
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of sulfonated ordered mesoporous carbon catalyst and its catalytic performance for esterification of free fatty acids in waste cooking oils†
Sun Naab,
Zhang Minhua ab,
Dong Xiuqinab and
Wang Lingtao*ab
ab,
Dong Xiuqinab and
Wang Lingtao*ab
aKey Laboratory for Green Chemical Technology of Ministry of Education, R&D Center for Petrochemical Technology, Tianjin University, Tianjin 300072, China. E-mail: ltwang@tju.edu.cn; Fax: +86-22-27406119; Tel: +86-22-27406119
bCollaborative Innovation Center of Chemical Science and Engineering, Tianjin, 300072, China
First published on 21st May 2019
Abstract
Sulfonated ordered mesoporous carbon (SO3H-OMC) solid acid catalysts from sucrose were prepared using hard-template method, and their catalytic performance as well as the deactivation mechanism for esterification of free fatty acids (FFAs) in waste cooking oils (WCOs) were evaluated. Effects of sulfonation time, sulfonation temperature and hard template structure type for the textural properties and acid properties of SO3H-OMC were systematically investigated by N2 adsorption–desorption, FT-IR, NH3-TPD, TEM and strong acid density analysis. The results indicated that, SO3H-OMC(s)-6-160 catalyst, which was prepared by using SBA-15 as hard template at sulfonation time of 6 h and sulfonation temperature of 160 °C, had well-ordered mesoporous structure and high –SO3H groups density (2.32 mmol g−1). Compared with SO3H-APC-6-160 catalyst, cation-exchange resin D072 and SO3H-OMC(k)-6-160 catalyst, it was found that the SO3H-OMC(s)-6-160 catalyst exhibited highest activity (FFAs conversion was 93.8%) and good stability for the FFAs esterification, attributed to its 2D-hexagonal channels and hydrophobic surface. The –SO3H groups being leached out of SO3H-OMC catalysts into the liquid phase (especially methanol) would be the main reason causing catalyst deactivation.
1. Introduction
Since the increasing price of petroleum and severe environmental pollutions of using fossil energy,1–3 biodiesel (fatty acids methyl esters, FAME), as a promising eco-friendly alternative fuel to replace traditional fuels, has drawn extensive attention for its biodegradability, nontoxicity and favorable combustion property.4However, due to the expenses of the raw materials like vegetable oil and animal fats are the main contributor to the cost of biodiesel production, biodiesel has not been widely promoted worldwide.5–7 In order to utilize economical raw materials, cheaply available waste cooking oils (WCOs) reused from restaurants and food plants are used.8,9 But the large numbers of free fatty acids (FFAs) existing in the WCOs will be consumed via the saponification reaction in the presence of alkaline catalyst, which makes it ineffective to remain in one-step trans-esterification to biodiesel production. Thus the pre-esterification of FFAs with methanol before normal trans-esterification should be carried out for the feedstock of high acid value.10–13 Generally, homogenous sulfuric acid, which is relatively cheap, exhibits high catalytic activity in the pre-esterification of FFAs, but it will lead to serious equipment corrosion, environment pollution and catalyst recycling difficulties.
To overcome these disadvantages, varieties of heterogeneous solid acid catalysts, such as molecular sieves,14–21 cation-exchange resins,22,23 and carbon-based solid acids,24,25 have been applied as alternative catalysts for the FFAs esterification. Among them, the sulfonated carbon-based catalysts26,27 are considered as the promising replacement for the liquid catalysts.
Recently, the ordered mesoporous carbon materials (OMC) have been widely studied.28 The typical strategy applied to prepare OMC involves the use of hard templates, such as SBA-15,29 KIT-6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 30,31 and MCM-48,29 with the purpose to obtain 2D-hexagonally packed mesopores or 3D-interconnected mesostructure for widely application.
30,31 and MCM-48,29 with the purpose to obtain 2D-hexagonally packed mesopores or 3D-interconnected mesostructure for widely application.
Currently, the –SO3H groups have been introduced to OMC materials with template SBA-15 by Peng et al.32 successfully, which could show the enhanced activity in the acid-catalyzed esterification of oleic acid with methanol. The sulfonic acid groups functionalized OMC have exhibited a broad application prospect for some large molecule reactions, like esterification reaction, owing to their well-defined pore structure, high specific surface area and inherent surface hydrophobicity.33,34 These unique features make the sulfonated OMC be benefit for long-chain FFAs molecules to access to their effective surface acidic sites, and inhibit the adsorption of byproduct water.
In this work, we prepared sulfonated ordered mesoporous carbon catalysts with sucrose as carbon precursor, SBA-15 and KIT-6 as hard template, respectively. The influences of sulfonation time, sulfonation temperature and hard template structure type on textual parameters, density of –SO3H groups, and catalytic activity were systematically studied. Additionally, in order to explore the effects of pore structure and surface property of catalyst on catalytic activity for FFAs esterification, the macroporous cation-exchange resin and amorphous porous carbon-based solid acid, which used the same carbon precursor and preparation conditions as the OMC materials, were evaluated. Besides, the stability of the catalyst and the reason of the catalyst deactivation had also been investigated.
