presence of both metallic Pd and PdO. The binding energy of peaks for Pd03d5/2 and Pd03d3/2 correspond to 335.5 and 340.6 eV, respectively, while the binding energy for Pd2+3d5/2 and Pd2+3d3/2 were at 337.8 and 341.9 eV, respectively.31 The transformation of valence state could be observed in Fig. S8a–c,† which was derived from XPS measurements. Moreover, the ratios for Pd0 and Pd2+ atoms in Pd@TS-1 and Pd@HTS-1 were approximately 2 and 1, respectively. On the basis of these results, we proposed a reaction mechanism for the synthetic process of the catalysts, subnano-sized Pd particles might be oxidated from Pd0 to Pd2+ to form PdO on the surface of the catalysts during reoxidation.
The catalytic performance of the TS-1 and HTS-1 encapsulated subnano-sized Pd/PdO hybrid in the direct synthesis of hydrogen peroxide from H2 and O2 were tested at ambient temperature without any promoters. Compared to the Pd supported by the active carbon, the selectivity of hydrogen peroxide was higher, the reason might be the formation of Ti–OOH32 and the confinement effect of the Pd encapsulated in the channel of the zeolite (Scheme 2). Both HTS-1 zeolite and Pd@zeolites showed significant amount of O2 adsorption according to the O2-TPD (Fig. S9†), which might be the reason for high activity/selectivity. The selectivity for hydrogen peroxide on Pd@TS-1-OR is lower than that on Pd@TS-1-O, while the degradation rate of hydrogen peroxide on Pd@TS-1-OR are higher than that on Pd@TS-1-O (Fig. 4 and Table S3†), which was attributed to the change in oxidation state from Pd2+ to Pd0 after reductive treatment, in agreement with previous reports.27,33 The selectivity of hydrogen peroxide over Pd@TS-1 increased after an oxidation/reduction/oxidation cycle, the reason might be the weaker adsorption of O2 and H2, the intermediate OOH and the production H2O2 and the suppression of H2O2 decomposition.20
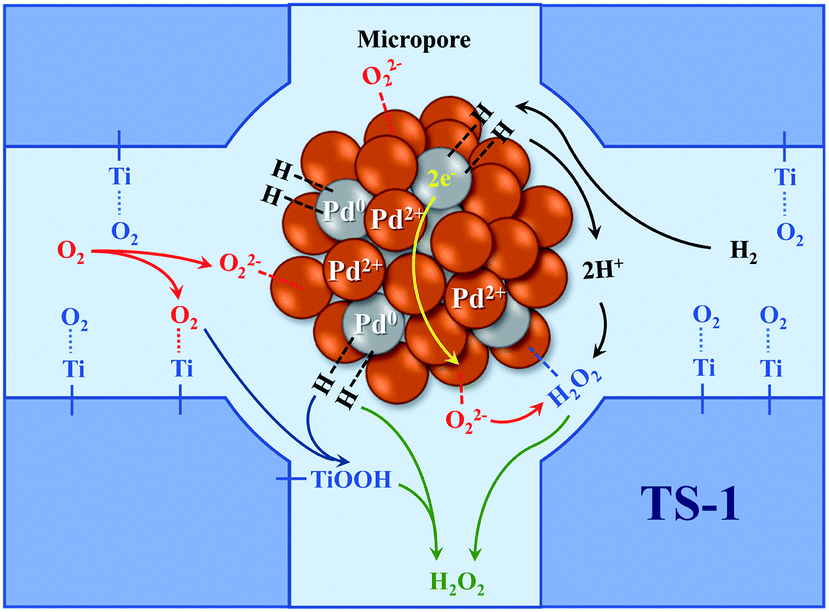 | ||
| Scheme 2 Schematic of the mechanism for DSHP by Pd@TS-1. | ||
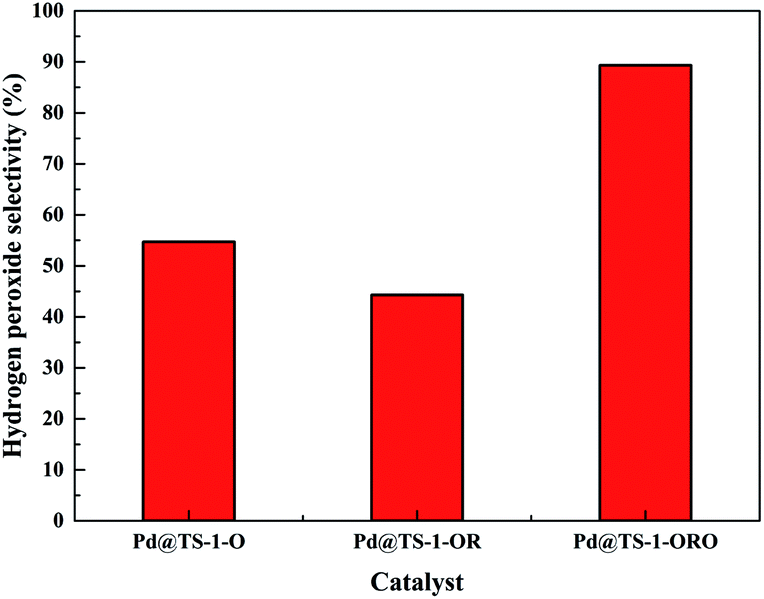 | ||
| Fig. 4 H2O2 selectivity of DSHP over Pd@TS-1 with different oxidation states for 5 min reaction. Reaction conditions (same as Fig. 5 and 6): H2/Ar (2.9 MPa) and air (1.35 MPa), 8.5 g solvent (2.9 g water, 5.6 g MeOH), 0.02 g catalyst, RT, 1200 rpm. | ||
