
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFixing CO2 into β-oxopropylcarbamates in neat condition by ionic gelation/Ag(I) supported on dendritic fibrous nanosilica†
Liping Chang*a,
Rahele Zhianib and
Seyed Mohsen Sadeghzadeh *b
*b
aSchool of Continuing Education of Xinxiang University, Xinxiang, Henan 453000, P. R. China. E-mail: xinxiangxueyuan@aliyun.com
bNew Materials Technology and Processing Research Center, Department of Chemistry, Islamic Azad University, Neyshabur Branch, Neyshabur, Iran. E-mail: seyedmohsen.sadeghzadeh@gmail.com
First published on 30th May 2019
Abstract
In this research, a novel approach to produce a novel nanocatalyst, AgBr supported on an ionic gelation (IG)-based nanomaterial, was developed through the aqueous coprecipitation approach for the dendritic fibrous nanosilica (DFNS) production and the wet impregnation method for IG coating, taking into account TPP as the cross-linking agent. In addition, DFNS/IG@Ag(I) had heterogeneous features that contributed to the quick improvement of the catalyst by filtration separation. The catalytic activity of DFNS/IG@Ag(I) was examined to synthesize β-oxopropylcarbamates through a multicomponent coupling of CO2, amines and propargyl alcohols in moderate conditions. The DFNS/IG@Ag(I) NPs were completely investigated by taking advantage of TG, TEM, FESEM and FT-IR spectroscopy analyses. The DFNS/IG@Ag(I) nanocatalyst indicated high stability in the reaction for five cycles without considerable loss of some properties like activity, which could be because of the high loading of IG on the catalyst surface.
Introduction
Algae exist in abundance in most kinds of aquatic environments. Among the different kinds of algae, spirulina is a type of blue-green, photosynthetic, multicellular, filamentous, as well as spiral-shaped microalgae. Spirulina filaments, easily separated from their medium, has 70% great quality protein, a good flavor, and great digestibility. Spirulina contains reactive functional groups like amino and hydroxyl, which are toxic, sensitive to pH, and biocompatible, and can be added to metals. Ionic gelation is one of the important approaches utilized to produce chitosan nanoparticles.1,2 The interpolation of NPs within a network comprising of amino and hydroxyl groups by utilizing a polyanionic cross-linker like tripolyphosphate or TPP3–7 facilitates the interplay of the NPs with the cationic spirulina through electrostatic interactions; at the same time, spirulina figures strong interactions with NPs due to its good chelating capability with metal ions. The advantage of a physical cross-linking factor or TPP over a chemical cross-linker (like glutaraldehyde) is the prohibition of the toxicity of reagents.In recent years, many studies have focused on the chemical fixation of CO2 due to its attributes such as low toxicity, cost efficiency, non-flammability, ubiquity and reproducibility, which greatly facilitate the making of C–N and C–C bonds in organic production. These properties of CO2 may be ascribed to its kinetic inertness and great stability in terms of thermodynamic properties. Recently, a significant approach has been obtained in this subject, which is the use of CO2 to produce different organic compounds such as amides, esters, alcohols, carbonates and carboxylic acids as well as carbamates. This has been favored by different efficient catalytic approaches. Considerable progress has been made, and several approaches have been developed in the case of the reaction of CO2 to produce products such as cyclic carbonates and urethanes as well as salicylic acid. One of these composites, namely, carbamates, is an important and diverse composite. Thus, utilizing carbon dioxide to develop an eco-friendly approach that efficiently yields carbamates is attractive.8–39 Among them, carbamates like β-oxopropylcarbamates have been vastly utilized in agronomy, organic production, and pharmaceutics. Most recently, β-oxopropylcarbamates were produced from CO2, secondary amines and propargylic alcohols. In addition, different catalysts such as ruthenium,40,41 Fe,42 Cu,43,44 Ag,45–50 and bicyclic guanidine51 have been explored in the multicomponent reaction. Cu and Ag have been illustrated to be the most impressive catalysts for this reaction. Silver compounds have been shown to be great transition metal catalysts with particular selectivity and they have considerable benefits over other catalysts.
Multicomponent reactions (MCRs) are one-pot reactions employing more than two starting materials, for example, 3, 4, …, 7 reactants, where most of the atoms of the starting materials are incorporated in the final product.52 Several descriptive tags are regularly attached to MCRs: they are atom economic, that is, the majority, if not all, of the atoms of the starting materials are incorporated in the product; they are efficient, that is, they efficiently yield the product since the product is formed in one-step instead of multiple sequential steps; they are convergent, that is, several starting materials combine in one reaction to form the product; they exhibit a very high bond-forming-index (BFI), that is, several non-hydrogen atom bonds are formed in one synthetic transformation.53 Therefore, MCRs are often a useful alternative to sequential multistep synthesis.
Many basic MCRs are name reactions, for example, Ugi,54 Passerini,55 van Leusen,56 Strecker,57 Hantzsch,58 and Biginelli,59 or one of their many variations. For example, in the Ugi reaction, the primary scaffold is mostly dictated by the type of the acid component (and to a less degree by the amine component), for example, carboxylic acid, carbonic acid, thiocarboxylic acids,60 HN3, H2O, H2S, HNCO, HNCS, and phenol, which is one of the few recent innovations regarding primary scaffold diversity in Ugi reactions,61 leading to α-acylaminocarboxamides, carbamates, α-acylaminothiocarbonamides, tetrazoles, α-aminoamides, α-aminothioamides, hydantoins, thiohydantoins and α-aminoarylamides, respectively.62 Additionally, since MCRs are often highly compatible with a range of unprotected orthogonal functional groups, on a second level, the scaffold diversity of MCR can be greatly enhanced by the introduction of orthogonal functional groups into the primary MCR product and allowing them to react in subsequent transformations, e.g. ring forming reaction. This two layered strategy has been extremely fruitful in the past, leading to a great manifold of scaffolds that are now routinely used in combinatorial and medicinal chemistry for drug discovery purposes.63
Recently, silica nanoparticles with a fibrous morphology have been widely studied by scholars after the invention of the dendritic fibrous nanosilica (DFNS). The dendritic fibrous nanosilica with a shrinkage morphology has been tested in solution to enhance the surface of the nanocatalyst compared to the surface of the usual cubic pore or hexagonal structured ordinary silica. In addition, dendritic fibrous nanosilica has the proper potential to be a support substance in drug delivery, isomerization and other applications. Moreover, varying the mesoporosity properties is one of the approaches utilized to solve the diffusion limitation difficulty; most catalysts, which are utilized to simplify isomerisation, have this issue.64,65 The dendritic fibrous nanosilica has indicated great potential in various utilizations such as Suzuki coupling,66 propane metathesis,67,68 propane metathesis for CO2 adsorbent,69 cumene hydrocracking, as well as alkane hydrogenolysis.70 Great surface area, high stability in the case of thermal behavior and high pore sizes of the dendritic fibrous nanosilica facilitate the high availability of a bulky mass of reactants at the energetic sites, which accordingly enhances the reaction rate and product formation. Moreover, some big pores on the external surfaces and small pores all over the structure are also beneficial as they act as carriers for genes and drugs. Herein, we have reported an ionic gelation related (DFNS/IG@Ag(I) NPs) catalytic apparatus, on which AgBr was supported, for the one-pot production of β-oxopropylcarbamates via the three-component reaction of propargylic alcohols, amines and CO2 at moderate conditions. Exclusively, this catalytic apparatus could be quickly recycled 5 times with the lowermost catalyst loading ever reported. These great catalytic performances are ascribed to the usage of the green ionic gelation that might act as a dual activator to both the substrates and CO2 (can be observed Scheme 1).
 | ||
| Scheme 1 Synthesis of β-oxopropylcarbamates in the presence of DFNS/IG@Ag(I) NPs. | ||
Experimental section
Materials and methods
High purity chemicals were procured from Fluka and Merck. An Electrothermal 9100 apparatus was utilized for the determination of uncorrected melting points in open capillaries. A VERTEC 70 spectrometer (Bruker) in transmission mode was used for the determination of FTIR spectra. Samples were pulverized and pelletized with spectroscopic grade KBr. The determination of the size and structure of the nanoparticles was done via a transmission electron microscope (TEM) (Phillips CM10) operated at 100 kV. The crystallographic structures of the nanoparticles were determined using powder X-ray diffraction (Bruker D8 Advance model) with Cu Kα radiation. Thermal gravimetric analysis (TGA) (NETZSCH STA449F3) was performed under a nitrogen atmosphere with a heating rate of 10 °C min−1. 1H and 13C NMR spectra were determined with a BRUKER DRX-300 AVANCE spectrometer and a BRUKER DRX-400 AVANCE spectrometer. The NMR spectra recorded for both elements were at 300.13 and 75.46 MHz, and 400.22 and 100.63 MHz, respectively. A Heraeus CHN-O-Rapid analyzer was used to perform elemental analyses for carbon, hydrogen and nitrogen. Thin layer chromatography (TLC) was conducted on silica gel polygram SILG/UV 254 plates for the determination of product purity and monitoring the reaction. A Shimadzu GCMS-QP5050 Mass Spectrometer was used to record the mass spectra.General procedure for the preparation of DFNS
2.5 g of tetraethyl orthosilicate was dissolved in a solution of 30 mL of cyclohexane and 1.5 mL of 1-pentanol. A stirred solution of 1 g of cetylpyridinium bromide and 0.6 g of urea in 30 mL of water was then added. The resulting mixture was continually stirred for 45 min at room temperature and then placed in a Teflon-sealed hydrothermal reactor and heated to 120 °C for 5 h. The silica formed was isolated by centrifugation, then washed with a mixture of deionized water and acetone, and dried in a drying oven. This material was then calcined at 500 °C for 6 h in air.General procedure for the preparation of DFNS/IG NPs
Dendritic fibrous nanosilica (50 mg) was diffused in an acetic acid solution using ultrasonication for 60 seconds. Spirulina (125 mg) was dissolved in a similar acetic acid solution and stirred at the temperature of 25 °C for 1 h. TPP solution (10.0 mL) was injected dropwise into the chitosan solution by utilizing a syringe pump at the volumetric flow rate of 1 mL min−1. After the complete addition of TPP, the mixture was stirred mechanically at 1000 rpm speed for 1 h. The formed product was gathered by filtration and then washed by using water at pH = 7, and dried in vacuum at the temperature of 25 °C for 24 h.General procedure for the catalytic synthesis of β-oxopropylcarbamates
AgBr (0.05 mmol), DFNS/IG NPs (10.0 mg), propargylic alcohols (5.0 mmol) and secondary amines (5 mmol) were mixed in a Schlenk tube equipped with a stir bar. After that, the apparatus was cleaned with carbon dioxide for more than two times, and the blend was stirred at the temperature of 50 °C and pressure of 15 bar with carbon dioxide for the desired time. When the reaction was complete, the mixture was partitioned by diethyl ether (3–15 mL). The upper layers were gathered and dried in vacuum to obtain the crude yield, which was purified by column chromatography using silica gel and petroleum ether/ethyl acetate (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1–20:1). For the recycling procedure, the catalyst was separated using filtration, washed with alcohol and dried with a pump.
