
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot one-step synthesis of Au@SiO2 core–shell nanoparticles and their shell-thickness-dependent fluorescent properties†
Shan Zhang,
Xiaochuan Xu,
Guoyu Zhang,
Bin Liu and
Jianhui Yang*
and
Jianhui Yang*
Key Laboratory of Synthetic and Natural Functional Molecule Chemistry (Ministry of Education), Shaanxi Key Laboratory of Physico-Inorganic Chemistry, College of Chemistry & Materials Science, Northwest University, Xi'an 710069, P. R. China. E-mail: jianhui@nwu.edu.cn; Fax: +86-29-81535026; Tel: +86-29-81535026
First published on 5th June 2019
Abstract
Herein, we report a one-pot one-step method for the preparation of Au@SiO2 core–shell nanoparticles (NPs) via a facile heating treatment of an alcoholic-aqueous solution of chloroauric acid (HAuCl4), 2-methylaminoethanol (2-MAE), cetyltrimethylammonimum bromide (CTAB), and tetraethylorthosilicate (TEOS). The size of the Au core and the thickness of the silica shell can be easily controlled by simply adjusting the volume of HAuCl4 and TEOS, respectively, which can hardly be achieved by other approaches. The as-prepared Au@SiO2 core–shell NPs exhibited shell-thickness-dependent fluorescent properties. The optimum fluorescence enhancement of fluorescein isothiocyanate (FITC) was found to occur at a silica shell thickness of 34 nm with an enhancement factor of 5.0. This work provides a new approach for the preparation of Au@SiO2 core–shell NPs and promotes their potential applications in ultrasensitive analyte detection, theranostics, catalysts and thin-film solar cells.
1 Introduction
Noble metal@oxide core–shell nanostructures have attracted much attention1–3 due to their unique properties and promising applications in catalysis,2 solar cells,4,5 biomedical materials,6 environmental remediation,7 and sensors.8 Among various noble metal@oxide core–shell nanostructures, gold@silica (Au@SiO2) core–shell nanoparticles (NPs) are probably most studied because of the synergetic properties of Au NPs and silica shells.9–11 Au NPs exhibit remarkable optical properties, such as surface plasmon resonance (SPR),12,13 surface-enhanced Raman scattering,14 plasmon-enhanced fluorescence (PEF),15,16 and are widely useful in the fields of sensing and biomedicine.17–19 The dense and porous silica shells offer attractive properties including optical transparency, versatile surface modification, high stability, and improved biocompatibility.20–23 More importantly, Au@SiO2 core–shell NPs provide access to easy surface modification of fluorescence molecules. The PEF effect strongly depends on two factors: one is the optimal distance between Au NPs and fluorophores to induce efficient excitation enhancement. The other is the spectral overlap between SPR spectra of Au NPs and the emission spectra of fluorophores to lead to emission enhancement. Silica can act as a tunable rigid spacer to control the gap distance for optimizing the fluorescence efficiency as well as SPR wavelength related with the size of Au NPs.15,16 Therefore, it is important to synthesize Au@SiO2 core–shell NPs with controllable Au core size and silica shell thickness.Significant efforts have been made to design Au@SiO2 core–shell NPs after the pioneering research by Liz-Marzan et al.9 The most common strategy for constructing Au@SiO2 core–shell NPs is through a two-step or multiple-step process. Au NPs as core were pre-synthesized and functionalized by certain ligands, such as (3-aminopropyl)trimethoxysilane, poly(vinyl pyrrolidone), and cetyltrimethylammonimum bromide (CTAB).9,24,25 Silica was then homogeneously coated on the surface of Au NPs via hydrolysis and condensation of tetraethylorthosilicate (TEOS) in an alkaline alcohol/water mixture. Au NPs are unstable and tended to aggregate during the coating process of silica, also results in the new nucleation and formation of silica NPs during this step-by-step procedure. Great progress has been made to overcome the obstacle for synthesizing Au@SiO2 core–shell NPs through a one-pot two-step strategy.26–28 It is desired to develop a facile, less time consuming and efficient strategy in the preparation of Au@SiO2 core–shell NPs.29
In this work, we present a one-pot one-step method for the preparation of Au@SiO2 core–shell NPs. As illustrated in Scheme 1, Au@SiO2 core–shell NPs are obtained via a facile heating treatment of the alcoholic-aqueous solution of chloroauric acid (HAuCl4), 2-methylaminoethanol (2-MAE), CTAB, and TEOS. Here, HAuCl4 was reduced by 2-MAE for the formation of Au NPs. The alkaline solution of 2-MAE could also serve as base-catalyzed hydrolysis and polymerization of TEOS to generate silica. CTAB acted as the stabilizer of Au NPs and the mesostructural template of silica. The size of Au cores could be tuned by changing the concentration of HAuCl4. The silica-shell thickness was controlled by adjusting the experimental conditions such as the volume of TEOS.
