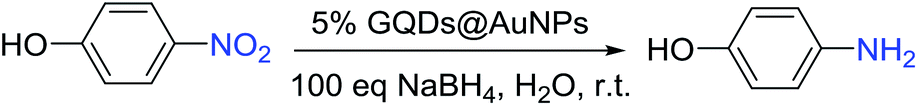DOI:
10.1039/C9RA02758K
(Paper)
RSC Adv., 2019,
9, 21215-21219
Synthesis of graphene quantum dot-stabilized gold nanoparticles and their application†
Received
12th April 2019
, Accepted 26th June 2019
First published on 9th July 2019
Abstract
Herein, we report an in situ synthesis of graphene quantum dots (GQDs), which have been synthesized from only starch and water and stabilize AuNPs in water. The construction of six gold nanocomposites, i.e. AuNPs 1–6, with sizes ranging from 13.4 nm to 32.6 nm, was accomplished by only mixing the GQDs and chloroauric acid in different amounts without any additional reductants and surfactants. HRTEM has confirmed that the AuNPs have been stabilized by the GQDs, and a core/shell AuNPs@GQD structure has formed. In addition, the as-synthesized AuNPs show excellent catalytic performance in the reduction of 4-nitrophenol, a pertinacious pollutant occurring in industrial wastewater.
1. Introduction
Green and facile original methods for the synthesis of transition metal nanoparticles (especially AuNPs) are still being extensively investigated because of multiple up-to-date applications of these NPs in biomedicine, sensors, electronic devices and catalysis.1 A number of natural or unnatural reductants (such as aminoboranes, citrate, hydrazine, sulfites, sodium borohydride, formaldehyde, alcohol, hydroxylamine, oxalic acid, hydrogen peroxide, ascorbic acid, dihydrogen, polyols and sugars) have been developed for the synthesis of AuNPs with different morphologies and attractive properties.2 Moreover, a large number of AuNP stabilizers are oxidizable by Au(III) without any additional reductants.3 However, the reduction mechanism is always unclear.
Graphene quantum dots (GQDs) are ultra-small fragments of graphene nanosheets with a two-dimensional lateral size (generally less than 100 nm). They are gaining extensive attraction due to their high chemical and thermal stability, low cytotoxicity, excellent solubility and biocompatibility, stable photoluminescence and high accessible surface area.4 Therefore, they have been widely used in bioimaging,5 photovoltaic devices,6 biosensing,7 and others.8 In general, GQDs are prepared by cleaving graphene, graphene oxide or carbon fibers under harsh conditions such as the use of a chemical oxidant (sulfuric acid or nitric acid) and a hydrothermal treatment.9 However, the purity of the GQDs is still affected by acid residues and metallic impurities. Therefore, it is still a challenge to develop an eco-friendly alternative method for the synthesis of GQDs. In 2018, a green and efficient one-pot hydrothermal synthesis of GQDs, with a narrow size distribution range from 2.25 to 3.50 nm, from only natural polymer starch and water was first reported by our group. In the synthetic process, at first, starch was mainly hydrolyzed to glucose, and it was subsequently converted into GQDs by cyclic condensation. Finally, the obtained GQDs were characterized by SEM, TEM, AFM, XPS, XRD, FTIR spectroscopy and Raman spectroscopy.10 Our group has a long-term interest in the synthesis of transition metal nanoparticles and their applications.11 Hence, in this study, we first report a green and facile method for the in situ synthesis of graphene quantum dot-stabilized AuNPs and their applications, where the GQDs have been obtained from only starch and water.10 The construction of the AuNPs 1–6 was accomplished by only mixing the GQDs and HAuCl4·3H2O in different amounts without any additional reductants and surfactants. Then, their efficiency as catalysts in the degradation of 4-nitrophenol upon reduction in water by sodium borohydride has been investigated by the kinetic studies including the determination of induction times and rate constants.
