
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrocatalytic oxidation of water by the immobilized [CuII(L-ala)(Phen)(H2O)]+ complex on a self-assembled NCS− modified gold electrode†
Abhinandan Mahanta ,
Koushik Barman‡
and
Sk Jasimuddin*
,
Koushik Barman‡
and
Sk Jasimuddin*
Department of Chemistry, Assam University, Silchar, Assam-788011, India. E-mail: sk.jasimuddin@aus.ac.in
First published on 1st August 2019
Abstract
Copper(II) complex [CuII(L-ala)(Phen)(H2O)]+ (L-ala = L-phenylalanine, phen = phenanthroline) was immobilized over a self-assembled NCS− modified gold electrode for the electrocatalytic oxidation of water. This surface anchored molecular complex can catalyze water oxidation reaction at a remarkably low overpotential of 327 mV with a current density of 0.5 mA cm−2 at neutral pH.
Oxidation of water is the key step for natural or artificial photosynthesis processes. In green plants, oxidation of water occurs at the Mn4CaO5 active site of the oxygen-evolving enzyme of photosystem II.1 Nature converts water to dioxygen very easily, but by artificial means this is highly challenging due to its highly demanding thermodynamic and kinetic nature.2 To overcome these problems several water oxidation catalysts have been developed. Despite much progress on water-oxidation catalysts, major improvements are necessary in several areas like lowering of the overpotential, increasing catalyst stability, enhancing efficiency and reducing the overall cost. In recent years, various homogeneous catalysts and electrocatalysts for the water oxidation reaction based on earth abundant metals such as manganese,3,4 iron,5,6 cobalt,7,8 nickel,9,10 and copper11,12 have been reported. Among them copper is an attractive choice due to its high abundance, rich coordination chemistry13 and vital role in bio-mimetic oxygen chemistry.14 Numerous copper complexes have been used as homogeneous water oxidation electrocatalysts. Mayer and co-workers reported for the first time a homogeneous copper catalyst [(bpy)Cu(μ-OH)]2(OAc)2 that could electrochemically oxidize water in basic media at an overpotential 750 mV with a turnover frequency of 100 s−1.15 Several copper complexes of the polyamide ligand have been shown their catalytic water oxidation property.16,17 F. Chen et al. reported a water soluble copper complex [L–CuII–OAc]− (where OAc = acetate, L = N,N′-2,6-dimethylphenyl-2,6-pyridinedicarboxamidate) could electrocatalyze water oxidation to evolve O2 in 0.1 M carbonate buffer (pH 10) with an onset overpotential of 650 mV.18 P. G. Barros and co-workers reported a copper(II)-complex with redox non-innocent tetradentate amidate acyclic ligands that can electrocatalyze the water oxidation process at pH 11.5 with overpotential of 700 mV and as the electron-donating capacity at the aromatic ring increases, the overpotential notably reduced down to around 170 mV.19 Apart from these mononuclear copper catalysts which were operated in basic conditions, an oxidatively stable dinuclear copper-based catalyst, [Cu2(BPMAN)(μ-OH)]3+ (BPMAN = 2,7-[bis(2-pyridylmethyl)aminomethyl]-1,8-naphthyridine) has been reported, which efficiently catalyzed water oxidation at a neutral pH without decomposition during long-term electrolysis.20 On the other hand, several copper species such as CuII-Gly,21 Cu(II)-1,2-ethylenediamine22 and [CuII(TPA)H2O]2+ (ref. 23) have been used as precursors for active heterogeneous copper oxide formation through electrodeposition in alkaline media and catalyzed the water oxidation process. A. Prevedello et al. reported a copper(II) species with tetraaza macrocyclic ligand that can acts as heterogeneous (active copper oxide layer) and homogeneous (molecular species) electrocatalyst in alkaline (pH = 9–12) and neutral media, respectively.24 All of these known water oxidation catalysts have some practical problems, for example molecule based homogeneous catalysts are prone to decomposition under moderate applied potentials. To overcome these issues, some alternative strategies have been adopted such as grafting of molecular catalysts onto an electrode surface via covalent attachment or by anchoring on the functionalized self-assembled monolayer modified electrode. Placing the catalyst at an interface reduces the amount of catalyst needed and may enhance the rate. Few reports are available on surface anchored molecular complexes of Ru,25 Ir,26 V,27 Ce28 that could efficiently electrocatalyze the water oxidation reaction, are durable for a long time and have an overpotential that is quite low (250–350 mV) compared to the homogeneous system. Still earth abundant metal containing molecular complex modified electrode systems are scarce.