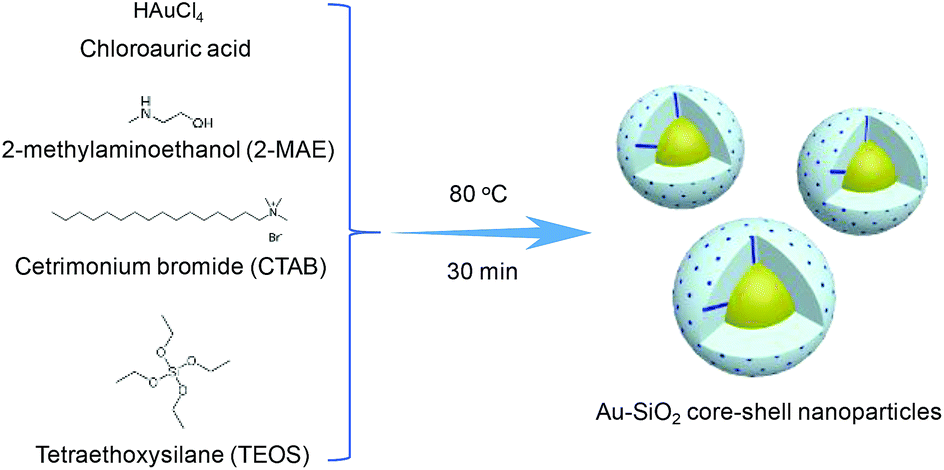 | ||
| Scheme 1 Schematic illustration of the preparation of Au@SiO2 core–shell NPs. | ||
2 Experimental section
2.1 Synthesis of Au@SiO2 core–shell NPs
All the chemicals used here were analytical grade without further purification. In a typical synthesis, 38 mL of deionized water, 6 mL of ethanol, 0.07 g of cetrimonium bromide (CTAB), 2 mL of 2-methylaminoethanol (0.133 mol L−1), 2 mL of chloroauric acid (HAuCl4·4H2O, 8.14 mmol L−1) and 0.10 mL of pure tetraethoxysilane (TEOS) were mixed in a 100 mL of beaker under ultrasonicating. The beaker was put into a heating mantle at 80 °C under stirring. The color of the solution rapidly changed from a light yellow to deep red within 2 to 3 min. The solution was kept at 80 °C for 30 min and then cooled down to room temperature. The products were then collected by washing twice by centrifugation at 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 min and finally dispersed in water.
000 rpm for 30 min and finally dispersed in water.
2.2 Synthesis of Au@SiO2-FITC NPs
0.003 g of Au@SiO2 core–shell NPs with different thicknesses were dispersed in 1 mL of deionized water respectively. 20 μL of glacial acetic acid and 10 μL of 3-aminopropyl-trimethoxysilane were added to each system under ultrasonic. After the solutions were stirred for 4 h, the samples were washed with ethanol for one time by centrifugation and dispersed in 0.5 mL ethanol. The dispersions mixed with 2 mL of fluorescein isothiocyanate (FITC, 0.002 g) solution in ethanol and then were stirred in the dark for 24 h. The final product was collected by centrifugation at 11000 rpm for 15 min. The obtained FITC-modified NPs were dispersed in 1 mL of water for further characterization.