2. Results and discussion
2.1 Synthesis of the AuNPs
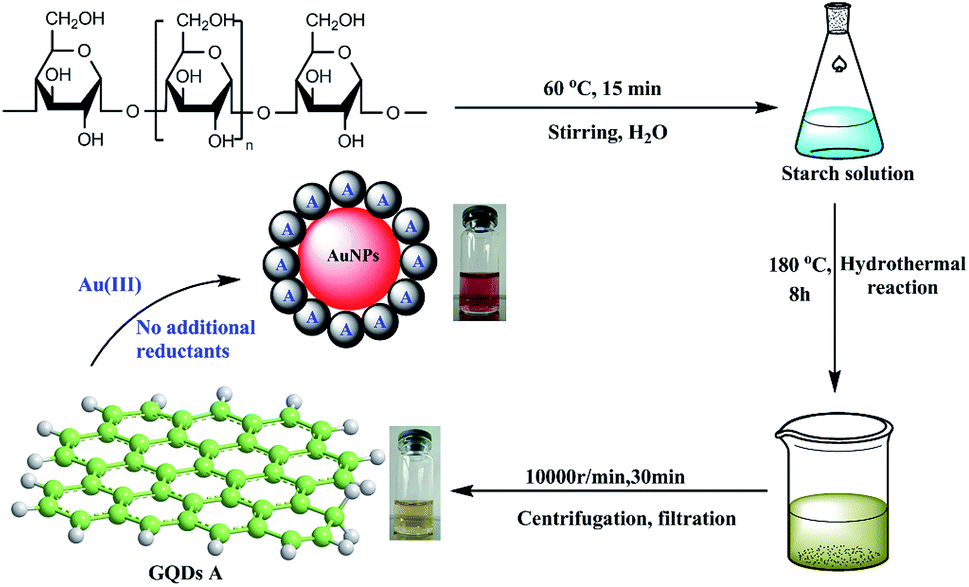
At first, the GQDs were synthesized from only commercial natural polymer starch and water using our previously reported method (Fig. 1).7 The obtained GQDs were well dispersed in water with a pale yellow color in sunlight, whereas the color would change to green under a 365 nm ultraviolet lamp (Fig. 2). This is owing to the characteristic unsaturated carbon–carbon double bond in GQDs. Transmission electron microscopy (TEM) also confirmed that the main diameter of the obtained GQDs ranged from 2.25 to 3.50 nm; then, HAuCl4·3H2O in different amounts (2.5 × 10−4 mmol, 0.625 × 10−3 mmol, 1.25 × 10−3 mmol, 2.5 × 10−3 mmol and 5.0 × 10−3 mmol) was added dropwise to the GQD solution at the same concentration at 0 °C, provoking an immediate formation of the claret (AuNP) color (Fig. 1), corresponding to the reduction of the cation to the zero-valent metal and the formation of the AuNPs 1–5. The AuNP-6 was synthesized at 30 °C using the same amount of HAuCl4·3H2O as used for AuNP-5. Furthermore, as a comparative experiment, the commercial natural polymer starch (instead of the GQDs) was tested under the same abovementioned condition. The result shows that the commercial natural polymer starch is insoluble in water at 0 °C or 30 °C and does not reduce gold(III) salts to AuNPs.
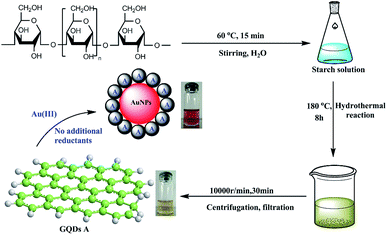 |
| | Fig. 1 The green synthesis of the AuNPs. | |
 |
| | Fig. 2 A TEM image of the GQDs and an image of the GQDs under a UV-vis lamp (inset). | |
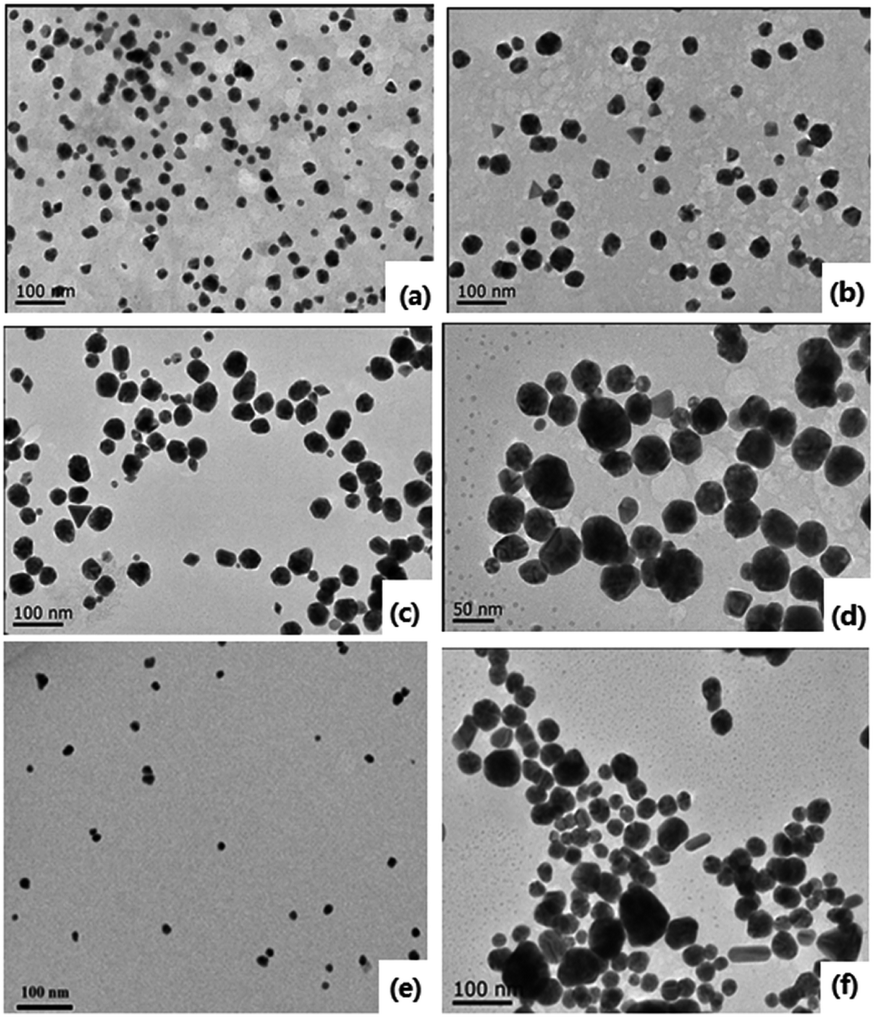
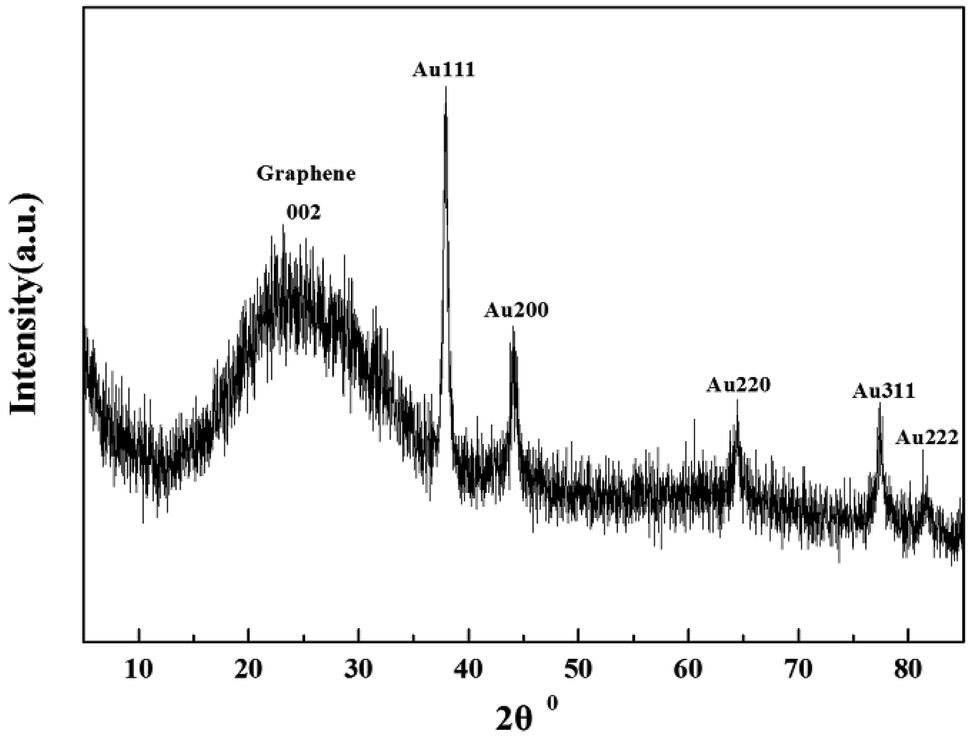
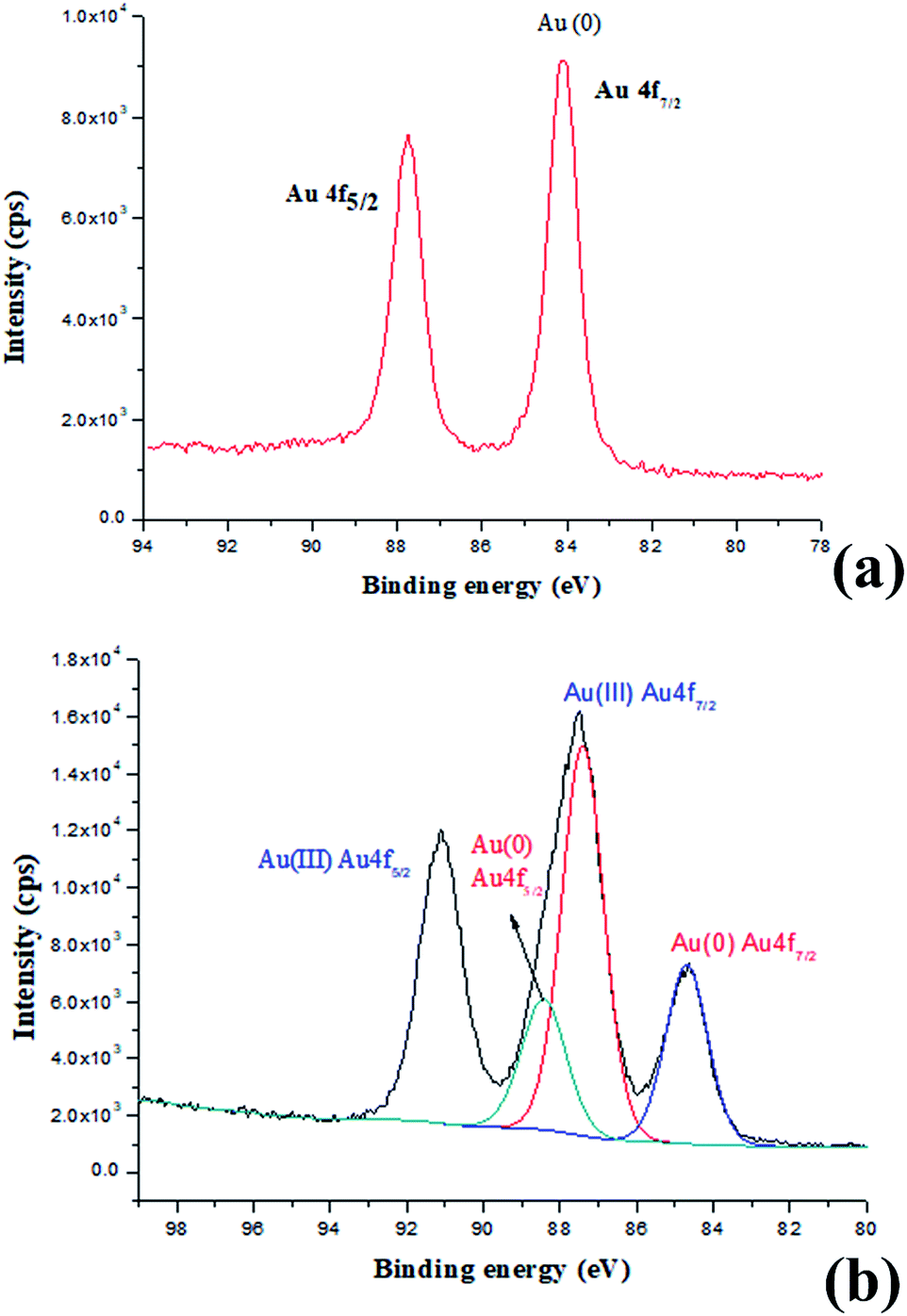
Indeed, the GQDs are also successful reductants for the reduction of gold(III) salts to AuNPs because the AuNPs 1–6 show a surface plasmon band (SPB) at approximately 530 nm, the characteristic peak of gold nanoparticles, in the observed UV-vis spectra (Table 1 and Fig. S1–S6 in ESI†). These AuNPs have been successfully stabilized via a net