:1–20:1). For the recycling procedure, the catalyst was separated using filtration, washed with alcohol and dried with a pump.
Results and discussion
The dendritic fibrous nanosilica was produced using a simple approach and then functionalized using the ionic gelation (IG) of TPP and spirulina. The produced DFNS/IG NPs were then characterized using various methods like TG analysis, TEM, BET, FESEM, and FT-IR spectroscopy. The morphology as well as the structure of the dendritic fibrous nanosilica and DFNS/IG NPs were well marked using TEM and FESEM analysis. The SEM image of the dendritic fibrous nanosilica nanoparticles indicates dandelion-like structures with diameters of around 300 nm and a wrinkled radial structure (as can be seen in Fig. 1a). In addition, this fiber (with a thickness of around 8.5 nm) originated from the center of the considered spheres and extended in all radial dimensions (can be seen in Fig. 1b). Moreover, the excessive extension of the wrinkled radial structures resulted in cone-shaped open pores. The images obtained from FESEM and TEM illustrate that the whole sphere was completely solid and consisted of fibers. In addition, this structure of open hierarchical channels and fibers were convenient for mass transfer in the case of reactants and enhanced the availability of active sites. The FESEM and TEM images of DFNS/IG NPs indicated that after correction, the morphology of the dendritic fibrous nanosilica did not vary (can be observed in Fig. 1c and d). TGA of DFNS/IG NPs was also performed at temperatures ranging from 25 °C to 800 °C for investigating the thermal stability, as shown in Fig. S1.† The weight loss at 239 °C was because of the removal of solvents existing on the surface of the dendritic fibrous nanosilica. The obtained weight loss for DFNS/IG NPs was 38 percent by weight. These results indicated the great grafting ability of the organic materials on dendritic fibrous nanosilica. | ||
| Fig. 1 (a) FESEM images of DFNS NPs; (b) TEM images of DFNS NPs; (c) FESEM images of DFNS/IG NPs; (d) TEM images of DFNS/IG NPs. | ||
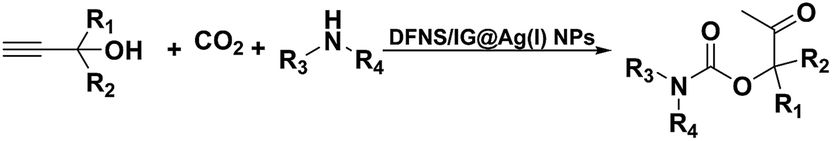
The analysis of nitrogen physisorption indicated that the BET specific surface area of the dendritic fibrous nanosilica and DFNS/IG NPs was around 678 and 209 m2 g−1, respectively. The reduction in the surface area of DFNS/IG in comparison with the dendritic fibrous nanosilica could be because of the supporting IG on the dendritic fibrous nanosilica. In addition, the decrease in the surface area in the case of DFNS/IG NPs was clearly due to the poor bicontinuous concentric IG morphology of the nanocatalyst. As can be seen in Fig. 2, the nitrogen adsorption–desorption isotherms of the dendritic fibrous nanosilica based catalysts indicated a type IV isotherm with an H1-type hysteresis loop, proposing the presence of mesopores. The corresponding pore sizes predicted from the desorption series of the nitrogen isotherm using the BJH procedure displayed a narrow pore size distribution that peaked at about 9 nm (as can be seen Table 1). The excellent mesopore size of dendritic fibrous nanosilica with great capacity may aid the loading of TPP and spirulina, which have comparatively high molecular sizes.
 | ||
| Fig. 2 Adsorption–desorption isotherms of the (a) DFNS NPs and (b) DFNS/IG NPs; (c) BJH pore size distributions of the DFNS NPs and (d) DFNS/IG NPs. | ||
| Catalysts | SBET (m2 g−1) | Va (cm3 g−1) | DBJH (nm) |
|---|---|---|---|
| DFNS | 679 | 3.3 | 9 |
| DFNS/IG | 210 | 1.4 | 6 |
FT-IR spectroscopy was used to specify the surface enhancement of the obtained catalyst (as seen in Fig. S2†). The FT-IR spectrum indicated peaks in the frequency range of about 3400–3300 cm−1 corresponding to the oxygen–hydrogen stretching vibration. This demonstrated the presence of carbohydrates and amino acids. The peaks in the frequency range of 3500–3300 cm−1 for the nitrogen–hydrogen stretching vibrations confirmed the presence of amines in proteins as well as lipids. The bands at 2972 and 2896 cm−1 were considered to belong to the carbon–hydrogen stretching of the aliphatic moieties. The peak at 1720 cm−1 was because of the stretching of the carbonyl group in the esters and amino acids. The conforming peak at 1642 cm−1 corresponding to the nitrogen–hydrogen bending vibration indicated the presence of the carbonyl β-unsaturated ketone amide. The peak existing at 1411 cm−1 confirmed the presence of methylene bending vibration. The bands at 1342 cm−1 and 1278 cm−1 indicated carbon–oxygen stretching and oxygen–hydrogen bending vibrations of the oxygen of alcohol, respectively. On functionalization with tripolyphosphate, the intensity of the peak at 1048 cm−1 enhanced and a new peak appeared at 882 cm−1, which was related to the phosphate and phosphoramide groups, and this proposed a reaction occurring between –P3O105− and –NH3+. The main variation in the IR spectra was the presence of another peak in the TPP bead spectrum at 1150 cm−1, probably due to the –P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration, determining the presence of phosphate groups.
O stretching vibration, determining the presence of phosphate groups.
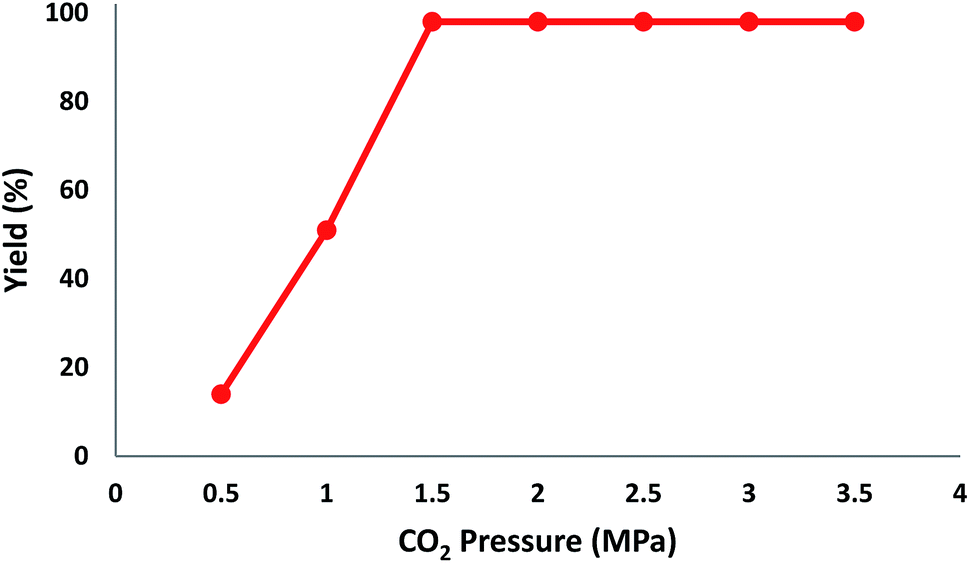
The reaction of CO2, 2-methylbut-3-yn-2-ol, and pyrrolidine was chosen as the base reaction to test the optimal catalytic system. Data shown in Table 2 indicates that the reaction could not be performed without catalysts at mild conditions. Moreover, no product was obtained in the presence of AgBr or DFNS/IG NPs alone (as seen in Table 2, entries 2–3). On this foundation, different Ag salts such as AgI, AgBr, and AgBF4, which could be used together with DFNS/IG NPs for the coupling reaction to occur and obtain moderate to great product yields, were successively examined (Table 2, entries 4–13). In comparison, AgBr exhibited the best catalytic efficiency. 2 mol% of AgBr in the presence of DFNS/IG NPs could efficiently catalyze the considered reaction (Table 2, entry 5).
| Entry | Catalytic system | Yieldb (%) | |
|---|---|---|---|
| a Reaction conditions: [Ag] (0.05 mmol), DFNS/IG NPs (5 mg), 2-methylbut-3-yn-2-ol (5 mmol), pyrrolidine (5 mmol), CO2 (1.5 MPa), 50 °C, 5 h.b GC yields [%]. | |||
| 1 | — | — | — |
| 2 | AgBr | — | — |
| 3 | — | DFNS/IG | — |
| 4 | AgCl | DFNS/IG | 89 |
| 5 | AgBr | DFNS/IG | 98 |
| 6 | AgI | DFNS/IG | 53 |
| 7 | AgOAC | DFNS/IG | 29 |
| 8 | Ag2O | DFNS/IG | 46 |
| 9 | Ag2CO3 | DFNS/IG | 54 |
| 10 | AgNO3 | DFNS/IG | 61 |
| 11 | AgBF4 | DFNS/IG | 28 |
| 12 | Ag2WO4 | DFNS/IG | 19 |
| 13 | AgPF6 | DFNS/IG | 53 |
In a related research to improve the reaction situation, it was determined that solvent-free situations were rather impressive than those utilizing solvents, considering the reaction time as well as the yield of products (Table 3). The vital role of temperature in the preparation of β-oxopropylcarbamate was examined. It was obvious that the reaction procedure was sensitive to the reaction temperature. For minimizing the reaction activation energy, the reactions were performed at 50 °C by taking advantage of the great efficiency of DFNS/IG NPs. Temperatures higher than 50 °C did not lead to changes in the reaction efficiency. In addition, Table 3 shows the influence of time on the production of β-oxopropylcarbamate. After 5 hours, the yield of β-oxopropylcarbamate product increased up to 98%, while further extension in time did not lead to increase in the product yield. Therefore, the appropriate time for the production of β-oxopropylcarbamate was 5 hours. Another influential parameter, which might be considered for this reaction, is the amount of DFNS/IG NPs for efficient catalysis to synthesise β-oxopropylcarbamate. The average efficiency of product formation was found by the addition of 3.0 mg of the catalyst to the reaction. The best yield was obtained when the reaction was performed in the presence of 5 mg of DFNS/IG NPs. It is significant that the reaction was not enhanced on using higher amounts of DFNS/IG NPs.
