
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid and halide compatible synthesis of 2-N-substituted indazolone derivatives via photochemical cyclization in aqueous media†
Hui-Jun Nieab,
An-Di Guob,
Hai-Xia Lina and
Xiao-Hua Chen *b
*b
aDepartment of Chemistry, Innovative Drug Research Center, College of Sciences Shanghai University, Shanghai, 200444, China
bChinese Academy of Sciences Key Laboratory of Receptor Research, Synthetic Organic & Medicinal Chemistry Laboratory, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China. E-mail: xhchen@simm.ac.cn
First published on 30th April 2019
Abstract
Indazolone derivatives exhibit a wide range of biological and pharmaceutical properties. We report a rapid and efficient approach to provide structurally diverse 2-N-substituted indazolones via photochemical cyclization in aqueous media at room temperature. This straightforward protocol is halide compatible for the synthesis of halogenated indazolones bearing a broad scope of substrates, which suggests a new avenue of great importance to medicinal chemistry.
The indazolone ring system constitutes the core structural element found in a large family of nitrogen heterocycles as exemplified by those shown in Scheme 1.1 Indazolone derivatives have been receiving much attention due to their promising pharmacological activities. Given the unique bioactive core skeleton, indazolone derivatives exhibit a wide range of biological and pharmaceutical properties such as antiviral and antibacterial activities (1–4),2 new prototypes for antichagasic drugs (5),3 antihyperglycemic properties (6),4 TRPV1 receptor antagonists for analgesics (7),5 anti-flammatory agents (8),6 angiotensin II receptor antagonists (9),7 highly potent CDKs inhibitors for anticancer (10)8 and so on.9 The privileged indazolone structures have high potential as core components for the development of related compounds leading to medicinal agents.
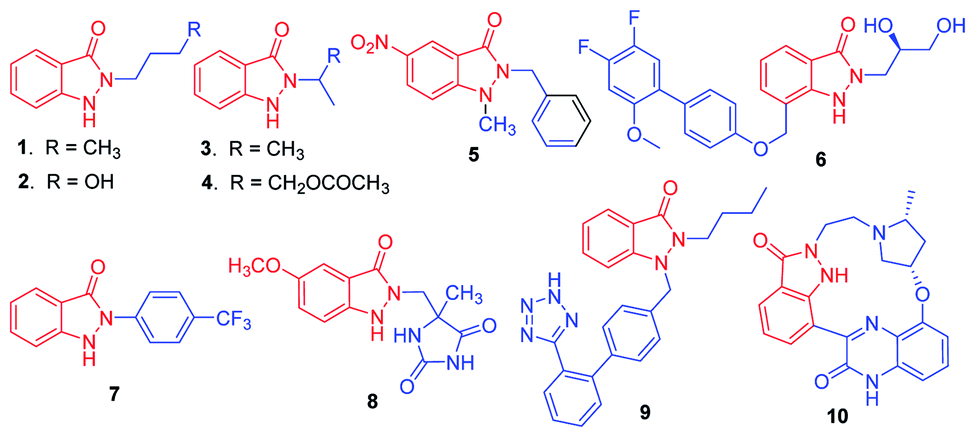 | ||
| Scheme 1 Biologically active molecules containing indazolone skeletons. | ||
Due to their versatility in pharmaceutical applications, many synthetic approaches have been developed for the construction of indazolone skeletons (Scheme 2), including CuO-mediated coupling of 2-haloarylcarboxylic acids with methylhydrazine,10 cyclization of N-aryl-o-nitrobenzamides through Ti(VI) reagent or Zn(II) reagent,11 Cu(I)-mediated intramolecular C–N bond formation or a base-mediated intramolecular SNAr reaction of 2-halobenzohydrazides,12 Cu(I)-catalyzed oxidative C–N cross-coupling and dehydrogenative N–N formation sequence,13 Rh-catalyzed C–H activation/C–N bond formation and Cu-catalyzed N–N bond formation between azides and arylimidates,14 Friedel–Crafts cyclization of N-isocyanates using Masked N-isocyanate precursors,15 PIFA-mediated intramolecular oxidative N–N bond formation by trapping of N-acylnitrenium intermediates,16 and recently reported reaction of o-nitrobenzyl alcohol with primary amines in basic conditions.17 These approaches are complementary providing avenue to access various substitution patterns,18 however most methods rely on the requirements for transition-metal catalysts. In fact, the procedures for synthesis indazolone skeletons from Friedel–Crafts cyclization of N-isocyanates and Davis–Beirut derived reaction still suffer from harsh reaction conditions such as high reaction temperature (i.e. more than 150 °C or 20 equiv. of KOH at 100 °C for 24 h).15,17b Very recently, one photochemical route was reported for preparation of indazolone skeletons from o-nitrobenzyl alcohols and primary amines,19 however, this approach still need long reaction time (24 hours) and halogen substituted substrate could not be compatible in the reaction conditions.19b Thus, the efficient and general methods tolerating a wide scope of readily available starting materials for synthesis of indazolones without a transition-metal catalyst involved are still in great demand.