effect by the GQDs and the organic starch residue. Moreover, TEM was used to confirm the morphology and the size of the AuNPs. However, note that the reducing capacity of our GQDs is remarkably similar to that of ferrocene.12 Thus, the reduction of chloroauric acid was slightly slower with the GQDs than that with the classic reducing agent sodium borohydride; this led to the formation of much larger AuNPs than those in the latter case as the sizes of the obtained AuNPs 1–5 were in the range between 13.4 and 32.6 nm with different amounts of chloroauric acid (Fig. 3a–e). This size range is specifically optimal and useful for the application of AuNPs in cell imaging and biomedical usage.13 Moreover, the size of these AuNPs was modulated by varying the reaction temperature. The AuNP-6 (30.0 nm) obtained at 30 °C was much larger than the AuNP-5 obtained at 0 °C (13.4 nm). This is probably due to Ostwald ripening that occurs at 30 °C following the Au atom nucleation, which in turn causes a decrease in the number of Au nuclei and the AuNP overgrowth.14 Moreover, the representative high-resolution transmission electron microscopy (HRTEM) image of AuNP-5 exhibits the Au (111), (311), (220) and (200) lattice fringe distances of 0.231 nm, 0.122 nm, 0.148 nm and 0.202 nm in the inner region, respectively. However, the lattice spaces (020) and (002) of the graphitic carbon are 0.250 nm and 0.357 nm, respectively, on the surface of the nanoparticles (Fig. S7 in ESI†); it is obvious that the inner region of the AuNPs has been stabilized by the GQDs, and as a result, a core/shell AuNPs@GQD structure is formed. More importantly, in the FTIR spectrum of the AuNPs-5 (Fig. S8†), the peaks at 1631 and 1402 cm−1 are related to the skeletal vibrations of the aromatic rings of the GQDs, whereas the weak peaks at 1053 and 2923 cm−1 are the stretching vibrations of the C–O and C–H bond in the aromatic rings, respectively, confirming the honeycomb lattice of the graphene structure in the AuNP-5. Moreover, the UV-vis absorption