2. Experimental
2.1 Materials
Pluronic P123 (EO20PO70EO20) and tetraethoxysilane (TEOS) were purchased from Sigma-Aldrich Corporation, USA. Other chemical reagents were purchased from Guangfu Chemical Corporation, China. All chemical reagents were analytical grade and directly used without any further purification.2.2 General synthesis procedures of mesoporous silica templates
Two types of mesoporous silica materials (i.e. SBA-15 and KIT-6 zeolites) were employed for the preparation of the OMC. The mesoporous SBA-15 silica zeolite was synthesized according to the procedure reported by Zhao et al.,35 while KIT-6 mesoporous silica zeolite was prepared by the procedure described by Kleitz et al.312.3 Preparation of sulfonated mesoporous carbon catalyst samples
The synthesis of OMC was performed by means of nanocasting.36 Two different templates were used, i.e. SBA-15 zeolite and KIT-6 zeolite. The mesoporous silica zeolite was impregnated with sucrose as the carbon precursor, and then carbonized under N2 atmosphere at 600 °C. The resultant carbon–silica composites were immersed in 48% HF aqueous solution at room temperature to remove the silica template completely. The OMC materials were obtained after filtration, washing and drying.The preparation of sulfonated OMC was carried out under N2 atmosphere protection. Initially, 0.5 g OMC was added into a PTFE-lined stainless steel hydrothermal autoclave containing 20 mL of concentrated sulfuric acid (98%). The mixtures were heated at heating rate of 5 °C min−1 to 120–180 °C for 2–8 h. The samples were washed repeatedly with distilled water, and then dried in a vacuum oven at 60 °C for 8 h. The sulfonated OMC samples thus obtained were labeled as SO3H-OMC(X)-t-Ts, where X = s, k, which were abbreviations for SBA-15 and KIT-6 zeolites, respectively, t was the sulfonation time (h), and Ts was the sulfonation temperature (°C).
2.4 Preparation of other solid acid catalyst samples
The sugar-derived solid acid catalysts were prepared by using sucrose as the carbon precursor, and labeled as SO3H-APC-6-160, where ‘APC’ was the abbreviation for amorphous porous carbons, 6 denoted the sulfonation time (6 h), and ‘160’ denoted the sulfonation temperature (160 °C). All operating parameters of carbonization and sulfonation were the same as the SO3H-OMC catalyst samples. The strongly acidic macroporous cation-exchange resin (D072) was also used for the comparison with above prepared sulfonated OMC catalysts.2.5 Characterization
The nitrogen adsorption–desorption measurement was performed on a Micromeritics Tristar 3000 type instrument at the temperature of −196 °C. The samples were degassed under vacuum at 200 °C for 12 h prior to measure. The specific surface area was calculated by Brunauer–Emmett–Teller (BET) method. The pore volume was taken at P/P0 = 0.99 assuming complete pore saturation. Ammonia temperature programmed desorption characterization (NH3-TPD) was performed on a Micromeritics autochemII 2920 type instrument under a mixed stream composed of 1 v% NH3 in Ar. The microscopic morphology and mesoporous channel structure of sulfonated mesoporous carbon catalysts were observed from the transmission electron microscopic images (TEM) taken by a FEI Tecnai G2 F20 type instrument operated at 400 kV. The identification of surface functional groups was carried out by the Fourier transform infrared (FT-IR) spectra, which were recorded on a Nicolet 6700 type spectrometer with 32 scanning times at 4 cm−1 resolution (from 400 cm−1 to 4000 cm−1). The content of strong acidic groups (i.e. –SO3H) on the catalyst surface was measured by the alkaline neutralization titration.37 The density measurement procedure for strong acidic groups was as follows: 0.1 g pre-dried catalyst sample and 1 g NaCl were dispersed in 30 mL deionized water. The suspensions were stirred for 2 h and filtered, then the filtrate was collected and titrated with 0.1001 M NaOH standard solution using methyl red indicator. The density of –SO3H groups could be calculated by the following formula (1):| D−SO3H = 0.1001 (V1 – V0)/mcat | (1) |
2.6 Catalytic performance measurement
In ordered to explore the effects of textual property and surface property on the catalytic activity of SO3H-OMC catalyst samples, the comparative research was conducted with two different kinds of solid acid catalyst samples (SO3H-APC-6-160 and D072). The catalytic esterification of free fatty acids (FFAs) with methanol was performed in a three-necked round-bottom flask, which was equipped with reflux condenser, magnetic stirrer and temperature adjustable water bath. In a typical procedure, the reaction was carried out at feed molar ratio 9:1 of methanol to FFAs, the usage amount of catalysts was of 10 wt%, and then the reaction system was maintained at temperature of 70 °C with the stirring rate of 800 rpm. After reaction proceeded for 6 h, the reaction was terminated and the reactant temperature was cooled to room temperature, and then kept static for 2 min. Subsequently, samples of oil phase were transferred out for the acid value analysis by the titration method,37 according to BS EN ISO 660 (National Standard of the People's Republic of China. GB/T5330-2005). The conversion of FFAs was calculated from acid value using formula (2):| FFAs conversion (%) = (1 − AVf/AVi) × 100% | (2) |
Recycling experiments were performed to investigate the stability of the sulfonated OMC catalysts (SO3H-OMC(s)-6-160). At the end of each reutilization cycle (each reaction was 6 h), the SO3H-OMC catalysts were filtered out from the reaction mixture, washed with anhydrous ethanol to remove the residual reactants from the catalyst surface, and then dried in vacuum at 100 °C for 2 h, finally the catalyst samples were stored in a desiccator to insulate water adsorption, and reused in the subsequent reaction runs. To eliminate the effect of the catalyst loss caused by filtration, the usage quantity of catalyst samples was reduced to 90% of that in previous cycle. To keep the same proportion between reactant and catalyst, the reactant amount should be also adjusted accordingly.