In the present article we have fabricated a gold electrode using copper(II)-complex, [Cu(L-phe)(Phen)(H2O)](ClO4) and NH4SCN through two step self-assembly process (Scheme 1) and characterized by spectral, electrochemical and microscopic process. The modified electrode is stable and efficiently oxidizes water to oxygen in neutral pH medium.
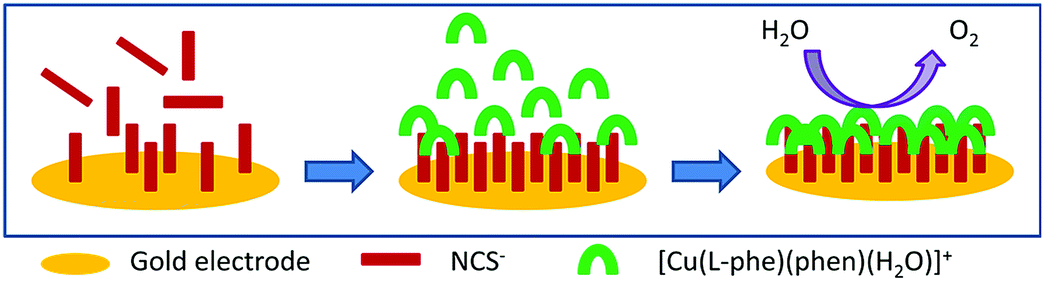 | ||
| Scheme 1 Electrode modification using self-assembly process. | ||
The stepwise modification of the gold electrode was monitored by using cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) (Fig. 1a and b). CV of 0.5 mM [Fe(CN)6]3−/4− (redox probe) in 0.1 M PBS (pH 7.0) at bare gold electrode shows a quasi-reversible couple (ΔE = 80 mV) with an anodic current density of 2.7 mA cm−2. After modification with NCS− on gold electrode surface, an irreversible redox couple for [Fe(CN)6]3−/4− was obtained with significant decrease (around 2.5 mA cm−2) of anodic current density. This result supports the formation of self-assembled layer of NCS− which retards the electron transfer process between electrode and the probe molecule. After immobilization of [Cu(L-phe)(phen)(H2O)]+ complex on NCS− modified gold electrode, the anodic current density was around 0.9 mA cm−2 indicates the electronic communication between Au electrode and [Fe(CN)6]3−/4− through CuII-complex and in-turn confirms the proper modification. EIS study clearly shows that the charge transfer resistance (Rct) of CuII-complex–SCN–Au electrode is less than SCN–Au electrode and support the CV results.
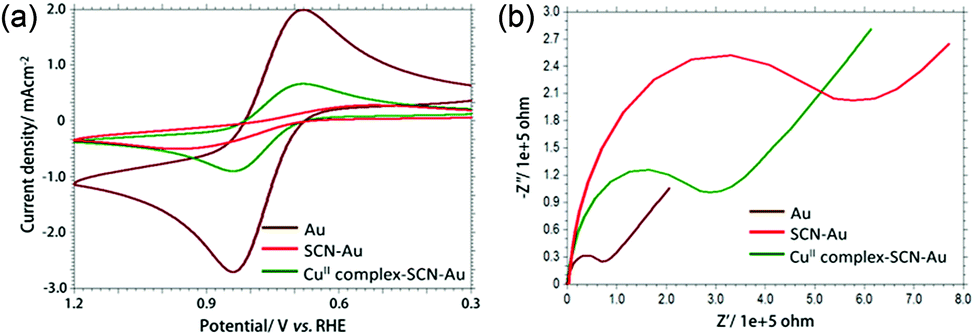 | ||
| Fig. 1 Overlaid cyclic voltammogram (a) and Nyquist plot (−Z′′ versus Z′) obtained from EIS experiment (b) of 0.5 mM K4[Fe(CN)6] in 0.1 M PBS (pH 7.0) at bare Au (brown curve), NCS-Au (red curve) and [Cu(L-phe)(phen)(H2O)]–SCN–Au (green curve). | ||
In order to confirm the fabrication of CuII-complex on SCN–Au modified electrode a comparative CV was taken for bare Au and [Cu(L-phe)(phen)(H2O)]–SCN–Au in 0.1 M PBS (Fig. S1†). An anodic peak at +0.84 V versus RHE was obtained and is due to the CuII/III oxidation couple29–31 and proves the presence of CuII-complex over SCN–Au electrode.