2.3 Characterization
Transmission electron microscopy (TEM) images were obtained from FEI F200 (200 kV). The samples for TEM images were prepared by drop-casting the nanoparticle dispersion onto carbon-coated copper grids. Scanning electron microscope (SEM) image was performed on FEI Quanta 400 FEG. X-ray diffraction (XRD) analysis was performed on an X-ray diffractometer (Bruker, D8 Advance) with Cu radiation (Kα = 1.54059 Å). The samples for SEM and XRD measurements were prepared by drying the nanoparticle dispersion on the silicon wafer. The products were dispersed into water and put into 1 cm quartz cell for UV-vis absorption and emission spectra measurements. UV-vis absorption spectra were recorded on a Lambda 40P UV-vis spectrometer. The emission spectra were taken on an F-4500 fluorescence spectrophotometer with the excited wavelength of 460 nm. Nitrogen adsorption–desorption isotherms were measured with TR2 Star 3020. The sample for the measurement was dried at 60 °C for 6 h and then calcined in vacuum from room temperature to 550 °C for 6 h at a rate of 1 °C min−1 to remove the CTAB template.3 Results and discussion
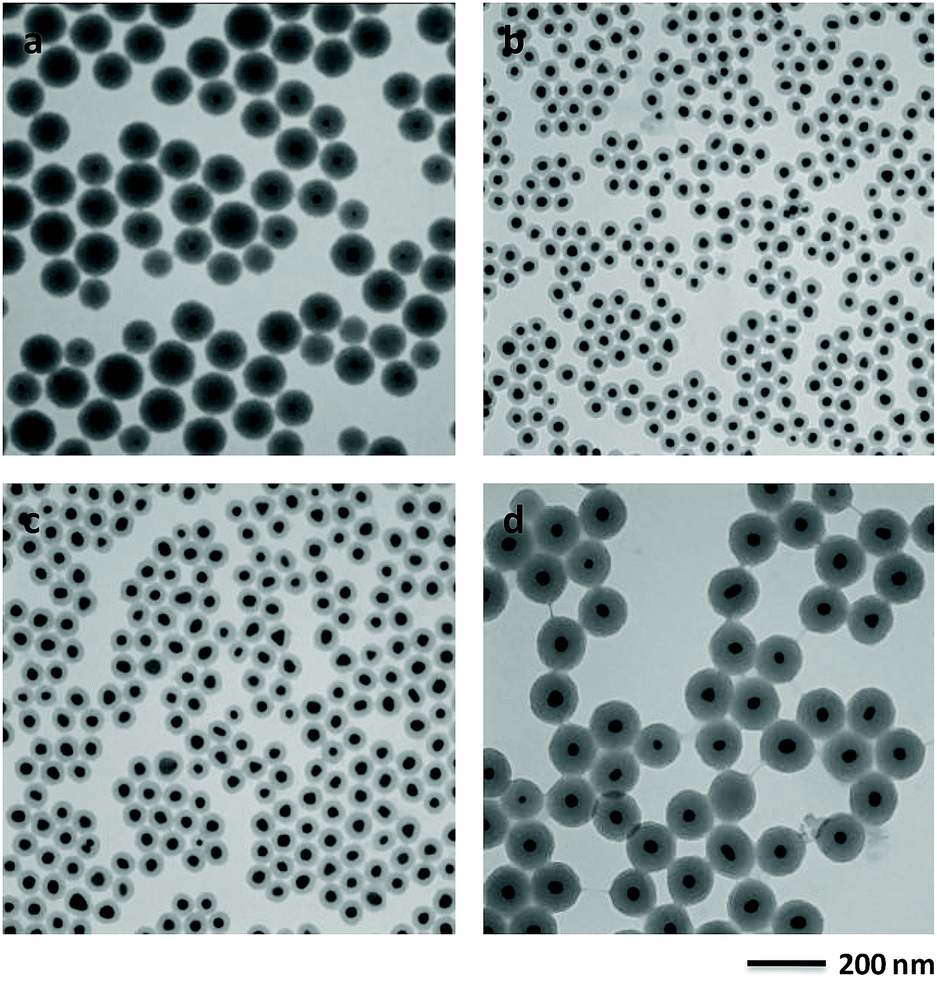
The phase and composition of the products were characterized by XRD in Fig. S1.† The XRD pattern (Fig. S1a†) of the product synthesized in the absence of TEOS by the typical synthesis shows the diffraction peaks of cubic face-centered phase metal gold (JCPDS card PDF#04-0784), which are indexed to the (111), (200), (220), (311) and (222) planes. For the XRD pattern (Fig. S1b†) of the as-prepared sample by the typical synthesis, there are five diffraction peaks, which match very well with that of Au NPs synthesized in the absence of TEOS. Also there is a broad peak between 20 and 25 degree which is assigned to the silica (JCPDS card PDF#38-0651).The morphology, structure and composition of the product were further investigated by SEM and TEM. Low-magnification of SEM (Fig. 1a) and TEM (Fig. 1b) images both confirmed the core–shell structure of the product with excellent monodispersity. The core size and shell thickness are 21.3 (Fig. S2a†) and 34.2 nm (Fig. S2b†), respectively. High-resolution TEM images (Fig. 1c and d) show that the shell layer has mesoporous structure and the interplane distance in core is 0.24 nm corresponded to the Au (111) plane. The nitrogen adsorption–desorption isotherms of the Au@SiO2 NPs demonstrate the representative type-IV curves with the surface area of 842.72 m2 g−1 and the pore size of 4.17 nm (Fig. S3†). HAADF image (Fig. 1e) shows the clear contrast in the central and outer region, further confirms that the particle adopts a core–shell structure. The elemental mappings (Fig. 1f–h) reveal that the core–shell nanostructure was composed of a Au core and a SiO2 shell, which also confirmed by XRD.