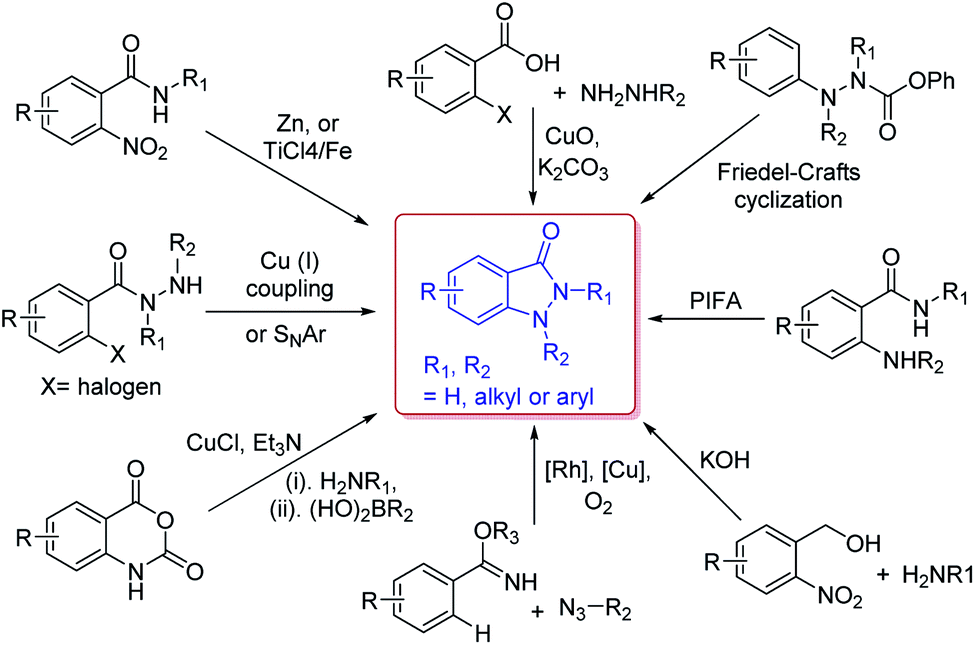 | ||
| Scheme 2 Representative approaches for the preparation of indazolone skeletons. | ||
o-Nitrobenzyl alcohol derivatives have shown many applications in material science and chemical biology area as a photolabile protecting group (Scheme 3a).20 Upon UV light-activation, o-nitrobenzyl alcohol derivatives generate corresponding aryl-nitroso compounds via photoisomerization.21 Based on the distinguishing feature of highly reactive of these photogenerated intermediates, we assumed that the reaction conditions would be crucial for the photoisomerization,20,22 thus the reactive intermediates should spontaneously and rapidly form indazolone structures via cyclization in the presence of primary amines in suitable reaction conditions (Scheme 3b). Herein, we report a rapid and efficient approach to provide structural diversity 2-N-substituted indazolones via photochemical cyclization in aqueous media at room temperature. This photochemical cyclization reaction is halide compatible for synthesis of halogen substituted indazolones, bearing a broad scope of substrates. This straightforward protocol for rapid construction of halogenated indazolone architectures suggests a new avenue of great importance to medicinal chemistry.
