
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient access to chiral dihydrobenzoxazinones via Rh-catalyzed hydrogenation†
Ziyi Chena,
Xuguang Yina,
Xiu-Qin Dong *a and
Xumu Zhangab
*a and
Xumu Zhangab
aKey Laboratory of Biomedical Polymers, Engineering Research Centre of Organosilicon Compounds & Materials, Ministry of Education, College of Chemistry and Molecular Sciences, Wuhan University, Wuhan, Hubei 430072, P. R. China. E-mail: xiuqindong@whu.edu.cn
bDepartment of Chemistry and Shenzhen Grubbs Institute, Southern University of Science and Technology, Shenzhen, Guangdong 518055, P. R. China
First published on 17th May 2019
Abstract
Rh/(S)-DTBM-SegPhos-catalyzed asymmetric hydrogenation of prochiral (Z)-2-(2-oxo-2H-benzo[b][1,4]oxazin-3(4H)-ylidene)acetate esters was successfully developed. A series of chiral dihydrobenzoxazinones were prepared through this efficient methodology with good to excellent results (up to >99% conversion, 93% yield and >99% ee), which are important motifs in the biologically active molecules.
Introduction
Chiral dihydrobenzoxazinones and derivatives are an important class of heterocycles, and are frequently found in biologically active molecules.1–4 For example, compound A is a potent anticholesteremic agent (Fig. 1).2 Compound B (Kinin B1) is used for the treatment of inflammation and pain in septicemia.3 Compound C is a pyruvate kinase activator, which can increase the lifetime of red blood cells.4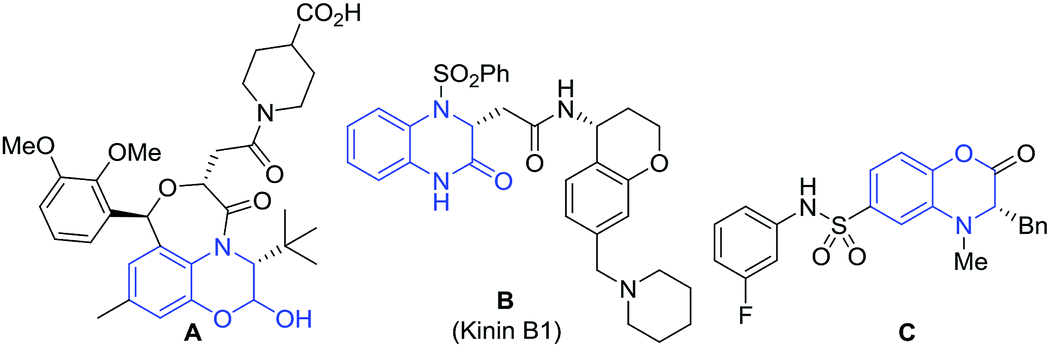 | ||
| Fig. 1 Examples of biologically active molecules containing chiral dihydrobenzoxazinone or related framework. | ||
Due to the great importance of chiral dihydrobenzoxazinones and derivatives, the investigation on the construction of these intriguing motifs has been an important research topic in organic synthesis. Therefore, enormous efforts were made to the development of efficient enantioselective methods, and some asymmetric synthetic methodologies have been established to access chiral dihydrobenzoxazinones and derivatives over the past decades.5–11 The asymmetric catalytic reduction of prochiral benzoxazinones and derivatives is one of the most important methods,5–7 including asymmetric hydrosilylation,5 asymmetric transfer hydrogenation,6 and asymmetric hydrogenation.3b,7 In addition, other efficient enantioselective methods were involved to prepare chiral dihydrobenzoxazinones and derivatives,4b,8–11 such as addition reaction of indoles or pyrroles with benzoxazinones,8 Rh-catalyzed asymmetric arylation of benzoxazinones and quinoxalinones with arylboroxines,4b dynamic kinetic resolution of α-bromo arylacetates in nucleophilic substitution with N-alkylated 2-aminophenols,9 asymmetric Mannich reaction of ketones with benzoxazinones,10 and organocatalytic reductive amination.11 Transition-metal-catalyzed asymmetric hydrogenation has been regarded as a straightforward and efficient method for the synthesis of chiral compounds with high atom-economic advantage.12 Based on our continuing research in the field of asymmetric hydrogenation, much attention were paid to the synthesis of chiral dihydrobenzoxazinones and derivatives through asymmetric hydrogenation. We herein successfully developed Rh-catalyzed asymmetric hydrogenation of (Z)-2-(2-oxo-2H-benzo[b][1,4]oxazin-3(4H)-ylidene)acetate esters using commercial (S)-DTBM-SegPhos ligand, affording a series of chiral dihydrobenzoxazinones with good to excellent results (Scheme 1, >99% conversion, 93% yield, >99% ee).