The productivity of DSHP over Pd@TS-1 increased with oxidation, reduction and reoxidation treatment in 30 minutes (Fig. 5 and Table S3†), demonstrated that PdO layer on monometallic Pd catalysts could suppress oxygen dissociation and H2O2 degradation,12 the appropriate PdO formed on the surface of the catalysts after reoxidation can optimize the H2O2 production. The hierarchical Pd@TS-1 (35010 mmol gPd−1 h−1) is remarkably higher than those of conventional Pd@TS-1 (3210 mmol gPd−1 h−1), the superior hydrogen peroxide production rate of Pd@HTS-1-ORO indicating that the Pd encapsulated by uniformed topology structure of TS-1 highly limited by the effect of pore-diffusion resistance.11 Compared to Pd@TS-1, it was noteworthy that Pd@HTS-1 with only 0.1 wt% Pd content and subnano size after oxidative treatments showed famous reaction activity without any promoters under mild condition, which could be mainly ascribed to the presence of internal diffusion limitation within encapsulated micropore zeolites. The micropore structure limited the use of Pd metal because a part of the Pd crystal surface was blocked by zeolite supports, the hydrogen and oxygen were restricted by the configurational diffusion of zeolite to the Pd surface. Moreover, the formed and desorption H2O2 was also constrained by the micropore and thereby resulted in prolonged residence time of the product leading to degradation of H2O2. The intracrystal diffusion no longer limited the mass transport process of the hierarchical zeolite due to the presence of additional porosity. Although the physical and structural properties (including the primary particle size, the properties of the external surface and so on) were different between Pd@HTS-1 and Pd@TS-1, we may still draw a conclusion that the excellent catalytic activity is mainly attributed to the presence of mesopore favours diffusion of both reactants and products to and off the active sites in micropores.
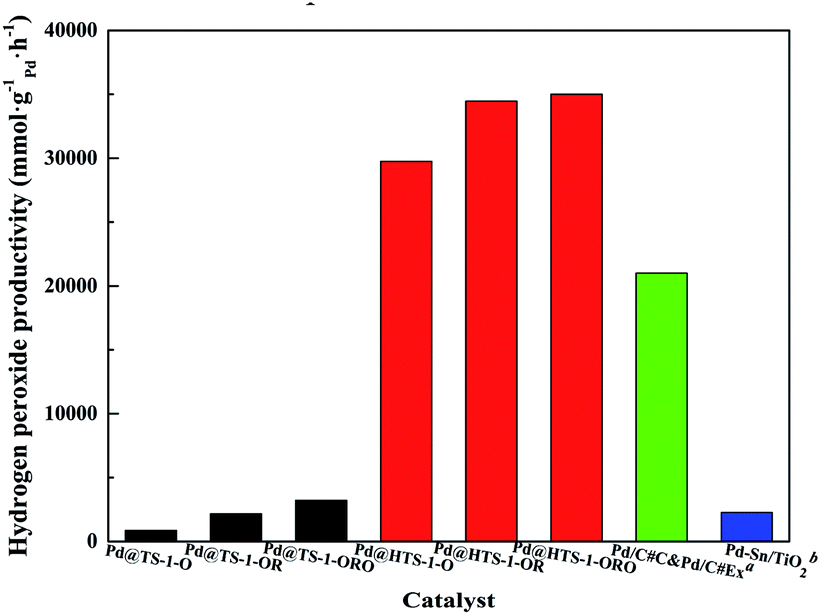 | ||
| Fig. 5 Macro reaction rate for H2O2 production over Pd@TS-1 and Pd@HTS-1. aPd/C#C&Pd/C#Ex from Young-Min Chung;34 bPd–Sn/TiO2 from Hutchings.29 | ||
The TON of H2O2 production at different reaction time over the six different Pd@TS-1 and Pd@HTS-1 catalysts were shown in Fig. 6. The TON increases with increasing reaction time, however, the slop of the TON–time curves (dTON/dt) seems decreased with increasing time, which revealed that the net productivity rate of hydrogen peroxide synthesis declined slightly with increasing time, especially for the Pd@HTS-1-OR at the reaction period of 30–60 min. The accumulative productivity of hydrogen peroxide slowed down, the reason might be the rapid decrease of hydrogen partial pressure in the medium and the ongoing H2O2 degradation.