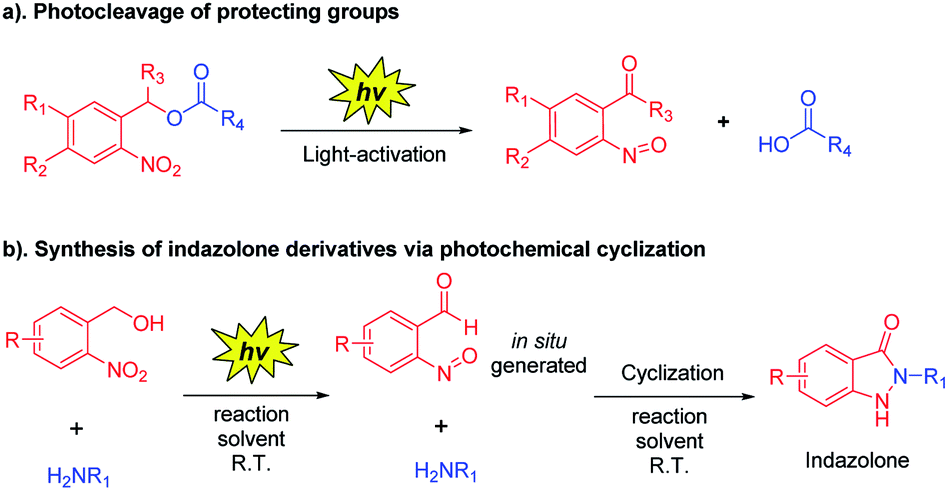 | ||
| Scheme 3 Synthesis of indazolone derivatives via photochemical cyclization. | ||
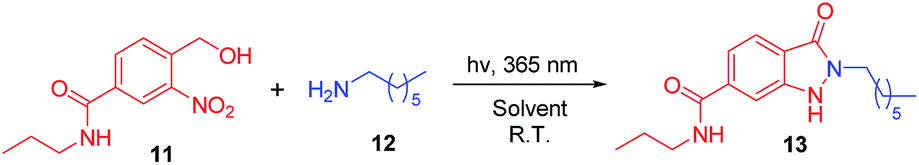
The initial investigation to develop a method for synthesis of indazolone derivatives via photochemical cyclization started with 4-(hydroxymethyl)-3-nitro-N-propylbenzamide 11 and heptan-1-amine 12 upon UV light-activation in methanol, smoothly leading to the formation of indazolone 13 in 52% yield (Table 1, entry 1) in 3 hours. A survey of different solvents such as THF, n-BuOH and acetonitrile revealed that acetonitrile was most suitable for the reaction, providing the best results than other solvents (entry 2–4). To our delight, the photochemical cyclization reaction gave better yield in an aqueous media (entry 5), while in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CH3CN/PBS aqueous mixture, the reaction afforded lower yield (entry 6). Interestingly, aqueous media of n-BuOH/H2O rapidly furnished the indozolone product with best yield (entry 7 vs. entry 8–14) after a survey of different aqueous media. The effectiveness of different light source was investigated (entry 7, 15–16), the light with 365 nm resulted in the best yield than others. Further exploration of temperature and the ratio of 11/12, the reaction provide product in a modest yields (entry 17–18). In addition, not much variation in the yield was observed (entry 19) by prolonging the reaction time. The reaction afforded with only 19% yield in 3 hours using the PBS as reaction media (entry 20). On the other hand, with the excess of primary amine 12 in the reaction system, we could get product with 46% yield (entry 21), possibly due to photogenerated intermediate aryl-nitroso is highly reactive and not stable. These results indicated that the reaction media and the ratio of 11/12 are important for the reaction. Taken together, these results demonstrated that the photochemical cyclization reaction rapidly provided 2-N-substituted indazolone with high yield only need 3 hours at the optimal conditions up on UV light-activation.
:1 CH3CN/PBS aqueous mixture, the reaction afforded lower yield (entry 6). Interestingly, aqueous media of n-BuOH/H2O rapidly furnished the indozolone product with best yield (entry 7 vs. entry 8–14) after a survey of different aqueous media. The effectiveness of different light source was investigated (entry 7, 15–16), the light with 365 nm resulted in the best yield than others. Further exploration of temperature and the ratio of 11/12, the reaction provide product in a modest yields (entry 17–18). In addition, not much variation in the yield was observed (entry 19) by prolonging the reaction time. The reaction afforded with only 19% yield in 3 hours using the PBS as reaction media (entry 20). On the other hand, with the excess of primary amine 12 in the reaction system, we could get product with 46% yield (entry 21), possibly due to photogenerated intermediate aryl-nitroso is highly reactive and not stable. These results indicated that the reaction media and the ratio of 11/12 are important for the reaction. Taken together, these results demonstrated that the photochemical cyclization reaction rapidly provided 2-N-substituted indazolone with high yield only need 3 hours at the optimal conditions up on UV light-activation.