With varying concentration of CuII-complex from 1.0 mM to 5.0 mM both the anodic and cathodic current were increased (Fig. S2a†). A plot of anodic current versus concentration of CuII-complex gives a linear regression equation Ipa (μA) = 0.221C (mM) + 1.511 (R2 = 0.998) and is shown in Fig. S2b.† This observation also certifies a successful immobilization of CuII-complex on self-assembled NCS− modified gold electrode. From the Fig. S3,† it can be seen that the current density increases linearly with the increasing square root of scan rate, following the linear regression equation, J (mA cm−2) = 0.0063 √ν (mV s−1) + 0.0247 (R2 = 0.996), thereby, indicating a diffusion controlled electron transfer process at [Cu(L-phe)(phen)(H2O)]–SCN–Au modified electrode.32
The modification process was also confirmed by using FE-SEM and EDX analysis. FE-SEM image (Fig. S4a and b†) shows the surface morphology of bare and copper complex modified gold electrode. Bare gold shows a smooth surface morphology whereas the CuII-complex–SCN–Au electrode shows nearly smooth surface and supports the film formation.
The EDX analysis (Fig. S4c†) and elemental mapping images (Fig. S5†) confirms the presence of Cu, N, O and S elements and supports the proper immobilization of CuII-complex on self-assembled NCS− modified gold electrode.
For further confirmation, FTIR Spectra of SCN–Au and CuII-complex–SCN–Au electrodes were recorded in the frequency range 450–4000 cm−1 (Fig. S6†). A sharp and broad peak at 2059 cm−1, a weak peak at 795 cm−1 and a peak at 480 cm−1 are assigned as the ν(CN), ν(CS) and δ(NCS), respectively and confirms that NCS− adsorbed on Au electrode surface and coordinated through N atom.33 After immobilization of Cu(II) complex over SCN–Au electrode surface the characteristic peaks for CN, CS and NCS are shifted to 2136 cm−1, 781 cm−1 and 476 cm−1, respectively and supports the bond formation between Au-NCS and Cu(II)-complex. Coordination of sulphur with copper of Cu(II)-complex is confirmed by the presence of new bands at around 575 cm−1 which is also assignable to Cu–N bond stretching for the Cu(II)-complex.34 Another indication for (Cu-S) bond is the presence of weak band at 456 cm−1.35 IR bands at around 3053 cm−1, 1653 cm−1 and 1595 cm−1 are due to of CH2, COO− and NH2 group, respectively36 and supports the presence of L-ala in the Cu(II)-complex. The peaks for O–H, N–H and C–H are observed at 3434, 2937 and 3343 cm−1, respectively. From the IR data it can be conclude that the gold electrode was properly modified with NCS− and [Cu(L-phe)(phen)(H2O)]+.
