
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient and sustainable laccase-catalyzed iodination of p-substituted phenols using KI as iodine source and aerial O2 as oxidant†
Mark Sdahl,
Jürgen Conrad,
Christina Braunberger and
Uwe Beifuss *
*
Bioorganische Chemie, Institut für Chemie, Universität Hohenheim, Garbenstr. 30, Stuttgart, D-70599, Germany. E-mail: ubeifuss@uni-hohenheim.de; Fax: +49 711 459 22951; Tel: +49 711 459 22171
First published on 21st June 2019
Abstract
The laccase-catalyzed iodination of p-hydroxyarylcarbonyl- and p-hydroxyarylcarboxylic acid derivatives using KI as iodine source and aerial oxygen as the oxidant delivers the corresponding iodophenols in a highly efficient and sustainable manner with yields up to 93% on a preparative scale under mild reaction conditions.
Introduction
The iodophenol moiety represents an important structural motif for many of the nearly 200 iodine-containing natural products known so far.1 Typical examples include the cytotoxic tyramine derivative iodocionin (I) from the Mediterranean ascidian Ciona edwardsii,2a the cytotoxic 6′-iodoaureol (II) from the sponge Smenospongia sp.,2b the antibiotic cyclic depsipeptide miuraenamide B (III) from the myxobacterial SMH-27-4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2c and the thyroid hormones triiodothyronine (IV)2d and thyroxin (V) (Fig. 1).2d The most prominent non-natural biologically active iodophenol is amiodarone (VI)3 which is widely used as an antiarrhythmic drug in the treatment of ventricular and supraventricular tachyarrhythmias. Iodoaromatics in general and iodophenols in particular are important substrates for Pd-catalyzed cross couplings,4 such as the Sonogashira-coupling,5a the carbonylative Sonogashira-coupling,5b the Negishi-coupling,5c the Suzuki–Miyaura-coupling5d and related reactions,5e Stille-,5f Hiyama-5g and Heck-couplings.5h Iodophenols can also be used as substrates for the preparation of organometallics, such as Grignard reagents, under suitable conditions.6 This is why the development of selective, efficient and environmentally benign methods for the preparation of iodoaromatics in general and iodophenols in particular is of the greatest importance.
2c and the thyroid hormones triiodothyronine (IV)2d and thyroxin (V) (Fig. 1).2d The most prominent non-natural biologically active iodophenol is amiodarone (VI)3 which is widely used as an antiarrhythmic drug in the treatment of ventricular and supraventricular tachyarrhythmias. Iodoaromatics in general and iodophenols in particular are important substrates for Pd-catalyzed cross couplings,4 such as the Sonogashira-coupling,5a the carbonylative Sonogashira-coupling,5b the Negishi-coupling,5c the Suzuki–Miyaura-coupling5d and related reactions,5e Stille-,5f Hiyama-5g and Heck-couplings.5h Iodophenols can also be used as substrates for the preparation of organometallics, such as Grignard reagents, under suitable conditions.6 This is why the development of selective, efficient and environmentally benign methods for the preparation of iodoaromatics in general and iodophenols in particular is of the greatest importance.
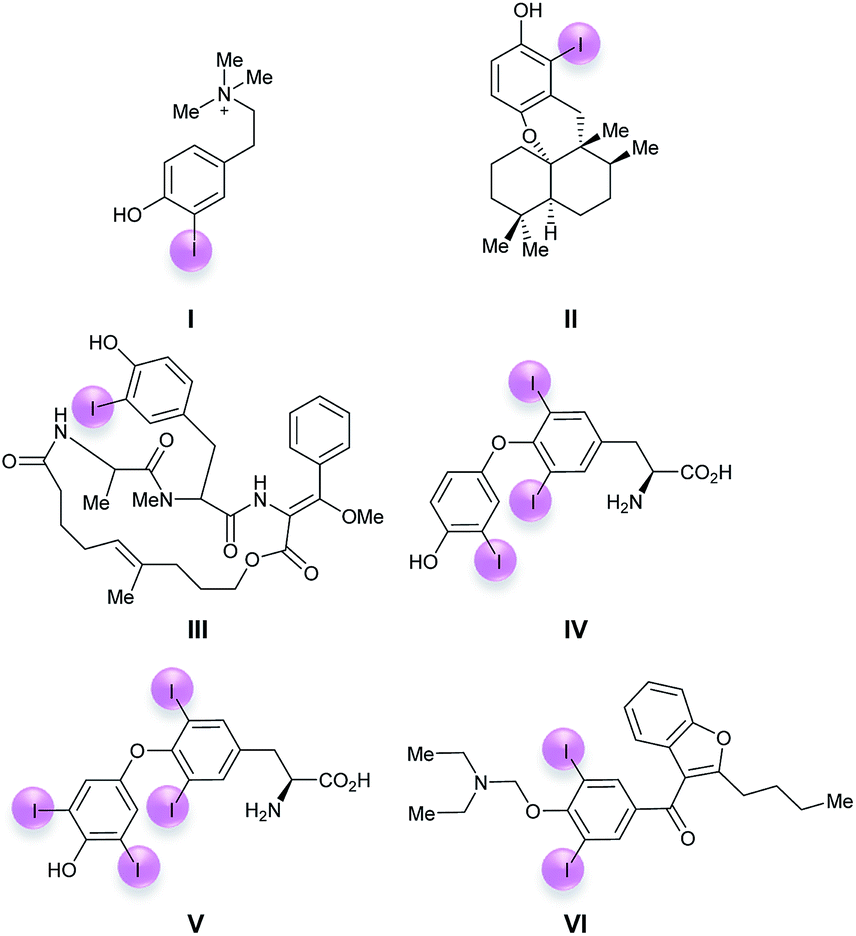 | ||
| Fig. 1 Important iodinated phenolic compounds. | ||
Over the years, a number of methods have been developed for the preparation of iodoaromatics.7 Without doubt, the most popular is the electrophilic aromatic substitution. Among the classical methods are also the Sandmeyer reaction,8 the ortho-lithiation/halogenation9 and the Hunsdiecker reaction.10 Recently, the synthesis of iodoaromatics has been achieved by methods which are based on sp2 C–H activation.7b
For the preparation of iodophenols the electrophilic aromatic substitution is also the most widely used method. For this purpose, phenols are reacted (a) with iodination agents like NIS,11a PyICl,11b TICA,11c BMPDCI,11d IPy2BF4,11e BTMA ICl2,11f I2,11g I2-amine complex,11h I2/AgNO3,11i or I2/KI,11j (b) with I2 in combination with an oxidant, such as (n-BuPPh3)2S2O8,12a HIO3,12b K2FeO4/K10,12c H2O2,12d O2/NaNO212e or TICA/SiO2,12f (c) with an iodide in combination with an oxidant such as NaOCl,13a NaClO2,13b H2O2,13c KClO3,13d NaIO4,13e oxone,13f KIO3,13g DMSO,13h H2SO413i or tert-butylhypochlorite.13j
Most of these methods have a number of serious disadvantages. Among them are the use of more than equimolar amounts of iodination agents and/or oxidants. Many of the reagents employed are acutely toxic, corrosive, explosive and oxidizing, while others are commercially not available, expensive or difficult to prepare. In addition, many iodinations have to be performed in highly volatile and/or toxic organic solvents. Most electrophilic aromatic substitutions with I2 or iodide as iodine source can only be run successfully in the presence of heavy metal reagents and/or strong oxidants. All methods which make use of I2 as iodine source suffer from the fact, that only one iodine atom ends up in the product of the electrophilic aromatic substitution while the other remains unused in the reaction medium. Clearly, this results in lower values of atom economy.14 Approaches which are based on the combination of I2 and iodides, respectively, as the iodine source, with sustainable oxidants, such as O212e or H2O2,12d,13c are often hampered by their restricted substrate scope12d,e or by the fact that they require highly acidic conditions.12e,13c As a result, there is great demand for iodination methods which are not only highly selective and efficient but also fulfill the requirements of sustainable chemistry in order to protect the environment.
Over the last few years, a keen interest in oxidative halogenations, which allow for the use of halides as halogen sources instead of the halogens themselves, has emerged.7d With respect to sustainability, transition metal- and enzyme-catalyzed transformations using H2O2 or O2 as oxidants are particularly attractive. Enzyme-catalyzed oxidative halogenations with H2O2 as oxidant are usually catalyzed by less specific heme- and vanadium-dependent haloperoxidases, while oxidative halogenations with O2 are mainly catalyzed by more substrate specific flavin-dependent halogenases and non-iron O2-dependant halogenases.15 In this context, studies towards the regioselective bromination and chlorination catalyzed by FAD-dependant tryptophan halogenases16a deserve to be mentioned since they can be performed on a preparative scale.16b In contrast, enzyme-catalyzed iodinations have received only marginal attention so far. It is known that oxidative iodinations can be catalyzed by lactoperoxidases,17a–c a chloroperoxidase17d and a horseradish peroxidase.17e However, as good as nothing is known concerning substrate specificity, substrate scope, selectivity, efficiency, scalability and sustainability of these reactions. First observations concerning the laccase-catalyzed oxidation of iodide to iodine can be traced back to the reports of Xu17f and Amachi et al.17g Later, Ihssen et al. have reported on the laccase-catalyzed iodination of phenolic compounds.17h However, a closer look at their results reveals that their method does not allow for the chemoselective iodination of phenols on a preparative useful scale. In most cases, the formation of the iodophenols was accompanied by the formation of products resulting from oxidative dimerization. Structure and yields of the iodinated products are difficult to evaluate since they were not isolated in pure form. Moreover, the scope of the method was not studied.