Another recycling experiment was conducted to explore the catalyst deactivation situation. Two experimental schemes were designed. Scheme-1, the SO3H-OMC(s)-6-160 catalysts were washed repeatedly with methanol for five times, and then the collected filtrate solution after each filtration was reacted with FFAs without the acidic catalysts; Scheme-2, the SO3H-OMC(s)-6-160 catalysts were washed repeatedly with FFAs for five times, and then the collected filtrate solution after each filtration was reacted with methanol without acidic catalysts. All other reaction conditions were the same as recycling experiments.
3. Results and discussion
3.1 Study on sulfonation conditions of mesoporous carbon-based solid acid catalysts
| Samples name | Specific surface area (m2 g−1) | Pore volume (cm3 g−1) | Density of –SO3H groups (mmol g−1) | ||||
|---|---|---|---|---|---|---|---|
| Stotal | Smicro | Smeso | Vtotal | Vmicro | Vmeso | ||
| SO3H-OMC(s)-2-160 | 542 | 109 | 433 | 0.86 | 0.08 | 0.78 | 2.02 |
| SO3H-OMC(s)-4-160 | 541 | 110 | 431 | 0.85 | 0.09 | 0.78 | 2.28 |
| SO3H-OMC(s)-6-160 | 537 | 116 | 421 | 0.85 | 0.09 | 0.76 | 2.32 |
| SO3H-OMC(s)-8-160 | 532 | 113 | 419 | 0.84 | 0.09 | 0.75 | 2.33 |
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SO bonds (1214 cm−1) appeared after the sulfonation treatment,38 which might be an evidence for the incorporation of –SO3H groups in the catalysts successfully. Additionally, with the sulfonation temperature increased from 120 °C to 180 °C, the intensity of characteristic absorption peaks for –SO3H groups also increased, indicating the promote influence of higher sulfonation temperature on the attachment of –SO3H groups on the surface of OMC(s).
SO bonds (1214 cm−1) appeared after the sulfonation treatment,38 which might be an evidence for the incorporation of –SO3H groups in the catalysts successfully. Additionally, with the sulfonation temperature increased from 120 °C to 180 °C, the intensity of characteristic absorption peaks for –SO3H groups also increased, indicating the promote influence of higher sulfonation temperature on the attachment of –SO3H groups on the surface of OMC(s).
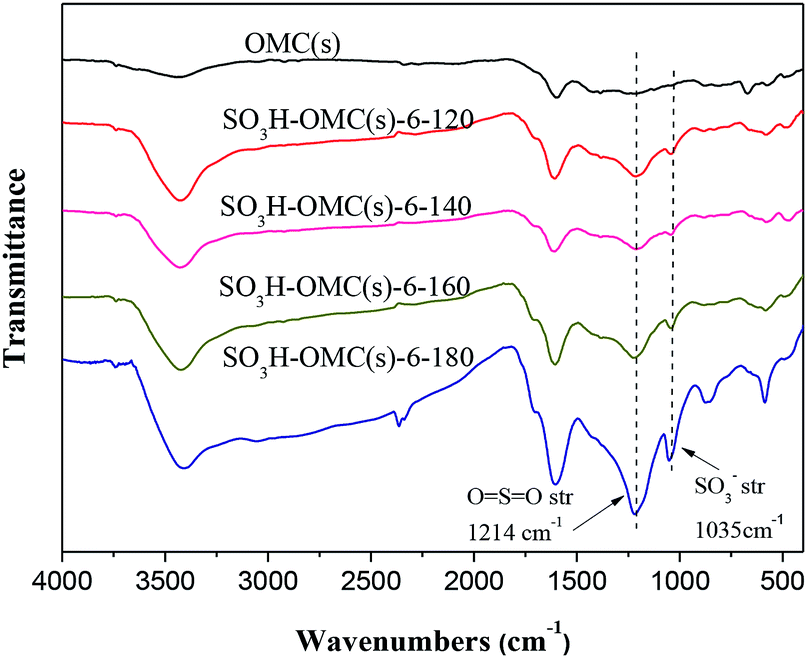 | ||
| Fig. 1 FT-IR spectra of OMC and sulfonated OMC samples prepared at different sulfonation temperature. | ||
Fig. 2 showed the NH3-TPD profiles of SO3H-OMC(s)-6-Ts catalyst samples prepared at the sulfonation temperature from 120 °C to 180 °C. With the programmed temperature rising from 150 °C to 550 °C, the broad NH3 desorption peak appeared at around 270 °C, indicating the presence of the medium strong acid sites on the surface of SO3H-OMC(s)-6-Ts samples. As with the rising of sulfonation temperature, the area of NH3-TPD desorption peak increased gradually, suggesting the increase in the number of –SO3H groups on surface. And no obvious offsets of NH3-TPD peak were observed, which implied that the sulfonation temperature did not effect on the acidic strength of surface acid sites.