|
||||
|---|---|---|---|---|
| Entry | Solvent | 11:12 |
Time (h) | Yieldf (%) |
| a Reaction conditions: heptan-1-amine (12, 0.3 mmol), 4-(hydroxymethyl)-3-nitro-N-propylbenzamide (11, 0.75 mmol), solvent 6 mL, exposed to UV lamp with 365 nm, at R.T.b UV lamp with 254 nm.c Using blue light.d The ratio of 11/12 = 1.5/1.e Reaction carried out at 50 °C.f Isolated yield. | ||||
| 1 | MeOH | 2.5:1 |
3 | 52 |
| 2 | THF | 2.5:1 |
3 | 58 |
| 3 | n-BuOH | 2.5:1 |
3 | 57 |
| 4 | CH3CN | 2.5:1 |
3 | 61 |
| 5 | CH3CN:H2O = 3:1 |
2.5:1 |
3 | 67 |
| 6 | CH3CN:PBS = 3:1 |
2.5:1 |
3 | 61 |
| 7 | n-BuOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :H2O = 3:1 :H2O = 3:1 |
2.5:1 |
3 | 82 |
| 8 | n-BuOH:PBS = 3:1 |
2.5:1 |
3 | 56 |
| 9 | MeOH:H2O = 3:1 |
2.5:1 |
3 | 38 |
| 10 | i-PrOH:H2O = 3:1 |
2.5:1 |
3 | 63 |
| 11 | tBuOH:H2O = 3:1 |
2.5:1 |
3 | 55 |
| 12 | THF:H2O = 3:1 |
2.5:1 |
3 | 49 |
| 13 | DMF:H2O = 3:1 |
2.5:1 |
3 | 45 |
| 14 | Dioxane:H2O = 3:1 |
2.5:1 |
3 | 67 |
| 15b | n-BuOH:H2O = 3:1 |
2.5:1 |
3 | <10 |
| 16c | n-BuOH:H2O = 3:1 |
2.5:1 |
3 | 22 |
| 17d | n-BuOH:H2O = 3:1 |
1.5:1 |
3 | 45 |
| 18e | n-BuOH:H2O = 3:1 |
2.5:1 |
3 | 28 |
| 19 | n-BuOH:H2O = 3:1 |
2.5:1 |
6 | 85 |
| 20 | PBS | 2.5:1 |
3 | 19 |
| 21 | n-BuOH:H2O = 3:1 |
1:2.5 |
3 | 46 |
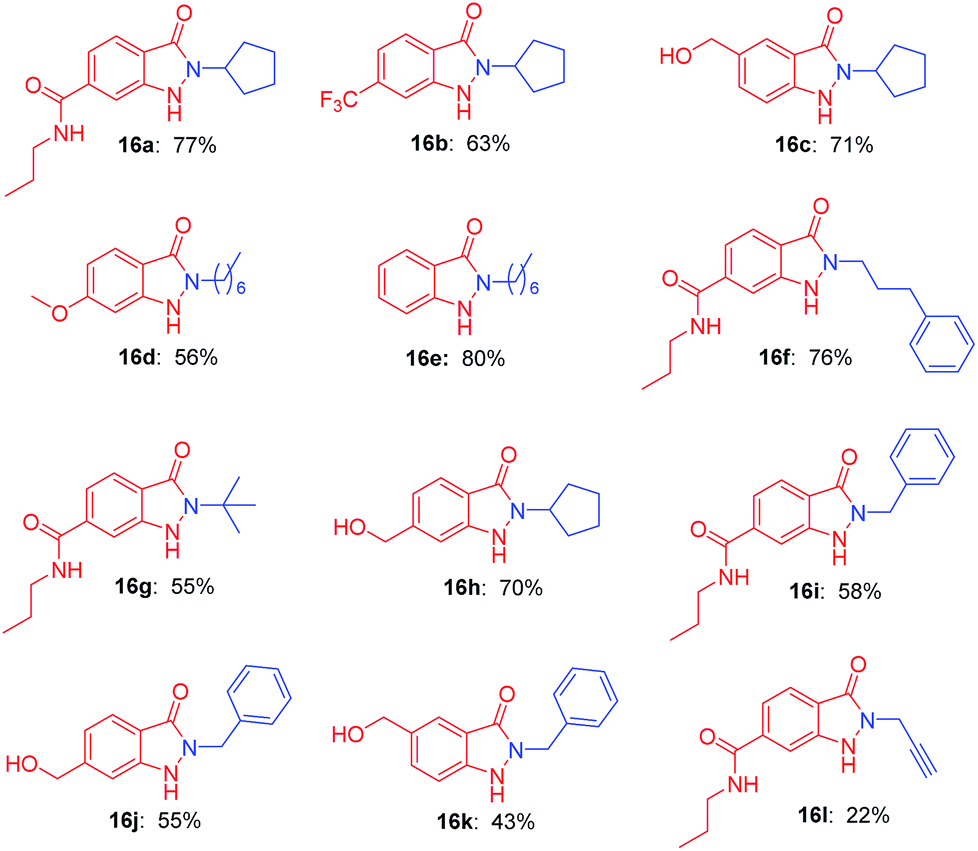
With the optimal conditions in hand, we investigated the generality for the scope of o-nitrobenzyl alcohols and primary amines (Table 2). In the photochemical cyclization reaction conditions, the substitution on the benzene ring was well tolerated. o-Nitrobenzyl alcohols with either electron-withdrawing groups (such as propylcarbamoyl, trifluoromethyl group) or electron-donating groups (such as methoxy, hydroxymethyl group) reacted with primary amines smoothly furnished indazolones in good yields (16a–16d, 56–77%). o-Nitrobenzyl alcohols participated in the reaction with various aliphatic amines afforded good yields (16e–16h, 55–80%). Bulky tert-butylamine reacted readily with the propylcarbamoyl substituted o-nitrobenzyl alcohol to give 16g in 55% yield, which is a significant advantage since this product cannot be accessed via N-alkylation of unsubstituted indazolones. Benzylic amine produced the desired products in a moderate yields (16i–16k, 43–58%). Finally propargylamine afforded indazolone 16l with 22% yield.