The electrocatalytic activity of the bare and modified gold electrodes for water oxidation were investigated using linear sweep voltammetry (LSV) in 0.1 M PBS at pH 7.0 in the potential window +0.6 to +2.0 V versus RHE (Fig. 2a). Oxidation of water was observed at higher potential ∼ +1.83 V versus RHE with current density 0.02 mA cm−2 and 0.01 mA cm−2 at bare and NCS− modified gold electrode, respectively. The anodic peak potential is shifted towards less positive potential +1.58 V versus RHE and at the same time current height is increased to 0.54 mA cm−2 when CuII-complex–SCN–Au electrode was used as working electrode. These results establish the electrocatalytic activity of CuII-complex–SCN–Au modified electrode towards the oxidation of water.37 The oxidation of water by the CuII-complex–SCN modified gold electrode shows remarkably low overpotential of around 327 mV at J = 0.5 mA cm−2 and onset overpotential of around 120 mV (J = 0.1 mA cm−2) in neutral PBS and the obtained result is comparable or in some cases quite better than the reported homogeneous Cu(II)-complex based systems or heterogeneous copper oxide films, copper foil etc. (Table S1†).
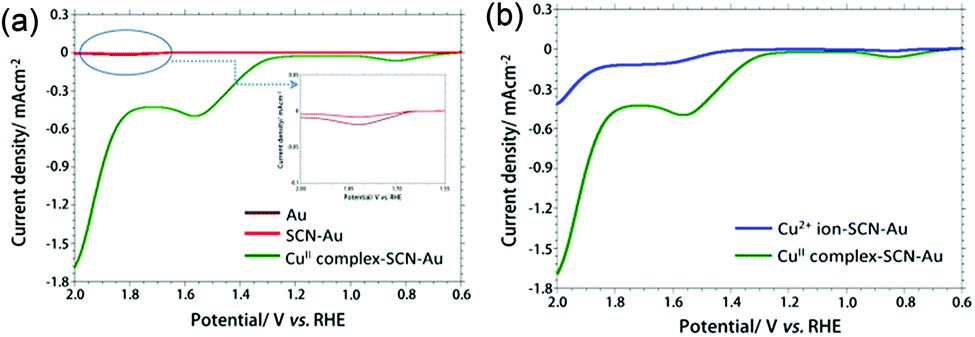 | ||
| Fig. 2 Overlaid LSV obtained at bare Au, SCN–Au and CuII-complex-NCS-Au electrode in 0.1 M PBS solution (pH 7.0) (a), overlaid LSV obtained at Cu2+ ion–SCN–Au electrode and CuII-complex–SCN–Au electrode in 0.1 M PBS solution (pH 7.0) (b). | ||
The electrocatalytic activity towards water oxidation at CuII-complex and Cu2+ ion immobilized SCN–Au electrodes was studied and shown in Fig. 2b. The LSV result shows that the anodic peak current is quite high (∼0.32 mA cm−2) and peak potential is less positive (∼0.15 V) in case of CuII-complex–SCN–Au than Cu2+–SCN–Au electrode suggest that the electrocatalytic activity towards water oxidation is higher in case of CuII-complex than Cu2+-ion modified electrode.
To confirm that the anodic peak at +1.58 V is solely due to the oxidation of water, LSV was performed using [Cu(L-phe)(phen)(H2O)]–SCN–Au electrode in the potential range of +0.6 to +2.0 V versus RHE in ultrapure CH3CN containing 0.1 M tetrabutylammonium perchlorate [Bu4N][ClO4] (pH = 7.0). No anodic peak was observed (Fig. S7a†), but upon addition of water a distinguished oxidative peak was appeared at +1.58 V versus RHE which proves that the water oxidation reaction taking place at the Cu(II)-complex modified electrode surface. It was also observed that with increasing water concentration (0.1–0.5 M) the anodic peak current was increased linearly (Fig. S7b†) which also confirms that the peak appeared at +1.58 V versus RHE is only due to the oxidation of water.38
Linear sweep voltammetry was carried out at low scan rate 5 mV s−1 in the applied potential range 260 mV to 280 mV in 0.1 M PBS at pH 7.0. The plot of logJ versus η (overpotential) produces a Tafel slope of 49 mV dec−1 (Fig. S8†) which indicates an excellent catalytic activity of the CuII-complex-NCS-Au electrode towards the oxidation of water.39