Laccases (benzenediol:oxygen oxidoreductase, EC 1.10.3.3) are enzymes which are produced by animals, plants, fungi and bacteria.18 Some are commercially available at a reasonable price. Laccases are known to catalyze a number of oxidations under mild reaction conditions in aqueous solvent systems at pH 3–8 using cheap and environmentally benign aerial oxygen as a sustainable oxidant.19 The substrate oxidation is accompanied by the reduction of O2 to H2O, which is the only byproduct of laccase-catalyzed reactions. Laccases with low (0.4–0.5 mV), medium (0.5–0.6 mV) and high (0.7–0.8 mV) redox potentials are known.20a–c By using laccase/mediator systems the substrate scope of laccase-catalyzed oxidations can be significantly widened.19,20d Among the transformations that can be catalyzed by laccases on a semi preparative or preparative scale are oxidations of several functional groups (CH3 → CHO,21a CH2OH → CHO,21b CH2OH → CO2H,21c CH2NH2 → CHO/CO2H21d), the transformation of 1,4-dihydropyridines to pyridines21e and oxidative couplings of phenols21f–i and related substrates21j as well as thiophenols.21k The oxidation of catechols and hydroquinones is also known. The resulting o- and p-benzoquinones can be intercepted in different reactions like 1,4-additions and Diels–Alder reactions. This approach provides not only access to simple 1,4-adducts22 but also to different carbo- and heterocycles.23
Here we show for the first time, that the laccase-catalyzed iodination of a wide range of p-substituted phenolic substrates delivers the iodinated products in a highly chemoselective manner on a preparative scale; the dimerization could be completely suppressed. Moreover, we will show that the laccase-catalyzed iodination can be developed to a sustainable iodination method.
Results and discussion
Reaction condition optimization
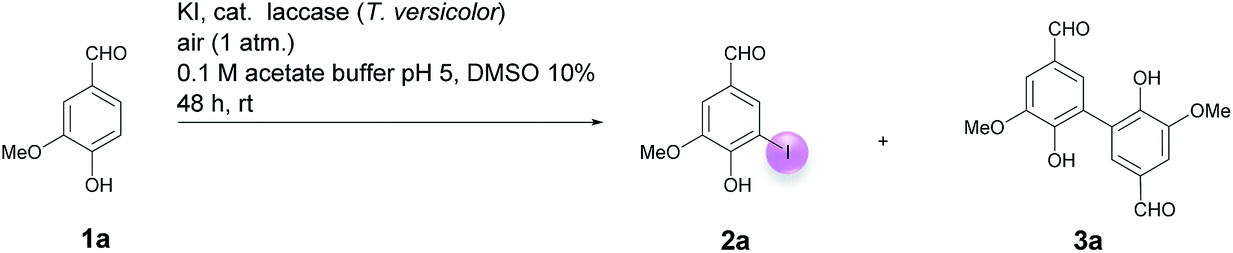
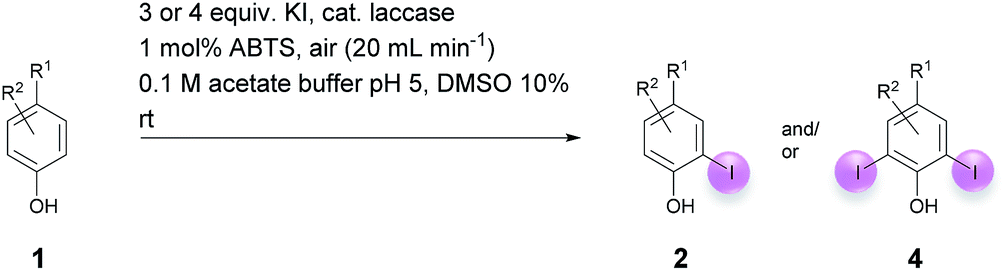
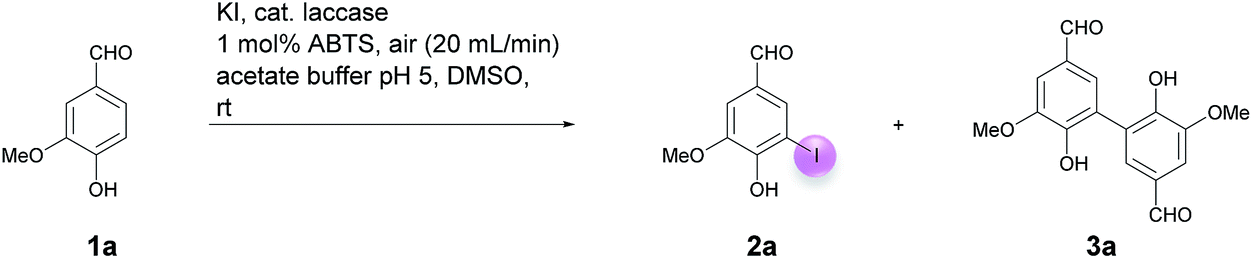
Motivated by our interest in using laccases as catalysts in preparative organic chemistry we wondered whether it would be possible to develop a selective, efficient, atom economic and sustainable iodination of aromatics on a preparative useful scale based on the laccase-catalyzed oxidation of alkali iodides to iodine using aerial oxygen as oxidant.The optimization of the reaction conditions was performed using the iodination of vanillin (1a) to 4-hydroxy-3-iodo-5-methoxybenzaldehyde (2a) as a model reaction since vanillin is a natural product that is manufactured from biomass by means of an established industrial process on a large scale.24 Against the background of our experience in the field of laccase-catalyzed transformations, equimolar amounts of 1a and KI were stirred with catalytic amounts (225 U) of T. versicolor laccase in the presence of aerial oxygen in acetate buffer (pH 5):DMSO = 9:1 for 48 h at rt (Table 1, entry 1). Under these conditions, the desired iodination product 2a was formed in only 7% yield. The main product was the dimer 6,6′-dihydroxy-5,5′-dimethoxy-[1,1′-biphenyl]-3,3′-dicarbaldehyde (divanillin) (3a). However, the formation of 3a was not surprising, since the laccase-catalyzed oxidative dimerization of 1a is known to deliver 3a.21f Under the conditions presented in Scheme 1, we isolated 3a in 91% yield (Scheme 1). To increase the yield of 2a, and to improve the 2a:3a ratio towards the formation of 2a, the reaction was run with 20 equiv. of KI. This resulted in an increase of the yield of 2a to 24% and an improvement of the 2a:3a ratio to 20:1 (Table 1, entry 2). To further suppress the oxidative dimerization, it was decided to keep the actual concentration of 1a in the reaction mixture as low as possible. This was achieved by continuous addition of 1a as a solution in DMSO (2 mmol 1a in 3 mL DMSO) over 24 h by using a syringe pump. As expected, the amount of dimer 3a in the crude product could be decreased considerably (2a:3a = 65:1); however, the isolated yield of 2a amounted to only 18% (Table 1, entry 3). Since it is assumed that the laccase undergoes partial iodination, which results in partial deactivation, parts of the laccase were added continuously during the reaction. Consequently, only 45 U laccase were added initially and the remaining laccase (180 U) was added dropwise via syringe pump during 24 h. This measure resulted in an increase of the yield of 2a to 34%; unfortunately, the ratio of 2a:3a decreased to 18:1 (Table 1, entry 4). It is well known that many laccase-catalyzed reactions give the products in much higher yields in the presence of a mediator.19,20d In earlier studies we have established that ABTS is a particularly suitable mediator for T. versicolor-catalyzed reactions.21e,k When the laccase-catalyzed iodination of 1a was run in the presence of 1 mol% ABTS, the yield of 2a increased to 46% and the 2a:3a ratio improved to 198:1 (Table 1, entry 5). Furthermore, it was found that the reaction volume has a decisive influence on the yield of 2a. When the buffer volume was doubled to 90 mL and the reaction was run in the absence of ABTS, the isolated yield of 2a could be improved to 75%. Unfortunately, this was accompanied by an increase of 3a in the crude product (2a:3a = 18:1) (Table 1, entry 6). However, when the transformation was performed in the presence of 1 mol% ABTS in 90 mL buffer, and the laccase as well as the substrate were added gradually, 2a could be isolated in 65% and the formation of 3a could be almost completely suppressed (2a:3a = 198:1) (Table 1, entry 7). Despite the fact that under these conditions the yield of 2a was 10% lower than in the absence of ABTS, all further experiments were performed in the presence of 1 mol% ABTS, since this measure guaranteed the effective suppression of 3a. A control experiment established that in the absence of laccase and in presence of 1 mol% ABTS neither 2a nor 3a were formed (Table 1, entry 8). The use of other cosolvents than DMSO (ethanol, acetone, ethyl acetate) and a phase transfer catalyst (Aliquat 336) had no positive impact on the yield of 2a.