peak intensity of the AuNPs versus reaction time for the AuNPs 1–6 was investigated. The result shows that the UV-vis absorption peak intensity of the AuNPs no longer increases after approximately 40 hours, indicating that the reductant (GQDs) has been used up (Fig. S9–S20 in ESI†). Furthermore, the crystalline nature of the AuNP-5 was determined using X-ray diffraction (XRD), where five Au diffraction peaks, i.e. Au (111), (200), (220), (311) and (222), as well as that of graphene (002) were observed in the 2θ range of 10–80°, which matched with the JCPDS card no. 75-0444 and 04-0784 (Fig. 4). To further verify the formation of the AuNPs, the X-ray photoelectron spectroscopy (XPS) spectra of AuNP 1 and 5 were obtained. The XPS spectra of 1 show the Au 4f peak positions at 84.0 and 87.6 eV. The binding energies of 84.0 eV and 87.6 eV for Au 4f7/2 and 4f5/2, respectively, are exclusively assigned to the zero-valent surface atoms of the AuNPs (Fig. 5a). In other words, the Au3+ ions were probably bonded and reduced by the –COOH and –OH groups of the GQDs, and further, the as-obtained AuNPs were stabilized by these –COOH and –OH groups. Au 4f was fitted with two pair of peaks for the Au 4f5/2 and 4f7/2 doublets in the XPS spectra of AuNP-5. The Au 4f7/2 peak positions were around 84.3 and 87.0 eV. These positions at 84.3 eV and 87.0 eV were assigned to Au(0) and Au(III), respectively (Fig. 5b). Obviously, chloroauric acid was not completely reduced by the GQDs under these reaction conditions as the amount of HAuCl4 gradually increased.15
Table 1 4-Nitrophenol reduction catalyzed by AuNPs in watera
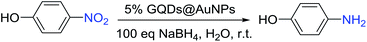
|
| Entry |
HAuCl4 (mmol) |
AuNPs |
T0c (s) |
kappd × 10−3 s−1 |
SPBe (nm) |
Df (nm) |
| 5.0 mol% AuNPs were used for catalyzing 4-nitrophenol reduction with NaBH4 being in excess (100/1). All the AuNPs 1–5 were synthesized at 0 °C except 6. The AuNP-6 was synthesized at 30 °C. Induction time. Rate constant. Surface plasmon band. Core size (from SEM or TEM) of the AuNPs. |