Fig. 3a shows the LSV at different scan rate ranging from 20–100 mV using [Cu(L-phe)(phen)(H2O)]–SCN–Au modified electrode in PBS solution (pH 7.0). A plot of normalized catalytic current (i/ν1/2) versus scan rates (ν) (Fig. 3b) gives an inverse relationship. This result indicates that a rate-limiting chemical step taking place prior to quick electron transfer to the electrode.40 It also confirms that the chemical rate determining step of the catalytic process is likely to be the O–O bond formation step.41
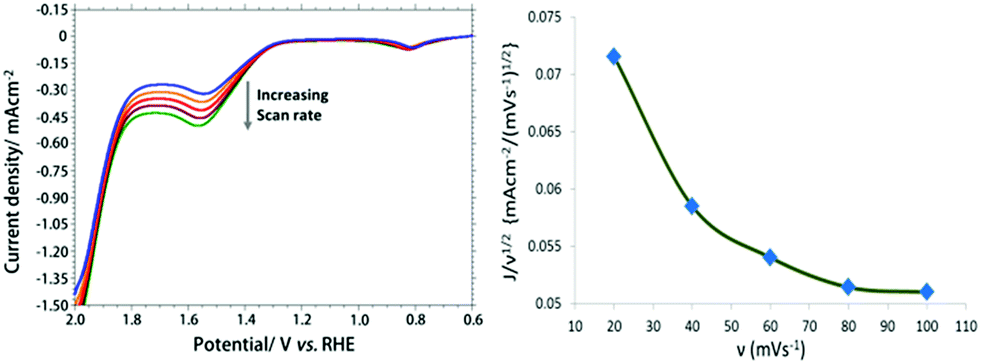 | ||
| Fig. 3 Overlaid LSV at CuII-complex–SCN–Au electrode in 0.1 M PBS (pH 7.0) with increasing scan rate in the range of 20–100 mV s−1 (a). A plot of normalized catalytic current versus scan rates (b). | ||
Fig. S9† illustrates the LSVs at CuII-complex–SCN–Au electrode with varying pH (6.0, 6.5, 7.0, 7.5, 8.0) of phosphate buffer solution. The anodic peak potentials were shifted towards less positive potential with increasing pH of the medium. The oxidation peak potential varies linearly with pH of the medium (Fig. S9b†) and follows the linear regression equation Epa (V) = −0.123pH + 2.502 (R2 = 0.997).
The slope of 0.123 V per pH shift indicating that 1e−/2H+ couple is involved in CuII-complex electrocatalyzed water oxidation reaction.42 The influence of pH on the peak current density (J) of water oxidation at the modified gold electrode (Fig. S10†) revealed that the J values were increased linearly up to 7.0 and then slowly decreased and thereafter decreased sharply. This observation indicates that pH 7.0 is the most effective pH for the oxidation of water by the CuII-complex modified electrode.
A plausible mechanism for the water oxidation over CuII-complex modified gold electrode is given in Scheme S1.† In the proposed mechanism, the catalytically active [CuIII(H2O)]+ complex on the gold electrode surface is formed after anodic oxidation of [CuII(H2O)] at +0.84 V versus RHE. Once formed, [CuIII(H2O)]+ oxidized H2O to O2 at +1.55 V versus RHE in neutral pH.41–43
The stability and oxygen generation capability of the CuII-complex–SCN–Au electrode was investigated using controlled potential electrolysis (CPE) at +1.58 V versus RHE using in 0.1 M PBS (pH 7.0) (Fig. 4a). The CPE experiment shows that the current density (J) rapidly declines to around 0.02 and 0.38 mA cm−2 within 20 seconds for the bare and the CuII-complex–SCN modified gold electrode, respectively and thereafter the current remains stable over the entire period of electrolysis.44 The obtained result agrees the high stability of the modified Au electrode during electrolysis. The stability of the complex modified electrode was also checked by using chronopotentiometry experiment for 60 minutes at a fixed current density of 0.51 mA cm−2 (Fig. S11†). A stable potential was obtained during two hour long electrolysis. This result also supports the high stability of CuII-complex–SCN modified electrode.