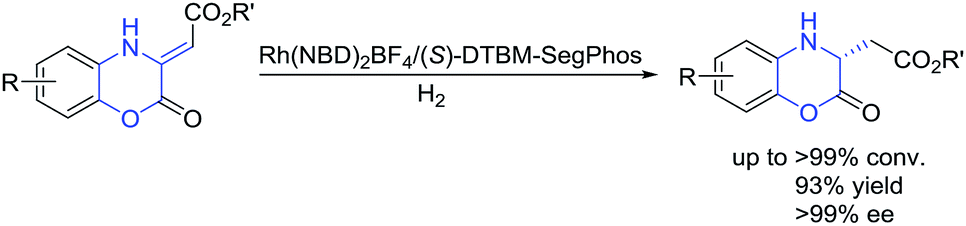 | ||
| Scheme 1 Preparation of chiral dihydrobenzoxazinones through Rh-catalyzed asymmetric hydrogenation. | ||
Results and discussion
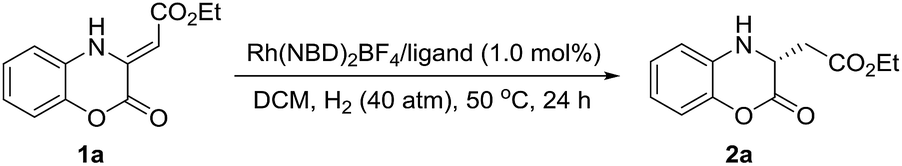
The initial investigation of Rh(NBD)2BF4-catalyzed asymmetric hydrogenation of model substrate ethyl (Z)-2-(2-oxo-2H-benzo[b][1,4]oxazin-3(4H)-ylidene)acetate 1a13 was started to evaluate a series of chiral diphosphine ligands (Fig. 2) under 40 atm H2 at 50 °C in CH2Cl2 for 24 h. As shown in Table 1, poor conversion and good enantioselectivity was obtained in the presence of easily available (R)-Binap (22% conversion, 87% ee, Table 1, entry 1). In addition, poor to moderate results were obtained with (RC, SP)-DuanPhos, (S, S)-f-Binaphane, (R, S)-JosiPhos, (S)-Binapine, ZhaoPhos, (S, S)-Ph-BPE and (S)-SegPhos as the ligand (3–62% conversions, 20–72% ee, Table 1, entries 2, 4–9). No reaction was observed using the (S, S)-Me-DuPhos as the ligand (Table 1, entry 3). To our delight, the ligand (S)-DTBM-SegPhos provided the promising reaction result with 73% conversion and 94% ee (Table 1, entry 10).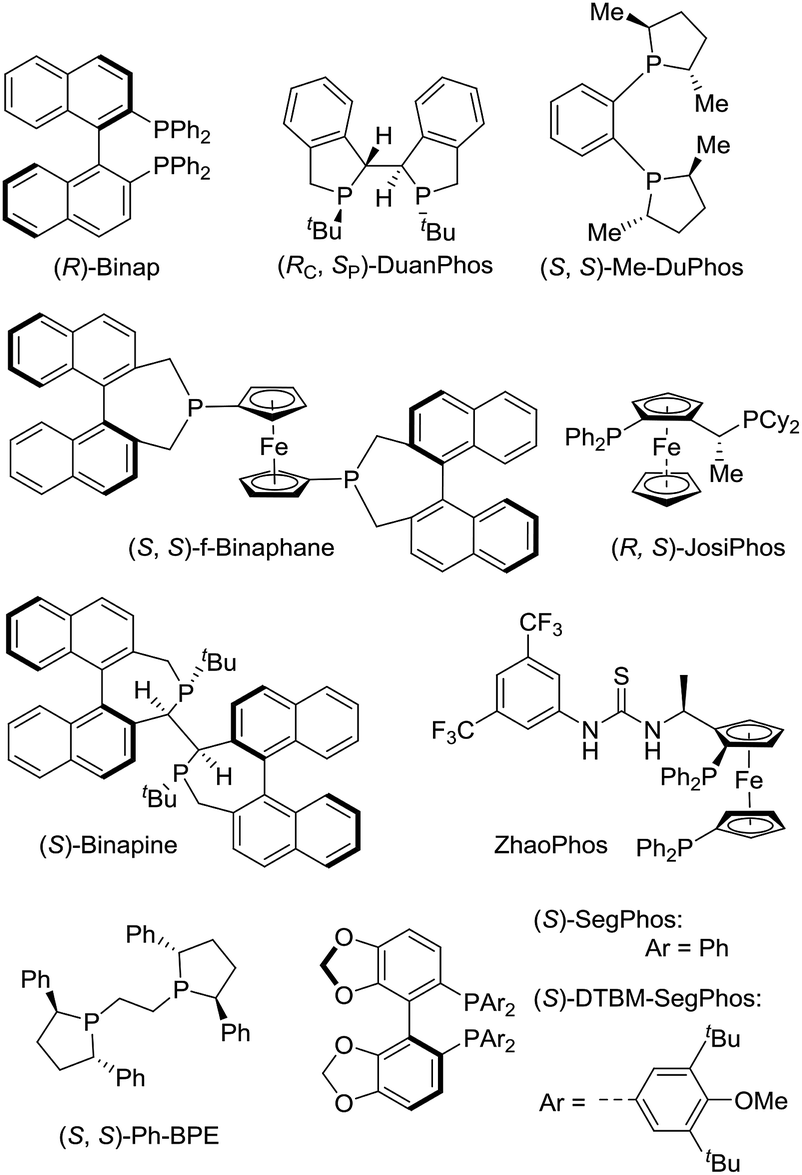 | ||
| Fig. 2 The structure of chiral diphosphine ligands. | ||
|
|||
|---|---|---|---|
| Entry | Ligand | Conv.b (%) | eec (%) |