| 1 |
2.5 × 10−4 |
1 |
540 |
1.9 |
527 |
19.4 |
| 2 |
0.625 × 10−3 |
2 |
0 |
3.9 |
530 |
29.0 |
| 3 |
1.25 × 10−3 |
3 |
0 |
5.8 |
533 |
29.5 |
| 4 |
2.5 × 10−3 |
4 |
0 |
6.4 |
531 |
32.6 |
| 5 |
5.0 × 10−3 |
5 |
0 |
9.3 |
538 |
13.4 |
| 6b |
5.0 × 10−3 |
6 |
0 |
9.2 |
533 |
30.0 |
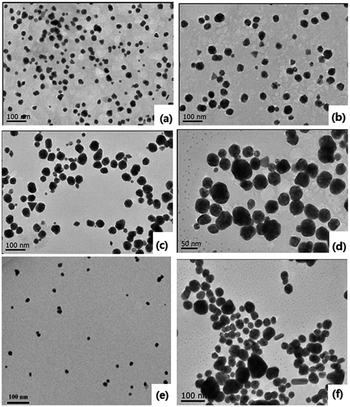 |
| | Fig. 3 TEM images of the AuNPs. (a)–(f) 1–6. | |
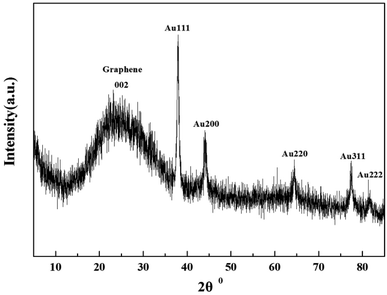 |
| | Fig. 4 XRD spectra of AuNPs-5. | |
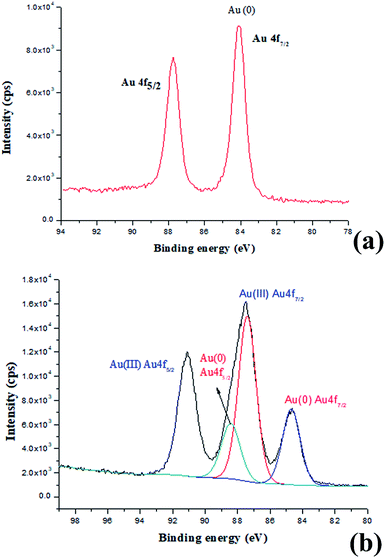 |
| | Fig. 5 (a) XPS spectra of AuNPs-1 and (b) XPS spectra of AuNPs-5. | |
2.2 Compared catalytic activities of 1–6 in the 4-nitrophenol reduction by NaBH4
4-Nitrophenol is the accessory substance of the procedures used for manufacturing pigments, pesticides, and pharmaceuticals in industry and hence regarded as one of the most highly poisonous and hazardous pollutants. The reduction of 4-nitrophenol to 4-aminophenol is extensively catalyzed by metal nanoparticles, and this reduction is well-known to be very sensitive to the nature of the metal nanoparticle surface.16 Moreover, it is very convenient to monitor the progress of the reduction of 4-nitrophenol to 4-aminophenol by UV-vis spectroscopic analysis due to their typical absorption peaks at λ = 400 nm (4-nitrophenol) and 300 nm (4-aminophenol).16c Therefore, to evaluate the catalysis capability of the synthesized AuNPs 1–6 as nanocatalysts, they were examined through the reduction of 4-nitrophenol (Table 1). The degradation of 4-nitrophenol has been carried out in the presence of 5.0 mol% AuNPs with 100 equiv of sodium borohydride in water at room temperature, and this large excess of sodium borohydride has been usually used in the literature to induce an apparent first-order kinetics and a relative lack of competitive sodium borohydride hydrolysis by water as a solvent. According to the experimental results, the reduction reaction did not need induction time in the presence of 5 mol% AuNPs except for the case of the AuNP-1 (Table 1, entries 1–6).