 | ||
| Fig. 1 (a) SEM, (b) TEM, (c and d) high-resolution TEM, (e) high angle annular dark field (HAADF) image combined with energy dispersive X-ray spectroscopy (EDX) mapping images of elemental Au (f), Si (g) and O (h) of Au@SiO2 core–shell NPs obtained by the typical synthesis. | ||
When the volume of HAuCl4 was decreased to half of that in the typical synthesis, the size of Au core decreased to 15.5 nm and the silica-shell thickness increased to 43.1 nm (Fig. 2a and S4†). When the volume of TEOS was decreased to 0.05 and 0.08 mL, the silica-shell thickness of Au@SiO2 core–shell NPs decreased to 12.6 and 20.8 nm (Fig. S5b and d†), respectively. If the volume of TEOS increased to 0.13 mL, the silica-shell thickness of Au@SiO2 core–shell NPs increased to 41.9 nm (Fig. S5f†). When the different volume of TEOS was used to synthesize Au@SiO2 core–shell NPs with various silica thicknesses, the size of Au core keeps similar around 20 nm (Fig. S2b, S5a, c and e†). The experimental conditions for Au@SiO2 core–shell NPs are summarized as shown in Table S1.† It demonstrated that the Au-core size and the silica-shell thickness of Au@SiO2 core–shell NPs could be easily controlled by adjusting the volume of HAuCl4 and TEOS, respectively.
 | ||
| Fig. 2 TEM image of Au@SiO2 core–shell NPs synthesized with 1 mL of 8.14 mmol L−1 HAuCl4 solution (a). TEM images of Au@SiO2 core–shell NPs obtained using 0.05 (b), 0.08 (c) and 0.13 mL (d) of TEOS, respectively. The synthetic condition is similar with the typical synthesis of Au@SiO2 core–shell NPs. | ||
2-MAE as an alkanolamine is employed here as a reducing agent during the formation of Au NPs.30 If there is no capping agent, irregular Au NPs with broad size distribution is formed by reduction of HAuCl4 using 2-MAE (Fig. S6†). CTAB formed micelles, which can be used as microreactors for synthesis of Au NPs with different shapes.28,31 Au NPs with the diameter of 19.2 nm could be obtained in the presence of CTAB (Fig. S7†). Herein, the beaker contained a mixture solution of HAuCl4, 2-MAE, CTAB and TEOS was put into a heating mental at 80 °C. The colour of the solution rapidly changed from a light yellow to deep red within 2 to 3 min. It means the formation of Au NPs due to the reducing of HAuCl4 by 2-MAE in the presence of CTAB at the first stage, which is confirmed by TEM (Fig. 3a). The sizes of Au NPs keep almost unchanged during the growing process of silica on their surfaces (Fig. 3b–d). It was suggested that Au NPs were formed through a fast nucleation and subsequent/consequent growth process within several minutes under relatively higher temperatures. The formation of monodisperse NPs is generally explained by the nucleation and growth model of LaMer.32 In this model, nucleation and growth are assumed to be two completely separate steps. The relative high temperature is necessary to obtain highly monodispersed NPs with a narrow size distribution. When the temperature is decreased to 60 °C, the nucleation occurs concurrently with the growth of NPs, which result in NPs with broad size distributions (Fig. S8†). If the volume of HAuCl4 was decreased to half of that in the typical synthesis, the size of Au core decreased to 15.5 nm (Fig. 2a) from 21.3 nm (Fig. 1) due to the lack feeding of Au atoms for the further growth of Au NPs. CTAB could stabilize Au NPs and also acted as the mesostructural template of silica on the surfaces of Au NPs.26 The aqueous solution of 2-MAE (pH = 11) could also serve as base-catalyzed hydrolysis and polymerization of TEOS to generate silica (Fig. S9†). The thickness of silica gradually increased as the reaction progressed (Fig. 3a–d). The amount of TEOS in solution is also critical to silica thickness (Fig. 1 and 2b–d). It was demonstrated that the reaction time or TEOS concentration can be used to control the silica thickness on Au NPs.