| Entry | Solvent | Temperature (°C) | Catalyst (mg) | Time (h) | Yielda (%) |
|---|---|---|---|---|---|
| a GC yields [%]. | |||||
| 1 | DMF | 70 | 9 | 6 | 33 |
| 2 | DMSO | 70 | 9 | 6 | 51 |
| 3 | Dioxane | 70 | 9 | 6 | 62 |
| 4 | CH3CN | 70 | 9 | 6 | 31 |
| 5 | CH2Cl2 | 70 | 9 | 6 | 52 |
| 6 | EtOAc | 70 | 9 | 6 | 65 |
| 7 | THF | 70 | 9 | 6 | 39 |
| 8 | Toluene | 70 | 9 | 6 | 54 |
| 9 | CHCl3 | 70 | 9 | 6 | 77 |
| 10 | n-Hexane | 70 | 9 | 6 | 22 |
| 11 | Benzene | 70 | 9 | 6 | 16 |
| 12 | CCl4 | 70 | 9 | 6 | 23 |
| 13 | Cyclohexane | 70 | 9 | 6 | 15 |
| 14 | H2O | 70 | 9 | 6 | 15 |
| 15 | EtOH | 70 | 9 | 6 | 39 |
| 16 | i-PrOH | 70 | 9 | 6 | 31 |
| 17 | MeOH | 70 | 9 | 6 | 20 |
| 18 | Solvent-free | 70 | 9 | 6 | 98 |
| 19 | Solvent-free | 70 | 7 | 6 | 98 |
| 20 | Solvent-free | 70 | 5 | 6 | 98 |
| 21 | Solvent-free | 70 | 3 | 6 | 73 |
| 22 | Solvent-free | 70 | 1 | 6 | 61 |
| 23 | Solvent-free | 70 | 7 | 5 | 98 |
| 24 | Solvent-free | 70 | 7 | 4 | 68 |
| 25 | Solvent-free | 70 | 7 | 3 | 41 |
| 26 | Solvent-free | 60 | 7 | 6 | 98 |
| 27 | Solvent-free | 50 | 7 | 6 | 98 |
| 28 | Solvent-free | 40 | 7 | 6 | 81 |
| 29 | Solvent-free | 30 | 7 | 6 | 76 |
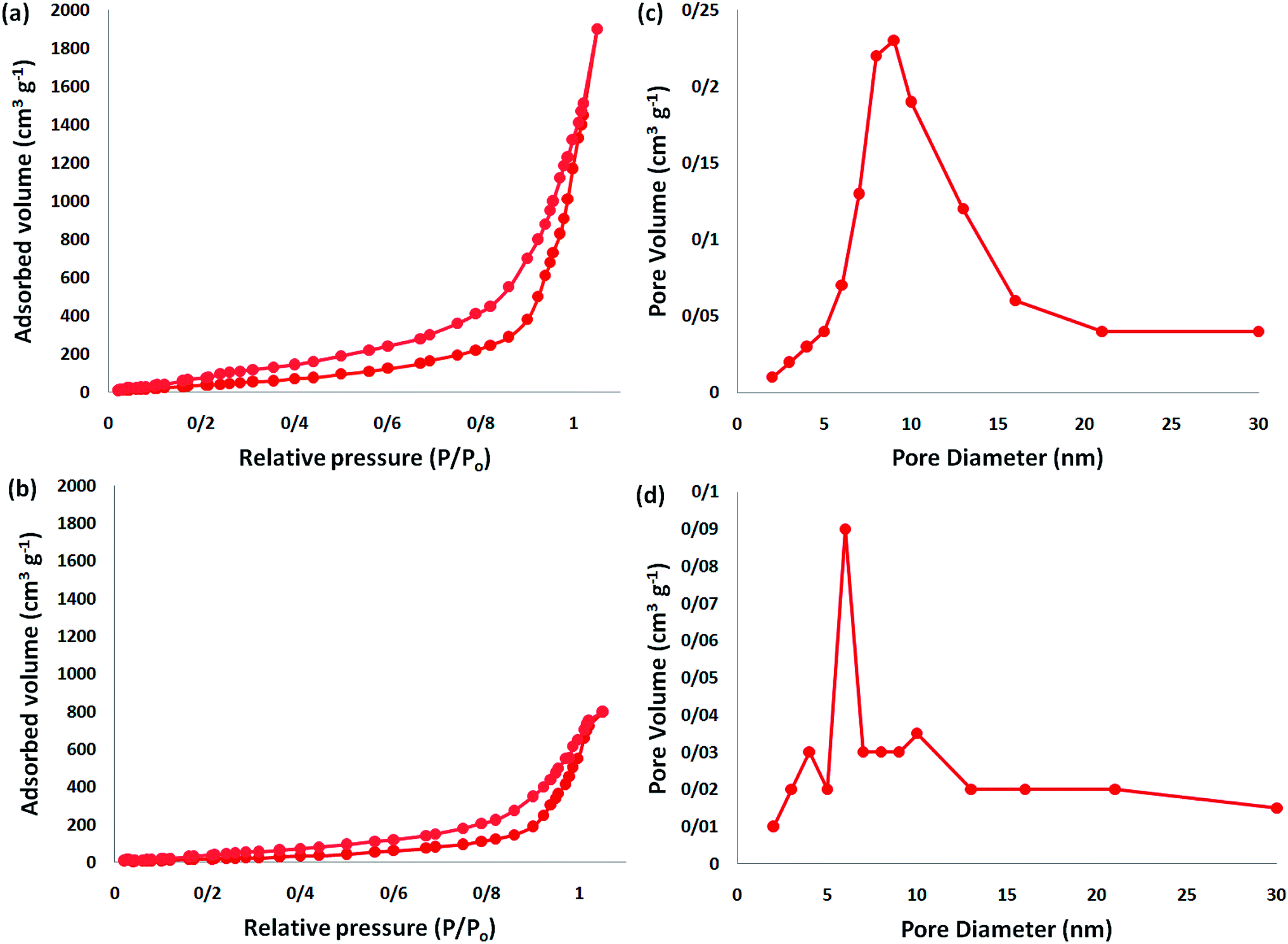
In addition, to enhance the production of β-oxopropylcarbamate, carbon dioxide pressure was studied. The optimal pressure of CO2 yielding the best production of DFNS/IG NPs would be specific, whereas the kinetics of the mass transfer reactions might change due to diffusion and reaction between CO2, 2-methylbut-3-yn-2-ol, and pyrrolidine. The influence of pressure was experimentally determined in the range from 0.5 to 3.5 MPa. The operation of DFNS/IG NPs enhanced strongly when the carbon dioxide pressure increased from 0.5 to 1.5 MPa, after which it sustainable in the pressure range of 1.5–3.5 MPa as shown in Fig. 3. These contrary facts determined the requirement for an optimum pressure of around 1.5 bar for the best production of β-oxopropylcarbamate.
 | ||
| Fig. 3 The effect of CO2 pressure on the synthesis of β-oxopropylcarbamate. | ||
Since the conditions of the reaction were improved, we then tested the substrate range by examining the reactivity of aryl or alkyl substituents with different secondary amines. The reaction of pyrrolidine with various terminal propargylic alcohols, directing the alkyl substituents in the propargylic position, occurred at the same condition and produced the corresponding carbamates with great yields (as observed in Table 4, entries 1–4). Commonly, tertiary alcohols were more active compared to primary alcohols and secondary alcohols, and simultaneously, indicated distinct reactivity with various substituent groups. The propargylic carbonate intermediate was quickly produced via the reaction of tertiary alcohols and carbon dioxide by the support of proper catalysts. Propargylic alcohols with cycloalkyl substitutions had lower reactivity (as observed in Table 4, entries 5 and 6), and the reason would be the influence of steric hindrance on the carbonate intermediate formation. In addition, other propargylic alcohols with unsaturated groups such as phenyl substitution showed a good yield of products after the addition of a certain amount of co-catalyst. Moreover, the secondary amines presented good reactivity. However, for the entries 15, slightly lower reactivity was found possibly due to the steric hindrance of methyl or cyclohexyl in amines during the nucleophilic attack to α-methylene carbonate.