|
||||||
|---|---|---|---|---|---|---|
| Entry | KI (equiv.) | Enzyme (U) | Mediator (mol%) | Buffer (mL) | 2a:3a |
Yield 2a (%) |
| a 2 mmol 1a were reacted. The yields of 2a refer to isolated yields, the ratio 2a:3a was determined by 1H NMR analysis of the crude product.b 1a in 3 mL DMSO was added during 24 h by syringe pump.c Initially, 45 U enzyme were added, additional enzyme (180 U) in 3 mL acetate buffer was added during 24 h by syringe pump. |
||||||
| 1 | 1 | 225 | — | 45 | 1:2 |
7 |
| 2 | 20 | 225 | — | 45 | 20:1 |
24 |
| 3b | 20 | 225 | — | 45 | 65:1 |
18 |
| 4c | 20 | 225 | — | 45 | 18:1 |
34 |
| 5 | 20 | 225 | ABTS (1) | 45 | 198:1 |
46 |
| 6 | 20 | 225 | — | 90 | 18:1 |
75 |
| 7b,c | 20 | 225 | ABTS (1) | 90 | 198:1 |
65 |
| 8 | 20 | — | ABTS (1) | 90 | — | — |
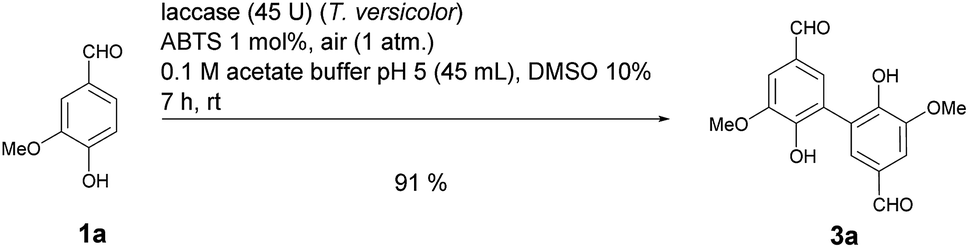 | ||
| Scheme 1 Laccase-catalyzed oxidative dimerization of vanillin (1a) to divanillin (3a). | ||
Table 2 summarizes the experiments performed to decrease the amounts of KI and laccase and to shorten the reaction time. Particularly gratifying was the observation that the yield of 2a can be increased by decreasing the amount of KI (Table 2, entries 1–4). With 3 equiv. of KI, the yield reaches its maximum (85%) (Table 2, entry 3). Even with only 1.5 equiv. KI, 2a was formed in 77% (Table 2, entry 4). With equimolar amounts of KI, however, the yield of 2a is only 11% (Table 2, entry 5). Further experiments proved that the amount of laccase can be reduced significantly from 225 U to 90 U without any loss of yield (Table 2, entry 6). Finally, it was revealed that the reaction time (48 h) can be shortened by a more effective air supply. When air was bubbled through the reaction solution at a rate of 20 mL min−1, the transformation was already finished after 15 h. Under these conditions, the yield of 2a amounted to 77% with 90 U laccase, and to 85% with a total amount of 135 U (Table 2, entries 7 and 8).
|
|||||
|---|---|---|---|---|---|
| Entry | Equiv. KI | Air | t [h] | Add. enzyme [U], [h] | Yield 2a (%) |
| a 2 mmol 1a were reacted in 90 mL buffer. The yields of 2a refer to isolated yields. Initially, 45 U laccase were added; additional laccase in 3 mL acetate buffer was added during the time given by syringe pump. Substrate in 3 mL DMSO was added by syringe pump during the same time the enzyme was added.b The ratio 2a:3a was determined by 1H NMR analysis of the crude product (198:1). |
|||||
| 1 | 20 | 1 atm | 48 | 180 (44) | 65 |
| 2 | 5 | 1 atm | 48 | 180 (44) | 72 |
| 3 | 3 | 1 atm | 48 | 180 (44) | 85 |
| 4 | 1.5 | 1 atm | 48 | 180 (44) | 77 |
| 5 | 1 | 1 atm | 48 | 180 (44) | 11 |
| 6 | 3 | 1 atm | 48 | 45 (44) | 86 |
| 7 | 3 | 20 mL min−1 | 15 | 45 (5) | 77 |
| 8 | 3 | 20 mL min−1 | 15 | 90 (5) | 85b |

As part of the optimization, the influence of the iodide source and the mediator was studied. It was established that the iodination cannot only be achieved with KI, but also with LiI, NaI, CsI and NH4I in comparable yields (81–87%) of 2a (Table 3, entries 1–5). In no case, the formation of 3a could be observed. The small yield differences suggest that the influence of the cation of the iodide source is negligible. In contrast, the mediator has a decisive influence on yield and selectivity. With none of the other mediators tested (violuric acid, 4-acetamido-TEMPO, 4-methoxy-TEMPO, HOBt, methyl syringate) comparable yields of 2a could be realized (Table 3, entries 6–10). In addition, it was found (TLC) that the formation of iodovanillin (2a) was accompanied by considerable amounts of the dimer 3a. Using 1 mol% HOBt as a mediator, this phenomenon was studied in some detail. The crude product analysis by 1H NMR showed that the 2a:3a ratio amounted to 2.3:1 (Table 3, entry 9). Finally, it was demonstrated that at 50 °C, which is close to the temperature optimum of many laccases,25 only traces of 2a were found. Experiments with laccases from other organisms, such as Agaricus bisporus and Pleurotus ostreatus, were also not effective.
|
|||
|---|---|---|---|
| Entry | Iodide source | Mediator | Yield 2a (%) |
| a 2 mmol 1a were reacted in 90 mL buffer. The yields of 2a refer to isolated yields. Initially, 45 U enzyme were added, additional laccase (90 U) in 3 mL acetate buffer was added during 5 h. Substrate in 3 mL DMSO was added by a second syringe pump during the same time the enzyme was added.b In addition to 2a, dimer 3a was detected (TLC). 2a and 3a were formed in a ratio of 2.3:1 as revealed by 1H NMR analysis of the crude product after filtration. |
|||
| 1 | KI | ABTS | 85 |
| 2 | LiI | ABTS | 81 |
| 3 | NaI | ABTS | 82 |
| 4 | CsI | ABTS | 87 |
| 5 | NH4I | ABTS | 81 |
| 6 | KI | Violuric acid | 22 |
| 7 | KI | 4-Acetamido-TEMPO | 33 |
| 8 | KI | 4-Methoxy-TEMPO | 29 |
| 9 | KI | HOBt | 21b |
| 10 | KI | Methyl syringate | 37 |
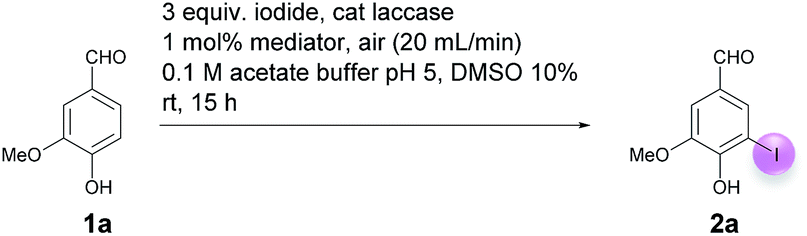
To summarize, the optimization studies showed that best results were obtained when 1a was reacted with 3 equiv. KI, catalytic amounts of laccase of T. versicolor (45 U + 90 U) and 1 mol% ABTS in acetate buffer (pH 5):DMSO = 9:1 at rt for 15 h while bubbling air through the reaction solution. In this way, 2a was isolated in 85% yield; the corresponding dimer 3a could not be detected (Table 3, entry 1). It should be mentioned that the model reaction can easily be upscaled from the 2 mmol to the 15 mmol scale. In doing so, 2a can easily be synthesized in gram amounts.
