DOI:
10.1039/C9RA02482D
(Paper)
RSC Adv., 2019,
9, 21591-21600
Retracted Article: Knockdown of TMPRSS11D inhibits the proliferation, migration and invasion of cervical cancer cells†
Received
2nd April 2019
, Accepted 1st July 2019
First published on 12th July 2019
Abstract
TMPRSS11D is a member of the type II transmembrane serine proteases (TTSPs) family that is implicated in the development and progression of several cancers. However, the biological roles of TMPRSS11D in cervical cancer have not been investigated. In the present study, we detected the expression levels of TMPRSS11D in human cervical cancer tissues and cell lines. The results showed that TMPRSS11D expression was significantly upregulated in cervical cancer tissues as compared to the adjacent normal tissues. Besides, TMPRSS11D was highly expressed in human cervical cancer cell lines. Then we knocked down TMPRSS11D in cervical cancer cell lines to evaluate the effects of TMPRSS11D knockdown on cervical cancer cells. The results showed that knockdown of TMPRSS11D significantly suppressed cell proliferation, migration and invasion in cervical cancer cell lines. Furthermore, the data revealed that TMPRSS11D knockdown prevented epithelial–mesenchymal transition (EMT), as proved by the increased E-cadherin expression, as well as decreased N-cadherin and fibronectin expressions. Additionally, knockdown of TMPRSS11D inhibited the activation of the PI3K/Akt pathway in cervical cancer cells. Furthermore, insulin-like growth factor-1 (IGF-1) treatment reversed the inhibitory effects of TMPRSS11D knockdown on cell proliferation and migration. Collectively, knockdown of TMPRSS11D exerted anti-tumor activity, at least in part, via inhibiting the PI3K/Akt pathway. These findings indicated that TMPRSS11D might serve as a novel therapeutic target for the treatment of cervical cancer.
1. Introduction
Cervical cancer is the fourth most common malignancy diagnosed in women with an estimated 570![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 new cases in 2018 representing 6.6% of all female cancers worldwide.1,2 More than 90% of cases of cervical cancer result from infection with the human papillomavirus (HPV).3,4 The HPV vaccine represents a revolutionary step forward in preventing cervical cancer. But, some vaccines are available only on a user-pays basis and therefore entail an out-of-pocket expense. This has contributed to the inequitable uptake.5 Currently, the management of patients with cervical cancer includes surgery or a concurrent chemoradiotherapy regimen.6 However, the 3-year to 5-year survival rate is <50% in many underdeveloped countries.1 Cervical cancer causes more than one quarter of a million deaths each year due to grossly deficient treatments in many developing countries.1 Until now, cervical cancer still remains one of the leading causes of cancer deaths in women. Previously, genetic mutation was thought to play a key role in the development of cervical cancer.7 But recent advances in the biology of cervical cancer have revealed that epigenetic alteration is widely observed in the carcinogenesis and metastasis of cervical cancer.7
000 new cases in 2018 representing 6.6% of all female cancers worldwide.1,2 More than 90% of cases of cervical cancer result from infection with the human papillomavirus (HPV).3,4 The HPV vaccine represents a revolutionary step forward in preventing cervical cancer. But, some vaccines are available only on a user-pays basis and therefore entail an out-of-pocket expense. This has contributed to the inequitable uptake.5 Currently, the management of patients with cervical cancer includes surgery or a concurrent chemoradiotherapy regimen.6 However, the 3-year to 5-year survival rate is <50% in many underdeveloped countries.1 Cervical cancer causes more than one quarter of a million deaths each year due to grossly deficient treatments in many developing countries.1 Until now, cervical cancer still remains one of the leading causes of cancer deaths in women. Previously, genetic mutation was thought to play a key role in the development of cervical cancer.7 But recent advances in the biology of cervical cancer have revealed that epigenetic alteration is widely observed in the carcinogenesis and metastasis of cervical cancer.7
Type II transmembrane serine proteases (TTSPs) are an identified family of proteolytic enzymes that are structurally defined by a single-pass transmembrane domain.8,9 The special localization allows these enzymes to mediate signal transduction between the intracellular and extracellular environment, thereby regulating various cellular responses. The TTSPs are found to be involved in a diverse range of physiological functions and play important roles in the development and homeostasis of various mammalian tissues.10 Additionally, aberrant expression of TTSP genes appears to be implicated in the development and progression of several human diseases, including cancer.10,11 TMPRSS11D belongs to the TTSP family and has been shown to be significantly correlated with several types of cancers, such as head and neck squamous cell carcinoma (HNSCC) and non-small cell lung cancer (NSCLC).12,13 However, the expression and biological roles of TMPRSS11D in cervical cancer remain largely unknown. Thus, in this study, we detected the expression of TMPRSS11D in human cervical cancer tissues and cell lines, and evaluated its roles in cervical cancer cells.