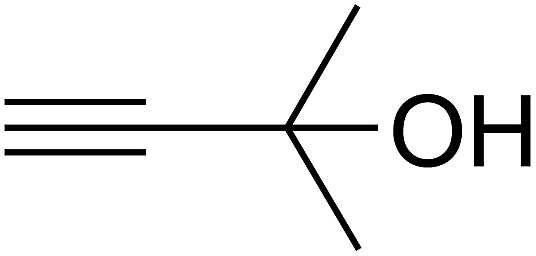
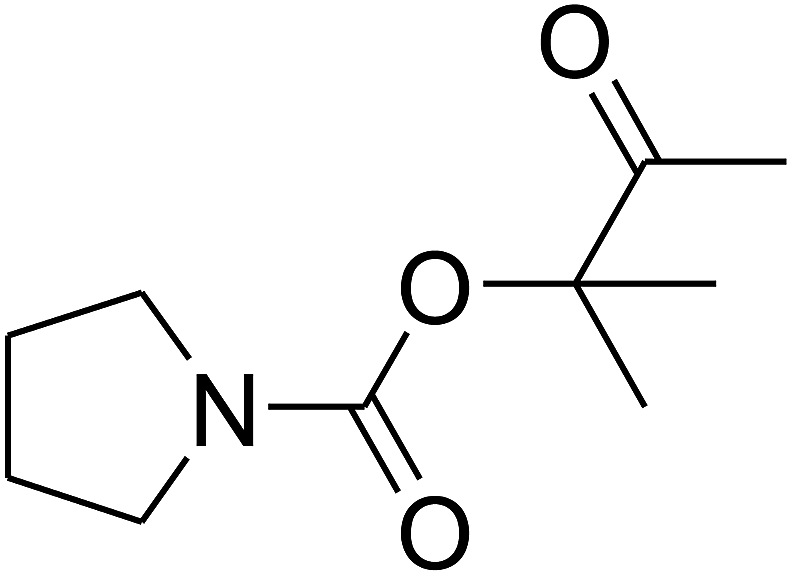
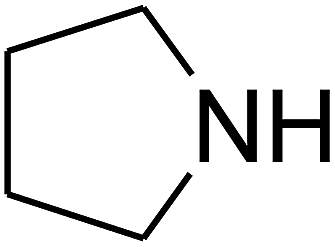
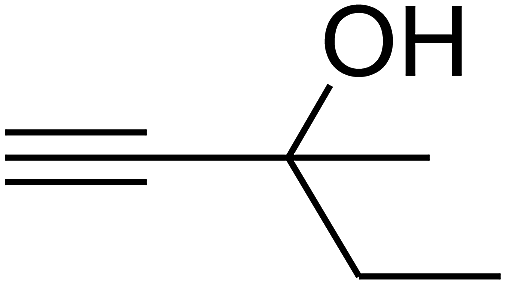
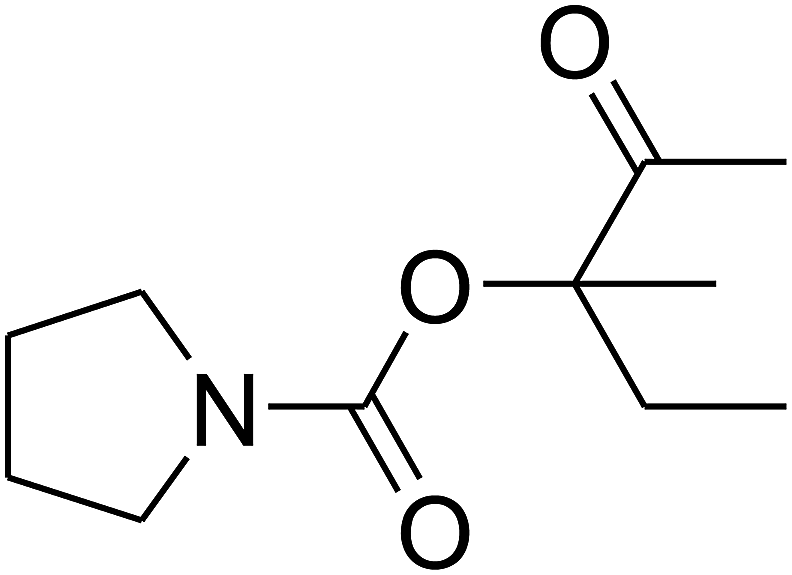
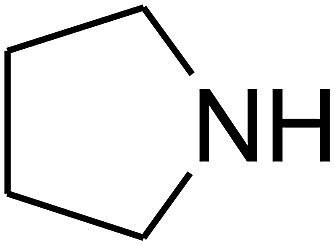
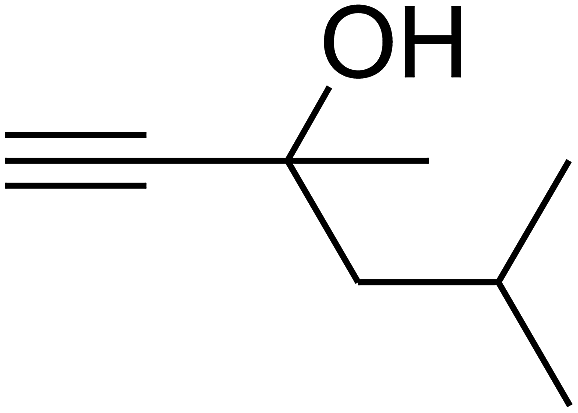
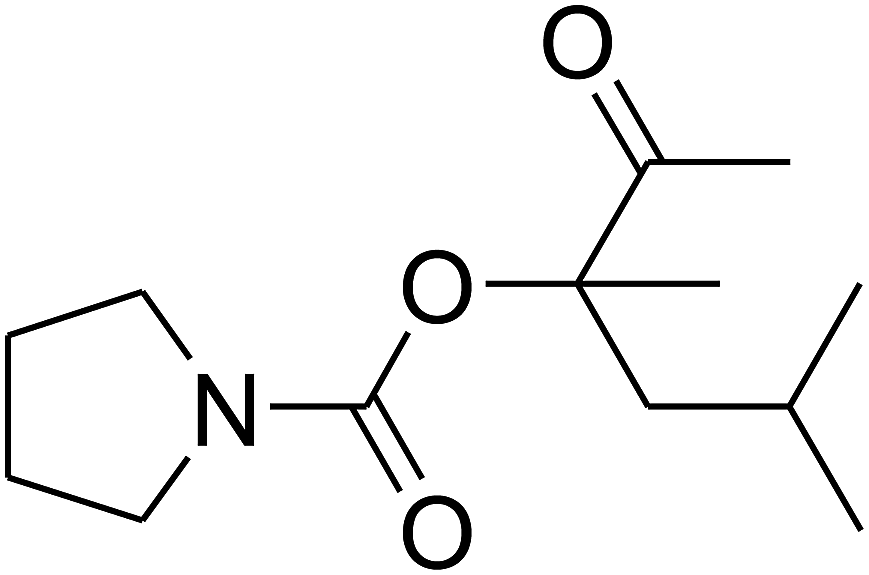
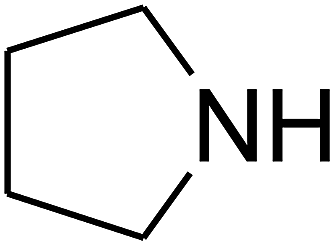
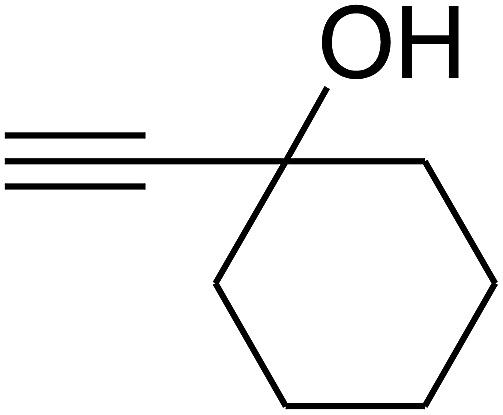
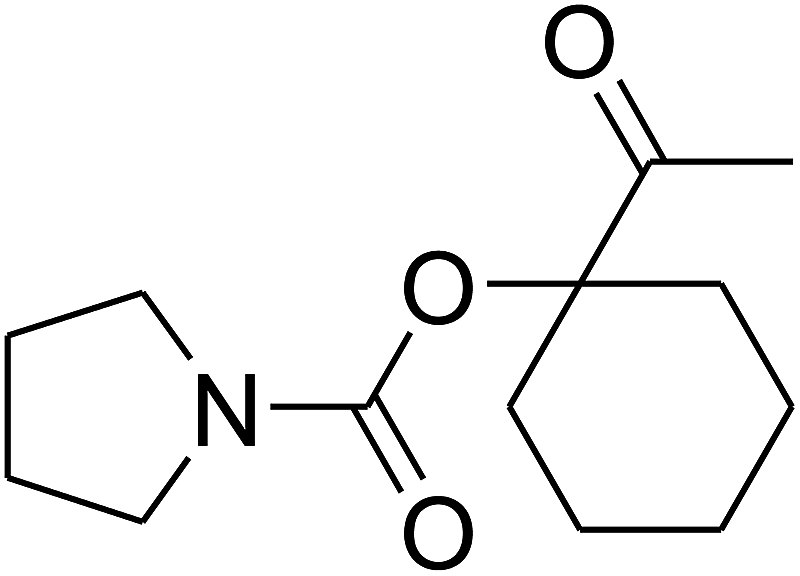
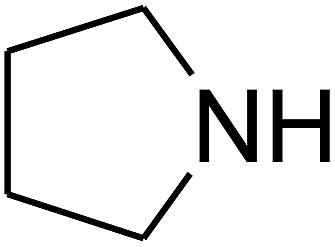
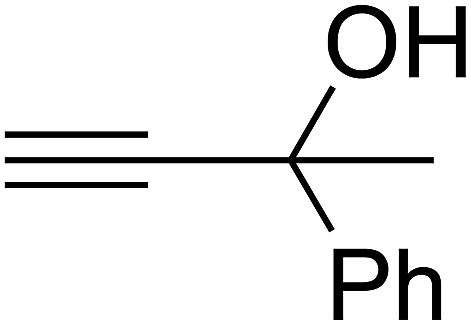
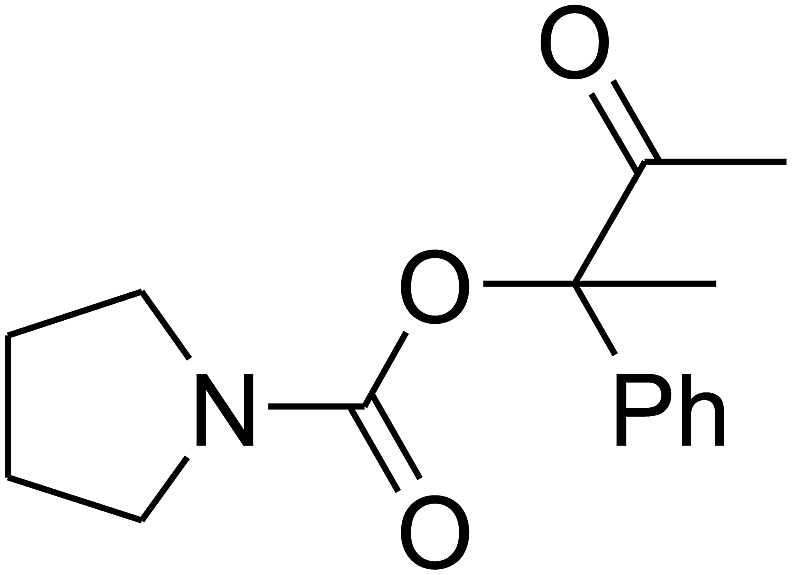
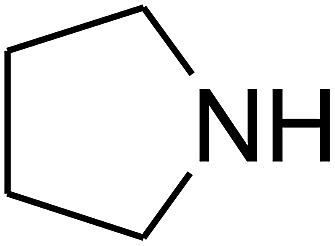
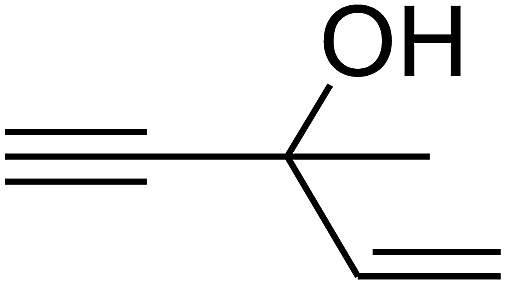
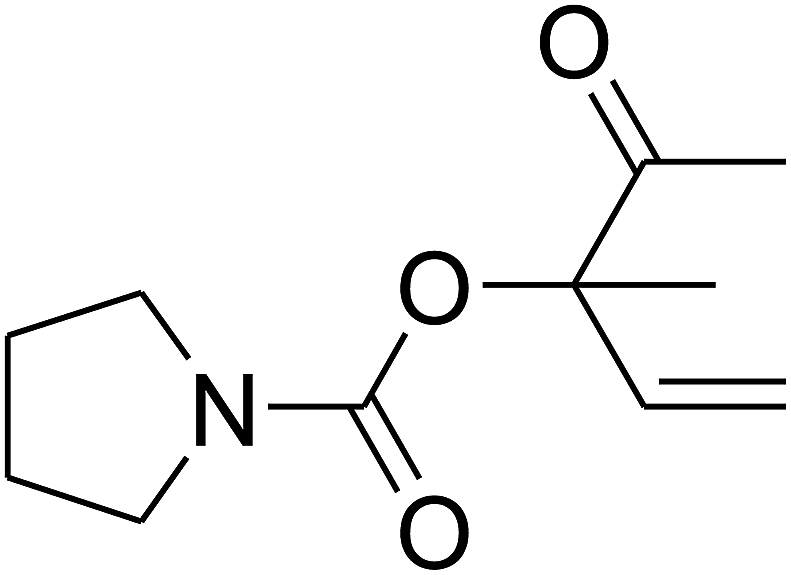
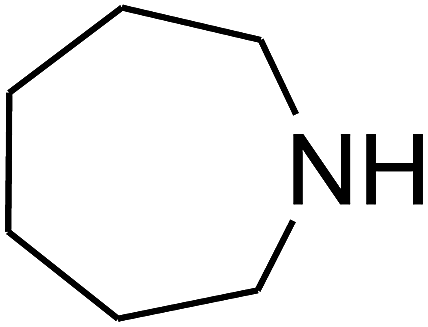
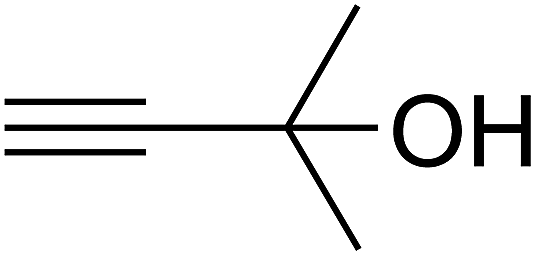
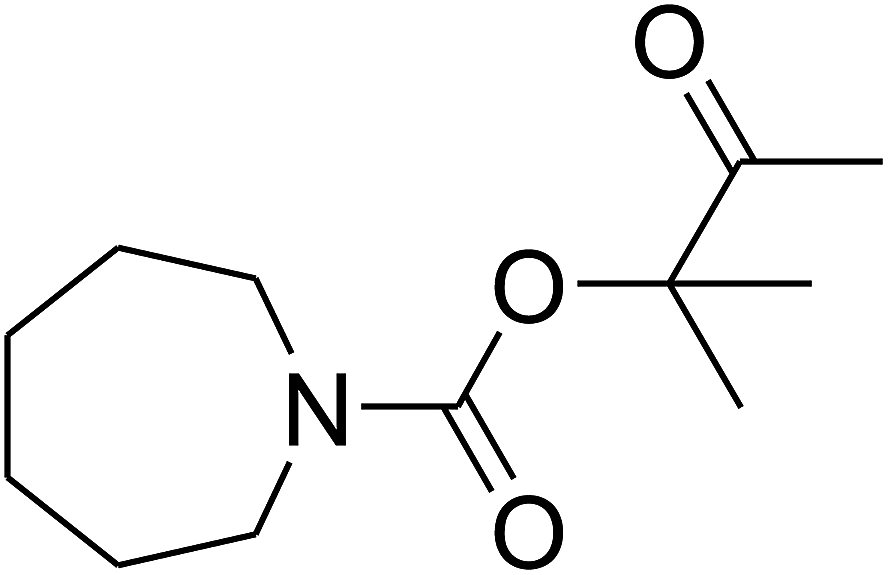
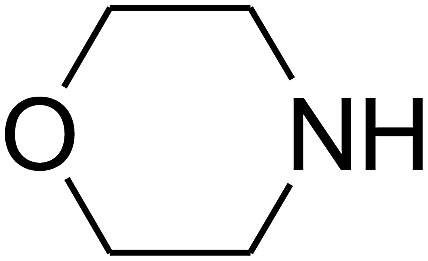
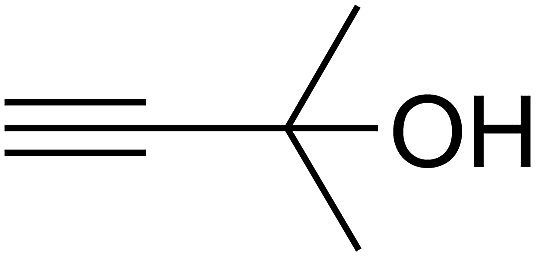
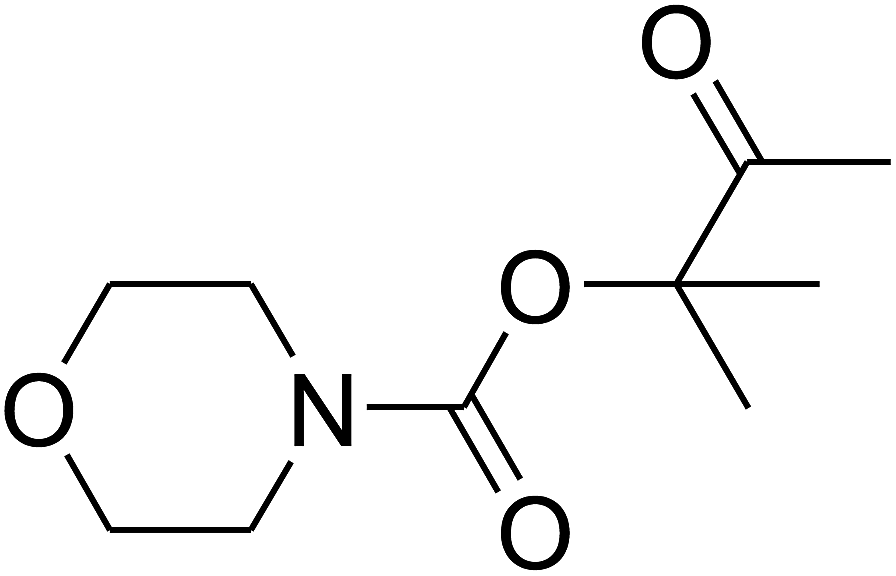
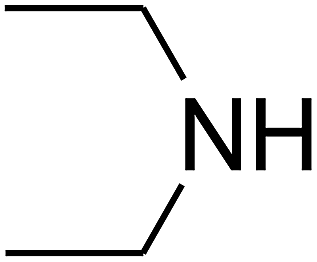
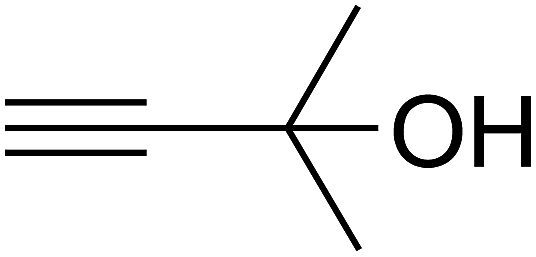
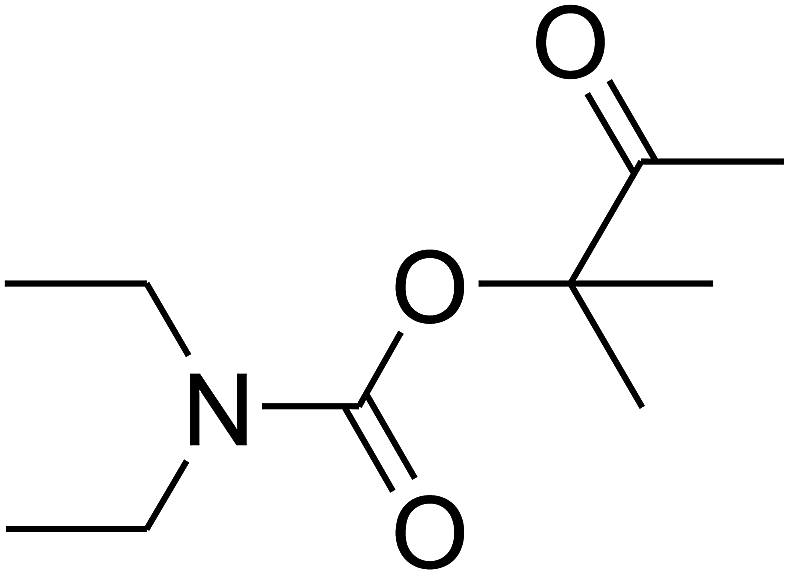
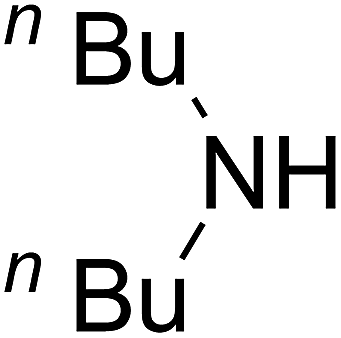
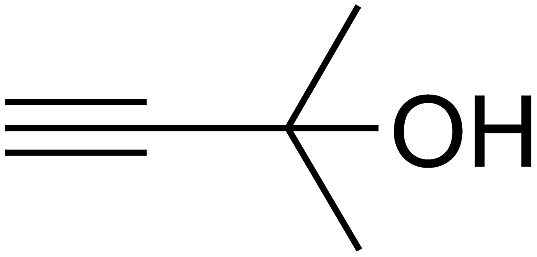
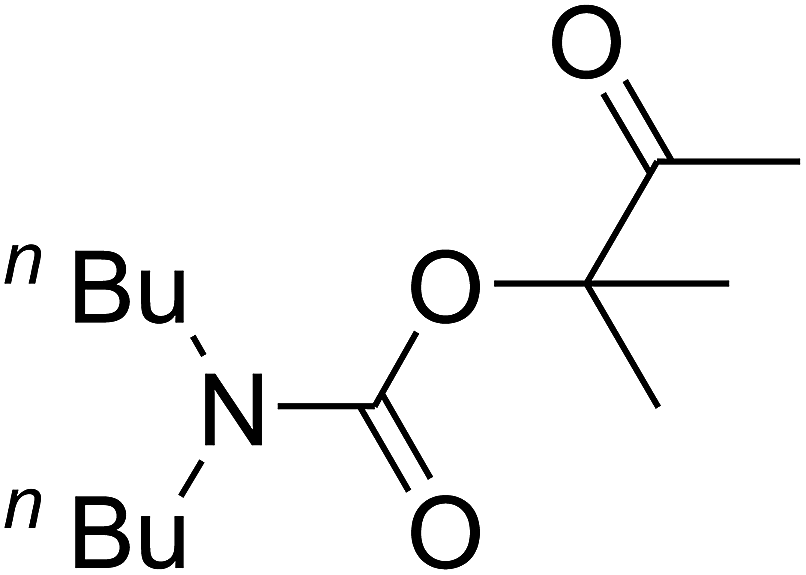