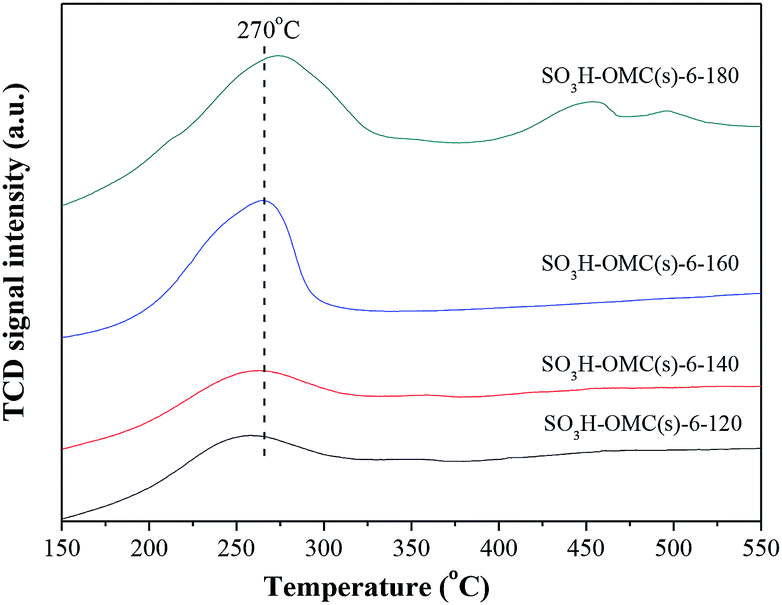 | ||
| Fig. 2 NH3-TPD profiles of sulfonated OMC samples prepared at different sulfonation temperature. | ||
The TEM images were presented in Fig. 3. It was proved that the ordered mesoporous morphology of SO3H-OMC(s)-6-Ts samples was finely maintained at the different sulfonation temperatures of 120 °C, 140 °C and 160 °C. However, the mesoporous framework structure of catalyst samples would be destroyed and collapsed obviously, when sulfonation temperature at 180 °C.
 | ||
| Fig. 3 TEM images of sulfonated OMC samples prepared at different sulfonation temperature. | ||
The data on the textural properties, the density of –SO3H groups and catalytic activity of SO3H-OMC catalyst samples prepared at different sulfonation temperature ranging from 120 °C to 180 °C were summarized in Table 2. It could be seen that the acid density of the SO3H-OMC(s)-6-Ts catalysts increased with the rising of sulfonation temperature, which was consistent with the characterization results observed from FTIR spectra and NH3-TPD profiles. When the treatment was at lower sulfonation temperature (120–160 °C), the specific surface area and the pore volume did not change obviously. In this temperature range, with the increasing of sulfonation temperature, the specific surface area and the pore volume of catalyst samples decreased slightly, which might be due to the –SO3H groups attached on the surface of OMC samples occupying the pore space. With the treatment at 180 °C, although the SO3H-OMC(s)-6-180 sample exhibited the highest density of –SO3H groups, the framework of it would be damaged, inducing distinct increase in specific surface area and pore volume, especially microporous specific surface area and pore volume.
| Samples name | Specific surface area (m2 g−1) | Pore volume (cm3 g−1) | Density of –SO3H groups (mmol g−1) | FFAs conv. (%) | ||||
|---|---|---|---|---|---|---|---|---|
| Stotal | Smicro | Smeso | Vtotal | Vmicro | Vmeso | |||
| SO3H-OMC(s)-6-120 | 563 | 109 | 454 | 0.89 | 0.08 | 0.81 | 1.92 | 89.6 |
| SO3H-OMC(s)-6-140 | 551 | 114 | 437 | 0.88 | 0.09 | 0.79 | 2.07 | 92.3 |
| SO3H-OMC(s)-6-160 | 537 | 116 | 421 | 0.85 | 0.09 | 0.76 | 2.32 | 93.8 |
| SO3H-OMC(s)-6-180 | 583 | 164 | 419 | 0.91 | 0.12 | 0.79 | 2.45 | 84.3 |
3.2 Catalytic activity evaluations
| Samples name | Specific surface area (m2 g−1) | Pore volume (cm3 g−1) | Density of –SO3H groups (mmol g−1) | FFAs conv. (%) | ||||
|---|---|---|---|---|---|---|---|---|
| Stotal | Smicro | Smeso | Vtotal | Vmicro | Vmeso | |||
| SO3H-OMC(s)-6-160 | 537 | 116 | 421 | 0.85 | 0.09 | 0.76 | 2.32 | 93.8 |
| SO3H-OMC(k)-6-160 | 612 | 138 | 474 | 0.90 | 0.11 | 0.79 | 2.09 | 92.8 |
| SO3H-APC-6-160 | 34 | — | — | 0.05 | — | — | 1.26 | 89.7 |
| D072 | — | — | — | — | — | — | 2.74 | 90.4 |
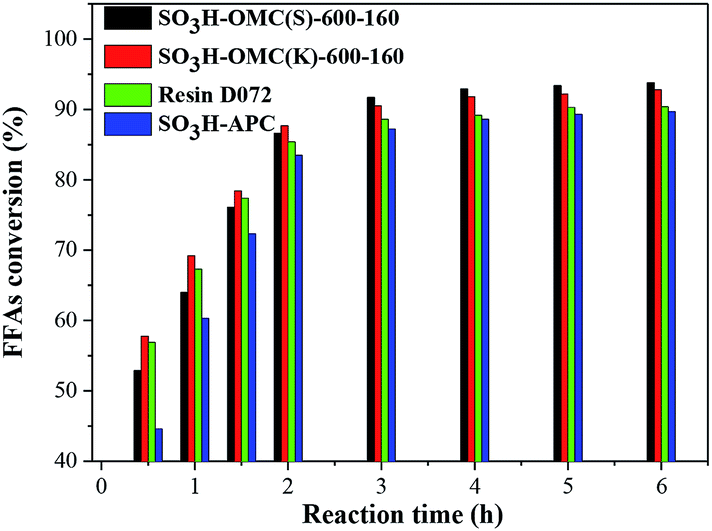 | ||
| Fig. 4 Catalytic performance of different catalysts for FFAs esterification. | ||