| a Reaction condition: substrate 1a (0.10 mmol), Rh(NBD)2BF4 (1.0 mol%), ligand (1.1 mol%), 1 mL DCM, H2 (40 atm), 50 °C, 24 h.b Determined by 1H NMR analysis.c Determined by HPLC analysis using a chiral stationary phase. DCM is CH2Cl2. NR = no reaction. NA = no available. | |||
| 1 | (R)-Binap | 22 | 87 |
| 2 | (RC, SP)-DuanPhos | 4 | 28 |
| 3 | (S, S)-Me-DuPhos | NR | NA |
| 4 | (S, S)-f-Binaphane | 5 | 49 |
| 5 | (R, S)-JosiPhos | 62 | 41 |
| 6 | (S)-Binapine | 22 | 20 |
| 7 | ZhaoPhos | 3 | 67 |
| 8 | (S, S)-Ph-BPE | 29 | 65 |
| 9 | (S)-SegPhos | 62 | 72 |
| 10 | (S)-DTBM-SegPhos | 73 | 94 |
The solvent played an important role in asymmetric catalytic reaction, and the Rh(NBD)2BF4/(S)-DTBM-SegPhos-catalyzed asymmetric hydrogenation of model substrate ethyl (Z)-2-(2-oxo-2H-benzo[b][1,4]oxazin-3(4H)-ylidene)acetate 1a was then carried out in different solvents. We found that moderate conversions and enantioselectivities were obtained in ethyl acetate, CHCl3 and iPrOH (40–66% conversions, 42–70% ee, Table 2, entries 1, 5, 9). Toluene, THF, 1,4-dioxane and DCE gave very poor enantioselectivities (Table 2, entries 2–4, 11). Although full conversion was achieved in TFE, moderate enantioselectivity was provided (>99% conversion, 56% ee, Table 2, entry 6). Trace conversions were observed in MeOH and EtOH (Table 2, entries 7–8). Among these solvents, DCM was still provided the highest enantioselectivity (94% ee, Table 2, entry 10).