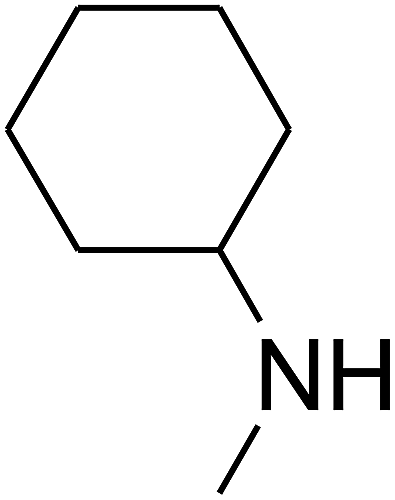
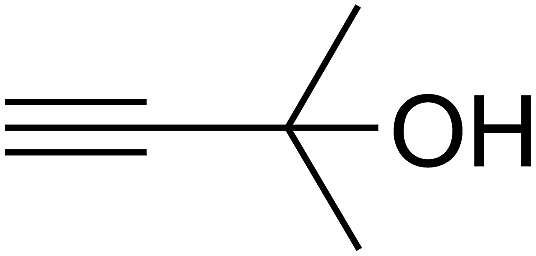
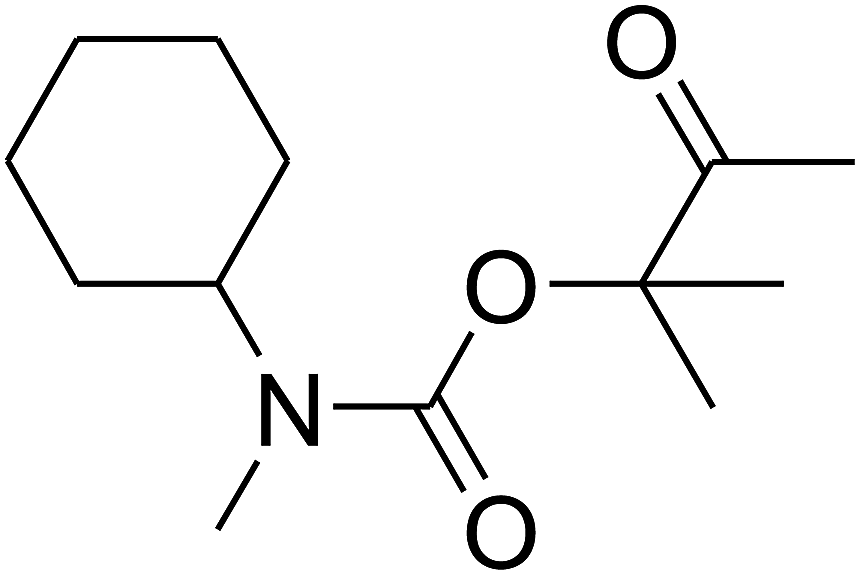
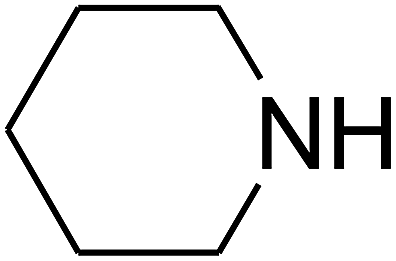
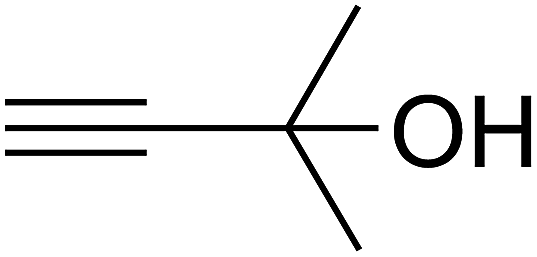
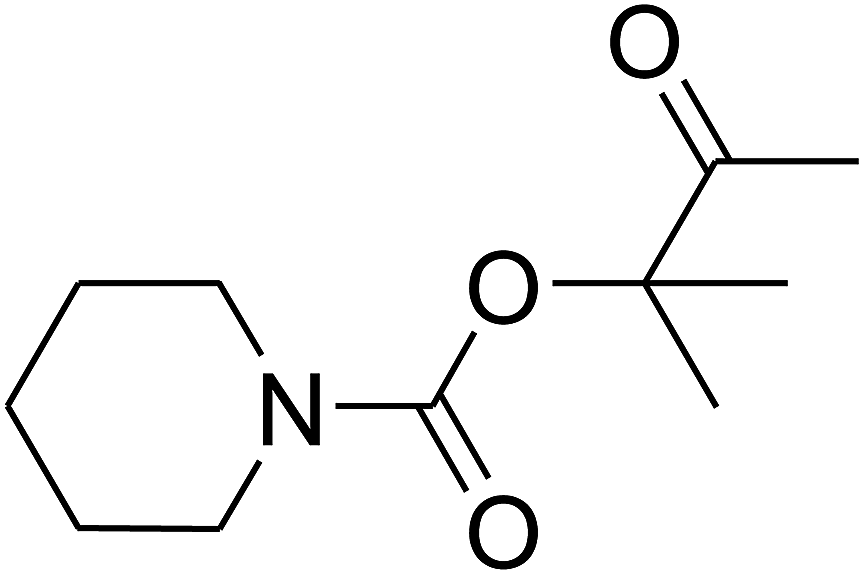
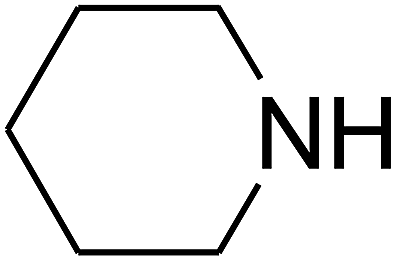
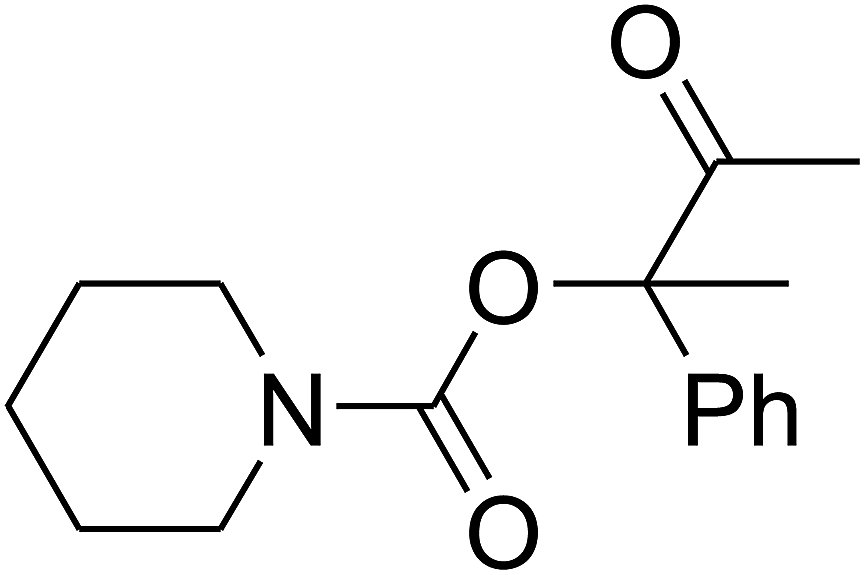
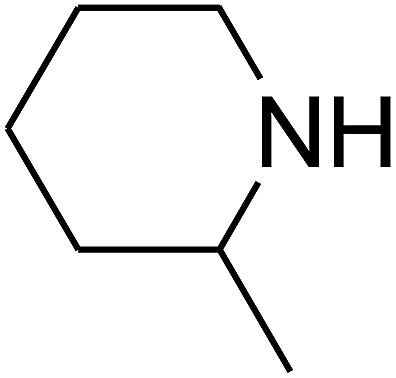
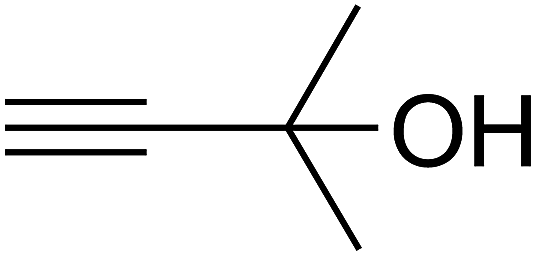
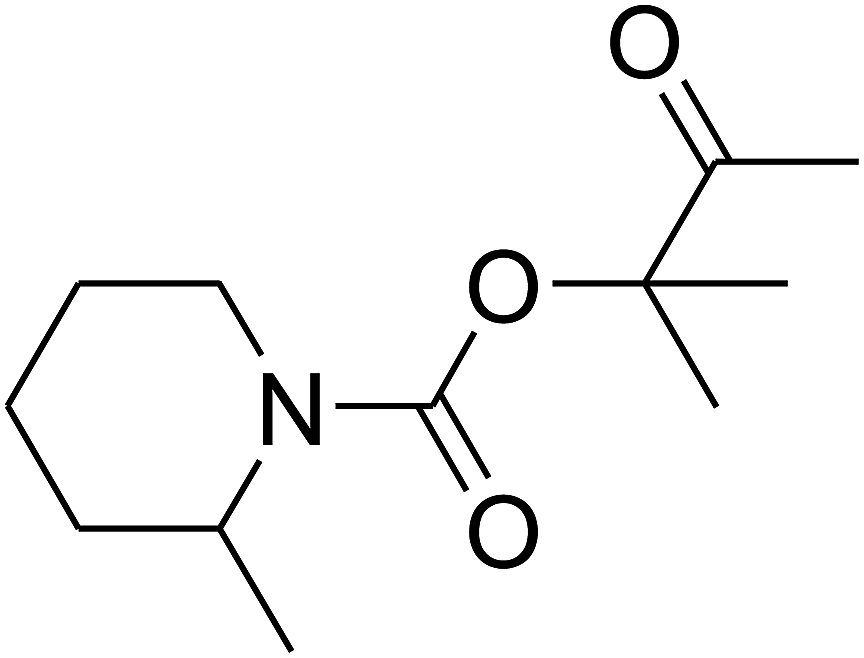
Based on the above yields, a proper mechanism for DFNS/IG@AgBr was determined and has been shown in Scheme 2. At first, spirulina and TPP simultaneously enable the –OH of the 2-methylbut-3-yn-2-ol and increase the nucleophilicity of the –OH to the carbon dioxide, which is trapped and activated by the synergistic effects of TPP and spirulina. Next, the species of Ag activated a triple bond to provide the connection between the charged oxygen and the carbon in the triple bond, causing the formation of the five-membered ring. Moreover, the catalyst was expelled by the five-membered ring, and the main free intermediate was formed. Finally, the amine attacked the carbonyl group in the intermediate, followed by the tautomerism of enol to ketone as well as the production of the corresponding β-oxopropylcarbamates.71–73