Substrate scope
Against this background, scope and limitations of the laccase-catalyzed iodination of phenolics were studied in greater detail (Tables 4–6). Initial experiments had revealed that 4-carbonyl- and 4-carboxyl-substituted phenols are the most suitable substrates. The reactions were performed under the conditions of Table 3, entry 1. If necessary, the reaction time and/or the amount of laccase was increased, and the addition rates of substrate and enzyme were adapted. Substrates with 2 potential iodination sites were reacted with 4 equiv. KI. Taking the successful iodination of 1a as a starting point, we set out to test a number of different 4-hydroxybenzaldehydes as substrates (Table 4). It was found that the 3-OMe group in 1a could be replaced easily with an OEt group (Table 4, entry 2) as well as different halogen atoms (Br, Cl) (Table 4, entries 3 and 4). The experiment with 1e demonstrates that substrates with an OMe group at C-2 can be iodinated in high yields, too (Table 4, entry 5). It is remarkable that with the unsubstituted 4-hydroxybenzaldehyde 1f the formation of the monoiodinated product could not be detected at all. Instead, the 3,5-diiodo derivative 4f was isolated in 70% (Table 4, entry 6). The laccase-catalyzed iodination is not restricted to benzene derivatives as the reaction with 4-hydroxy-1-naphthaldehyde (1g) clearly proves. The comparative low yield of 2g might be due to the tendency of the substrate for polymerization (Table 4, entry 7). Polymerization was also dominant in reactions with phenols carrying no aldehyde group in p-position, such as 4-bromo-2-methoxyphenol, 4-chloro-2-methoxyphenol, 2-methoxy-4-methylphenol, 4-methylphenol and 2,3-dihydroxybenzaldehyde. When the aldehyde group in p-position was replaced with an allyl group and an alkylidene group,21g resp., the exclusive formation of dimers was observed. Benzaldehydes carrying no hydroxyl group in p-position, such as 3,4-dimethoxybenzaldehyde and the o-hydroxyl substituted benzaldehydes 2-hydroxy-1-naphthaldehyde, 2-hydroxy-3-methoxybenzaldehyde, 2-hydroxybenzaldehyde, 5-bromo-2-hydroxybenzaldehyde and 5-chloro-2-hydroxybenzaldehyde, either didn't react at all or the iodinated product was only formed in traces. Next, a selection of p-hydroxyarylketones was employed as substrates (Table 5). The vanillin derivative 1-(4-hydroxy-3-methoxyphenyl)ethan-1-one, i.e., acetovanillon (1h) could be transformed into the monoiodinated compound 1-(4-hydroxy-3-iodo-5-methoxy-phenyl)ethan-1-one (2h) in 73% without any problems (Table 5, entry 1). When the methoxy group in 1h was replaced with a methyl group, i.e. 1i, the yield of the monoiodo product 2i was even higher (87%) (Table 5, entry 2). However, when the methoxy group was replaced with an electron withdrawing methoxycarbonyl group, i.e. 1j, the yield of 2j dropped dramatically to only 11% (Table 5, entry 3). C-2 substituted p-hydroxyarylmethylketones, such as 1k, could be iodinated easily as well. However, due to two free o-positions adjacent to the hydroxyl group, the formation of both the monoiodinated and the diiodinated products 2k and 4k took place. Fortunately, the ratio of the products 2 and 4 can be influenced by the reaction conditions. With 4 equiv. KI, the transformation of 1k delivered 2k and 4k with 73% and 13%, resp. When 1k was reacted with 1.1 equiv. KI, the yield of the monoiodinated 2k could be raised to 78% while the diiodo compound 4k was formed in only 4% (Table 5, entry 4). Using the transformation of 1l as an example, it was demonstrated that the 2:4 ratio can also be shifted in direction towards the diiodo product 4. With 4 equiv. of KI, 2l and 4l were isolated with 23% and 64%, respectively. However, when the amount of KI was raised to 6 equiv., the formation of the monoiodinated product 2l could be suppressed completely. Under these conditions, 4l was isolated in 84% (Table 5, entry 5). In analogy to 4-hydroxybenzaldehyde 4f, the transformation of 1-(4-hydroxyphenol)-propan-1-one (1m) exclusively delivered the diiodo compound in 75% (Table 5, entry 6). Other substrates with 2 potential iodination sites (1n–p) produced mixtures of mono- and diiodo products 2 and 4 when reacted with 4 equiv. KI (Table 5, entries 7–9). Finally, it was studied whether the laccase-catalyzed iodination can also be applied to p-hydroxy-benzoic acid derivatives (Table 6). For this purpose, iodination reactions were performed with benzoic acid esters 1q and 1r, the benzoic acid amide 1s and the benzonitrile 1t as substrates. In all cases, the expected products, i.e. 2q, 2r and 4r, 4s and 2t, were obtained (Table 6, entries 1–4). It came as a surprise that in contrast to the methylbenzoate 1r the corresponding benzyl 4-hydroxybenzoate and phenyl 4-hydroxybenzoate did not undergo the laccase-catalyzed iodination. The method presented here allows the iodination of a wide range of p-hydroxycarbonyl and p-hydroxyarylcarboxyl compounds under mild reaction conditions. By developing suitable reaction conditions, the oxidative coupling could be suppressed completely. Substrates with one iodination site produced the monoiodo products with yields up to 87%. When substrates with two potential iodination sites were used, in some cases the disubstituted products (1f, 1m, 1s) were formed selectively. In other cases (1k, 1l, 1n–p, 1r) mixtures of mono- and diiodo products were observed. By variation of the reaction conditions (amount of enzyme and KI, buffer volume, reaction time, addition rate of enzyme and substrate) the ratio of monoiodo and diiodo products could decisively be influenced.
|
||||
|---|---|---|---|---|
| Entry | 1 | Laccase (U) | Time (h) | Yield product (%) |
| a 2 mmol 1 were reacted in 90 mL buffer. The yields of 2 and 4 refer to isolated yields. Initially, 45 U enzyme were added, additionally laccase in buffer was added during 5 h by syringe pump. Substrate in 3 mL DMSO was added by a second syringe pump during the same time the enzyme was added. Substrates with one iodination site were reacted with 3 equiv. KI, substrates with 2 iodination sites were reacted with 4 equiv. KI.b Additional enzyme and substrate were added during 15 h. | ||||