|
|||
|---|---|---|---|
| Entry | Solvent | Conv.b (%) | eec (%) |
| a Reaction condition: substrate 1a (0.10 mmol), Rh(NBD)2BF4 (1.0 mol%), (S)-DTBM-SegPhos (1.1 mol%), 1 mL solvent, H2 (40 atm), 50 °C, 24 h.b Determined by 1H NMR analysis.c Determined by HPLC analysis using a chiral stationary phase. THF is tetrahydrofuran. TFE is trifluoroethanol. DCE is dichloroethane. | |||
| 1 | EtOAc | 64 | 66 |
| 2 | Toluene | 32 | 27 |
| 3 | THF | 75 | 16 |
| 4 | 1,4-Dioxane | 50 | 6 |
| 5 | CHCl3 | 40 | 70 |
| 6 | TFE | >99 | 56 |
| 7 | MeOH | Trace | NA |
| 8 | EtOH | Trace | NA |
| 9 | iPrOH | 66 | 42 |
| 10 | DCM | 73 | 94 |
| 11 | DCE | 64 | 22 |

In order to obtain high conversion and excellent enantioselectivity, the ratio of mixture of CH2Cl2 and TFE was inspected (Table 3). When the volumetric ratio of TFE and DCM is 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, the best results can be afforded with 96% conversion and 95% ee (Table 3, entry 5). In addition, several metal precursors were investigated in this asymmetric hydrogenation. Moderate conversion and excellent enantioselectivity was achieved with [Rh(COD)Cl]2 as metal precursor (72% conversion, 90% ee, Table 3, entry 7). [Ir(COD)Cl]2 gave poor reactivity and moderate enantioselectivity (37% conversion, 73% ee, Table 3, entry 8). There was no reaction in the presence of Ni(OAc)2 (Table 3, entry 9). To our delight, nearly the same reaction result can be achieved when the pressure of H2 was decreased from 40 atm to 20 atm (96% conversion, 97% ee, Table 3, entry 10).
:1, the best results can be afforded with 96% conversion and 95% ee (Table 3, entry 5). In addition, several metal precursors were investigated in this asymmetric hydrogenation. Moderate conversion and excellent enantioselectivity was achieved with [Rh(COD)Cl]2 as metal precursor (72% conversion, 90% ee, Table 3, entry 7). [Ir(COD)Cl]2 gave poor reactivity and moderate enantioselectivity (37% conversion, 73% ee, Table 3, entry 8). There was no reaction in the presence of Ni(OAc)2 (Table 3, entry 9). To our delight, nearly the same reaction result can be achieved when the pressure of H2 was decreased from 40 atm to 20 atm (96% conversion, 97% ee, Table 3, entry 10).