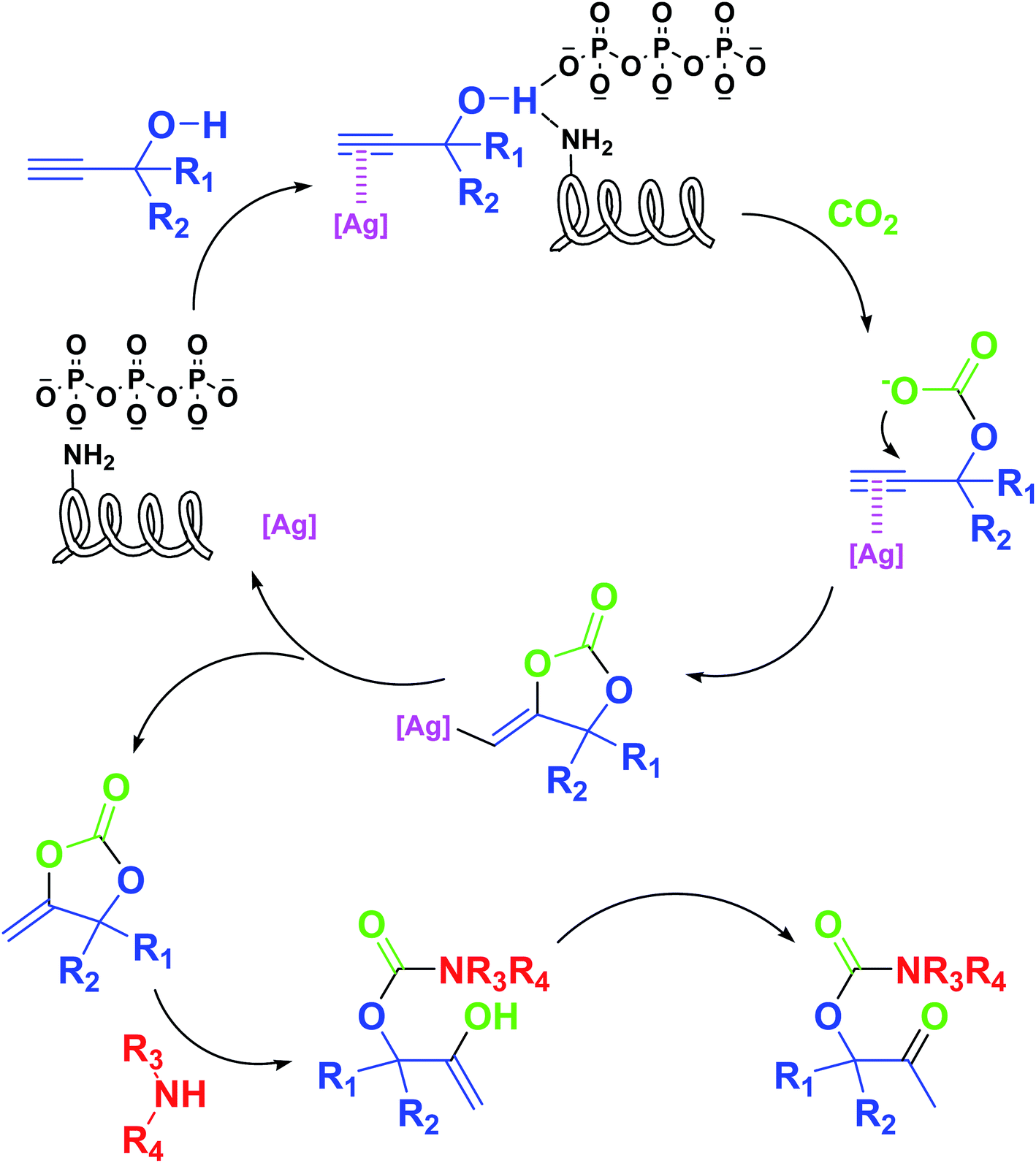 | ||
| Scheme 2 Proposed catalytic mechanism of the DFNS/IG@AgBr system. | ||
It can be noted that the heterogeneous quality of DFNS/IG NPs enables its comfortable recovery from the reaction environment. The activity of the recycled DFNS/IG NP catalysts under suitable conditions was investigated. After the completion of the reaction, the catalyst was isolated using filtration, washed with alcohol and dried with a pump. The synthesized DFNS/IG NPs were recovered for five consecutive cycles without experiencing any significant loss in the catalytic activity (as seen in Fig. 4). The sustainability of this catalyst was because of the stability of the catalyst structure. The notable capability of the DFNS/IG morphology may be ascribed to the cellulose fibers that were mighty enough to effectively conduct the reaction by preventing IG compression and abandoning IG during the process of reaction.
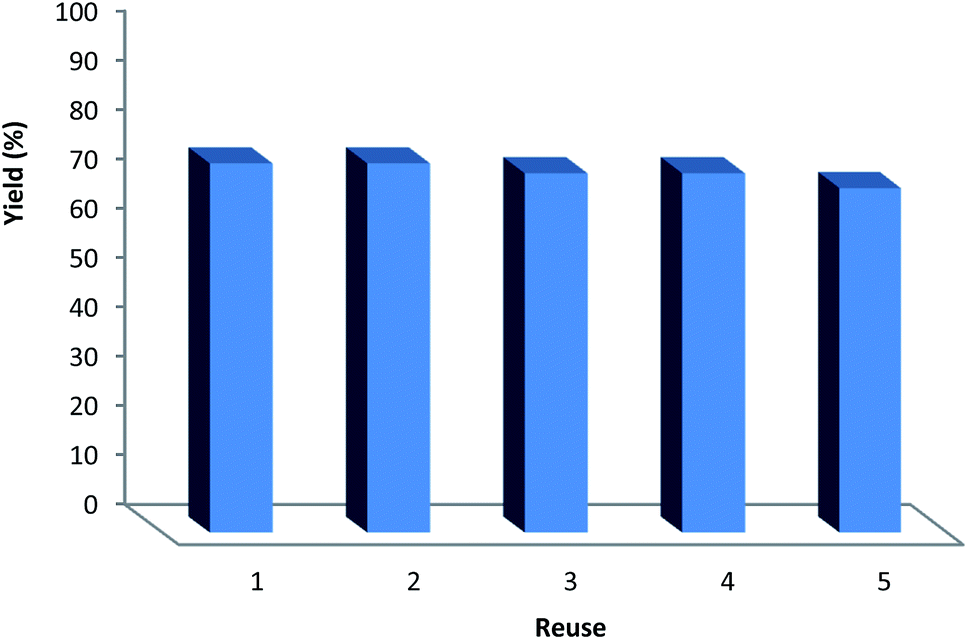 | ||
| Fig. 4 The reusability of the catalyst in the synthesis of β-oxopropylcarbamates. | ||
Conclusions