|
|---|
| a Reaction conditions: primary amines (15, 0.3 mmol), o-nitrobenzyl alcohols derivatives (14, 0.75 mmol), solvent 6 mL, isolated yield. |
 |
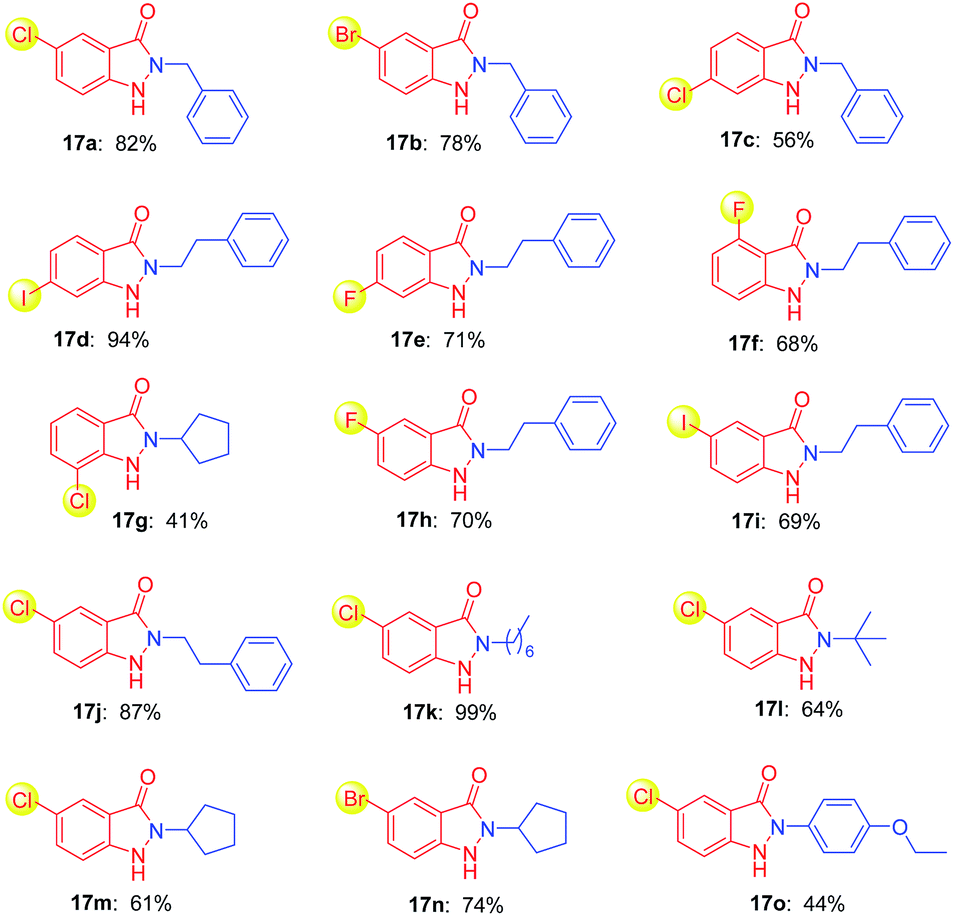
Many drug candidates and drugs are halogenated structures. In drug discovery, insertion of halogen atoms on hit or lead compounds was predominantly performed, with the aim to exploit their steric effects and structure–activity relationship, to form halogen bonds in ligand–target complexes, to optimize the ADME/T property.23 Given the versatility of halogen atom on bioactive molecules, we next investigated the halogen substituted o-nitrobenzyl alcohols as starting materials for construction of indazolone skeletons (Table 3). To our delight, the reaction of the chloride substituted o-nitrobenzyl alcohols with benzylic amine provided indazolones in good yields (17a, 17c) as well as bromide substituted o-nitrobenzyl alcohol (17b). Variation of the iodine, chloride and fluoride substituents on the ring of the o-nitrobenzyl alcohol also rapidly and efficiently afforded modest to good yields (17d–17i). We then performed the reaction of a variety of primary amines including aliphatic amines and aromatic amine. Various aliphatic amines gave the corresponding alkyl indazolones in good yields (17j–17n). We were delighted to find that aromatic amine (4-ethoxyaniline) gave 17o in 44% yield. In contrast, the aromatic amine gave low yield in recently reported Davis–Beirut derived reaction17b as well as aniline failed to give product in the recently reported photochemical approach (see ESI, Fig. S1†).19b Of note, in the recently reported photochemical approach, the reaction of chloride substituted o-nitrobenzyl alcohol with alkylamine gave indazolone with low yield, possibly because photocleavage of aryl halide bond is involved in that reaction conditions.19b These outcomes are significant in view of the challenges in construction of indazolone skeletons, in which additional halogen substitution on substrates is incompatible for indazolones synthesis.10,19b Importantly, our reaction condition is compatible with halide substrates, suggests a new protocol of importance to photochemical reactions, in which dehalogenation of aryl halide is known to be radical-mediated and exist in some reaction conditions.24
|
|---|
| a Reaction conditions: primary amines (15, 0.3 mmol), o-nitrobenzyl alcohols derivatives (14, 0.75 mmol), solvent 6 mL, isolated yield. |
 |