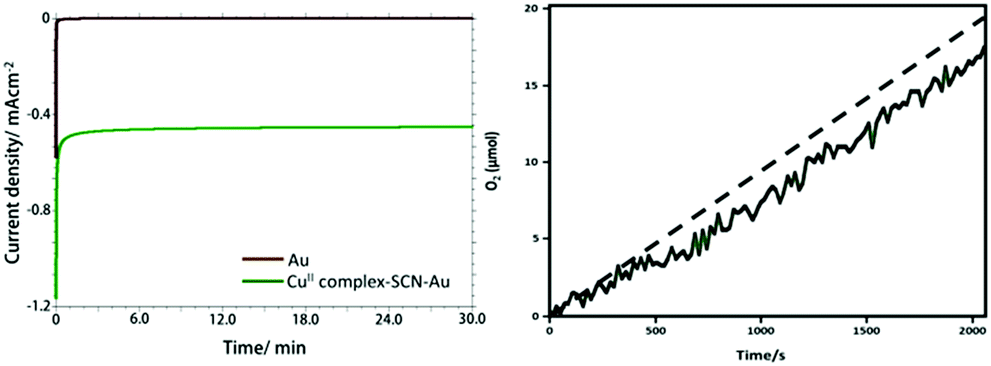 | ||
| Fig. 4 Bulk electrolysis with (green curve) and without (brown curve) the CuII-complex catalyst over the gold electrode in 0.1 M PBS at 1.58 V versus RHE (a). O2 evolution during controlled potential electrolysis of water at CuII-complex–SCN–Au electrode. Dotted line denotes the theoretical O2 evolution with 100% efficiency (b). | ||
During controlled potential electrolysis the generated oxygen in the head space of the gas tight electrochemical cell was monitored using a fluorescent probe. The concentration of the evolved oxygen was increased nearly linear fashion (Fig. 4b). It gives around 17 μmol of oxygen within 30 minutes of electrolysis with a Faraday efficiency of 96%. The theoretical yield of generated oxygen was calculated by assuming that the obtained current is due to four electron oxidation of water.45
The long term stability of the Cu(II)-complex modified electrode was explored by LSV measurement in 0.1 M PBS at 15 days intervals (Fig. S12†) and almost similar LSV responses were obtained after 15th and 30th day (relative standard deviation was 0.03%). Thus, it can be concluded that the electrocatalytic activity of the CuII-complex-NCS modified electrode does not suffer from dissolution and remained active.37 For further confirmation of stability of the catalyst, long term bulk electrolysis experiment was performed at +1.58 V for around eight hours (Fig. S13†) and a constant catalytic current was obtained over the entire period of electrolysis. This observation also establishes the robustness of the system. To check the surface morphology and durability of the modified electrode, FE-SEM and EDX analysis was done after the bulk electrolysis experiment (Fig. S14†). No considerable change of surface morphology was observed by comparing with the SEM image of the electrode surface before electrolysis (Fig. S4b†) and the presence of different elements such as Cu, N, O and S in the EDX spectrum (Fig. S14b†) support the stability of the catalyst.
In summary, [Cu(L-phe)(phen)(H2O)]–SCN–Au electrode was developed, characterized and applied for the electrocatalytic oxidation of water in neutral pH. The newly developed electrode was able to oxidize water at an impressively low overpotential 327 mV with a current density of 0.54 mA cm−2. Tafel slope of 49 mV dec−1 indicates excellent catalytic activity of the CuII-complex towards water oxidation reaction. During 30 minutes of electrolysis, 17 μmol of oxygen was produced with a Faraday efficiency of 96%. The electrode material was chip and the modified electrode was highly stable, reusable and may help for the development of commercial water oxidation catalysts.
Conflicts of interest
There are no conflicts to declare.References
- Y. Umena, K. Kawakami, J. R. Shen and N. Kamiya, Nature, 2011, 473, 5 CrossRef PubMed.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724 CrossRef CAS.
- C. W. Cady, R. H. Crabtree and G. W. Brudvig, Coord. Chem. Rev., 2008, 252, 444 CrossRef CAS PubMed; E. A. Karlsson, B. L. Lee, T. Akermark, E. V. Johnston, M. D. Karkas, J. Sun, O. Hansson, J. E. Backvall and B. Akermark, Angew. Chem., Int. Ed., 2011, 50, 11715 (Angew. Chem., 2011, 123, 11919) CrossRef PubMed.