The results show that a novel nanocatalyst called DFNS/IG NPs was synthesized. This study determined that DFNS/IG@AgBr NPs have considerable features like enhanced catalytic performance, excellent porosity, and high chemical stability for the production of β-oxopropylcarbamates via the three-component coupling reaction of CO2, propargylic alcohols and amines. Moreover, the first order dependency of the reaction was determined by catalyst loading, and based on the obtained results, a likely mechanistic route was found. In addition, we investigated the reusability of the DFNS/IG NPs, and the best results propose it to be an inexpensive catalyst to produce quinazolines.Conflicts of interest
There are no conflicts to declare.References
- A. Vonshak, A. Abeliovich, S. Boussiba, S. Arad and A. Richmond, Biomass, 1982, 2, 175–185 CrossRef.
- G. Torzillo, P. Bernardini and J. Masojídek, J. Phycol., 1998, 34, 504–510 CrossRef CAS.
- E. Guibal, Prog. Polym. Sci., 2005, 30, 71–109 CrossRef CAS.
- D. W. Cho, B. H. Jeon, C. M. Chon, F. W. Schwartz, Y. J. Jeong and H. C. Song, J. Ind. Eng. Chem., 2015, 28, 60–66 CrossRef CAS.
- D. Akin, A. Yakar and U. Gündüz, Water Environ. Res., 2015, 87, 425–436 CrossRef CAS PubMed.