As shown in Fig. 4, when the esterification was catalyzed by the cation-exchange resin D072, the FFAs conversion was higher than those catalyzed by SO3H-OMC(s)-160 catalysts at the initial stage, because its larger pore size would be favorable to the fast diffusion of long-chain FFAs molecules onto the surface acidic active sites. The reaction gradually reached to an equilibrium state with reaction time up to 4 h, and showed a lower FFAs conversion (89.3%). The relatively low catalytic activity of D072 should be attributed to its poor hydrophobicity. The surface acid active sites were easily combined with the byproduct water and further resulted in the deactivation of parts of the active sites.22,24
Compared with both of SO3H-APC-6-160 and D072, it could be seen (Fig. 4) that an obvious enhancement of the catalytic performance for the two sulfonated OMC catalyst samples. It was mainly because the effective adsorption of long chain fatty acid molecules could be facilitated in their regular mesoporous channels,39 and the acid active sites could be effectively inhibit from byproduct water, owing to the hydrophobicity of OMC surface.32,33,40
In addition, the SO3H-OMC(s) and SO3H-OMC(k) samples were prepared by hard template SBA-15 and KIT-6, respectively. It could be also seen from Table 3 and Fig. 4 that, although with a lower density of –SO3H groups, the FFAs conversion catalyzed by SO3H-OMC(k)-6-160 was higher than that catalyzed by SO3H-OMC(s)-6-160 at the initial stage. This phenomenon could be attributed to their difference in special mesoporous structure. The SO3H-OMC(k)-6-160 catalyst sample had 3D-cubic arrangement of branched rods,31 which could provide abundant diffusion channels and higher specific surface area for the adsorption of reactant molecules (as shown in Table 3), and thus its catalytic activity was higher at the initial stage of esterification. When the reaction time increased to 4 h, the catalytic activity of the SO3H-OMC(k)-6-160 sample would be exceeded by the SO3H-OMC(s)-6-160 sample gradually. It would be explained that the large organic molecules, such as free fatty acid methyl ester and FFAs, were inclined to stagger together and then block the intersection of the 3D channel, preventing the smoothly diffusion of the reactants and products. The enhancement in catalytic performance of the SO3H-OMC(s)-6-160 sample at this stage could be attributed to its higher density of –SO3H groups and the aligned mesoporous channels (2D-hexagonal), which could favor the diffusion of larger organic molecules, and were difficultly blocked.
These results could be attributed to the advantages of the SO3H-OMC(s) catalyst as follows: (a) Compared with the homogeneous catalysts, the SO3H-OMC(s) catalyst of lower corrosion was easy to reuse. (b) Compared with strongly acidic resins, the SO3H-OMC(s) catalyst with hydrophobic surface would effectively prevent the catalyst deactivation caused by the hydration of acid sites on catalyst surface with byproduct water. (c) Compared with amorphous porous carbon-based solid acid catalysts, the SO3H-OMC(s) catalyst with larger specific surface area and higher acid density had ordered and aligned 2D-hexagonal channels, which were favorable for adsorption and diffusion of long-chain molecules.