- S. Kal, L. A. Mensah and P. H. Dinolfo, Inorg. Chim. Acta, 2014, 423, 201 CrossRef CAS.
- M. K. Coggins, M. T. Zhang, A. K. Vannucci, C. J. Dares and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 5531 CrossRef CAS PubMed.
- M. Okamura, M. Kondo, R. Kuga, Y. Kurashige, T. Yanai, S. Hayami, V. K. Praneeth, M. Yoshida, K. Yoneda, S. Kawata and S. Masaoka, Nature, 2016, 530, 465 CrossRef CAS PubMed.
- H. A. Younus, N. Ahmad, A. H. Chughtai, M. Vandichel, M. Busch, K. V. Hecke, M. Yusubov, S. Song and F. Verpoort, ChemSusChem, 2017, 10, 862 CrossRef CAS PubMed.
- H. Chen, Z. Sun, X. Liu, A. Han and P. Du, J. Phys. Chem. C, 2015, 119, 8998 CrossRef CAS.
- Y. Han, Y. Wu, W. Lai and R. Cao, Inorg. Chem., 2015, 54, 5604 CrossRef CAS PubMed.
- M. Zhang, M. T. Zhang, C. Hou, Z. F. Ke and T. B. Lu, Angew. Chem., Int. Ed., 2014, 53, 13042 (Angew. Chem., 2014, 126, 13258) CrossRef CAS PubMed.
- K. J. Fisher, K. L. Materna, B. Q. Mercado, R. H. Crabtree and G. W. Brudvig, ACS Catal., 2017, 7, 3384 CrossRef CAS; C. Lu, J. Wang and Z. Chen, ChemCatChem, 2016, 8, 2165 CrossRef.
- K. J. Fisher, K. L. Materna, B. Q. Mercado, R. H. Crabtree and G. W. Brudvig, ACS Catal., 2017, 7, 3384 CrossRef CAS.
- K. D. Karlin and J. Zubieta, Copper coordination chemistry: Biochemical & Inorganic perspectives, Adenine Press, New York, 1983 Search PubMed.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. K. Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659 CrossRef CAS PubMed; R. Van Eldik, Advances in Inorganic Chemistry, Academic Press, 2006, vol. 56 Search PubMed.
- S. M. Barnett, K. I. Goldberg and J. M. Mayer, Nat. Chem., 2012, 4, 498 CrossRef CAS PubMed.
- M.-T. Zhang, Z. F. Chen, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2013, 135, 2048 CrossRef CAS PubMed.
- P. G. Barros, I. F. Ardoiz, S. Drouet, J. B. Buchholz, F. Maseras and A. Llobet, J. Am. Chem. Soc., 2015, 137, 6758 CrossRef PubMed.
- F. Chen, N. Wang, H. Lei, D. Guo, H. Liu, Z. Zhang, W. Zhang, W. Lai and R. Cao, Inorg. Chem., 2017, 56, 13368 CrossRef CAS PubMed.
- P. G. Barros, I. F. Ardoiz, S. Drouet, J. B. Buchholz, F. Maseras and A. Llobet, J. Am. Chem. Soc., 2015, 137, 6758 CrossRef PubMed.
- X. J. Su, M. Gao, L. Jiao, R. Z. Liao, P. E. M. Siegbahn, J. P. Cheng and M. T. Zhang, Angew. Chem., Int. Ed., 2015, 54, 4909 CrossRef CAS PubMed.
- C. Lu, J. Wang and Z. Chen, ChemCatChem, 2016, 8, 2165 CrossRef CAS.
- C. Lu, J. Du, X.-J. Su, M.-T. Zhang, X. Xu, T. J. Meyer and Z. Chen, ACS Catal., 2016, 6, 77 CrossRef CAS.
- X. Liu, H. Jia, Z. Sun, H. Chen, P. Xu and P. Du, Electrochem. Commun., 2014, 46, 1 CrossRef CAS.