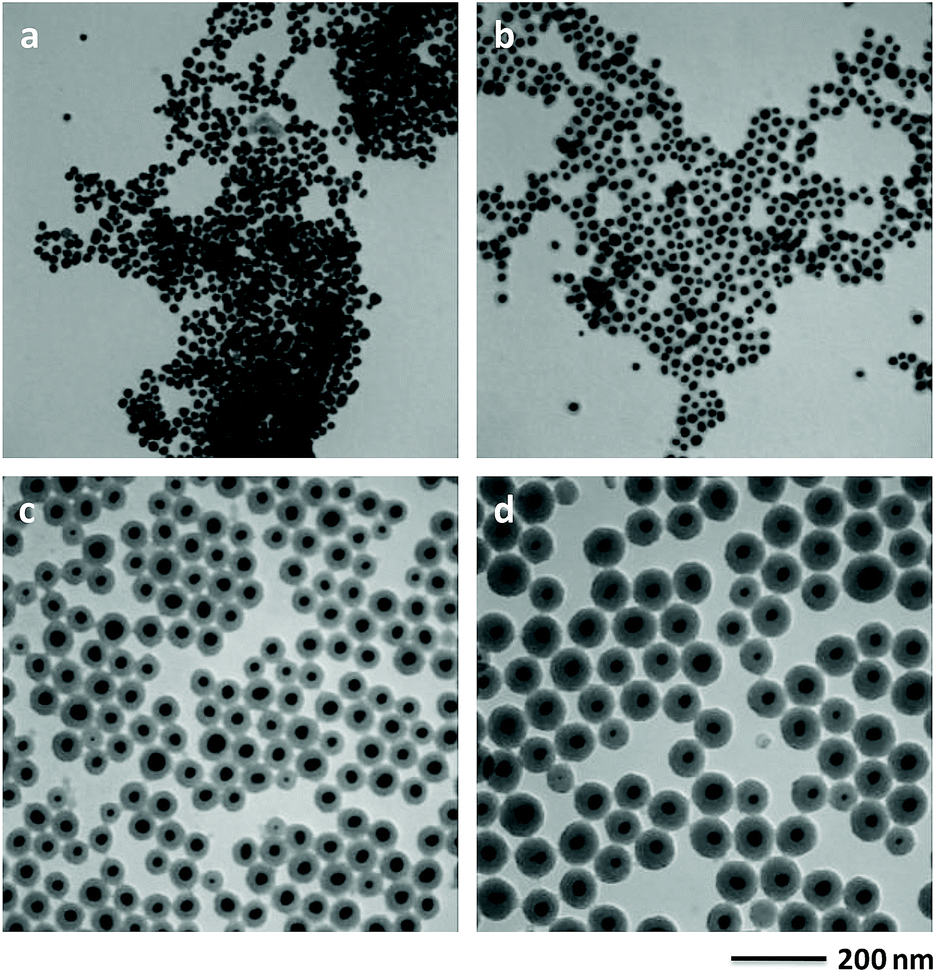 | ||
| Fig. 3 TEM images of the intermediate product collected after (a) 3 min, (b) 5 min, (c) 10 min and (d) 20 min of reaction. | ||
It is well known that Au NPs show SPR absorption in the visible to near infrared range. The SPR peaks are strongly related with the size and shape of Au NPs, core–shell structures, or the surrounding medium.12,13 As shown in Fig. 4a, Au NPs (Fig. S7†) synthesized in the absent of TEOS show a typical SPR peaks at 521 nm. When the surface of Au NPs coated with silica shell, the SPR peaks of Au@SiO2 core–shell NPs is red-shift from 521 to 528 nm due to an increase in the local refractive index of the surrounding medium. The position of the peak is not sensitive and slightly red-shift to 532 nm as the increase of shell thickness of silica from 12.6 to 41.9 nm. The variation of the absorption peak with silica-shell thickness is shown in Fig. 4b, which is consistent with previous report.9
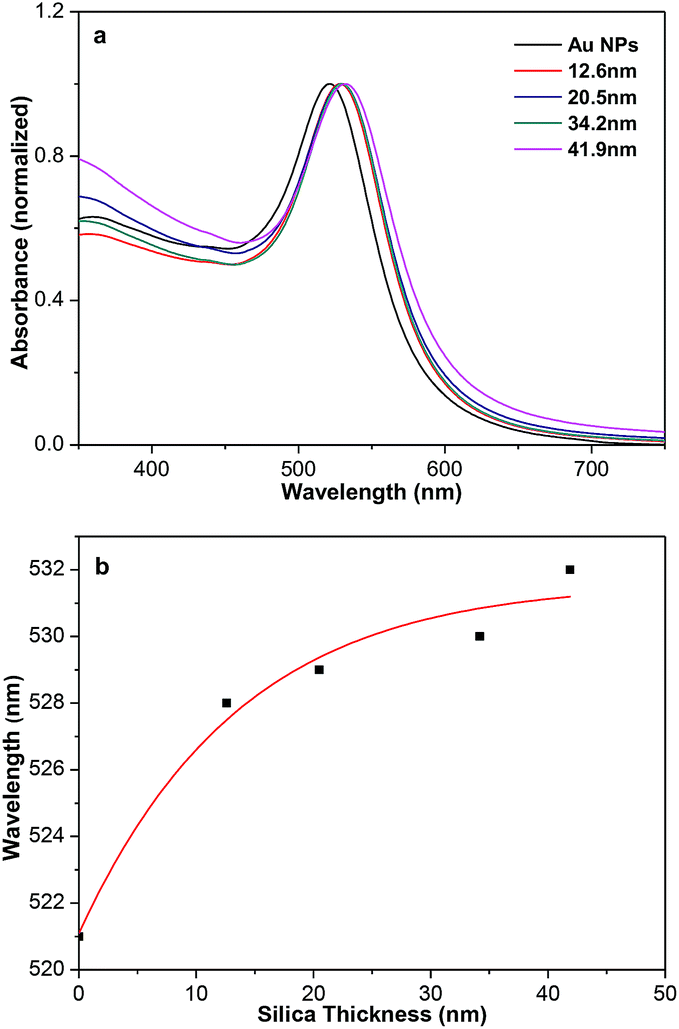 | ||
| Fig. 4 (a) Normalized UV-vis absorption spectra of aqueous dispersions of Au and Au@SiO2 core–shell NPs. (b) Variation of the absorption peak with silica-shell thickness. | ||
The plasmonic-fluorescence enhancement or quenching is closely related with the gold-fluorophore distance.15,16 The distances between the fluorophores and Au NPs cores could be tuned by the range of silica shell thicknesses used. Fluorescein isothiocyanate (FITC) was chosen as the fluorophore to demonstrate the ability to regulate the fluorescence because it could be covalently