3.3 Investigation on catalyst deactivation
A series of esterification recycling tests were carried out to evaluate the stability of SO3H-OMC(s)-6-160 catalyst, and the results were shown in Table 4. The FFAs conversion was gradually decreased from 93.8% to 87.3% during the first three use cycles, and then remained almost constant (ca. 85%) for the next three reuse cycles. According to Fig. S1,† it could be seen that the density of –SO3H groups had a strong linear relationship with the FFAs conversion. And the deactivation of FFAs esterification might be due either to the loss of surface acidic activity sites or to the parts of channel blockage by large molecules during the reaction.| Reuse times of catalyst samples | Specific surface area (m2 g−1) | Pore volume (cm3 g−1) | Density of –SO3H groups (mmol g−1) | FFAs conv. (%) | ||||
|---|---|---|---|---|---|---|---|---|
| Stotal | Smicro | Smeso | Vtotal | Vmicro | Vmeso | |||
| Fresh | 537 | 116 | 421 | 0.85 | 0.09 | 0.76 | 2.32 | 93.8 |
| First reuse | 517 | 112 | 405 | 0.81 | 0.09 | 0.72 | 2.18 | 90.1 |
| Second reuse | 496 | 113 | 386 | 0.77 | 0.08 | 0.69 | 2.07 | 87.3 |
| Third reuse | 487 | 110 | 377 | 0.76 | 0.08 | 0.68 | 2.02 | 85.9 |
| Fourth reuse | 485 | 113 | 372 | 0.75 | 0.08 | 0.67 | 1.98 | 85.5 |
| Fifth reuse | 484 | 108 | 378 | 0.75 | 0.08 | 0.67 | 1.97 | 85.2 |
In order to explore the reasons for catalyst deactivation, the data on textural properties and the density of –SO3H groups attached on the surface of reused SO3H-OMC(s)-6-160 catalysts were measured and the results were listed in Table 4. After reused six times, it could be observed that, the microporous specific surface area changed slightly, and the mesoporous specific surface area of SO3H-OMC(s)-6-160 catalyst was decreased by 50 m2 g−1, probably due to the plugging of mesopores. Additionally, the trend of the density of –SO3H groups on surface was much in agreement with that of FFAs conversion in the catalytic performance, suggesting the loss of the surface –SO3H groups could result in the deactivation of catalysts.
Fig. 5 showed the catalytic esterification activity between reactants and solution obtained from washing catalysts, using the method of Scheme-1 and Scheme-2 described in 2.6 Section. As could be seen from Fig. 5, the esterification between methanol and FFAs could proceed self-catalysis (with FFAs conversion of 15.7%). It was obvious that the FFAs conversions of two schemes were much higher than that of spontaneous esterification reaction, which suggested that of the partial active –SO3H groups on the catalyst surface were leached out, and dissolved in the washing solvent during agitation and immersion. The FFAs conversion comparison of Scheme-1 and Scheme-2 to self-catalysis esterification reaction could reflect the catalytic activity of the –SO3H groups which were dissolved in methanol and FFAs as washing solvent, respectively. On the fifth usage, the catalytic activities of the two schemes were close the spontaneous esterification reaction, suggesting no additional losing of –SO3H groups from the surface of the SO3H-OMC(s)-6-160 catalyst sample.
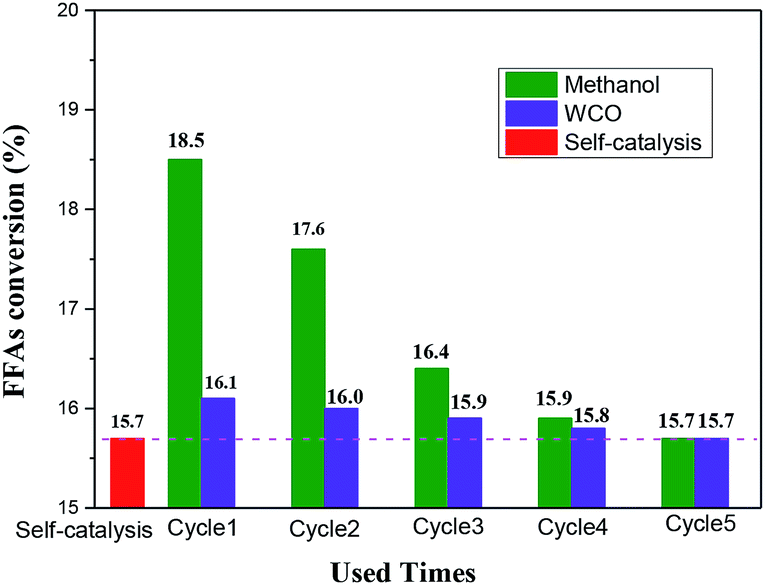 | ||
| Fig. 5 Catalytic esterification activity between reactants and solution obtained from washing catalyst. | ||
In this work, the catalytic activity of losing –SO3H groups could be the summation of catalytic activities of the –SO3H groups dissolved in above two different kinds of washing solvent. And the decline value of catalytic activity in Table 4 could be denoted by the difference between adjacent FFAs conversions (labeled as ΔX in Fig. 6).