- A. Prevedello, I. Bazzan, N. D. Carbonare, A. Giuliani, S. Bhardwaj, C. Africh, C. Cepek, R. Argazzi, M. Bonchio, S. Caramori and M. Robert, Chem.–Asian J., 2016, 11, 1281 CrossRef CAS PubMed.
- Z. Chen, J. J. Concepcion, J. W. Jurss and T. J. Meyer, J. Am. Chem. Soc., 2009, 131, 15580 CrossRef CAS PubMed.
- K. E. deKrafft, C. Wang, Z. Xie, X. Su, B. J. Hinds and W. Lin, ACS Appl. Mater. Interfaces, 2012, 4, 608 CrossRef CAS PubMed.
- K. Barman and S. Jasimuddin, Catal. Sci. Technol., 2015, 5, 5100 RSC.
- S. Garain, K. Barman, T. K. Sinha, S. Jasiumddin, J. Haeberle, K. Henkel, D. Schmeisser and D. Mandal, ACS Appl. Mater. Interfaces, 2016, 8, 21294 CrossRef CAS PubMed.
- M.-T. Zhang, Z. Chen, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2013, 135, 2048 CrossRef CAS PubMed.
- J. S. Pap, L. Szyrwiel, D. Srankó, Z. Kerner, S. Bartosz, S. Zbigniew and M. W. law, Chem. Commun., 2015, 51, 6322 RSC.
- J. S. Pap and Ł. Szyrwiel, Comments Inorg. Chem., 2017, 37, 59 CrossRef CAS.
- A. J. Bard and F. R. Faulkner, Electrochemical Methods, Fundamentals and Applications, Wiley, New York, 2nd edn, 2001 Search PubMed.
- M. Kabesova and J. Gazo, Chem. Zvesti, 1980, 34, 800 CAS; K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, John Wiley & Sons, 4th edn, 1986 Search PubMed.
- R. E. Cramer, D. M. Ho, W. V. Doorne, J. A. Ibers, T. Norton and M. Kashiwagi, Inorg. Chem., 1981, 20, 2457 CrossRef CAS; E. B. Seena and M. R. P. Kurup, Polyhedron, 2007, 26, 829 CrossRef.
- M. Bron and R. Holze, J. Electroanal. Chem., 1995, 385, 105 CrossRef.
- M. E. Mohamed and A. M. A. Mohammed, Int. Lett. Chem., Phys. Astron., 2013, 10, 1 Search PubMed.
- K. Barman and S. Jasimuddin, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2013, 52, 217 Search PubMed.
- M. Zhang, M. de Respinis and H. Frei, Nat. Chem., 2014, 6, 362 CrossRef CAS PubMed.
- Y. Gao, H. Chen, L. Ye, Z. Lu, Y. Yao, Y. Wei and X. Chen, Chin. J. Catal., 2018, 39, 479 CrossRef CAS.
- P. Zanello, in Inorganic Electrochemistry: Theory, Practice, and Application, The Royal Society of Chemistry, Cambridge, U.K., 2003 Search PubMed.
- F. Chen, N. Wang, H. Lei, D. Guo, H. Liu, Z. Zhang, W. Zhang, W. Lai and R. Cao, Inorg. Chem., 2017, 56, 13368 CrossRef CAS PubMed.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2007, 107, 5004 CrossRef PubMed.
- D. Lukács, Ł. Szyrwiel and J. S. Pap, Catalysts, 2019, 9, 83 CrossRef CAS; J. S. Pap, Ł. Szyrwiel, D. Srankó, Z. Kerner, B. Setner, Z. Szewczuk and W. Malinka, Chem. Commun., 2015, 51, 6322 RSC.
- K. Barman and S. Jasimuddin, Catal. Sci. Technol., 2015, 5, 5100 RSC.
- C. Lu, J. Wang and Z. Chen, ChemCatChem, 2016, 8, 2165 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, synthesis and characterization of Cu(II) complex, electrode modification, figures, probable mechanism, table and references etc. See DOI: 10.1039/c9ra02547b |
| ‡ At present in the Department of Chemistry, The University of Utah, Salt Lake City, Utah 84112-0850, USA |
| This journal is © The Royal Society of Chemistry 2019 |