|
|||||
|---|---|---|---|---|---|
| Entry | Metal precursor | Solvent | H2 (atm) | Conv.b (%) | eec (%) |
| a Reaction condition: substrate 1a (0.10 mmol), metal precursor (1.0 mol%), (S)-DTBM-SegPhos (1.1 mol%), 1 mL solvent, H2, 50 °C, 24 h.b Determined by 1H NMR analysis.c Determined by HPLC analysis using a chiral stationary phase. | |||||
| 1 | Rh(NBD)2BF4 | TFE/DCM = 1:2 |
40 | 61 | 94 |
| 2 | Rh(NBD)2BF4 | TFE/DCM = 1:4 |
40 | 59 | 93 |
| 3 | Rh(NBD)2BF4 | TFE/DCM = 1:8 |
40 | 66 | 96 |
| 4 | Rh(NBD)2BF4 | TFE/DCM = 2:1 |
40 | 56 | 91 |
| 5 | Rh(NBD)2BF4 | TFE/DCM = 4:1 |
40 | 96 | 95 |
| 6 | Rh(NBD)2BF4 | TFE/DCM = 8:1 |
40 | 90 | 95 |
| 7 | [Rh(COD)Cl]2 | TFE/DCM = 4:1 |
40 | 72 | 90 |
| 8 | [Ir(COD)Cl]2 | TFE/DCM = 4:1 |
40 | 37 | 73 |
| 9 | Ni(OAc)2 | TFE/DCM = 4:1 |
40 | NR | NA |
| 10 | Rh(NBD)2BF4 | TFE/DCM = 4:1 |
20 | 96 | 97 |
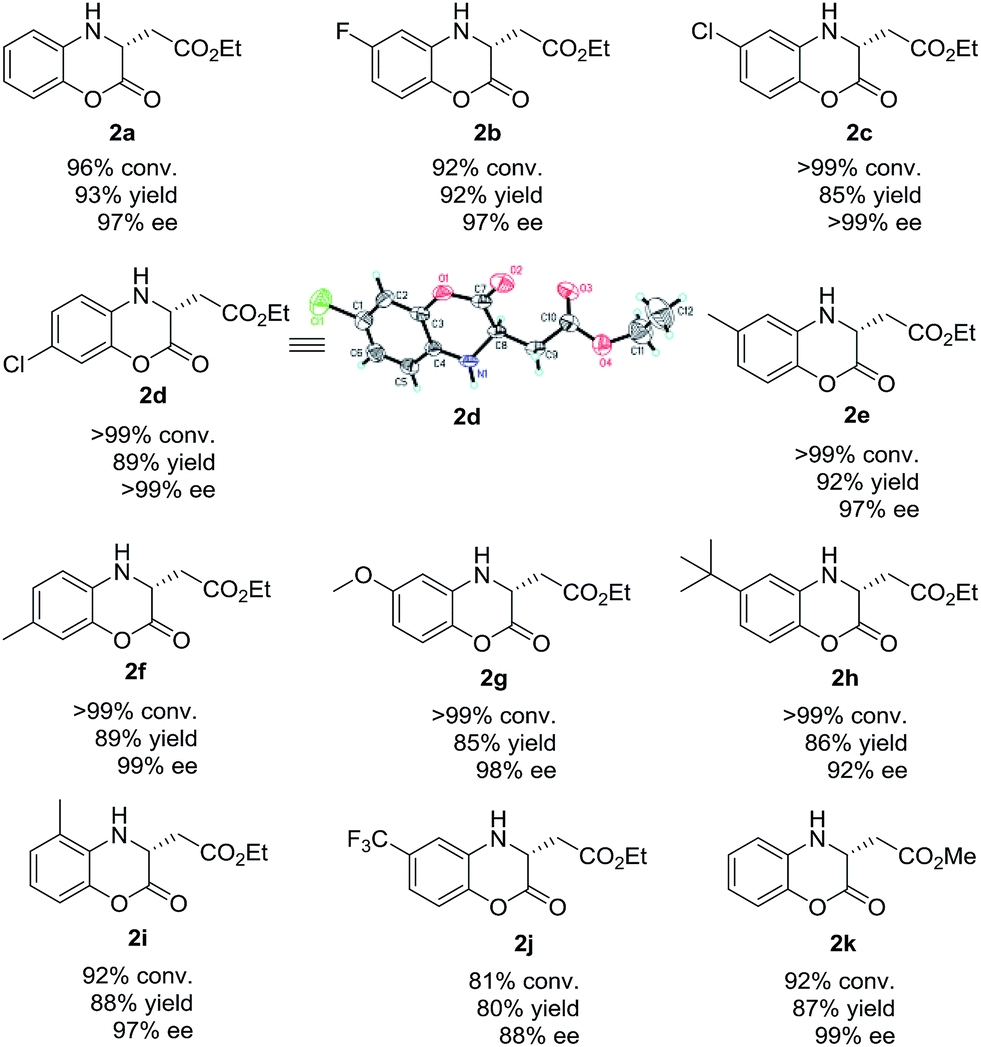
After establishing the optimized reaction conditions, we focused our attention on the exploration of the substrate scope generality of this Rh-catalyzed asymmetric hydrogenation of various prochiral (Z)-2-(2-oxo-2H-benzo[b][1,4]oxazin-3(4H)-ylidene)acetate esters. As listed in Table 4, the Rh-catalyzed asymmetric hydrogenation of a series of (Z)-2-(2-oxo-2H-benzo[b][1,4]oxazin-3(4H)-ylidene)acetate esters could proceed smoothly, affording the desired hydrogenation products chiral dihydrobenzoxazinones (2a–2k) with good to excellent results (81% to >99% conversions, 80–93% yields, 88% to >99% ee). The substrates (Z)-2-(2-oxo-2H-benzo[b][1,4]oxazin-3(4H)-ylidene)acetate esters bearing electron-withdrawing (1b–1d, 1j) or electron-donating (1e–1i) substituted groups on the benzo ring worked well in this asymmetric hydrogenation. In addition, we found that the position of substituted group on the benzo ring had little effect on the reactivity and enantioselectivity. Moreover, the ester group was well tolerated