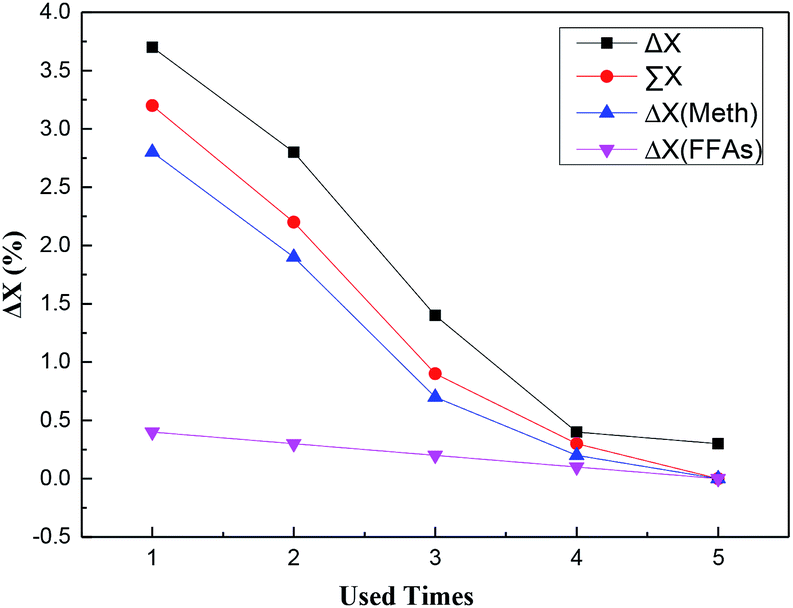 | ||
| Fig. 6 Deactivation analysis of SO3H-OMC(s)-6-160 catalyst sample ΔX (FFAs): the difference between adjacent FFAs conversion catalyzed by the –SO3H groups dissolved in FFAs as washing solvent; ΔX (Meth): the difference between adjacent FFAs conversion catalyzed by the –SO3H groups dissolved in methanol as washing solvent; ∑X: FFAs conversion catalyzed by the –SO3H groups dissolved in the two different kinds of washing solvent; ΔX: the difference between adjacent FFAs conversions in Table 4. | ||
As demonstrated in Fig. 6, with the usage times increasing, the catalytic activity of losing –SO3H groups decreased gradually until disappeared completely, which indicated the decreased amount of –SO3H groups leached out from the catalyst surfaces into the reaction solvent. Additionally, with the same usage time, the difference was very small when comparing the catalytic activity of the losing –SO3H groups with the decline value of the catalytic activity, and the latter was slightly larger than the former (shown in Fig. 6). Hence, it could be concluded that the loss of surface active sites (–SO3H groups) should mainly result in the catalyst deactivation in the esterification, affecting the activity and stability of catalysts.
Fig. 6 also showed that the catalytic activity of Scheme-1 was considerably higher than that of Scheme-2, accounting for a larger proportion in the catalytic activity of the losing –SO3H groups. The reason that aroused above-mentioned phenomenon could be partially attributed to the different usage amount of the solvents (molar ratio of methanol to FFAs was 9:1). More importantly, due to the stronger polarity of methanol than oils and fatty acids, the methanol as solvent could easily promote the formation of a local polar environment,19 resulting in the –SO3H groups being leached out of catalysts and the decrease of the catalytic activity.
4. Conclusions
In this work, sulfonated ordered mesoporous carbon (SO3H-OMC) solid acid catalysts with high density of –SO3H groups were obtained at the sulfonation time of 6 h and the sulfonation temperature of 160 °C. Compared to the sulfonated amorphous porous carbon-based solid acid catalyst and the cation-exchange resin D072, the SO3H-OMC catalysts exhibited relatively high activity for the FFAs esterification (FFAs conversion was 93.8%), which could be attributed to its high specific surface area, regular mesoporous channels and hydrophobic surface. The SO3H-OMC(s) catalyst with 2D-hexagonal channels exhibited a higher catalytic activity than the SO3H-OMC(k) catalyst with 3D-interconnected channels, attributed to its aligned pore structure which could favour large molecular diffusion and prevent channel blockage. Through series of deactivation experiments and analysis, it was found that, the –SO3H groups being leached out of SO3H-OMC catalysts into the liquids (especially into the reactant methanol with stronger polarity) would be the main reason causing catalyst deactivation during the FFAs esterification.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support from the Natural Science Foundation of China (No. 21104035).References
- F. R. Ma and M. A. Hanna, Bioresour. Technol., 1999, 70, 1–15 CrossRef CAS
.
- N. Yusuf, S. K. Kamarudin and Z. Yaakub, Energy Convers. Manage., 2011, 52, 2741–2751 CrossRef CAS
- P. S. Nigam and A. Singh, Prog. Energy Combust. Sci., 2011, 37, 52–68 CrossRef CAS
- M. J. Niu and X. J. Kong, RSC Adv., 2015, 5, 27273–27277 RSC
- G. P. Hammond, S. Kallu and M. C. McManus, Appl. Energy, 2008, 85, 506–515 CrossRef CAS
- S. K. M. Rozi, S. Bakhshaei, N. S. A. Manan and S. Mohamad, RSC Adv., 2016, 6, 87719–87729 RSC
- J. Yan, Appl. Energy, 2009, 86, S1–S10 CrossRef
- A. N. Phan and M. P. Tan, Fuel, 2008, 87, 3490–3496 CrossRef CAS
- M. A. M. Ali, R. M. Yunus, C. K. Cheng and J. Gimbun, RSC Adv., 2015, 5, 76743–76751 RSC
- M. Canakc