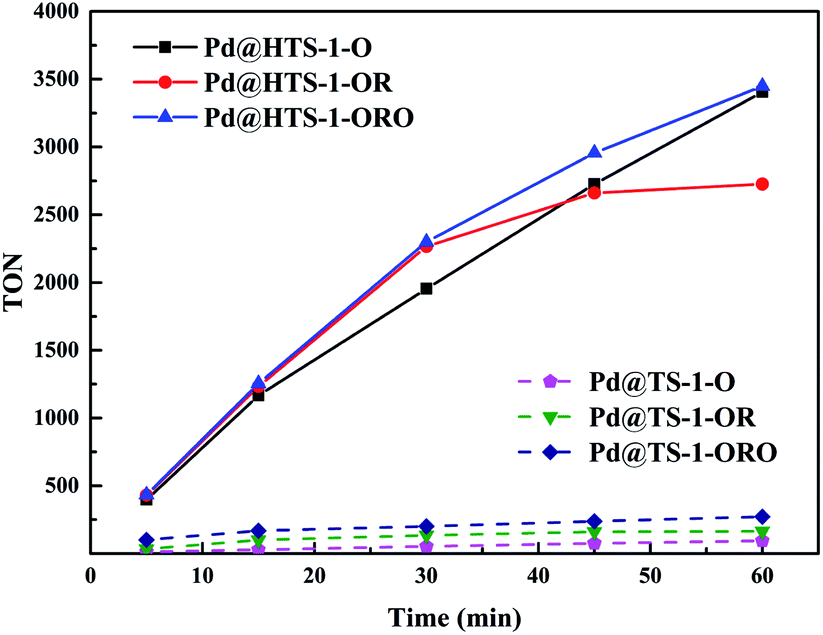 | ||
| Fig. 6 The TON of H2O2 production with different reaction time over Pd@TS-1 and Pd@HTS-1 catalysts. TON (turnover number) = mol (H2O2)/mol (surface Pd). | ||
In summary, successful encapsulation of subnano-size Pd metal particles within titanium silicate (TS-1) voids was achieved via the mercaptosilane-assisted DGC synthesis method. The subnano-size Pd nanoparticles encapsulated in HTS-1 zeolites exhibited superior thermal stability after the oxidation/reduction/oxidation heat treatment process adjusting Pd/PdO hybrid owing to the embedding confinement. The synthesized high-efficiency Pd@HTS-1-ORO showed the famous hydrogen peroxide synthesis productivity, a hydrogen peroxide production rate as high as about 35010 mmol H2O2 gPd−1 h−1. Our strategy brings about a finely tailored method to control particle size down to the subnano level and eliminate the diffusion inside metal encapsulated microporous zeolites, which is advantageous for catalytic activity and selectivity in direct synthesis of hydrogen peroxide. Thus, our approach opens up the possibility that the titanium-containing zeolites encapsulated noble metal catalyst can be extended further to selective oxidation reactions with H2O2 generated in situ from H2 and O2.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors were grateful for financial support from the National Science foundation of China (No. 21506189) and Zhejiang Provincial Natural Science Foundation of China (No. LY17B060008).Notes and references
- J. M. Camposmartin, G. Blancobrieva and J. L. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962 CrossRef CAS PubMed.
- S. Chinta and J. H. Lunsford, J. Catal., 2004, 225, 249 CrossRef CAS.
- J. K. Edwards and G. J. Hutchings, Angew. Chem., Int. Ed., 2010, 47, 9192 CrossRef PubMed.
- N. M. Wilson, D. T. Bregante, P. Priyadarshini and D. W. Flaherty, RSC Adv., 2017, 29, 122 Search PubMed.
- G. Blanco-Brieva, M. Montiel-Argaiz, F. Desmedt, P. Miquel, J. M. Campos-Martin and J. L. G. Fierro, RSC Adv., 2016, 6, 99291 RSC.
- H. Henkel and W. Weber, US Pat., 1108752, 1914.
- P. Tian, L. Ouyang, X. Xu, C. Ao, X. Xu, R. Si, X. Shen, M. Lin, J. Xu and Y.-F. Han, J. Catal., 2017, 349, 30 CrossRef CAS.