in this catalytic system. When the ethyl ester group was changed to methyl ester group, the substrate methyl (Z)-2-(2-oxo-2H-benzo[b][1,4]oxazin-3(4H)-ylidene)acetate (1k) was hydrogenated with high conversion and excellent enantioselectivity (92% conversion, 87% yield and 99% ee).
|
|---|
| a Reaction condition: substrate 1 (0.10 mmol), Rh(NBD)2BF4 (1.0 mol%), (S)-DTBM-SegPhos (1.1 mol%), 1 mL solvent, H2 (20 atm), 50 °C, 24 h. Conversion was determined by 1H NMR analysis. Yield is isolated yield. ee was determined by HPLC analysis using a chiral stationary phase. The configuration of 2d was determined by X-ray analysis.14 |
 |

Conclusions
In conclusion, the Rh/(S)-DTBM-SegPhos-catalyzed asymmetric hydrogenation of a variety of prochiral (Z)-2-(2-oxo-2H-benzo[b][1,4]oxazin-3(4H)-ylidene)acetate esters was successfully realized. This efficient methodology afforded chiral dihydrobenzoxazinones with good to excellent results (81% to >99% conversions, 80–93% yields, 88% to >99% ee), which are important and unique building blocks in the biologically active molecules.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (Grant No. 21432007, 21502145), Wuhan Morning Light Plan of Youth Science and Technology (Grant No. 2017050304010307), Shenzhen Nobel Prize Scientists Laboratory Project (Grant No. C17783101) and the Fundamental Research Funds for Central Universities (Grant No. 2042018kf0202). The Program of Introducing Talents of Discipline to Universities of China (111 Project) is also appreciated.Notes and references
- (a) D. V. Smil, S. Manku, Y. A. Chantigny, S. Leit, A. Wahhab, R. Deziel, T. P. Yan, M. Fournel, C. Maroun, Z. Li, A.-M. Lemieux, A. Nicolescu, J. Rahil, S. Lefebvre, A. Panetta, J. M. Besterman, S. Lefebvre, A. Panetta, J. M. Besterman and R. Déziel, Bioorg. Med. Chem. Lett., 2009, 19, 688–692 CrossRef CAS PubMed; (b) P. E. Mahaney, M. B. Webb, F. Ye, J. P. Sabatucci, R. J. Steffan, C. C. Chadwick, D. C. Harnish and E. J. Trybulski, Bioorg. Med. Chem., 2006, 14, 3455–3466 CrossRef CAS PubMed; (c) D.-S. Su, M. K. Markowitz, R. M. DiPardo, K. L. Murphy, C. M. Harrell, S. S. O'Malley, R. W. Ransom, R. S. L. Chang, S. Ha, F. J. Hess, D. J. Pettibone, G. S. Mason, S. Boyce, R. M. Freidinger and M. G. Bock, J. Am. Chem. Soc., 2003, 125, 7516–7517 CrossRef CAS PubMed; (d) F. Grant, S. Bartulis, L. Brogley, M. S. Dappan, R. Kasar, A. Khan, M. Neitzel, M. A. Pleiss, E. D. Thorsett, J. Tucker, M. Ye and J. Hawkinson, PCT Int. Appl., WO 2003093245 A1, 2003; (e) Z. Jones, R. Groneberg, M. Drew and C. T. Eary, US Pat., 20050282812 A1, 2005; (f) D.-S. Su and M. G. Bock, U.S. Pat. Appl. Publ. US 20050020591 A1, 2005; (g) J. Ilaš, P. Š. Anderluh, M. S. Dolenc and D. Kikelj, Tetrahedron, 2005, 61, 7325–7348 CrossRef; (h) C. J. Abraham, D. H. Paull, M. T. Scerba, J. W. Grebinski and T. Lectka, J. Am. Chem. Soc., 2006, 128, 13370–13371 CrossRef CAS PubMed; (i) M. Rueping, M. Stoeckel, E. Sugiono and T. Theissmann, Tetrahedron, 2010, 66, 6565–6568 CrossRef CAS.
- (a) C. M. Hayward and D. A. Scully, US Pat., US 6207664 B1, 2001; (b) T. Tanaka, I. Mizota, K. Umezu, A. Ito and M. Shimizu, Heterocycles, 2017, 95, 830–843 CrossRef CAS.
- (a) J. J. Chen, W. Qian, K. Biswas, V. N. Viswanadhan, B. C. Askew, S. Hitchcock, R. W. Hungate, L. Arik and E. Johnson, Bioorg. Med. Chem. Lett., 2008, 18, 4477–4481 CrossRef CAS PubMed; (b) M.-W. Chen, Z. Deng, Q. Yang, J. Huang and Y. Peng, Org. Chem. Front., 2019, 6, 746–750 RSC.
- (a) S. S.-S. Michael, WO2012151440, 2012; (b) X. Zhang, B. Xu and M.-H. Xu, Org. Chem. Front., 2016, 3, 944–948 RSC.
- (a) A. V. Maikov, K. Vranková, S. Stončius and P. Kočovský, J. Org. Chem., 2009, 74, 5839–5849 CrossRef PubMed; (b) Z.-Y. Xue, Y. Jiang, X.-Z. Peng, W.-C. Yuan and X.-M. Zhang, Adv. Synth. Catal., 2010, 352, 2132–2136 CrossRef CAS.