![[i with combining dot above]](https://www.rsc.org/images/entities/char_0069_0307.gif) , Bioresour. Technol., 2007, 98, 183–190 CrossRef PubMed
, Bioresour. Technol., 2007, 98, 183–190 CrossRef PubMed - S. Semwal and A. K. Arora, Bioresour. Technol., 2011, 102, 2151–2161 CrossRef CAS PubMed
- L. T. Wang, X. Q. Dong, H. X. Jiang, G. M. Li and M. H. Zhang, Fuel Process. Technol., 2014, 128, 10–16 CrossRef CAS
- L. T. Wang, X. Q. Dong, H. X. Jiang, G. M. Li and M. H. Zhang, Catal. Commun., 2014, 56, 164–167 CrossRef CAS
- A. A. Costa, P. R. S. Braga, J. L. de Macedo, J. A. Dias and S. C. L. Dias, Microporous Mesoporous Mater., 2012, 147, 142–148 CrossRef
- K. H. Chung and B. G. Park, J. Ind. Eng. Chem., 2009, 15, 388–392 CrossRef CAS
- S. Ajaikumar and A. Pandurangan, J. Mol. Catal. A: Chem., 2007, 266, 1–10 CrossRef CAS
- J. K. Satyarthi, D. Srinivas and P. Ratnasamy, Energy Fuels, 2010, 24, 2154–2161 CrossRef CAS
- J. A. Melero, L. F. Bautista, G. Morales, J. Iglesias and R. Sanchez-Vazquez, Chem. Eng. J., 2010, 161, 323–331 CrossRef CAS
- D. H. Zuo, J. Lane, D. Culy, M. Schultz, A. Pullar and M. Waxman, Appl. Catal., B, 2013, 129, 342–350 CrossRef CAS
- C. Pirez, J. M. Caderon, J. P. Dacquin, A. F. Lee and K. Wilson, ACS Catal., 2012, 2, 1607–1614 CrossRef CAS
- I. K. Mbaraka and B. H. Shanks, J. Catal., 2005, 229, 365–373 CrossRef CAS
- J. Y. Park, Z. M. Wang, D. K. Kim and J. S. Lee, Renewable Energy, 2010, 35, 614–618 CrossRef CAS
- R. Tesser, M. D. Serio, L. Casale, L. Sannino, M. Ledda and E. Santacesaria, Chem. Eng. J., 2010, 161, 212–222 CrossRef CAS
- L. T. Wang, X. Q. Dong, H. X. Jiang, G. M. Li and M. H. Zhang, Bioresour. Technol., 2014, 158, 392–395 CrossRef CAS PubMed
- T. Masakazu, T. Atsushi, O. Mai, J. N. Kondo, H. Shigenobu, D. Kazunari and H. Michikazu, Nature, 2005, 438, 178 CrossRef PubMed
- W. Y. Lou, M. H. Zong and Z. Q. Duan, Bioresour. Technol., 2008, 99, 8752–8758 CrossRef CAS PubMed
- J. A. Maciá-Agulló, M. Sevilla, M. A. Diez and A. B. Fuertes, Chemsuschem, 2010, 3, 1352–1354 CrossRef PubMed
- W. Xin and Y. H. Song, RSC Adv., 2015, 5, 83239–83285 RSC
- S. Jarczewski, M. Drozdek, P. Michorczyk, C. Cuadrado-Collados, J. Gandara-Loe, J. Silvestre-Albero and P. Kuśtrowski, Microporous Mesoporous Mater., 2018, 271, 262–272 CrossRef CAS
- V. Duraisamy, S. Palanivel, R. Thangamuthu and S. M. S. Kumar, ChemistrySelect, 2018, 3, 11864–11874 CrossRef CAS
- F. Kleitz, S. H. Choi and R. Ryoo, Chem. Commun., 2003, 9, 2136–2137 RSC
- L. Peng, A. Philippaerts, X. X. Ke, J. Van Noyen, F. De Clippel, G. Van Tendeloo, P. A. Jacobs and B. F. Sels, Catal. Today, 2010, 150, 140–146 CrossRef CAS
- X. Q. Wang, R. Liu, M. M. Waje, Z. W. Chen, Y. S. Yan, K. N. Bozhilov and P. Y. Feng, Chem. Mater., 2007, 19, 2395–2397 CrossRef CAS
- R. Liu, X. Q. Wang, X. Zhao and P. Y. Feng, Carbon, 2008, 46, 1664–1669 CrossRef CAS
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS PubMed
- C. Yu, J. Fan, B. Tian, D. Zhao and G. D. Stucky, Adv. Mater., 2010, 14, 1742–1745 CrossRef
- B. Chang, F. Jie, Y. Tian and X. Dong, Appl. Catal., A, 2012, 437–438, 149–154 CrossRef CAS
- S. Satoshi, N. Kiyotaka, K. Masaaki, Y. Daizo, K. Hideki, H. Shigenobu and H. Michikazu, J. Am. Chem. Soc., 2008, 130, 12787–12793 CrossRef PubMed
- X. Q. Dong, Y. Jiang, W. B. Shan and M. H. Zhang, RSC Adv., 2016, 6, 17118–17124 RSC
- F. J. Liu, K. Huang, A. M. Zheng, F. S. Xiao and S. Dai, ACS Catal., 2018, 8, 372–391 CrossRef CAS
- A. A. Kiss, F. Omota, A. C. Dimian and G. Rothenberg, Top. Catal., 2006, 40, 141–150 CrossRef CAS
- M. Di Serio, R. Tesser, M. Dimiccoli, F. Cammarota, M. Nastasi and E. Santacesaria, J. Mol. Catal. A: Chem., 2005, 239, 111–115 CrossRef CAS
- M. H. Zhang, A. X. Sun, Y. L. Meng, L. T. Wang, H. X. Jiang and G. M. Li, Catal. Surv. Asia, 2015, 19, 61–67 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02546d |
| This journal is © The Royal Society of Chemistry 2019 |