grafted on the surface of 3-aminopropyl-trimethoxysilane modified Au@SiO2 core–shell NPs.33,34 The amount of FITC molecules covalently linked on the surface of Au@SiO2 core–shell NPs should be almost the same as that in SiO2-FITC NPs (blank sample) through UV-vis absorption spectra (Fig. S10†). Fig. 5a shows the shell-thickness-dependent fluorescent spectra of Au@SiO2 core–shell NPs with 20 nm Au cores. The enhancement factor was plotted against the silica thickness (Fig. 5b), which shows the maximum enhancement of 5.0 at about 34 nm thickness of silica. Deviation from the optimum silica thickness resulted in a decrease enhancement factor, which is consistent very well with previous reports.35–37 The SPR bonds of Au@SiO2 core–shell NPs around 520 nm overlap well with the emission peak of FITC. The enhancement is attributed to the enhanced radiative decay rate, which causes increased fluorescence quantum yield and shortened fluorescence lifetime. Also, nonradiative energy transfer from the fluorescence of FITC to the metallic surface, which generally quenches its fluorescence. The metal-enhanced and quenched fluorescence comes as a result of competition between the distance-dependent mechanisms.35
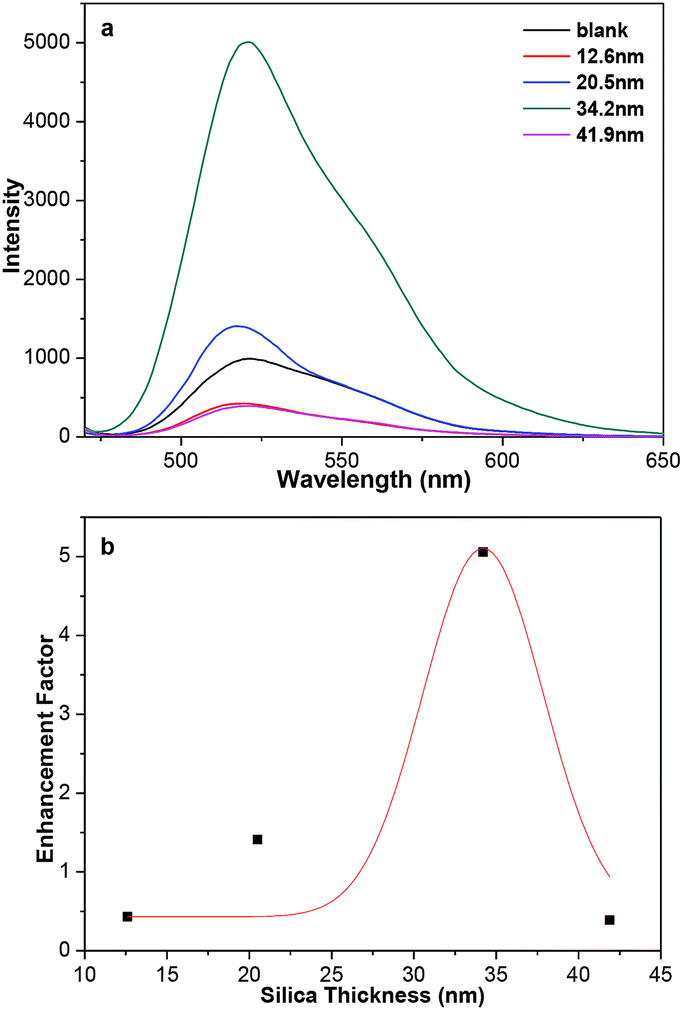 | ||
| Fig. 5 (a) Fluorescent emission spectra of Au@SiO2-FITC NPs with different thickness of silica shell. (b) The plot of the enhancement factor of the emission at 520 nm verse silica thickness. The red curve represents Gaussian fit to this data. | ||
4 Conclusions