- (a) M. Rueping, A. P. Antonchick and T. Theissmann, Angew. Chem., Int. Ed., 2006, 45, 6751–6755 CrossRef CAS PubMed; (b) F. Shi, W. Tan, H.-H. Zhang, M. Li, Q. Ye, G.-H. Ma, S.-J. Tu and G. Li, Adv. Synth. Catal., 2013, 355, 3715–3726 CrossRef CAS; (c) Y. Zhang, R. Zhao, R. L.-Y. Bao and L. Shi, Eur. J. Org. Chem., 2015, 3344–3351 CrossRef CAS; (d) X. Zhang, A. Kormos and J. Zhang, Org. Lett., 2017, 19, 6072–6075 CrossRef CAS PubMed.
- (a) Q.-A. Chen, M.-W. Chen, C.-B. Yu, L. Shi, D.-S. Wang, Y. Yang and Y.-G. Zhou, J. Am. Chem. Soc., 2011, 133, 16432–16435 CrossRef CAS PubMed; (b) Q.-A. Chen, K. Gao, Y. Duan, Z.-S. Ye, L. Shi, Y. Yang and Y.-G. Zhou, J. Am. Chem. Soc., 2012, 134, 2442–2448 CrossRef CAS PubMed; (c) L.-Q. Lu, Y. Li, K. Junge and M. Beller, J. Am. Chem. Soc., 2015, 137, 2763–2768 CrossRef CAS PubMed; (d) J. Wang, Z.-H. Zhu, M.-W. Chen, Q.-A. Chen and Y.-G. Zhou, Angew. Chem., Int. Ed., 2019, 58, 1813–1817 CrossRef CAS PubMed.
- (a) T. Kano, R. Takechi, R. Kobayashi and K. Maruoka, Org. Biomol. Chem., 2014, 12, 724–727 RSC; (b) H. Lou, Y. Wang, E. Jin and X. Lin, J. Org. Chem., 2016, 81, 2019–2026 CrossRef CAS PubMed.
- E. Youk, W. Park and Y. S. Park, Synth. Commun., 2018, 48, 1331–1337 CrossRef CAS.
- M. Viji, J. Sim, S. Li, H. Lee, K. Oh and J.-K. Jung, Adv. Synth. Catal., 2018, 360, 4464–4469 CrossRef CAS.
- R. I. Storer, D. E. Carrera, Y. Ni and D. W. C. MacMillan, J. Am. Chem. Soc., 2006, 128, 84–86 CrossRef CAS PubMed.
- Selected examples: (a) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029–3070 CrossRef CAS PubMed; (b) S. J. Roseblade and A. Pfaltz, Acc. Chem. Res., 2007, 40, 1402–1411 CrossRef CAS PubMed; (c) G. Erre, S. Enthaler, K. Junge, S. Gladiali and M. Beller, Coord. Chem. Rev., 2008, 252, 471–491 CrossRef CAS; (d) T. L. Church and P. G. Andersson, Coord. Chem. Rev., 2008, 252, 513–531 CrossRef CAS; (e) J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, Chem. Rev., 2011, 111, 1713–1760 CrossRef CAS PubMed; (f) D.-S. Wang, Q.-A. Chen, S.-M. Lu and Y.-G. Zhou, Chem. Rev., 2012, 112, 2557–2590 CrossRef CAS PubMed; (g) J.-H. Xie and Q.-L. Zhou, Acta Chim. Sin., 2012, 70, 1427–1438 CrossRef CAS; (h) J. J. Verendel, O. Pàmies, M. Diéguez and P. G. Andersson, Chem. Rev., 2014, 114, 2130–2169 CrossRef CAS PubMed; (i) Z. Zhang, N. A. Butt and W. Zhang, Chem. Rev., 2016, 116, 14769–14827 CrossRef CAS PubMed; (j) S.-F. Zhu and Q.-L. Zhou, Acc. Chem. Res., 2017, 50, 988–1001 CrossRef CAS PubMed.
- G. Choudhary and R. K. Peddinti, Green Chem., 2011, 13, 3290–3299 RSC.
- The X-ray crystal data of compound 2d has been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 1905706.†.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR spectra of compounds. CCDC 1905706. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra02694k |
| This journal is © The Royal Society of Chemistry 2019 |