| 1 | a | 45 + 90 | 15 | 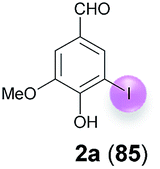 |
| 2 | b | 45 + 90 | 15 | 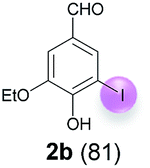 |
| 3 | c | 45 + 90 | 15 | 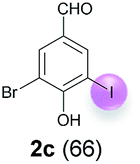 |
| 4 | d | 45 + 90 | 48 | 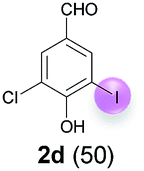 |
| 5 | e | 45 + 90 | 15 | 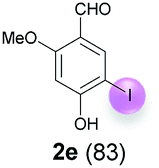 |
| 6b | f | 45 + 300 | 144 | 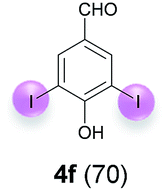 |
| 7 | g | 45 + 90 | 15 | 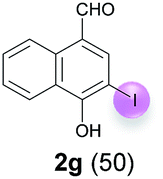 |
|
||||
|---|---|---|---|---|
| Entry | 1 | Laccase (U) | Time (h) | Yield products (%) |
| a 2 mmol 1 were reacted in 90 mL buffer. The yields of 2 and 4 refer to isolated yields. Initially, 45 U enzyme were added, additionally laccase in buffer was added during 5 h by syringe pump. Substrate in 3 mL DMSO was added by a second syringe pump during the same time the enzyme was added. Substrates with one iodination site were reacted with 3 equiv. KI, substrates with 2 iodination sites were reacted with 4 equiv. KI.b Initially, 1.1 equiv. KI and the substrate were added as solids. No DMSO was used. 180 U enzyme were added during 12 h.c 180 mL buffer and 6 equiv. KI were used. 300 U additional enzyme and substrate were added during 5 h.d Additional enzyme and substrate were added during 30 h.e Additional enzyme and substrate were added during 20 h. | ||||
| 1 | h | 45 + 90 | 15 | 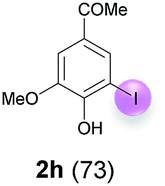 |
| 2 | i | 45 + 90 | 15 | 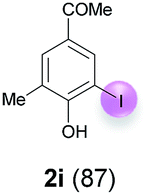 |
| 3 | j | 45 + 180 | 168 | 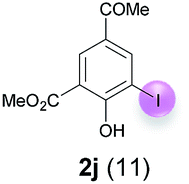 |
| 4 | k | 45 + 180 | 15 | 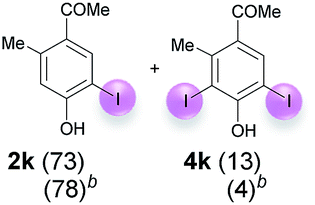 |
| 5 | l | 45 + 225 | 24 | 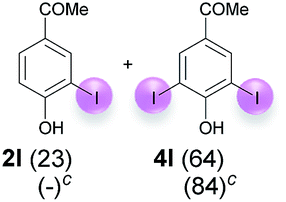 |
| 6d | m | 45 + 360 | 168 | 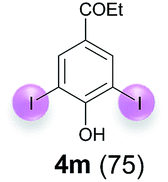 |
| 7 | n | 45 + 180 | 60 | 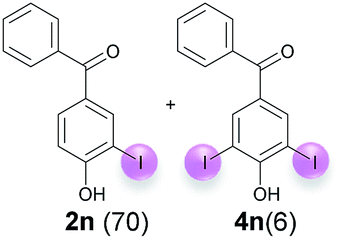 |
| 8e | o | 45 + 360 | 120 | 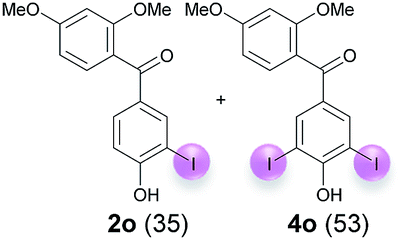 |
| 9 | p | 45 + 90 | 24 | 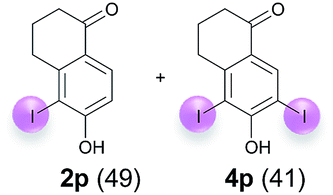 |
|
||||
|---|---|---|---|---|
| Entry | 1 | Laccase (U) | Time(h) | Yield products (%) |
| a 2 mmol 1 were reacted in 90 mL buffer. The yields of 2 and 4 refer to isolated yields. Initially, 45 U enzyme were added, additionally laccase in buffer was added during 5 h by syringe pump. Substrate in 3 mL DMSO was added by a second syringe pump during the same time the enzyme was added. Substrates with one iodination site were reacted with 3 equiv. KI, substrates with 2 iodination sites were reacted with 4 equiv. KI.b Additional enzyme and substrate were added during 10 h.c Additional enzyme and substrate were added during 30 h. | ||||
| 1 | q | 45 + 90 | 36 | 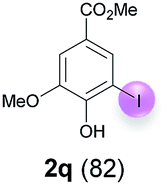 |
| 2b | r | 45 + 180 | 120 | 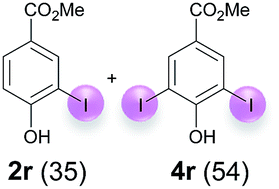 |
| 3c | s | 45 + 500 | 48 | 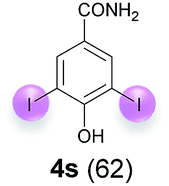 |
| 4 | t | 45 + 270 | 24 | 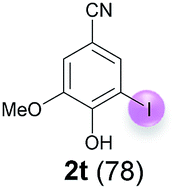 |
Reaction mechanism
A plausible equation for the laccase-catalyzed iodination of phenolics with KI as the iodine source and oxygen as the terminal oxidant is presented in Scheme 2. From the equation it is clear that only 1 equiv. of KI is required for the iodination and that 1 equiv. KOH is formed during the course of the reaction which results in a pH rise of the reaction solution. It is assumed that the method can be regarded as an electrophilic aromatic substitution of the p-hydroxyarylcarbonyls and p-hydroxyarylcarboxyls with elemental iodine as the electrophile (Scheme 3). For simplicity, it is assumed that I2 acts as iodination agent. However, it cannot be excluded that I3− is the actual active iodine species, because I3− is formed rapidly in aqueous solutions containing iodide and iodine.26 The mechanism is supported by the observation that the regioselectivity of the iodination is governed by the well-known substituent effects of the electrophilic aromatic substitution. The molecular iodine required as electrophile is generated in situ by the laccase-catalyzed oxidation of KI using oxygen as oxidant. Xu has demonstrated that the laccase-catalyzed oxidation of iodide can be enhanced by ABTS.17f Consequently, it is assumed that in the first step 2 molecules ABTS undergo oxidation to their corresponding radical cations. The two electrons released from ABTS reduce one oxygen atom to H2O. The ABTS radical cations on the other hand oxidize two iodides to I2. The molecular iodine formed in turn reacts with the phenolic substrate Ar–H to produce the iodinated product Ar–I as well as HI. It should be highlighted that in contrast to typical electrophilic substitutions with I2 as reagent, the HI generated during the electrophilic aromatic substitution is not wasted, but undergoes a laccase-catalyzed reoxidation to I2. This allows a significant reduction of the amount of the iodine source and an improvement of the atom economy from 72 to 85% (Scheme 4). The atom economy of the laccase-catalyzed reaction can be further improved by replacing KI with NaI, LiI or NH4I (Table 3, entries 2, 3 and 5).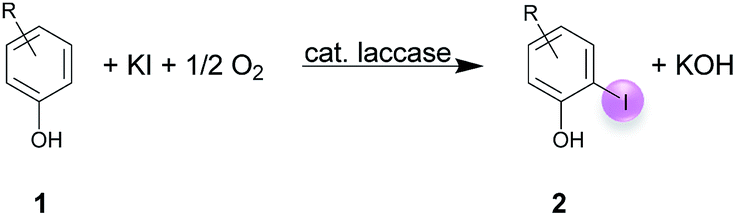 | ||
| Scheme 2 Chemical equation of the laccase-catalyzed iodination. | ||
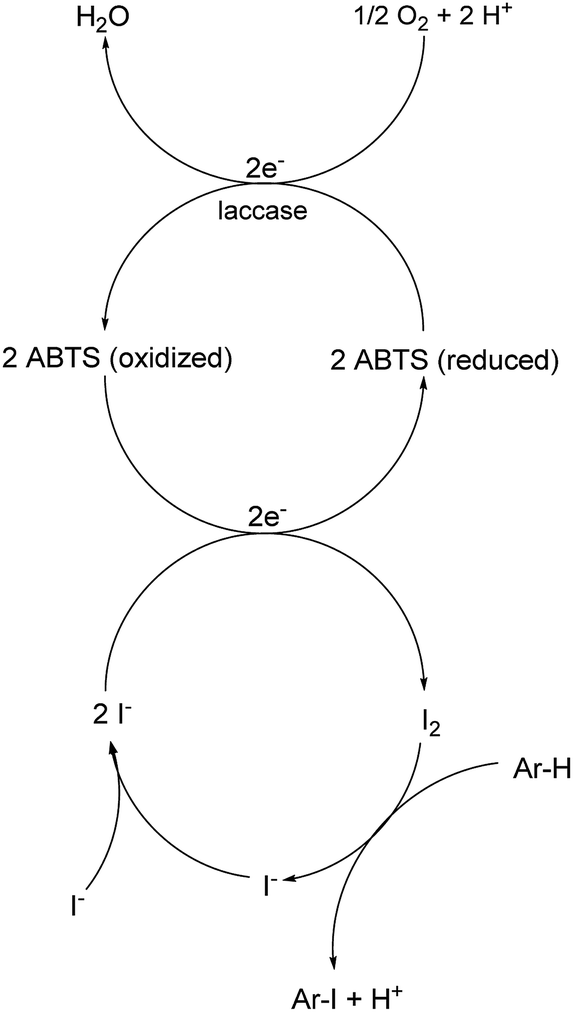 | ||
| Scheme 3 Proposed mechanism of the laccase-catalyzed iodination. | ||
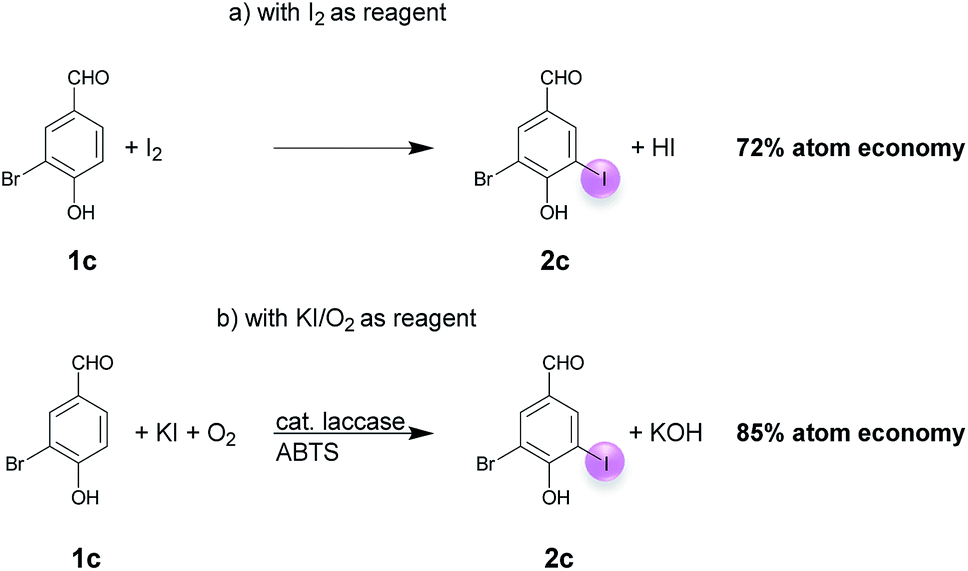 | ||
| Scheme 4 Atom economy of the laccase-catalyzed iodination of 1c with (a) I2 and (b) KI/O2. | ||
Even if most of the iodinations were run with a considerable excess of KI (3 equiv.) (Tables 4–6) we have demonstrated that the monoiodination can also be performed successfully, when the amount of KI is reduced from 3 to 1.5 equiv. (Table 2). In one case, the monoiodination could be achieved with as little as 1.1 equiv. KI (Table 5, entry 4). Further benefits of this method are as follows: it is based on using (a) a biocatalyst which can be obtained from renewable materials, (b) KI as an easy to handle iodine source and (c) aerial oxygen as the oxidant to generate I2 from KI. Furthermore, the reactions can be performed under extremely mild reaction conditions, i.e. in an aqueous solvent system (pH 5) at room temperature.