In summary, we have demonstrated a facile one-pot one-step method for the preparation of Au@SiO2 core–shell NPs. The size of Au core and the thickness of silica shell can be easily controlled by properly adjusting the initial reaction solution which can be hardly achieved by other approaches. The as-prepared Au@SiO2 core–shell NPs exhibited the shell-thickness-dependent fluorescent properties. The optimum fluorescence enhancement of FITC was found to occur at a silica shell thickness of 34 nm with an enhancement factor of 5.0. This work provides a new approach for the preparation of Au@SiO2 core–shell NPs and promotes their potential applications in ultrasensitive analyte detection, theranostics, catalysts and thin-film solar cells.11Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21501141 and 21673172) and the Education Commission of Shaanxi Province (No. 14JS092). J. Yang thanks Cyrus Tang Foundation (China) for Tang Scholar.Notes and references
- R. G. Chaudhuri and S. Paria, Chem. Rev., 2012, 112, 2373 CrossRef PubMed.
- G. Li and Z. Tang, Nanoscale, 2014, 6, 3995 RSC.
- S. Liu, M. D. Regulacio, S. Y. Tee, Y. W. Khin, C. P. Teng, L. D. Koh, G. Guan and M.-Y. Han, Chem. Rec., 2016, 16, 1965 CrossRef CAS PubMed.
- X. Liu, J. Iocozzia, Y. Wang, X. Cui, Y. Chen, S. Zhao, Z. Li and Z. Lin, Energy Environ. Sci., 2017, 10, 402 RSC.
- P. Rai, Sustainable Energy Fuels, 2019, 3, 63 RSC.
- A. Burns, H. Ow and U. Wiesner, Chem. Soc. Rev., 2006, 35, 1028 RSC.
- P. Rai, S. M. Majhi, Y.-T. Yu and J.-H. Lee, RSC Adv., 2015, 5, 76229 RSC.
- K. Mondal and A. Sharma, RSC Adv., 2016, 6, 83589 RSC.