With developed and optimized protocol, we rapid synthesis of indazolones 1 and 2 with antiviral and antibacterial activities with good to excellent yields,2 in aqueous media at room temperature for 3 hours (Scheme 4), respectively. This rapid and efficient access to the privileged indazolone architectures will have great usefulness in medicinal chemistry.
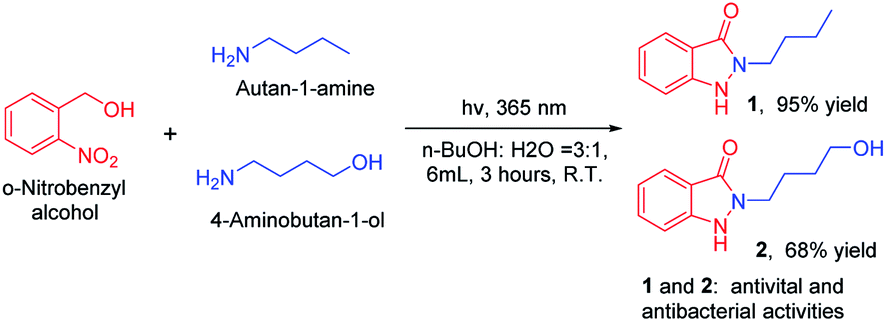 | ||
| Scheme 4 Straightforward synthesis of indazolones (1 and 2). Reaction conditions: primary amines (0.3 mmol), o-nitrobenzyl alcohol (0.75 mmol), isolated yield. | ||
Finally, we could detect the aryl-nitroso compound as photogenerated intermediate on UPLC-MS analysis (see ESI, Fig. S2†). The proposed mechanism for this photochemical cyclization begins by generation of the aryl-nitroso compound which can rapidly undergo cyclization with primary amines, subsequent for dehydration and tautomerization (see ESI, Fig. S3†).17
In summary, we report a photochemical cyclization approach to provide 2-N-substituted indazolones up to 99% yield with structural diversity from the reaction of o-nitrobenzyl alcohols and primary amines in aqueous media at room temperature. This photochemical cyclization reaction is rapid and halide compatible for synthesis of halogenated indazolones, bearing a broad scope of substrates, while previous reported photochemical reaction has met less success.19b In addition, our reaction condition is compatible with halide substrates, suggests a new protocol of importance to photochemical reactions, in which photocleavage of aryl halide bonds is exist in some reaction conditions. The current transformation enabling rapid and efficient access to the privileged indazolone architectures has great usefulness in medicinal chemistry and diversity-oriented synthesis, thus will provide promising candidates for chemical biology research and drug discovery.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation of China (NSFC Grants 21602235), Natural Science Foundation of Shanghai (16ZR1442800), Shanghai Rising-Star program and Chinese Academy of Science.References
- (a) S. G. Zhang, C. G. Liang and W. H. Zhang, Molecules, 2018, 23, 2783 CrossRef PubMed; (b) I. Denya, S. F. Malan and J. Joubert, Expert Opin. Ther. Pat., 2018, 28, 441 CrossRef CAS PubMed; (c) A. Thangadurai, M. Minu, S. Wakode, S. Agrawal and B. Narasimhan, Med. Chem. Res., 2012, 21, 1509–1523 CrossRef CAS; (d) M. J. Haddadin, W. E. Conrad and M. J. Kurth, Mini-Rev. Med. Chem., 2012, 12, 1293 CrossRef CAS PubMed; (e) J. Y. Dong, Q. J. Zhang, Z. T. Wang, G. Huang and S. S. Li, ChemMedChem, 2018, 13, 1490 CrossRef CAS PubMed; (f) D. D. Gaikwad, A. D. Chapolikar, C. G. Devkate, K. D. Warad, A. P. Tayade, R. P. Pawar and A. J. Domb, Eur. J. Med. Chem., 2015, 90, 707 CrossRef CAS PubMed.