Towards sustainable laccase-catalyzed iodinations
The next goal was the development of the laccase-catalyzed iodination towards a truly sustainable synthetic method. Despite of numerous advantages, the laccase-catalyzed iodinations presented in Tables 4–6 cannot be regarded as truly sustainable since the amount of waste produced is still too high. For example, the E-factor27 of the transformation of 1a to 2a amounts to 31.9 kg kg−1 (Table 7, entry 1). The unfavorable E-factors are due to (a) the use of an excess of KI (3 equiv. KI for substrates with one iodination site and 4 equiv. for substrates with two iodination sites) and (b) the use of large volumes of solvent. This is why we embarked on further development towards a synthetic method, which fulfills all relevant requirements for a green reaction. To reduce the E-factor, a number of options were available. Among them were the reduction of (a) the amount of laccase, (b) the excess of KI, (c) the DMSO concentration, (d) the molarity of the buffer and (e) the chemoselectivity (iodination versus oxidative coupling) and on buffer volume itself. However, the focus of our optimizations, was not only the improvement of the E-factor, but also on high chemical yield. A number of observations proved to be decisive for the optimization process which is summarized in Table 7. It was found that (a) the amount of KI can be reduced from 3 to 2 and 1.5 equiv., resp., (Table 7, entries 5, 6 and 11–15), (b) the concentration of DMSO can be reduced from 10 vol% to 1.25 vol% (Table 7, entries 3, 4, 13 and 14) (in some cases the cosolvent could be completely omitted) and (c) the buffer volume can be reduced by 50% (Table 7, entries 1–3). Furthermore, it was found that the dimer formation can be reduced to a minimum by increasing the molarity of the acetate buffer (Table 7, entries 7–10). In this context, the relationship between pH and the proportion of dimer should be mentioned. It was found that an increase of the pH of the reaction mixture during the course of the reaction resulted in an increase of the proportion of the dimer 3a in the crude product (Table 7, entries 7–10). To keep the proportion of the dimer as low as possible it is necessary to adjust the pH value around pH = 5. Furthermore, it had to be taken into account that the reduction of (a) the amount of KI to 1.5 equiv., (b) DMSO to 1.25 vol% and (c) the buffer volume to half of its initial volume (45 mL) could only be achieved when the amount of laccase was increased to 270 U and the addition rate of enzyme as well as substrate was increased to 15 h. Considering all of the above aspects, the E-factor for the transformation of 1a to 2a can be reduced from 31.9 kg kg−1 (Table 7, entry 1) to 4.6 kg kg−1 (Table 7, entry 14). Under these conditions, 2a was isolated in 77% and the 2a/3a ratio amounted to 198:1. A further improvement of the E-factor to 2.9 kg kg−1 was possible when the reaction was performed in the complete absence of DMSO (Table 7, entry 15). Under these conditions, the yield did increase to 83%; however, the 2a/3a ratio worsened to 31:1. Further measures to increase sustainability but not necessarily the E-factor of the model reaction related to the reaction mixture workup included the complete renunciation of organic solvents during workup, a substantial renunciation of purification by column chromatography and the replacement of Na2S2O7 – which was used to remove residual I2 from the reaction mixture – with ascorbic acid (Table 7, entries 14, 15 and Table 8). Additional information and calculations concerning the greenness and efficiency (TON, TOF, STY) of selected reactions are presented in the ESI.† A control experiment in absence of the laccase with I2 clearly revealed that under these conditions the yield is considerable lower than under the conditions of the laccase-catalyzed reaction with KI as iodination agent (Table 7, entry 16).
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Laccase (U) | Substrate + enzyme addition t (h) | KI (equiv.) | Time (h) | DMSO (vol%) | Buffer (mol L−1) | Buffer vol. (mL) | Isolated yield 2a (%) | 2a:3a |
ΔpH | E-factor |
| a 2 mmol substrate were reacted. 45 U laccase were added initially, additional laccase and substrate were added separately and simultaneously during the time given via syringe pump. The ratio of 2a and 3a was determined after filtration and drying of the crude product via 1H NMR.b Isolated yield after filtration.c Enzyme was added during 15 h via syringe pump and substrate was added manually as a solid.d 0.75 equiv. I2, no laccase and ABTS was used. | |||||||||||
| 1 | 45 + 90 | 5 | 3 | 15 | 10 | 0.1 | 90 | 85 | 198:1 |
31.9 | |
| 2 | 45 + 90 | 5 | 3 | 15 | 10 | 0.1 | 45 | 62 | 38:1 |
19.5 | |
| 3 | 45 + 135 | 5 | 3 | 20 | 10 | 0.1 | 45 | 85 | 48:1 |
13.9 | |
| 4 | 45 + 225 | 5 | 3 | 20 | 2.5 | 0.1 | 45 | 59 | 20:1 |
9.6 | |
| 5 | 45 + 225 | 10 | 3 | 20 | 2.5 | 0.1 | 45 | 63 | 65:1 |
8.9 | |
| 6 | 45 + 225 | 10 | 2 | 20 | 2.5 | 0.1 | 45 | 70 | 31:1 |
6.6 | |
| 7 | 45 + 90 | 5 | 3 | 20 | 10 | 0.05 | 45 | 64 | 20:1 |
+2.3 | 18.5 |
| 8 | 45 + 90 | 5 | 3 | 20 | 10 | 0.1 | 45 | 66 | 26:1 |
+2.2 | 18.3 |
| 9 | 45 + 90 | 5 | 3 | 20 | 10 | 0.2 | 45 | 58 | 98:1 |
+1.2 | 21.9 |
| 10 | 45 + 90 | 5 | 3 | 20 | 10 | 0.5 | 45 | 43 | 198:1 |
+0.5 | 33.8 |
| 11 | 45 + 225 | 10 | 2 | 20 | 2.5 | 0.2 | 45 | 77 | 48:1 |
6.6 | |
| 12 | 45 + 225 | 10 | 1.5 | 20 | 2.5 | 0.2 | 45 | 68 | 65:1 |
7.0 | |
| 13 | 45 + 225 | 15 | 1.5 | 20 | 2.5 | 0.2 | 45 | 67 | 198:1 |
7.2 | |
| 14 | 45 + 225 | 15 | 1.5 | 24 | 1.25 | 0.2 | 45 | 77b | 198:1 |
4.6 | |
| 15 | 45 + 225 | 10c | 1.5 | 24 | 0 | 0.2 | 45 | 83b | 31:1 |
2.9 | |
| 16d | 0 | 15 | — | 24 | 1.25 | 0.2 | 45 | 47d | 198:1 |
6.7 | |
|
|||||
|---|---|---|---|---|---|
| Entry | 1 | Method | Yield product (%) | 2a:3a |
E-factor |
| a 2 mmol substrate were reacted in 45 mL buffer. Initially, 45 U enzyme were added, additionally laccase (225U) in buffer (562 μL) was added during 15 h by syringe pump. Method A: substrate in 562 μL DMSO was added by a second syringe pump during the same time the enzyme was added. Method B: substrate was added during 10 h as a solid. The yields refer to yields after filtration and washing of the crude product with water. Ratio of 2a:3a was analyzed via 1H NMR after filtration and drying of the crude product.b 72 h reaction time.c 255 U laccase were added during 36 h.d 48 h reaction time. |
|||||
| 1 | a | A | 77 (2a) | 198:1 |
4.63 |
| 2 | b | A | 80 (2b) | 98:1 |
4.14 |
| 3b | c | B | 90 (2c) | 198:1 |
2.22 |
| 4c | e | B | 90 (2e) | 198:1 |
2.55 |
| 5d | i | B | 92 (2i) | 198:1 |
2.49 |
| 6 | k | B | 93 (2k) | 198:1 |
2.44 |

To prove the usefulness of the conditions developed, a number of substrates were iodinated according to the conditions given in Table 7, entries 14 and 15 (Table 8).
Substrates with a tendency for polymerization (1a, 1b) were reacted according to Table 7, entry 14 (Method A). Under these conditions, the iodination of 1b produced 2b with high selectivity and 80% yield (Table 8, entry 2). The E-factor for this transformation was 4.14 kg kg−1. A number of substrates with no tendency to dimerization (1c, 1e, 1i, 1k) were reacted under the conditions of Table 7, entry 15 (Method B) to deliver the monoiodinated products 2c, 2e, 2i and 2k in remarkably high yields, with outstanding selectivities and remarkably low E-factors (Table 8, entries 3–6).
Conclusions
In summary, a simple-to-perform and sustainable enzyme-catalyzed method for the selective and efficient iodination of p-hydroxyarylcarbonyl- and p-hydroxyarylcarboxylic acid derivatives on a preparative scale has been developed. It relies on the use of easy to handle KI as iodine source which undergoes laccase-catalyzed oxidation to iodine by using safe and cheap aerial oxygen as oxidant. The reactions can be performed in an aqueous solvent system under mild reaction conditions. The iodinated products which arise from electrophilic aromatic substitutions could be isolated with yields up to 93%. Depending on the substitution pattern of the substrates either monoiodinated or diiodinated products are formed. The use of KI instead of I2 as iodination agent allows the increase of the atom economy from 72 to 85%. By proper choice of the reaction conditions the competing oxidative phenolic coupling can be completely suppressed. Further optimization measures such as decreasing the amounts of KI and laccase, optimizing the buffer system, completely dispensing with organic solvents during workup, using ascorbic acid to destroy residual iodine and avoiding chromatographic purification enabled us to develop the laccase-catalyzed reaction towards a sustainable iodination method.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Mr Mario Wolf (University of Hohenheim) for recording NMR spectra and Dr Alevtina Mikhael, Dr Claudia Oellig (University of Hohenheim), and Mr Joachim Trinkner (University of Stuttgart) for recording mass spectra.Notes and references
- (a) L. Wang, X. Zhou, M. Fredimoses, S. Liao and Y. Liu, RSC Adv., 2014, 4, 57350–57376 RSC; (b) G. W. Gribble, Naturally Occurring Organohalogen Compounds - A Comprehensive Update, Springer, Wien, 2010 CrossRef.