- L. M. Liz-Marzan, M. Giersig and P. Mulvaney, Langmuir, 1996, 12, 4329 CrossRef CAS.
- A. Guerrero-Martinez, J. Perez-Juste and L. M. Liz-Marzan, Adv. Mater., 2010, 22, 1182 CrossRef CAS.
- C. Hanske, M. N. Sanz-Ortiz and L. M. Liz-Marzan, Adv. Mater., 2018, 30, 1707003 CrossRef.
- S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 4212 CrossRef CAS.
- S. Eustis and M. A. El-Sayed, Chem. Soc. Rev., 2006, 35, 209 RSC.
- Y. Wang, B. Yan and L. Chen, Chem. Rev., 2013, 113, 1391 CrossRef CAS PubMed.
- R. Jiang, B. Li, C. Fang and J. Wang, Adv. Mater., 2014, 26, 5274 CrossRef CAS PubMed.
- J.-F. Li, C.-Y. Li and R. F. Aroca, Chem. Soc. Rev., 2017, 46, 3962 RSC.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293 CrossRef CAS PubMed.
- D. A. Giljohann, D. S. Seferos, W. L. Daniel, M. D. Massich, P. C. Patel and C. A. Mirkin, Angew. Chem., Int. Ed., 2010, 49, 3280 CrossRef CAS PubMed.
- M. Li, S. K. Cushing and N. Wu, Analyst, 2015, 140, 386 RSC.
- T. Ung, L. M. Liz-Marzan and P. Mulvaney, J. Phys. Chem. B, 2001, 105, 3441 CrossRef CAS.
- D. Tarn, C. E. Ashley, M. Xue, E. C. Carnes, J. I. Zink and C. J. Brinker, Acc. Chem. Res., 2013, 46, 792 CrossRef CAS PubMed.
- J. Feng, C. Gao and Y. Yin, Nanoscale, 2018, 10, 20492 RSC.
- H. Kang, J. T. Buchman, R. S. Rodriguez, H. L. Ring, J. He, K. C. Bantz and C. L. Haynes, Chem. Rev., 2019, 119, 664 CrossRef CAS PubMed.
- C. Graf, D. L. J. Vossen, A. Imhof and A. van Blaaderen, Langmuir, 2003, 19, 6693 CrossRef CAS.
- P. Botella, A. Corma and M. T. Navarro, Chem. Mater., 2007, 19, 1979 CrossRef CAS.
- J. Chen, R. Zhang, L. Han, B. Tu and D. Zhao, Nano Res., 2013, 6, 871 CrossRef CAS.
- C. Wu, Z.-Y. Lim, C. Zhou, W. G. Wang, S. Zhou, H. Yin and Y. Zhu, Chem. Commun., 2013, 49, 3215 RSC.
- J.-T. Song, X.-S. Zhang, M.-Y. Qin and Y.-D. Zhao, Dalton Trans., 2015, 44, 7752 RSC.
- R. Liu and R. D. Priestley, J. Mater. Chem. A, 2016, 4, 6680 RSC.
- https://www.chemicalbook.com/ChemicalProductProperty_EN_CB2745059.htm.
- J. Perez-Juste, L. M. Liz-Marzan, S. Carnie, D. Y. C. Chan and P. Mulvaney, Adv. Funct. Mater., 2004, 14, 571 CrossRef CAS.
- J. Park, J. Joo, S. G. Kwon, Y. Jang and T. Hyeon, Angew. Chem., Int. Ed., 2007, 46, 4630 CrossRef CAS PubMed.
- Y. Zhang, X. Kong, B. Xue, Q. Zeng, X. Liu, L. Tu, K. Liu and H. Zhang, J. Mater. Chem. C, 2013, 1, 6355 RSC.
- P. D. Nallathamby, J. Hopf, L. E. Irimata, T. L. McGinnity and R. K. Roedera, J. Mater. Chem. B, 2016, 4, 5418 RSC.
- H. Li, J. Kang, J. Yang and B. Wu, J. Phys. Chem. C, 2016, 120, 16907 CrossRef CAS.
- H. Li, J. Kang, J. Yang and B. Wu, J. Alloys Compd., 2016, 673, 283 CrossRef CAS.
- H. Li, J. Yang, Q. Deng, S. Dou, W. Zhao, C. Lin and X. Liu, Sci. China Mater., 2018, 61, 401 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02543j |
| This journal is © The Royal Society of Chemistry 2019 |