- (a) A. Roth, S. Ott, K. M. Farber, T. A. Palazzo, W. E. Conrad, M. J. Haddadin, D. J. Tantillo, C. E. Cross, J. P. Eiserich and M. J. Kurth, Bioorg. Med. Chem., 2014, 22, 6422 CrossRef CAS PubMed; (b) J. Leis and C. Carter, US Pat., US20140179637 A1, 2014.
- (a) A. Montero-Torres, M. C. Vega, Y. Marrero-Ponce, M. Rolon, A. Gomez-Barrio, J. A. Escario, V. J. Aran, A. R. Martinez-Fernandez and A. Meneses-Marcel, Bioorg. Med. Chem., 2005, 13, 6264 CrossRef CAS PubMed; (b) M. C. Vega, M. Rolon, A. Montero-Torres, C. Fonseca-Berzal, J. A. Escario, A. Gomez-Barrio, J. Galvez, Y. Marrero-Ponce and V. J. Aran, Eur. J. Med. Chem., 2012, 58, 214 CrossRef CAS PubMed.
- Y. M. Qian, D. Bolin, K. Conde-Knape, P. Gillespie, S. Hayden, K. S. Huang, A. R. Olivier, T. Sato, Q. Xiang, W. Y. Yun and X. L. Zhang, Bioorg. Med. Chem. Lett., 2013, 23, 2936 CrossRef CAS PubMed.
- S. R. Fletcher, E. McIver, S. Lewis, F. Burkamp, C. Leech, G. Mason, S. Boyce, D. Morrison, G. Richards, K. Sutton and A. B. Jones, Bioorg. Med. Chem. Lett., 2006, 16, 2872 CrossRef CAS PubMed.
- W. S. Yu, Z. Y. Guo, P. Orth, V. Madison, L. Chen, C. Y. Dai, R. J. Feltz, V. M. Girijavallabhan, S. H. Kim, J. A. Kozlowski, B. J. Lavey, D. S. Li, D. Lundell, X. D. Niu, J. J. Piwinski, J. Popovici-Muller, R. Rizvi, K. E. Rosner, B. B. Shankar, N. Y. Shih, M. A. Siddiqui, J. Sun, L. Tong, S. Umland, M. K. C. Wong, D. Y. Yang and G. W. Zhou, Bioorg. Med. Chem. Lett., 2010, 20, 1877 CrossRef CAS PubMed.
- A. Cappelli, C. Nannicini, A. Gallelli, G. Giuliani, S. Valenti, G. Mohr, M. Anzini, L. Mennuni, F. Ferrari, G. Caselli, A. Giordani, W. Peris, F. Makovec, G. Giorgi and S. Vomero, J. Med. Chem., 2008, 51, 2137 CrossRef CAS PubMed.
- N. Kawanishi, T. Sugimoto, J. Shibata, K. Nakamura, K. Masutani, M. Ikuta and H. Hirai, Bioorg. Med. Chem. Lett., 2006, 16, 5122 CrossRef CAS PubMed.
- (a) H. Wang, H. Y. Han and D. D. Von Hoff, Cancer Res., 2006, 66, 9722 CrossRef CAS PubMed; (b) M. H. Norman, G. C. Rigdon, F. Navas and B. R. Cooper, J. Med. Chem., 1994, 37, 2552 CrossRef CAS PubMed; (c) K. A. M. Abouzid and H. S. El-Abhar, Arch. Pharmacal Res., 2003, 26, 1 CrossRef CAS; (d) P. Bruneau, C. Delvare, M. P. Edwards and R. M. Mcmillan, J. Med. Chem., 1991, 34, 1028 CrossRef CAS PubMed.
- D. Vina, E. del Olmo, J. L. Lopez-Perez and A. San Feliciano, Org. Lett., 2007, 9, 525 CrossRef CAS PubMed.
- G. Dou and D. Shi, J. Comb. Chem., 2009, 11, 10737 Search PubMed.