- (a) A. Aiello, E. Fattorusso, C. Imperatore, M. Menna and W. E. G. Müller, Mar. Drugs, 2010, 8, 285–291 CrossRef CAS PubMed; (b) H. Prawat, C. Mahidol, W. Kaweetripob, S. Wittayalai and S. Ruchirawat, Tetrahedron, 2012, 68, 6881–6886 CrossRef CAS; (c) T. Iizuka, R. Fudou, Y. Jojima, S. Ogawa, S. Yamanaka, Y. Inukai and M. Ojika, J. Antibiot., 2006, 59, 385–391 CrossRef CAS PubMed; (d) G. A. Brent, J. Clin. Invest., 2012, 122, 3035–3043 CrossRef CAS PubMed.
- G. V. Naccarelli, D. L. Wolbrette, H. M. Patel and J. C. Luck, Curr. Opin. Cardiol., 2000, 15, 64 CrossRef CAS PubMed.
- For reviews, see: (a) D. Roy and Y. Uozumi, Adv. Synth. Catal., 2018, 360, 602–625 CrossRef CAS; (b) A. de Meijere, S. Bräse and M. Oestreich, Metal-Catalyzed Cross-Coupling Reactions and More, Wiley-VCH, Weinheim, 2014 CrossRef; (c) C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed; (d) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS PubMed.
- (a) R. Wang, S. Mo, Y. Lu and Z. Shen, Adv. Synth. Catal., 2011, 353, 713–718 CrossRef CAS; (b) H. Miao and Z. Yang, Org. Lett., 2000, 2, 1765–1768 CrossRef CAS PubMed; (c) A. J. Ross, H. L. Lang and R. F. W. Jackson, J. Org. Chem., 2010, 75, 245–248 CrossRef CAS PubMed; (d) N. Iranpoor, H. Firouzabadi, A. Safavi, S. Motevalli and M. M. Doroodmand, Appl. Organomet. Chem., 2012, 26, 417–424 CrossRef CAS; (e) K. Takami, H. Yorimitsu, H. Shinokubo, S. Matsubara and K. Oshima, Org. Lett., 2001, 3, 1997–1999 CrossRef CAS PubMed; (f) D. B. Pacardo, M. Sethi, S. E. Jones, R. R. Naik and M. R. Knecht, ACS Nano, 2009, 3, 1288–1296 CrossRef CAS PubMed; (g) C. Singh, A. P. Prakasham, M. K. Gangwar, R. J. Butcher and P. Ghosh, ACS Omega, 2018, 3, 1740–1756 CrossRef CAS; (h) T. d. A. Fernandes, B. Gontijo Vaz, M. N. Eberlin, A. J. M. da Silva and P. R. R. Costa, J. Org. Chem., 2010, 75, 7085–7091 CrossRef CAS PubMed.
- F. Kopp, A. Krasovskiy and P. Knochel, Chem. Commun., 2004, 2288–2289 RSC.
- For reviews, see: (a) N. L. Sloan and A. Sutherland, Synthesis, 2016, 48, 2969–2980 CrossRef; (b) W. Hao and Y. Liu, Beilstein J. Org. Chem., 2015, 11, 2132–2144 CrossRef CAS PubMed; (c) V. V. K. M. Kandepi and N. Narender, Synthesis, 2012, 44, 15–26 CrossRef CAS; (d) A. Podgoršek, M. Zupan and J. Iskra, Angew. Chem., Int. Ed., 2009, 48, 8424–8450 CrossRef PubMed; (e) S. Stavber, M. Jereb and M. Zupan, Synthesis, 2008, 10, 1487–1513 CrossRef; (f) E. B. Merkushev, Synthesis, 1988, 12, 923–937 CrossRef.
- C. Galli, Chem. Rev., 1988, 88, 765–792 CrossRef CAS.
- V. Snieckus, Chem. Rev., 1990, 90, 879–933 CrossRef CAS.
- R. G. Johnson and R. K. Ingham, Chem. Rev., 1956, 56, 219–269 CrossRef CAS.
- (a) P. Bovonsombat, J. Leykajarakul, C. Khan, K. Pla-on, M. M. Krause, P. Khanthapura, R. Ali and N. Doowa, Tetrahedron Lett., 2009, 50, 2664–2667 CrossRef CAS; (b) S. V. Khansole, S. S. Mokle, M. A. Sayyed and Y. B. Vibhute, J. Chin. Chem. Soc., 2008, 55, 871–874 CrossRef CAS; (c) R. da S. Ribeiro, P. M. Esteves and M. C. S. de Mattos, J. Braz. Chem. Soc., 2008, 19, 1239–1243 CrossRef CAS; (d) A. Deshmukh, B. Gore, H. V. Thulasiram and V. P. Swamy, RSC Adv., 2015, 5, 88311–88315 RSC; (e) J. Barluenga, J. M. Gonzalez, M. A. Garcia-Martin, P. J. Campos and G. Asensio, J. Org. Chem., 1993, 58, 2058–2060 CrossRef CAS; (f) S. Kajigaeshi, T. Kakinami, H. Yamasaki, S. Fujisaki, M. Kondo and T. Okamoto, Chem. Lett., 1987, 16, 2109–2112 CrossRef; (g) C. Krishna Prasad and P. V. S. Machi Raju, J. Appl. Chem., 2014, 4, 1460–1467 Search PubMed; (h) L. C. R. M. da Frota, R. C. P. Canavez, S. L. da S. Gomes, P. R. R. Costa and A. J. M. da Silva, J. Braz. Chem. Soc., 2009, 20, 1916–1920 CrossRef CAS; (i) M. S. Yusubov, E. N. Tveryakova, E. A. Krasnokutskaya, I. A. Perederyna and V. V. Zhdankin, Synth. Commun., 2007, 37, 1259–1265 CrossRef CAS; (j) R. Francke, G. Schnakenburg and S. R. Waldvogel, Eur. J. Org. Chem., 2010, 2357–2362 CrossRef CAS.
- (a) H. Tajik, A. A. Esmaeili, I. Mohammadpoor-Baltork, A. Ershadi and H. Tajmehri, Synth. Commun., 2003, 33, 1319–1323 CrossRef CAS; (b) A. T. Shinde, S. B. Zangade, S. B. Chavan, A. Y. Vibhute, Y. S. Nalwar and Y. B. Vibhute, Synth. Commun., 2010, 40, 3506–3513 CrossRef CAS; (c) H. Keipour, M. A. Khalilzadeh, B. Mohtat, A. Hosseini and D. Zareyee, Chin. Chem. Lett., 2011, 22, 1427–1430 CrossRef CAS; (d) M. Jereb, M. Zupan and S. Stavber, Chem. Commun., 2004, 2614–2615 RSC; (e) J. Iskra, S. Stavber and M. Zupan, Tetrahedron Lett., 2008, 49, 893–895 CrossRef CAS; (f) B. Akhlaghinia and M. Rahmani, J. Braz. Chem. Soc., 2010, 21, 3–6 CrossRef CAS.
- (a) K. J. Edgar and S. N. Falling, J. Org. Chem., 1990, 55, 5287–5291 CrossRef CAS; (b) L. Lista, A. Pezzella, A. Napolitano and M. d'Ischia, Tetrahedron, 2008, 64, 234–239 CrossRef CAS; (c) N. Narender, K. S. K. Reddy, K. V. V. K. Mohan and S. J. Kulkarni, Tetrahedron Lett., 2007, 48, 6124–6128 CrossRef CAS; (d) R. Sathiyapriya and R. J. Karunakaran, Synth. Commun., 2006, 36, 1915–1917 CrossRef CAS; (e) L. Emmanuvel, R. K. Shukla, A. Sudalai, S. Gurunath and S. Sivaram, Tetrahedron Lett., 2006, 47, 4793–4796 CrossRef CAS; (f) K. V. V. Krishna Mohan, N. Narender and S. J. Kulkarni, Tetrahedron Lett., 2004, 45, 8015–8018 CrossRef CAS; (g) S. Adimurthy, G. Ramachandraiah, P. K. Ghosh and A. V. Bedekar, Tetrahedron Lett., 2003, 44, 5099–5101 CrossRef CAS; (h) S. Song, X. Sun, X. Li, Y. Yuan and N. Jiao, Org. Lett., 2015, 17, 2886–2889 CrossRef CAS PubMed; (i) M. A. Pasha and Y. Y. Myint, Synth. Commun., 2004, 34, 2829–2833 CrossRef CAS; (j) T. Kometani, D. S. Watt, T. Ji and T. Fitz, J. Org. Chem., 1985, 50, 5384–5387 CrossRef CAS.
- B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259–281 CrossRef CAS.
- (a) J. Latham, E. Brandenburger, S. A. Shepherd, B. R. K. Menon and J. Micklefield, Chem. Rev., 2018, 118, 232–269 CrossRef CAS PubMed; (b) V. Agarwal, Z. D. Miles, J. M. Winter, A. S. Eustáquio, A. A. El Gamal and B. S. Moore, Chem. Rev., 2017, 117, 5619–5674 CrossRef CAS PubMed; (c) C. Schnepel and N. Sewald, Chem.–Eur. J., 2017, 23, 12064–12086 CrossRef CAS PubMed; (d) V. Weichold, D. Milbredt and K.-H. van Pée, Angew. Chem., Int. Ed., 2016, 55, 6374–6389 CrossRef CAS PubMed.