Only the catalysis by the AuNP-1 was found to be slow and required the induction time of 540 s due to the reorganization of the catalyst surface; this issue of induction time requirement was rectified by the stabilization of the AuNP-1 with excess GQDs that generated a partial steric inhibition for the substrate to access the surface of the AuNPs (Fig. 4a). Thus, the AuNP-1 was used in 5% mmol only, providing kapp = 1.9 × 10−3 (Table 1, entry 1). The reaction rate kapp of the corresponding AuNPs increases in the order of 1 < 2 < 3 < 4 < 5 simultaneously as the amount of chloroauric acid increases during the process of the synthesis (Fig. S21–S32 in ESI†). According to this rule, we also tried to synthesize AuNPs with high amount of chloroauric acid. Unfortunately, the AuNPs produced were unstable and immediately aggregated due to this concentration increase. Interestingly, the AuNP-6 showed the same reaction rate as AuNP-5, i.e. kapp = 9.2 × 10−3, although the size of 6 (30.0 nm) was almost two times that of 5 (13.4 nm). It is clear that the AuNP-5 shows the highest reaction rate among all the synthesized AuNPs (Table 1, entry 5), and this is consistent with the smallest size of 13.4 nm of the AuNP-5. Moreover, the GQDs alone do not catalyze the degradation of 4-nitrophenol under these conditions (Fig. S33 in ESI†). In addition, an overall mechanistic model of this reaction is shown and has been discussed in Fig. S34 in ESI.†
3. Conclusion
Herein, a green and facile method for the in situ synthesis of graphene quantum dot (GQD)-stabilized Au nanoparticles (AuNPs) in water was developed. It first involves the successful synthesis of GQD from only starch and water. The construction of the AuNPs 1–6 nanocomposites was accomplished by only mixing the GQDs and chloroauric acid in different amounts without any additional reductants and surfactants. The TEM images show that the sizes of the AuNPs 1–6 are between 13.4 and 32.6 nm. This size range is specifically optimal and useful for the application of AuNPs in cell imaging and biomedical usage. Furthermore, the HRTEM showed that the AuNPs (inner) were stabilized by GQDs, and consequently, a core/shell AuNPs@GQDs structure had emerged. The reducing driving force of these GQDs is much weaker than that of other strong reducing agents such as NaBH4, whereas it is remarkably similar to that of ferrocene. Both the GQDs and ferrocene reduce Au(III) with a modest driving force, which results in the formation of much larger AuNPs than those obtained using the strong reductant NaBH4. Furthermore, the catalytic efficiency of these different nanocomposites in the degradation of 4-nitrophenol by sodium borohydride in water was demonstrated, and these composites were compared by investigating their 4-nitrophenol reduction kinetics via the determination of induction times and rate constants. In conclusion, these GQD@AuNP nanocomposites show wide potential applications in the cell imaging, sensing, and catalysis fields.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support received from the National Natural Science Foundation of China (Grant No. 21805166 and 21606145), Natural Science Foundation of Hubei Province (Grant No. 2018CFB386), the 111 Project of Hubei Province (Grant No. 2018-19-1), State Key Laboratory of Coordination Chemistry Foundation of Nanjing University (No. SKLCC1811) and China Three Gorges University is gratefully acknowledged.