- V. R. Choudhary, C. Samanta and T. V. Choudhary, Catal. Commun., 2007, 8, 1310 CrossRef CAS.
- H. Lee, S. Kim, D.-W. Lee and K.-Y. Lee, Catal. Commun., 2011, 12, 968 CrossRef CAS.
- M. G. Seo, S. Kim, H. E. Jeong, D. W. Lee and K. Y. Lee, Appl. Catal., A, 2016, 511 Search PubMed.
- M. Seo, D. W. Lee, S. S. Han and K. Y. Lee, ACS Catal., 2017, 7, 3039 CrossRef CAS.
- D. Farrusseng and A. Tuel, New J. Chem., 2016, 40, 3933 RSC.
- S. Yook, C. K. Han, Y. G. Kim, W. Choi and M. Choi, ACS Sustainable Chem. Eng., 2017, 5, 1208 CrossRef CAS.
- Y. Zuo, M. Liu, T. Zhang, L. Hong, X. Guo, C. Song, Y. Chen, P. Zhu, C. Jaye and D. Fischer, RSC Adv., 2015, 5, 17897 RSC.
- G. Xiong, Q. Jia, Y. Cao, L. Liu and Z. Guo, RSC Adv., 2017, 7, 24046 RSC.
- E. J. Beckman, Green Chem., 2003, 5, 332 RSC.
- X. Feng, D. Chen and X. G. Zhou, RSC Adv., 2016, 6, 44050 RSC.
- M. Santonastaso, S. J. Freakley, P. J. Miedziak, G. L. Brett, J. K. Edwards and G. J. Hutchings, Org. Process Res. Dev., 2014, 18, 1455 CrossRef CAS.
- G. Li, J. Edwards, A. F. Carley and G. J. Hutchings, Catal. Commun., 2007, 8, 247 CrossRef CAS.
- A. M. Joshi, W. N. Delgass and K. T. Thomson, J. Phys. Chem. B, 2005, 109, 22392 CrossRef CAS PubMed.
- I. Moreno, N. F. Dummer, J. K. Edwards, M. Alhumaimess, M. Sankar, R. Sanz, P. Pizarro, D. P. Serrano and G. J. Hutchings, Catal. Sci. Technol., 2013, 3, 2425 RSC.
- G. Wu, J. Xiao, L. Zhang, W. Wang, Y. Hong, H. Huang, Y. Jiang, L. Lia and C. Wang, RSC Adv., 2016, 6, 101071 RSC.
- E. Jorda, A. Tuel, R. Teissier and J. Kervennal, Zeolites, 1997, 19, 238 CrossRef CAS.
- A. C. Alba-Rubio, J. L. G. Fierro, L. León-Reina, R. Mariscal, J. A. Dumesic and M. López Granados, Appl. Catal., B, 2017, 202, 269 CrossRef CAS.
- B. Wang, M. Lin, X. Peng, B. Zhu and X. Shu, RSC Adv., 2016, 6, 44963 RSC.
- X. Liu, C. Yang, Y. Wang, Y. Guo, Y. Guo and G. Lu, Chem. Eng. J., 2014, 243, 192 CrossRef CAS.
- S. Melada, R. Rioda, F. Menegazzo, F. Pinna and G. Strukul, J. Catal., 2006, 239, 422 CrossRef CAS.
- F. Wang, C. Xia, S. P. de Visser and Y. Wang, J. Am. Chem. Soc., 2019, 141, 901 CrossRef CAS PubMed.
- S. J. Freakley, Q. He, J. H. Harrhy, L. Lu, D. A. Crole, D. J. Morgan, E. N. Ntainjua, J. K. Edwards, A. F. Carley and A. Y. Borisevich, Science, 2016, 351, 956 CrossRef PubMed.
- S. Goel, S. I. Zones and E. Iglesia, J. Am. Chem. Soc., 2014, 136, 15280 CrossRef CAS PubMed.
- A. F. Sierra-Salazar, W. S. J. Li, M. Bathfield, A. Ayral, S. Abate, T. Chave, S. I. Nikitenko, V. Hulea, S. Perathoner and P. Lacroix-Desmazes, Catal. Today, 2017, 306, 16 CrossRef.
- B. Silvia, B. Francesca, D. Alessandro and L. Carlo, Phys. Chem. Chem. Phys., 2007, 9, 4854 RSC.
- J. Pritchard, M. Piccinini, R. Tiruvalam, Q. He, N. Dimitratos, J. A. Lopezsanchez, D. J. Morgan, A. F. Carley, J. K. Edwards and C. J. Kiely, Catal. Sci. Technol., 2013, 3, 308 RSC.
- S. Lee and Y. M. Chung, Mater. Lett., 2019, 234, 58 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02452b |
| This journal is © The Royal Society of Chemistry 2019 |