- (a) S. Tanimori, Y. Ozaki, Y. Lesaki and M. Kirihata, Synlett, 2008, 13, 1973 CrossRef; (b) S. Tanimori, Y. Kobayashi, Y. Iesaki, Y. Ozaki and M. Kirihata, Org. Biomol. Chem., 2012, 10, 1381 RSC; (c) W. J. Wang, J. H. Chen, Z. C. Chen, Y. F. Zeng, X. J. Zhang, M. Yan and A. S. C. Chan, Synthesis, 2016, 48, 3551 CrossRef CAS.
- S. Liu, L. Xu and Y. Wei, J. Org. Chem., 2019, 84, 1596 CrossRef CAS PubMed.
- D. G. Yu, M. Suri and F. Glorius, J. Am. Chem. Soc., 2013, 135, 8802 CrossRef CAS PubMed.
- E. B. Elkaeed, J. An and A. M. Beauchemin, J. Org. Chem., 2017, 82, 9890 CrossRef CAS PubMed.
- A. Correa, I. Tellitu, E. Dominguez and R. SanMartin, J. Org. Chem., 2006, 71, 3501 CrossRef CAS PubMed.
- (a) M. J. Kurth, M. M. Olmstead and M. J. Haddadin, J. Org. Chem., 2005, 70, 1060 CrossRef CAS PubMed; (b) J. S. Zhu, N. Kraemer, M. E. Shatskikh, C. J. Li, J. H. Son, M. J. Haddadin, D. J. Tantillo and M. J. Kurth, Org. Lett., 2018, 20, 4736 CrossRef CAS PubMed.
- Other selected approaches for the preparation of indazolone skeletons see: (a) C. J. Li, T. Y. Zhang, Z. Zeng, X. J. Liu, Y. F. Zhao, B. Zhang and Y. Q. Feng, Org. Lett., 2012, 14, 479 CrossRef CAS PubMed; (b) J. S. Chen, P. Chen, C. Song and J. Zhu, Chem. –Eur. J., 2014, 20, 14245 CrossRef CAS PubMed; (c) S. W. Park, H. Choi, J. H. Lee, Y. J. Lee, J. M. Ku, S. Y. Lee and T. G. Nam, Arch. Pharmacal Res., 2016, 39, 302 CrossRef CAS PubMed; (d) E. Schutznerova and V. Krchnak, J. Org. Chem., 2016, 81, 3585 CrossRef CAS PubMed.
- (a) Light-triggered covalent assembly of gold nanoparticles with indazolone structure formation in aqueous solution see: J. P. Lai, Y. Y. Xu, X. Mu, X. L. Wu, C. Li, J. S. Zheng, C. L. Wu, J. B. Chen and Y. B. Zhao, Chem. Commun., 2011, 47, 3822 RSC; (b) During the preparation of this manuscript, a photochemical preparation of indazolone derivatives was reported: J. S. Zhu, N. Kraemer, C. J. Li, M. J. Haddadin and M. J. Kurth, J. Org. Chem., 2018, 83, 15493 CrossRef CAS PubMed.
- (a) H. Zhao, E. S. Sterner, E. B. Coughlin and P. Theato, Macromolecules, 2012, 45, 1723 CrossRef CAS; (b) P. Klan, T. Solomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119 CrossRef CAS PubMed.
- Y. V. Il'ichev, M. A. Schworer and J. Wirz, J. Am. Chem. Soc., 2004, 126, 4581 CrossRef PubMed.
- (a) M. Rasmusson, R. Lindh, N. Lascoux, A. N. Tarnovsky, M. Kadi, O. Kuhn, V. Sundstrom and E. Akesson, Chem. Phys. Lett., 2003, 367, 759 CrossRef CAS; (b) L. Li, W. B. Liu, H. Y. Zeng, X. Y. Mu, G. Cosa, Z. T. Mi and C. J. Li, J. Am. Chem. Soc., 2015, 137, 8328 CrossRef CAS PubMed.
- (a) M. Z. Hernandes, S. M. T. Cavalcanti, D. R. M. Moreira, W. F. de Azevedo and A. C. L. Leite, Curr. Drug Targets, 2010, 11, 303 CrossRef CAS PubMed; (b) R. Wilcken, M. O. Zimmermann, A. Lange, A. C. Joerger and F. M. Boeckler, J. Med. Chem., 2013, 56, 1363 CrossRef CAS PubMed.
- (a) I. Ghosh, T. Ghosh, J. I. Bardagi and B. Konig, Science, 2014, 346, 725 CrossRef CAS PubMed; (b) M. Haring, R. Perez-Ruiz, A. Jacobi von Wangelin and D. D. Diaz, Chem. Commun., 2015, 51, 16848 RSC; (c) K. Chen, P. He, S. Zhang and P. Li, Chem. Commun., 2016, 52, 9125 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02466b |
| This journal is © The Royal Society of Chemistry 2019 |