2. Materials and methods
2.1 Patient samples
All experiments were performed in accordance with the guidelines of The First Affiliated Hospital of Xi'an Jiaotong University (Xi'an, China), and experiments were approved by the ethics committee of The First Affiliated Hospital of Xi'an Jiaotong University. A total of 23 paired cervical cancer and adjacent normal tissues were collected from patients with cervical cancer who underwent surgery from February 2016 to May 2017 at The First Affiliated Hospital, Xi'an Jiaotong University. All patients had provided informed consents for this research. The clinical information of cervical cancer patients was shown in Table 1.
Table 1 The clinical information of cervical cancer patients
| |
No. patients (n = 23) |
| Age |
| ≤45 |
7 |
| >45 |
16 |
|
| FIGO stage |
| Early stage |
10 |
| Advanced stage |
13 |
|
| Histological grade |
| Well |
2 |
| Moderate |
6 |
| Poor |
15 |
|
| Lymph node metastasis |
| Negative |
9 |
| Positive |
14 |
2.2 Cell culture
Human cervical cancer cell lines (ME180, SiHa, CaSki, and HeLa) and normal cervical epithelial cell line (H8) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco Laboratories, Grand Island, NY, USA) or RPMI-1640 medium (Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco) and 0.1% penicillin–streptomycin (Sigma-Aldrich, St. Louis, MO, USA). All cells were grown in a humidified incubator with 5% CO2 at 37 °C. For the insulin-like growth factor-1 (IGF-1) treatment groups, cells were treated with 100 ng mL−1 IGF-1 (PHG0078; Invitrogen, Carlsbad, CA, USA) for 24 h.
2.3 Cell transfection
Small interfering RNA (siRNA) targeting TMPRSS11D (si-TMPRSS11D) and negative control siRNA (si-NC) were obtained from GenePharma (Shanghai, China). CaSki and HeLa cells were seeded at a density of 1 × 104 cells per well in a 96-well microplate and incubated for 24 h. Then si-TMPRSS11D or si-NC (30 nM) was transfected into the cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. After 48 h post siRNA transfection, transfection efficiency was tested using qRT-PCR and Western blot. The experiment was performed in triplicate.
2.4 Quantitative real-time PCR (qRT-PCR)
Total RNA from clinical tissues and cultured cells was extracted using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. The isolated RNA was used for complementary DNA (cDNA) synthesis using a high capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA). PCR amplification was performed using SYBR Premix DimerEraser (Takara, Dalian, China) with the following condition: denaturation at 95 °C for 10 min; followed by 30 cycles 95 °C for 60 s, 57 °C for 30 s, 72 °C for 30 s; final extension at 72 °C for 5 min. TMPRSS11D expression was normalized to that of the housekeeping gene β-actin. The primer sequences were: TMPRSS11D forward, 5′-TAC ACA GGA ATA CAG GAC TT-3′ and reverse 5′-CTC ACA CCA CTA CCA TCT-3′; β-actin forward, 5′-ATCA CCAT TGGC AATG AGCG-3′ and reverse 5′-TTGA AGGT AGTT TCGT GGAT-3′. The experiment was performed in triplicate.