- D. H. Kim, D. E. Nikles and C. S. Brazel, Materials, 2010, 3, 4051–4065 CrossRef CAS PubMed.
- A. Jain, K. Thakur, G. Sharma, P. Kush and U. K. Jain, Carbohydr. Polym., 2016, 137, 65–74 CrossRef CAS PubMed.
- T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107(6), 2365–2387 CrossRef CAS PubMed.
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3(1), 43–81 RSC.
- K. Huang, C. L. Sun and Z. J. Shi, Chem. Soc. Rev., 2011, 40(5), 2435–2452 RSC.
- C. Das Neves Gomes, O. Jacquet, C. Villiers, P. Thuery, M. Ephritikhine and T. Cantat, Angew. Chem., Int. Ed. Engl., 2012, 51(1), 187–190 CrossRef CAS PubMed.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- B. H. Xu, J. Q. Wang, J. Sun, Y. Huang, J. P. Zhang, X. P. Zhang and S. J. Zhang, Green Chem., 2015, 17(1), 108–122 RSC.
- X. F. Liu, X. Y. Li, C. Qiao and L. N. He, Synlett, 2018, 29(05), 548–555 CrossRef CAS.
- C. Song, Catal. Today, 2006, 115(1–4), 2–32 CrossRef CAS.
- J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Angew. Chem., Int. Ed., 2016, 55(26), 7296–7343 CrossRef CAS PubMed.
- A. Pinaka and G. C. Vougioukalakis, Coord. Chem. Rev., 2015, 288, 69–97 CrossRef CAS.
- C. Maeda, Y. Miyazaki and T. Ema, Catal. Sci. Technol., 2014, 4(6), 1482 RSC.
- D. Yu, S. P. Teong and Y. Zhang, Coord. Chem. Rev., 2015, 293–294, 279–291 CrossRef CAS.
- Y. Yuan, C. Chen, C. Zeng, B. Mousavi, S. Chaemchuen and F. Verpoort, ChemCatChem, 2017, 9, 882–887 CrossRef CAS.
- I. Tommasi, Catalysts, 2017, 7(12), 380 CrossRef.
- A. Goeppert, M. Czaun, J. P. Jones, G. K. Surya Prakash and G. A. Olah, Chem. Soc. Rev., 2014, 43(23), 7995–8048 RSC.
- J. Schneidewind, R. Adam, W. Baumann, R. Jackstell and M. Beller, Angew. Chem., 2017, 56(7), 1890 CrossRef CAS PubMed.
- S. Kar, J. Kothandaraman, A. Goeppert and G. S. Prakash, J. CO2 Util., 2018, 23, 212–218 CrossRef CAS.
- P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1995, 95(2), 259–272 CrossRef CAS.
- L. Zhang, J. Cheng, T. Ohishi and Z. Hou, Angew. Chem., Int. Ed. Engl., 2010, 49(46), 8670–8673 CrossRef CAS PubMed.
- T. Lehtinen, E. Efimova, P. L. Tremblay, S. Santala, T. Zhang and V. Santala, Bioresour. Technol., 2017, 243, 30–36 CrossRef CAS PubMed.
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12(9), 1514 RSC.
- Y. Yuan, Y. Xie, C. Zeng, D. Song, S. Chaemchuen, C. Chen and F. Verpoort, Catal. Sci. Technol., 2017, 7(14), 2935–2939 RSC.
- Y. Yuan, Y. Xie, D. Song, C. Zeng, S. Chaemchuen, C. Chen and F. Verpoort, Appl. Organomet. Chem., 2017, 31(12), 3867 CrossRef.
- S. L. Peterson, S. M. Stucka and C. J. Dinsmore, Org. Lett., 2010, 12(6), 1340–1343 CrossRef CAS PubMed.
- E. Vessally, R. Mohammadi, A. Hosseinian, L. Edjlali and M. Babazadeh, J. CO2 Util., 2018, 24, 361–368 CrossRef CAS.
- Y. Peng, J. Liu, C. Qi, G. Yuan, J. Li and H. Jiang, Chem. Commun., 2017, 53(18), 2665–2668 RSC.
- Z. Zhihua, X. Shumei and H. Liangnian, Acta Phys.-Chim. Sin., 2017, 34(8), 838–844 Search PubMed.
- P. Adams and F. A. Baron, Chem. Rev., 1965, 65(5), 567–602 CrossRef CAS.
- A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96(3), 951–976 CrossRef CAS PubMed.
- M. M. Heravi and V. Zadsirjan, Tetrahedron: Asymmetry, 2013, 24(19), 1149–1188 CrossRef CAS.
- J. M. Ahn, J. C. Peters and G. C. Fu, J. Am. Chem. Soc., 2017, 139(49), 18101–18106 CrossRef CAS.
- F. Shi, Y. Deng, T. SiMa, J. Peng, Y. Gu and B. Qiao, Angew. Chem., Int. Ed. Engl., 2003, 42(28), 3257–3260 CrossRef CAS PubMed.
- Y. Sasaki and P. H. Dixneuf, J. Org. Chem., 1987, 52, 4389–4391 CrossRef CAS.
- C. Bruneau and P. H. Dixneuf, Tetrahedron Lett., 1987, 28, 2005–2008 CrossRef CAS.
- T. J. Kim, K. H. Kwon, S. C. Kwon, J. O. Baeg and S. C. Shim, J. Organomet. Chem., 1990, 389, 205–217 CrossRef CAS.
- H. S. Kim, J. W. Kim, S. C. Kwon, S. C. Shim and T. J. Kim, J. Organomet. Chem., 1997, 545, 337–344 CrossRef.
- X. D. Li, X. D. Lang, Q. W. Song, Y. K. Guo and L. N. He, Chin. J. Org. Chem., 2016, 36, 744–751 CrossRef CAS.
- C. Qi, L. Huang and H. Jiang, Synthesis, 2010, 2010, 1433–1440 CrossRef.
- Q. W. Song, B. Yu, X. D. Li, R. Ma, Z. F. Diao, R. G. Li, W. Li and L. N. He, Green Chem., 2014, 16, 1633–1638 RSC.
- Q. W. Song, W. Q. Chen, R. Ma, A. Yu, Q. Y. Li, Y. Chang and L. N. He, ChemSusChem, 2015, 8, 821–827 CrossRef CAS.
- Q. W. Song, Z. H. Zhou, H. Yin and L. N. He, ChemSusChem, 2015, 8, 3967–3972 CrossRef CAS.
- K. Sekine and T. Yamada, Chem. Soc. Rev., 2016, 45, 4524–4532 RSC.
- Q. W. Song, P. Liu, L. H. Han, K. Zhang and L. N. He, Chin. J. Chem., 2018, 36, 147–152 CrossRef CAS.
- N. D. Ca, B. Gabriele, G. Ruffolo, L. Veltri, T. Zanetta and M. Costa, Adv. Synth. Catal., 2011, 353, 133–146 CrossRef.
- C. G. Neochoritis, T. Zhao and A. Dömling, Chem. Rev., 2019, 119, 1970–2042 CrossRef CAS PubMed.
- A. Dömling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083–3135 CrossRef PubMed.
- I. Ugi, R. Meyr, U. Fetzer and C. Steinbrückner, Angew. Chem., 1959, 71, 386–388 Search PubMed.
- M. Gazz, Chim. Ital., 1921, 51, 126 Search PubMed.
- D. Van Leusen and A. M. Van Leusen, Org. React. (N.Y.), 2003, 57, 419 Search PubMed.
- A. Strecker, Ann. Chem. Pharm., 1854, 91, 349 CrossRef.
- A. Hantzsch, Chem. Ber., 1881, 14, 1637 CrossRef.
- (a) P. Biginelli, Chem. Ber., 1891, 24, 1317 CrossRef; (b) P. Biginelli, Chem. Ber., 1891, 24, 2962 CrossRef; (c) C. O. Kappe, Tetrahedron, 1993, 49, 6937 CrossRef CAS.
- S. Heck and A. Dömling, Synlett, 2000, 2000, 424–426 CrossRef.
- (a) L. El Kaïm, L. Grimaud and J. Oble, Angew. Chem., Int. Ed., 2005, 44, 7961–7964 CrossRef PubMed; (b) L. El Kaïm and L. Grimaud, Mol. Diversity, 2010, 14, 855–867 CrossRef PubMed.
- I. Ugi and C. Steinbrückner, Angew. Chem., 1960, 72, 267 CrossRef CAS.
- (a) A. Dömling, Chem. Rev., 2006, 106, 17–89 CrossRef PubMed; (b) W. Wang and A. J. Dömling, Comb. Chem., 2009, 11, 403–409 CrossRef CAS.
- S. M. Sadeghzadeh, Microporous Mesoporous Mater., 2016, 234, 310–316 CrossRef CAS.
- A. Maity and V. Polshettiwar, ChemSusChem, 2017, 10, 3866–3913 CrossRef CAS.
- U. Patil, A. Fihri, A. H. Emwas and V. Polshettiwar, Chem. Sci., 2012, 3, 2224–2229 RSC.
- V. Polshettiwar, J. ThivolleCazat, M. Taoufik, F. Stoffelbach, S. Norsic and J. M. Basset, Angew. Chem., Int. Ed., 2011, 50, 2747–2751 CrossRef CAS.
- A. Fihri, D. Cha, M. Bouhrara, N. Almana and V. Polshettiwar, ChemSusChem, 2012, 5, 85–89 CrossRef CAS PubMed.
- U. Patil, A. Fihri, A. H. Emwas and V. Polshettiwar, Chem. Sci., 2012, 3, 2224–2229 RSC.
- A. Fihri, M. Bouhrara, U. Patil, D. Cha, Y. Saih and V. Polshettiwar, ACS Catal., 2012, 2, 1425–1431 CrossRef CAS.
- D. Song, D. Li, X. Xiao, C. Cheng, S. Chaemchuen, Y. Yuan and F. Verpoort, J. CO2 Util., 2018, 27, 217–222 CrossRef CAS.
- Q. W. Song, Z. H. Zhou, H. Yin and L. N. He, ChemSusChem, 2015, 8, 3967–3972 CrossRef CAS.
- Q. N. Zhao, Q. W. Song, P. Liu, K. Zhang and J. Hao, ChemistrySelect, 2018, 3, 6897–6901 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02680k |
| This journal is © The Royal Society of Chemistry 2019 |