Notes and references
-
(a) M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS;
(b) G. Yue, S. Su, N. Li, M. Shuai, X. Lai, D. Astruc and P. Zhao, Coord. Chem. Rev., 2016, 311, 75–84 CrossRef CAS;
(c) C. Gautier and T. Burgi, ChemPhysChem, 2009, 10, 483–492 CrossRef CAS;
(d) J. Zheng, C. Zhou, M. Yu and J. Liu, Nanoscale, 2012, 4, 4073–4083 RSC;
(e) P. Ghosh, G. Han, M. De, C. Kyu, K. Vincent and M. Rotello, Adv. Drug Delivery Rev., 2008, 60, 1307–1315 CrossRef CAS;
(f) R. A. Sperling, P. R. Gil, F. Zhang, M. Zanellaa and W. J. Parak, Chem. Soc. Rev., 2008, 37, 1896–1908 RSC;
(g) R. Sardar, A. M. Funston, P. Mulvaney and R. W. Murray, Langmuir, 2009, 25, 13840–13851 CrossRef CAS;
(h) D. A. Giljohann, D. S. Seferos, W. L. Daniel, M. D. Massich, P. C. Patel and C. A. Mirkin, Angew. Chem., Int. Ed., 2010, 49, 3280–3294 CrossRef CAS;
(i) W. Zhou, X. Gao, D. Liu and X. Chen, Chem. Rev., 2015, 115, 10575–10636 CrossRef CAS;
(j) X.-Q. Qiao, Z.-W. Zhang, F.-Y. Tian, D.-F. Hou, Z.-F. Tian, D.-S. Li and Q.-C. Zhang, Cryst. Growth Des., 2017, 17, 3538–3547 CrossRef CAS;
(k) J. J. Du, C. Chen, Y. L. Gan, R. H. Zhang, C.-Y. Yang and X. W. Zhou, Energy, 2014, 39, 17634–17637 CAS;
(l) Z. Kang, X. Tan, X. Li, T. Xiao, L. Zhang and J. Lao, Phys. Chem. Chem. Phys., 2016, 18, 1992–1997 RSC.
- H. Kang, J. T. Buchman, R. S. Rodriguez, H. L. Ring, J. He, K. C. Bantz and C. L. Haynes, Chem. Rev., 2019, 119, 664–699 CrossRef CAS.
-
(a) P. Zhao, N. Li and D. Astruc, Coord. Chem. Rev., 2013, 257, 638–665 CrossRef CAS;
(b) N. Li, P. Zhao and D. Astruc, Angew. Chem., Int. Ed., 2014, 53, 1756–1789 CrossRef CAS PubMed.
-
(a) X. M. Li, M. C. Rui and J. Z. Song, Adv. Funct. Mater., 2015, 25, 4929–4947 CrossRef CAS;
(b) M. Bacon, S. J. Bradley and T. Nann, Part. Part. Syst. Charact., 2014, 31, 415–428 CrossRef CAS;
(c) L. S. Li and X. Yan, J. Phys. Chem. Lett., 2010, 1, 2572–2576 CrossRef CAS;
(d) H. Tetsuka, R. Asahi, A. Nagoya, K. Okamoto, I. Tajima, R. Ohta and A. Okamoto, Adv. Mater., 2012, 24, 5333–5338 CrossRef CAS;
(e) N. Suzuki, Y. Wang, P. Elvati, Z.-B. Qu, K. Kim, S. Jiang, E. Baumeister, J. Lee, B. Yeom, J. H. Bahng, J. Lee, A. Violi and N. A. Kotov, ACS Nano, 2016, 10, 1744–1755 CrossRef CAS PubMed;
(f) J. Peng, W. Gao, B. K. Gupta, Z. Liu, R. R. Aburto, L. Ge, L. Song, L. B. Alemany, X. Zhan, G. Gao, S. A. Vithayathil, B. A. Kaipparettu, A. A. Marti, T. Hayashi, J.-J. Zhu and P. M. Ajayan, Nano Lett., 2012, 12, 844–849 CrossRef CAS;
(g) C. X. Guo, H. B. Yang, Z. M. Sheng, Z. S. Lu, Q. L. Song and C. M. Li, Angew. Chem., Int. Ed., 2010, 49, 3014–3017 CrossRef CAS;
(h) L. A. Ponomarenko, F. Schedin, M. I. Katsnelson, R. Yang, E. W. Hill, K. S. Novoselov and A. K. Geim, Science, 2008, 320, 356–358 CrossRef CAS;
(i) F. Liu, M.-H. Jang, H. D. Ha, J.-H. Kim, Y.-H. Cho and T. S. Seo, Adv. Mater., 2013, 25, 3657–3662 CrossRef CAS.
-
(a) W. S. Kuo, H. H. Chen, S. Y. Chen, C. Y. Chang, P. C. Chen, Y. I. Hou, Y. T. Shao, H. F. Kao, C. L. Lilian Hsu, Y. C. Chen, S. J. Chen, S. R. Wu and J. Y. Wang, Biomaterials, 2017, 120, 185–194 CrossRef CAS;
(b) W. Chen, J. Shen, G. Lv, D. Li, Y. Hu, C. Zhou, X. Liu and Z. Dai, ChemistrySelect, 2019, 4, 2898–2902 CrossRef CAS.
- K. A. Ritter and J. W. Lyding, Nat. Mater., 2009, 8, 235–242 CrossRef CAS.