- (a) J. T. Payne, M. C. Andorfer and J. C. Lewis, Angew. Chem., Int. Ed., 2013, 52, 5271–5274 CrossRef CAS PubMed; (b) M. Frese and N. Sewald, Angew. Chem., Int. Ed., 2015, 54, 298–301 CrossRef CAS PubMed.
- (a) H. S. Schiller, E. F. Schlenk, K. Gosney and W.-J. Chen, Anal. Lett., 1976, 9, 419–428 CrossRef CAS; (b) A. L. Hubbard and Z. A. Cohn, J. Cell Biol., 1972, 55, 390–405 CrossRef CAS PubMed; (c) J. I. Thorell and B. G. Johansson, Biochim. Biophys. Acta, Protein Struct., 1971, 251, 363–369 CrossRef CAS; (d) A. Taurog and E. M. Howells, J. Biol. Chem., 1966, 241, 1329–1339 CAS; (e) J. Pommier, L. Sokoloff and J. Nunez, Eur. J. Biochem., 1973, 38, 497–506 CrossRef CAS PubMed; (f) F. Xu, Appl. Biochem. Biotechnol., 1996, 59, 221–230 CrossRef CAS; (g) M. Seki, J. Oikawa, T. Taguchi, T. Ohnuki, Y. Muramatsu, K. Sakamoto and S. Amachi, Environ. Sci. Technol., 2013, 47, 390–397 CrossRef CAS PubMed; (h) J. Ihssen, M. Schubert, L. Thöny-Meyer and M. Richter, PLoS One, 2014, 9, e89924 CrossRef PubMed.
- For reviews, see: (a) K. Agrawal, V. Chaturvedi and P. Verma, Bioresour. Bioprocess, 2018, 5, 4 CrossRef; (b) O. V. Morozova, G. P. Shumakovich, M. A. Gorbacheva, S. V. Shleev and A. I. Yaropolov, Biochemistry, 2007, 72, 1136–1150 CAS; (c) A. M. Mayer and R. C. Staples, Phytochemistry, 2002, 60, 551–565 CrossRef CAS PubMed; (d) E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Chem. Rev., 1996, 96, 2563–2606 CrossRef CAS PubMed; (e) C. F. Thurston, Microbiology, 1994, 140, 19–26 CrossRef CAS.
- For reviews, see: (a) M. D. Cannatelli and A. J. Ragauskas, Chem. Rec., 2017, 17, 122–140 CrossRef CAS PubMed; (b) M. Mogharabi and M. A. Faramarzi, Adv. Synth. Catal., 2014, 356, 897–927 CrossRef CAS; (c) S. Witayakran and A. J. Ragauskas, Adv. Synth. Catal., 2009, 351, 1187–1209 CrossRef CAS; (d) A. Kunamneni, S. Camarero, C. García-Burgos, F. J. Plou, A. Ballesteros and M. Alcalde, Microb. Cell Fact., 2008, 7, 32 CrossRef PubMed.
- (a) P. Schneider, M. B. Caspersen, K. Mondorf, T. Halkier, L. K. Skov, P. R. Østergaard, K. M. Brown, S. H. Brown and F. Xu, Enzyme Microb. Technol., 1999, 25, 502–508 CrossRef CAS; (b) F. Xu, W. Shin, S. H. Brown, J. A. Wahleithner, U. M. Sundaram and E. I. Solomon, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1996, 1292, 303–311 CrossRef; (c) B. R. M. Reinhammar, Biochim. Biophys. Acta, Bioenerg., 1972, 275, 245–259 CrossRef CAS; (d) A. I. Cañas and S. Camarero, Biotechnol. Adv., 2010, 28, 694–705 CrossRef PubMed.
- (a) A. Potthast, T. Rosenau, C.-L. Chen and J. S. Gratzl, J. Org. Chem., 1995, 60, 4320–4321 CrossRef CAS; (b) A. Potthast, T. Rosenau, C. L. Chen and J. S. Gratzl, J. Mol. Catal. A: Chem., 1996, 108, 5–9 CrossRef CAS; (c) P. Galletti, M. Pori, F. Funiciello, R. Soldati, A. Ballardini and D. Giacomini, ChemSusChem, 2014, 7, 2684–2689 CrossRef CAS PubMed; (d) P. Galletti, F. Funiciello, R. Soldati and D. Giacomini, Adv. Synth. Catal., 2015, 357, 1840–1848 CrossRef CAS; (e) H. T. Abdel-Mohsen, J. Conrad and U. Beifuss, Green Chem., 2012, 14, 2686–2690 RSC; (f) U. Krings, V. Esparan and R. G. Berger, Flavour Fragrance J., 2015, 30, 362–365 CrossRef CAS; (g) M.-A. Constantin, J. Conrad and U. Beifuss, Green Chem., 2012, 14, 2375–2379 RSC; (h) B. Pickel, M.-A. Constantin, J. Pfannstiel, J. Conrad, U. Beifuss and A. Schaller, Angew. Chem., Int. Ed., 2010, 49, 202–204 CrossRef CAS PubMed; (i) F. d'Acunzo, C. Galli and B. Masci, Eur. J. Biochem., 2002, 269, 5330–5335 CrossRef PubMed; (j) I. Effenberger, B. Zhang, L. Li, Q. Wang, Y. Liu, I. Klaiber, J. Pfannstiel, Q. Wang and A. Schaller, Angew. Chem., Int. Ed., 2015, 54, 14660–14663 CrossRef CAS PubMed; (k) H. T. Abdel-Mohsen, K. Sudheendran, J. Conrad and U. Beifuss, Green Chem., 2013, 15, 1490–1495 RSC.
- (a) H. T. Abdel-Mohsen, J. Conrad and U. Beifuss, Green Chem., 2014, 16, 90–95 RSC; (b) S. Hajdok, J. Conrad and U. Beifuss, J. Org. Chem., 2012, 77, 445–459 CrossRef CAS PubMed; (c) K. W. Wellington, P. Steenkamp and D. Brady, Bioorg. Med. Chem., 2010, 18, 1406–1414 CrossRef CAS PubMed.
- (a) H. T. Abdel-Mohsen, J. Conrad, K. Harms, D. Nohr and U. Beifuss, RSC Adv., 2017, 7, 17427–17441 RSC; (b) S. Suljić, J. Pietruszka and D. Worgull, Adv. Synth. Catal., 2015, 357, 1822–1830 CrossRef; (c) M. D. Cannatelli and A. J. Ragauskas, J. Mol. Catal. B: Enzym., 2015, 119, 85–89 CrossRef CAS; (d) S. Emirdağ-Öztürk, S. Hajdok, J. Conrad and U. Beifuss, Tetrahedron, 2013, 69, 3664–3668 CrossRef; (e) H. T. Abdel-Mohsen, J. Conrad and U. Beifuss, J. Org. Chem., 2013, 78, 7986–8003 CrossRef CAS PubMed; (f) H. Leutbecher, G. Greiner, R. Amann, A. Stolz, U. Beifuss and J. Conrad, Org. Biomol. Chem., 2011, 9, 2667–2673 RSC; (g) H. Leutbecher, M.-A. Constantin, S. Mika, J. Conrad and U. Beifuss, Tetrahedron Lett., 2011, 52, 605–608 CrossRef CAS; (h) H. Leutbecher, S. Hajdok, C. Braunberger, M. Neumann, S. Mika, J. Conrad and U. Beifuss, Green Chem., 2009, 11, 676–679 RSC; (i) S. Hajdok, J. Conrad, H. Leutbecher, S. Strobel, T. Schleid and U. Beifuss, J. Org. Chem., 2009, 74, 7230–7237 CrossRef CAS PubMed; (j) S. Hajdok, H. Leutbecher, G. Greiner, J. Conrad and U. Beifuss, Tetrahedron Lett., 2007, 48, 5073–5076 CrossRef CAS; (k) S. Witayakran, A. Zettili and A. J. Ragauskas, Tetrahedron Lett., 2007, 48, 2983–2987 CrossRef CAS; (l) H. Leutbecher, J. Conrad, I. Klaiber and U. Beifuss, Synlett, 2005, 20, 3126–3130 Search PubMed; (m) U. T. Bhalerao, C. Muralikrishna and B. R. Rani, Tetrahedron, 1994, 50, 4019–4024 CrossRef CAS.
- M. Fache, B. Boutevin and S. Caillol, ACS Sustainable Chem. Eng., 2016, 4, 35–46 CrossRef CAS.
- P. J. Strong and H. Claus, Crit. Rev. Environ. Sci. Technol., 2011, 41, 373–434 CrossRef.
- V. T. Calabrese and A. Khan, J. Phys. Chem. A, 2000, 104, 1287–1292 CrossRef CAS.
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC and references cited therein..
Footnote |
| † Electronic supplementary information (ESI) available: Experimental data and copies of the 1H NMR and 13C NMR spectra. See DOI: 10.1039/c9ra02541c |
| This journal is © The Royal Society of Chemistry 2019 |