2.5 Western blot analysis
Total protein of CaSki and HeLa cells were prepared using a protein extraction kit (Millipore, Billerica, MA, USA) according to the manufacturer's instructions. Protein contents were determined using a BCA protein kit (A53227; Thermo Scientific, Waltham, MA, USA). Equal amounts of protein samples (50 μg) were separated on 12% SDS-PAGE electrophoresis. Then the samples were transferred to Immobilon PVDF membrane (Millipore) and subsequently blocked with 5% skim milk in TBST buffer for 1 h at room temperature. Proteins were probed with different primary antibodies against TMPRSS11D (1:500; ab127031; Abcam, Cambridge, MA, USA), E-cadherin (1:500; ab1416; Abcam), N-cadherin (1:1000; ab18203; Abcam), fibronectin (1:1000; ab2413; Abcam), PI3K (1:500; #4292; Cell Signaling Technology, Boston, MA, USA), p-PI3K (1:500; #4228; Cell Signaling Technology), Akt (1:1000; #9272; Cell Signaling Technology), p-Akt (Ser473) (1:1000; #4060; Cell Signaling Technology), and β-actin (1:3000; #4970; Cell Signaling Technology) overnight at 4 °C, followed by incubation with horseradish peroxidase-conjugated secondary antibody (1:3000; sc-2030; Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 1 h at room temperature. Bands were visualized using an enhanced chemiluminescence (ECL) kit (35055; Thermo Scientific) according to the manufacturer's protocol. The experiment was performed in triplicate.
2.6 Cell proliferation assay
Cell Counting Kit-8 (CCK-8; ck04-500; Dojindo, Kumamoto, Japan) was used to assess the proliferation of the CaSki and HeLa cells according to the manufacturer's instructions. In brief, cells with different transfections were plated into 96-well plates at a density of 5 × 103 cells per well and cultured for 24, 48, 72 and 96 h, respectively. Then, 10 μL of CCK-8 was added in to the culture medium, followed by an incubation for another 1 h. The absorbance value (OD) at 450 nm was measured with a spectrophotometric plate reader (Bio-Rad, Hercules, CA, USA). The experiment was performed in triplicate.
2.7 Cell apoptosis assay
Cell apoptosis was detected using flow cytometry method with an Annexin V-FITC Apoptosis Detection Kit (BMS500FI-20; Invitrogen). Briefly, the transfected cells were rinsed twice with PBS, followed by staining with Annexin V-FITC and propidium iodide (PI) for 20 min at room temperature. Finally, cells were analyzed by fluorescence-activated cell sorting (FACS) on a flow cytometer (Becton Dickinson, San Jose, CA, USA). The experiment was performed in triplicate.
2.8 Transwell migration and invasion assay
Cell migration and invasion were detected using transwell assay using 24-transwell chambers (Costar, Bodenheim, Germany). For the migration assay, the CaSki and HeLa cells (4 × 105 cells per mL) were resuspended in serum-free DMEM. The upper chambers were loaded with 100 μL cell suspension, while the lower chambers were filled with 500 μL DMEM containing 15% FBS. Following incubation for 24 h, the cells stayed on the upside of the filters were removed using a cotton swab. Cells stayed on the underside of the filters were fixed with 4% paraformaldehyde for 15 min and stained with 0.1% crystal violet for 10 min. The average number of cells in six randomly selected fields was counted. For the invasion assay, the same procedures described above were used, except that the filters were pre-coated with were Matrigel (354480; BD Biosciences, Franklin Lakes, NJ, USA) to form a genuine reconstituted basement membrane. The experiment was performed in triplicate.
2.9 Statistical analysis
The data are expressed as the mean ± standard deviation (SD), and analyzed using the software GraphPad Prism version 6.0 (GraphPad Software, San Diego, CA, USA). Student's t-test (between two different groups) or one-way ANOVA (among more than two groups) was used to analyze the statistical difference. P values less than 0.05 indicate a statistically significant difference.
3. Results
3.1 TMPRSS11D was highly expressed in human cervical cancer tissues and cell lines
First, we evaluated the expression pattern of TMPRSS11D in clinical samples. As shown in Fig. 1A, TMPRSS11D mRNA levels in cervical cancer tissues were significantly higher than that in the adjacent normal tissues. The results of Western blot analysis showed that the protein expression of TMPRSS11D was also increased in cervical cancer tissues (Fig. 1B). Next, we tested the expression of TMPRSS11D in four human cervical cancer cell lines including ME180, SiHa, CaSki, and HeLa cells. Results in Fig. 1C and D indicated that TMPRSS11D is highly expressed in cervical cancer cell lines, especially in CaSki and HeLa cells.
 |
| | Fig. 1 TMPRSS11D was upregulated in human cervical cancer tissues and cell lines. (A and B) TMPRSS11D mRNA expressions in cervical cancer tissues and adjacent normal tissues. (C and D) The expression levels of TMPRSS11D at both mRNA and protein in four human cervical cancer cell lines. n = 3. *p < 0.05. | |
3.2 Knockdown of TMPRSS11D inhibited the proliferation of cervical cancer cells
In order to further evaluate the role of TMPRSS11D, TMPRSS11D was knocked down in CaSki and HeLa cells through transfection with si-TMPRSS11D. The results of qRT-PCR and Western blot analysis suggested that TMPRSS11D was successfully knocked down in CaSki and HeLa cells (Fig. 2A–D). CCK-8 assay suggested that knockdown of TMPRSS11D markedly reduced cell proliferation of CaSki and HeLa cells compared with si-NC transfected cells (Fig. 2E and F).
 |
| | Fig. 2 Cell proliferation of cervical cancer cells was decreased after si-TMPRSS11D transfection. (A–D) TMPRSS11D expression in CaSki and HeLa cells after transfection with si-TMPRSS11D or si-NC. (E and F) Cell proliferation of CaSki and HeLa cells were detected using CCK-8 assay. n = 4. *p < 0.05. | |
3.3 Knockdown of TMPRSS11D promotes the apoptosis of cervical cancer cells
Then, we examined the effect of TMPRSS11D knockdown on cell apoptosis. The results of flow cytometry-based Annexin V-FITC/PI double staining indicated that as compared with the si-NC group, knockdown of TMPRSS11D significantly prompted the apoptosis of CaSki and HeLa cells, respectively (Fig. 3A and B).
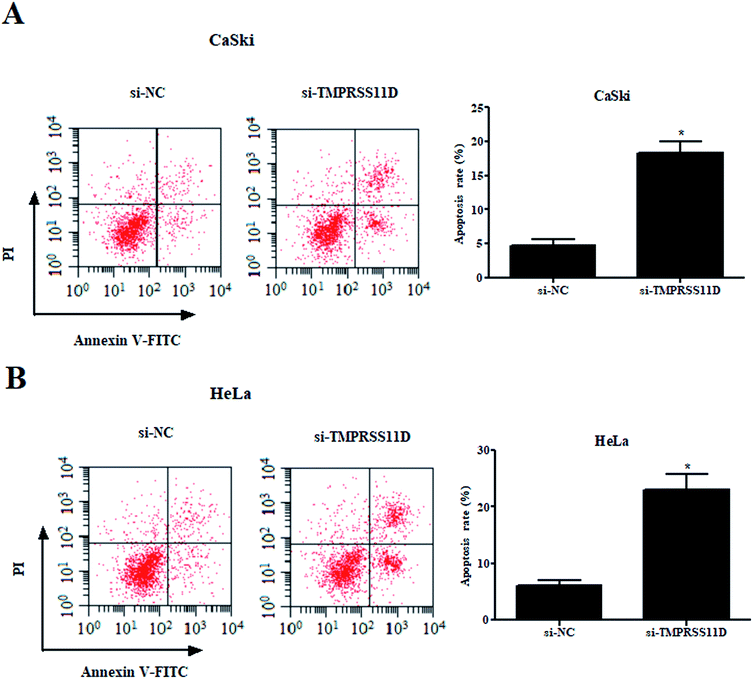 |
| | Fig. 3 Knockdown of TMPRSS11D promotes the apoptosis of cervical cancer cells. Cell apoptosis were detected through PI staining and the Annexin V method after transfection with si-TMPRSS11D or si-NC, followed by flow cytometry. (A) Figure was obtained from CaSki cells; (B) figure was obtained from HeLa cells. n = 4. *p < 0.05. | |
3.4 Knockdown of TMPRSS11D suppressed the migration and invasion of cervical cancer cells
Compared with the si-NC transfected cells, a significant decrease of cell migration was observed in si-TMPRSS11D transfected CaSki and HeLa cells (Fig. 4A and B). In addition, TMPRSS11D knockdown also caused dramatic reduction in cell invasion in both CaSki and HeLa cells (Fig. 4C and D).
 |
| | Fig. 4 Cell migration and invasion of cervical cancer cells were reduced after si-TMPRSS11D transfection. Cell migration and invasion were measured using transwell assays at 24 h post transfection. (A and B) Cell migration of cervical cancer cells. (C and D) Cell invasion of cervical cancer cells. n = 4. *p < 0.05. | |
3.5 Knockdown of TMPRSS11D prevented the epithelial–mesenchymal transition (EMT) phenotype in cervical cancer cells
To evaluate the effect of TMPRSS11D knockdown on EMT phenotype, the expression levels of marker proteins including E-cadherin, N-cadherin, and fibronectin were measured using Western blot analysis. As shown in Fig. 5A and B, TMPRSS11D knockdown resulted in increase in E-cadherin expression, as well as decrease in N-cadherin and fibronectin expressions in both CaSki and HeLa cells, suggesting that TMPRSS11D knockdown prevented EMT phenotype.
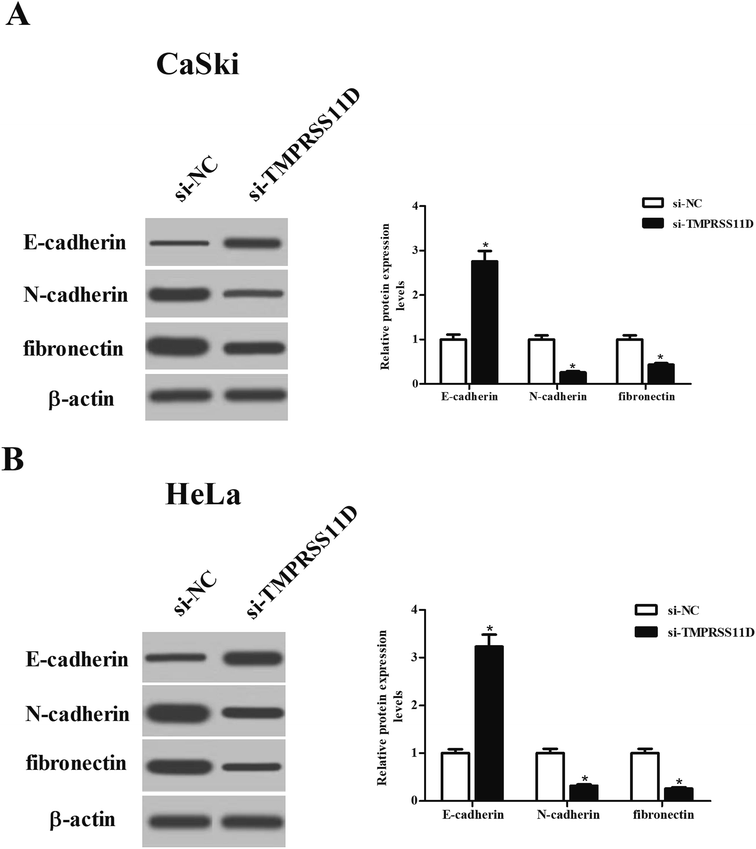 |
| | Fig. 5 EMT phenotype of cervical cancer cells was prevented by si-TMPRSS11D transfection. After 24 h post transfection, the expression levels of EMT markers including E-cadherin, N-cadherin, and fibronectin in CaSki (A) and HeLa cells (B) were measured using Western blot analysis. n = 4. *p < 0.05. | |
3.6 Knockdown of TMPRSS11D repressed the activation of PI3K/Akt pathway in cervical cancer cells
To investigate the effect of TMPRSS11D knockdown on PI3K/Akt pathway, the expression levels of PI3K, p-PI3K, Akt, and p-Akt were measured using Western blot analysis. As indicated in Fig. 6A, the expression levels of p-PI3K and p-Akt were markedly decreased in HeLa cells after transfection with si-TMPRSS11D. Then the cells were treated with IGF-1, a PI3K/Akt agonist, to activate PI3K/Akt pathway. Fig. 6B proved that IGF-1 treatment induced cell proliferation of si-TMPRSS11D transfected HeLa cells. Furthermore, IGF-1 treatment reversed the suppressive effects of TMPRSS11D knockdown on cell migration of HeLa cells (Fig. 6C).
 |
| | Fig. 6 Inhibitory effects of TMPRSS11D knockdown were mediated by suppression of PI3K/Akt pathway in cervical cancer cells. (A) Expression levels of PI3K, p-PI3K, Akt, and p-Akt were measured using Western blot analysis at 24 h post transfection. (B) Cell proliferation of HeLa cells after IGF-1 treatment. (C) Cell migration of HeLa cells after IGF-1 treatment. n = 3. *p < 0.05 vs. si-NC group; #p < 0.05 vs. si-TMPRSS11D group. | |
4. Discussion
TMPRSS11D, also known as human airway trypsin-like protease (HAT), belongs to the differentially expressed in squamous cell carcinoma (DESC) subfamily.14 TMPRSS11D triggers specific responses, notably in epithelial cells, including inflammatory cell recruitment, inflammatory cytokine production, and anticoagulant processes.14 Previous studies have demonstrated that it plays possible roles in respiratory physiology and disease.14,15 In recent years, TMPRSS11D is observed to be associated with the progression and development of various cancers. TMPRSS11D has been found to be downregulated in the HNSCC cancerous samples when compared with the control samples.12 TMPRSS11D expression exhibits positive association with the overall survival of patients with HNSCC, indicating that it may be used as potential tumor marker and prognostic indicator for HNSCC. Besides, expression levels of TMPRSS11D are significantly higher in NSCLC tumorous tissues than that in adjacent normal tissues. High TMPRSS11D protein expression in tumorous tissues is correlated with TNM stage and poor overall survival in NSCLC patients.12 In addition, TMPRSS11D protein is consistently expressed on the cell surface of differentiated epithelial cells in healthy cervical and esophageal epithelia. But TMPRSS11D expression is significantly reduced or lost in late stages of cervical and esophageal cancers. These findings suggest that TMPRSS11D may potentially be a marker for clinical grading and assessment of prognosis in patients with squamous cell carcinomas.16
However, the role of TMPRSS11D in cervical cancer has not been fully understood. In the present study, we found that TMPRSS11D expression was significantly higher in cervical cancer tissues than that in the adjacent normal tissues. Moreover, TMPRSS11D was highly expressed in human cervical cancer cell lines. It is well known that tumor cells are characterized by malignant proliferation, migration and invasion.17,18 Therefore, we knocked down TMPRSS11D in cervical cancer cell lines by transfection with si-TMPRSS11D to evaluate the effects of TMPRSS11D knockdown on cell proliferation, migration and invasion. The results showed that knockdown of TMPRSS11D significantly suppressed cell proliferation, migration and invasion in cervical cancer cell lines, implying that knockdown of TMPRSS11D exhibited anti-tumor activity. EMT is a process by which epithelial cells lose their cell polarity and cell–cell adhesion, and gain migratory and invasive properties to become mesenchymal stem cells.19 It has been proven that EMT is crucial for the initiation of metastasis in cancer progression.20 Epithelial cells express high levels of E-cadherin, whereas mesenchymal cells express N-cadherin, fibronectin and vimentin.20,21 Thus, E-cadherin, N-cadherin, fibronectin and vimentin are usually used as biomarkers of EMT process. In this study, we revealed that E-cadherin expression was increased, while N-cadherin and fibronectin expressions were decreased in si-TMPRSS11D transfected cervical cancer cell lines, suggesting that TMPRSS11D knockdown prevented EMT phenotype.
Emerging evidences have denoted that there are multiple pathways implicated in cervical carcinogenesis, such as RAF/MEK/ERK, PI3K/Akt, and Wnt/β-catenin pathways.22 These pathways have emerged as promising directed therapeutic targets in the treatment of this malignancy. The PI3K/Akt pathway integrates signals from growth factors and cytokines, thereby regulating diverse cellular functions including cell growth, differentiation, proliferation, apoptosis, and survival, as well as angiogenesis and cellular metabolism.23 PI3K/Akt pathway is frequently deregulated in human cancers including cervical cancer.23–26 Targeting PI3K/Akt pathway offers a promising perspective for cervical cancer therapy.27 For example, Zhang et al. reported that lncRNA ANRIL promoted carcinogenesis in cervical cancer through the PI3K/Akt pathway.28 Our results showed that knockdown of TMPRSS11D repressed the activation of PI3K/Akt pathway in cervical cancer cells. Treatment with IGF-1 abolished the inhibitory effects of si-TMPRSS11D on cell proliferation and migration. The results suggested that si-TMPRSS11D exerted its roles, at least in part, via inhibiting PI3K/Akt pathway.
5. Conclusion
From our data, we concluded that TMPRSS11D was up-regulated in cervical cancer tissues and cell lines. Knockdown of TMPRSS11D suppressed cell proliferation, migration, invasion and EMT process, at least in part, via inhibiting PI3K/Akt pathway in cervical cancer cells. TMPRSS11D may represent a novel therapeutic target for cervical cancer therapies.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Acknowledgements
This study was supported by the project of Shaanxi Administration of Traditional Chinese Medicine in China (No. 15-LC044).
References
- W. Small Jr, M. A. Bacon, A. Bajaj, L. T. Chuang, B. J. Fisher, M. M. Harkenrider, A. Jhingran, H. C. Kitchener, L. R. Mileshkin, A. N. Viswanathan and D. K. Gaffney, Cancer, 2017, 123, 2404–2412 CrossRef.
- S. Pimple, G. Mishra and S. Shastri, Curr. Opin. Obstet. Gynecol., 2016, 28, 4–10 CrossRef PubMed.
- A. Goodman, BMJ, 2015, 350, h2372 CrossRef PubMed.
- P. Tsikouras, S. Zervoudis, B. Manav, E. Tomara, G. Iatrakis, C. Romanidis, A. Bothou and G. Galazios, J. BUON, 2016, 21, 320–325 Search PubMed.
- L. Y. Lee and S. M. Garland, F1000Research, 2017, 6, 866–876 Search PubMed.
- L. Denny, Discov. Med., 2012, 14, 125–131 Search PubMed.
- J. Fang, H. Zhang and S. Jin, Tumor Biol., 2014, 35, 5083–5093 CrossRef CAS.
- T. H. Bugge, T. M. Antalis and Q. Wu, J. Biol. Chem., 2009, 284, 23177–23181 CrossRef CAS.
- J. D. Hooper, J. A. Clements, J. P. Quigley and T. M. Antalis, J. Biol. Chem., 2001, 276, 857–860 CrossRef CAS.
- R. Szabo and T. H. Bugge, Int. J. Biochem. Cell Biol., 2008, 40, 1297–1316 CrossRef CAS.
- S. L. Webb, A. J. Sanders, M. D. Mason and W. G. Jiang, Front. Biosci., 2011, 16, 539–552 CrossRef CAS.
- X. Cao, Z. Tang, F. Huang, Q. Jin, X. Zhou and J. Shi, Oncotarget, 2017, 8, 12812–12819 Search PubMed.
- M. J. Duhaime, K. O. Page, F. A. Varela, A. S. Murray, M. Z. Silverman, G. L. Zoratti and K. List, J. Cell. Physiol., 2016, 231, 1476–1483 CrossRef CAS.
- A. Menou, J. Duitman, P. Flajolet, J. M. Sallenave, A. A. Mailleux and B. Crestani, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2017, 312, L657–L668 CrossRef.
- M. Chokki, S. Yamamura, H. Eguchi, T. Masegi, H. Horiuchi, H. Tanabe, T. Kamimura and S. Yasuoka, Am. J. Respir. Cell Mol. Biol., 2004, 30, 470–478 CrossRef CAS.
- M. J. Duhaime, K. O. Page, F. A. Varela, A. S. Murray, M. E. Silverman, G. L. Zoratti and K. List, J. Cell. Physiol., 2016, 231, 1476–1483 CrossRef CAS PubMed.
- Q. Du, Y. Zhang, X. X. Tian, Y. Li and W. G. Fang, Oncol. Rep., 2009, 22, 659–665 CAS.
- S. F. Tavazoie, C. Alarcon, T. Oskarsson, D. Padua, Q. Wang, P. D. Bos, W. L. Gerald and J. Massague, Nature, 2008, 451, 147–152 CrossRef CAS.
- D. Kong, Y. Li, Z. Wang and F. H. Sarkar, Cancers, 2011, 3, 716–729 CrossRef.
- A. Moustakas and A. G. de Herreros, Mol. Oncol., 2017, 11, 715–717 CrossRef.
- S. Lamouille, J. Xu and R. Derynck, Nat. Rev. Mol. Cell Biol., 2014, 15, 178–196 CrossRef CAS.
- J. Manzo-Merino, A. Contreras-Paredes, E. Vazquez-Ulloa, L. Rocha-Zavaleta, A. M. Fuentes-Gonzalez and M. Lizano, Arch. Med. Res., 2014, 45, 525–539 CrossRef CAS.
- A. Bahrami, M. Hasanzadeh, S. M. Hassanian, S. ShahidSales, M. Ghayour-Mobarhan, G. A. Ferns and A. Avan, J. Cell. Biochem., 2017, 118, 4163–4169 CrossRef CAS.
- S. A. Danielsen, P. W. Eide, A. Nesbakken, T. Guren, E. Leithe and R. A. Lothe, Biochim. Biophys. Acta, 2015, 1855, 104–121 CAS.
- P. Toren and A. Zoubeidi, Internet J. Oncol., 2014, 45, 1793–1801 CrossRef CAS.
- S. X. Yang, E. Polley and S. Lipkowitz, Cancer Treat. Rev., 2016, 45, 87–96 CrossRef CAS.
- J. Wu, C. Chen and K. N. Zhao, Curr. Cancer Drug Targets, 2013, 13, 143–156 CrossRef CAS.
- D. Zhang, G. Sun, H. Zhang, J. Tian and Y. Li, Biomed. Pharmacother., 2017, 85, 511–516 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02482d |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a