-
(a) X. Niu, Y. Zhong, R. Chen, F. Wang, Y. Liu and D. Luo, Sens. Actuators, B, 2018, 255, 1577–1581 CrossRef CAS;
(b) Q. Lu, W. Wei, Z. Zhou, Z. Zhou, Y. Zhang and S. Liu, Analyst, 2014, 139, 2404–2410 RSC;
(c) W. Lv, Y. Ju, Y. Chen and X. Chen, Int. J. Hydrogen Energy, 2018, 43, 10334–10340 CrossRef CAS;
(d) Y. Zhua, S. Lu, A. G. Manoharia, X. Dong, F. Chen, W. Xu, Z. Shi and C. Xu, J. Electroanal. Chem., 2017, 796, 75–81 CrossRef.
-
(a) X. Zhou, Y. Zhang, C. Wang, X. Wu, Y. Yang, B. Zheng, H. Wu, S. Guo and J. Zhang, ACS Nano, 2012, 6, 6592–6599 CrossRef CAS;
(b) J. P. Melo, P. L. Ríos, P. Povea, C. Morales-Verdejo and M. B. Camarada, ACS Omega, 2018, 3, 7278–7287 CrossRef CAS.
- W. Chen, G. Lv, W. Hu, D. Li, S. Chen and Z. Dai, Nanotechnol. Rev., 2018, 7, 157–185 CAS.
- W. Chen, D. Li, L. Tian, W. Xiang, T. Wang, W. Hu, Y. Hu, S. Chen, J. Chen and Z. Dai, Green Chem., 2018, 20, 4438–4442 RSC.
-
(a) X. Liu and D. Astruc, Coord. Chem. Rev., 2018, 359, 112–126 CrossRef CAS;
(b) X. Liu and D. Astruc, Adv. Mater., 2017, 29, 1605305 CrossRef;
(c) X. Liu, J. Ruiz and D. Astruc, Chem. Commun., 2017, 53, 11134–11137 RSC;
(d) X. Liu and D. Astruc, Adv. Synth. Catal., 2018, 360, 3426–3459 CrossRef CAS;
(e) X. Liu and J.-R. Hamon, Coord. Chem. Rev., 2019, 389, 94–118 CrossRef CAS;
(f) X. Liu, D. Gregurec, J. Irigoyen, A. Martinez, S. Moya, R. Ciganda, P. Hermange, J. Ruiz and D. Astruc, Nat. Commun., 2016, 7, 13152 CrossRef CAS;
(g) X. Liu, J. Ruiz and D. Astruc, J. Inorg. Organomet. Polym., 2018, 28, 399–406 CrossRef CAS;
(h) D. Astruc, C. Deraedt, R. Djeda, C. Ornelas, X. Liu, A. Rapakousiou, J. Ruiz, Y. Wang and Q. Wang, Molecules, 2018, 23, 966 CrossRef;
(i) X. Liu, Y. Huang, X. Meng, J. Li, D. Wang, Y. Chen, D. Tang and B. Chen, Synlett, 2019, 30, 1026–1036 CrossRef CAS;
(j) J. Yan, G. Liu, N. Li, N. Zhang and X. Liu, Eur. J. Inorg. Chem., 2019, 2806–2810 CrossRef CAS.
-
(a) R. Ciganda, J. Irigoyen, D. Gregurec, R. Hernandez, S. Moya, C. Wang, J. Ruiz and D. Astruc, Inorg. Chem., 2016, 55, 6361–6363 CrossRef CAS;
(b) R. Ciganda, H. Gu, R. Hernández, A. Escobar, A. Martínez, L. Yate, S. Moya, J. Ruiz and D. Astruc, Inorg. Chem., 2017, 56, 2784–2791 CrossRef CAS;
(c) D. Astruc, Eur. J. Inorg. Chem., 2017, 6–29 CrossRef CAS.
- K. Saha, S. S. Agasti, C. Kim, X. N. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS.
- Y. Bia and G. Lu, Chem. Commun., 2008, 6402–6404 RSC.
- M. Yamashita, H. Ohashi, Y. Kobayashi, Y. Okaue, T. Kurisaki, H. Wakita and T. Yokoyama, J. Colloid Interface Sci., 2008, 319, 25–29 CrossRef CAS.
-
(a) A. A. Pal and T. Pal, Chem. Commun., 2015, 51, 9410–9431 RSC;
(b) P. Hervés, M. Pérez-Lorenzo, L. M. Liz-Marzàn, J. Dzubiella, Y. Lu and M. Ballauff, Chem. Soc. Rev., 2012, 41, 5577–5587 RSC;
(c) P. Zhao, X. Feng, D. Huang, G. Yang and D. Astruc, Coord. Chem. Rev., 2015, 287, 114–136 CrossRef CAS;
(d) C. Deraedt, L. Salmon, S. Gatard, R. Ciganda, R. Hernandez, J. Ruiz and D. Astruc, Chem. Commun., 2014, 50, 14194–14196 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02758k |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab