
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective hydrodeoxygenation of p-cresol as a model for coal tar distillate on Ni–M/SiO2 (M = Ce, Co, Sn, Fe) bimetallic catalysts
Liuyi Pan ab,
Yulong Hea,
Menglong Niuc,
Yong Dana and
Wenhong Li*a
ab,
Yulong Hea,
Menglong Niuc,
Yong Dana and
Wenhong Li*a
aSchool of Chemical Engineering, Northwest University, Xi'an, Shaanxi 710069, P. R. China. E-mail: liwenhong@nwu.edu.cn
bCollege of Chemistry & Chemical Engineering, Baoji University of Arts and Sciences, Baoji 721013, Shaanxi, China
cCollege of Chemistry & Chemical Engineering, Xi'an Shiyou University, Xi'an, Shaanxi Province 710065, P. R. China
First published on 5th July 2019
Abstract
Ni–M/SiO2 with different binary metals M (M = Ce, Co, Sn, Fe) prepared by an incipient impregnation method was used in the hydrodeoxygenation (HDO) of low-temperature coal tar distillate, which is rich in phenolic compounds. p-Cresol, as a model compound of the distillate, was used to evaluate the activity and selectivity of BTX products on the series of reduced Ni–M/SiO2 catalysts in a fixed bed reactor. The properties of the catalysts were characterized by N2 adsorption–desorption, ICP-AES, XRD, H2-TPR, and XPS. Benzene and toluene as the direct deoxygenation (DDO) products and cyclohexane and methylcyclohexane as the hydrogenolysis (HYD) products were detected to evaluate the selectivity of the path in the deoxygenation process. In this series of catalysts, the order of reactivity was Ni–Ce > Ni–Sn > Ni–Co > Ni–Fe > monometallic Ni. Meanwhile, the addition of Ce and Co loaded in the Lewis acid sites of the catalyst affected the electron distribution of nickel atom and its atomic arrangement on the surface of the carrier. Compared to monometallic Ni, the DDO path become dominant on Ni–Ce and Ni–Co and the selectivity for BTX products increased from 58.8% to 77.4% and 71.1%, respectively. The binary metal Sn, unlike the former two metals, formed a Ni3Sn crystal form with Ni, which resulted in significant enhancement of the HYD path while obviously increasing the reactivity.
1 Introduction
The production of fuels such as gasoline and diesel by the hydrogenation of coal tar has been frequently reported and has been industrialized.1–3 In low-temperature coal tar, more than 90% of the distillate below 230 °C consists of phenolic compounds,4,5 mainly monocyclic phenols, which account for more than 60% of the total phenols.6 In the process of hydrotreating, these compounds often lead to the formation of large amounts of naphthenes and water; thus, it is not only difficult to obtain high-octane fuel, but the catalyst life is adversely affected.7 Moreover, if the intent is to use the hydrogenation product downstream to produce BTX, a dehydrogenation reforming process is also necessary.8 Therefore, the selective hydrodeoxygenation of monocyclic phenolic compounds enriched in coal tar can improve the quality of fuel oil and can also benefit the production of BTX products.9,10Hydrodeoxygenation of monocyclic phenolic compounds usually involves two basic reaction pathways. One involves breaking C–O bonds to obtain aromatics, called direct deoxygenation (DDO); the other proceeds via prehydrogenation of the aromatic rings, leading to alicyclic hydrogenation dehydrogenation (HYD).11,12 The traditional catalyst used in hydrotreating coal tar is generally a sulfided catalyst which contains γ-Al2O3 as a carrier and is loaded with active metals such as Ni, W, Co, and Mo.13–15 The sulfurized CoMo/γ-Al2O3 catalysts used in studies on phenol hydrodeoxygenation possess both high reactivity and high selectivity but are deactivated easily.16,17 Senol18 studied HDO experiments of phenol on sulfided CoMo/Al2O3 and NiMo/Al2O3 catalysts. The results showed that the main hydrodeoxygenation product of phenol on the sulfided Ni–Mo catalyst was alicyclics, while the product on Co–Mo was mainly benzene; also, the catalyst deactivation was mainly due to coking and loss of sulfur atoms. Viljava19 observed obvious coke deactivation of CoMo/Al2O3 catalyst during the HDO of guaiacol, especially at high reaction temperatures.
Supported noble metal catalysts are also common in HDO.17,20,21 Gutierrez22 studied the HDO of p-cresol on zirconia-supported mono- and bimetallic noble metal (Rh, Pd, Pt) catalysts. They found that the noble metal catalysts simultaneously showed higher catalytic activity and lower carbon deposition compared with the conventional sulfided CoMo catalyst. As new hydroprocessing catalysts, the application of transition metal phosphides in HDO has attracted great attention in recent years. Especially, metal–nickel phosphide catalysts show superior reactivity and sulfur resistance.23–26 Zhao27 prepared a series of metal phosphide catalysts, including Ni2P/SiO2, Fe2P/SiO2, MoP/SiO2, Co2P/SiO2 and WP/SiO2, for hydrodeoxygenation of guaiacol. The results showed that Ni2P/SiO2 has the highest reactivity, and the products were mainly phenol and benzene. In contrast, commercial CoMoS/Al2O3 showed little activity and was deactivated quickly. Metal-reduced catalysts are widely used in HDO processes, especially those loaded with nickel.28,29 In general, a small amount of a binary metal, such as La,30 Mo,31 Cu,32,33 or Fe,33 is considered to improve stability and reactivity.
Various types of catalysts have been studied in the hydrodeoxygenation reactions of phenol, guaiacol, anisole et al. as bio-oil models. However, phenolic compounds in coal tar have different species and distributions than those in bio-oil; these compounds mainly consist of methylphenol, dimethylphenol and higher phenols (such as naphthol and indophenol). Few studies have been reported on the selective hydrodeoxygenation of these phenolic compounds to obtain BTX products via the DDO pathway. Especially, research about the effects of different Ni–M binary metals on the hydrodeoxygenation pathway of alkyl phenols is rare. In the present study, a series of Ni–M/SiO2 bimetallic catalysts (M = Ce, Co, Sn, Fe) were prepared by an incipient-wetness impregnation method. p-Cresol, a monocyclic phenolic compound with high content in the fraction of coal tar, was selected as a model compound to evaluate the deoxidation and transalkylation during the hydrogenation process. The fixed bed hydrogenation reaction was carried out in the temperature range of 330 °C to 390 °C to evaluate the activity and product selectivity of the catalysts, and the catalysts were also characterized by BET, XRD, H2-TPR, NH3-TPD and XPS.
2 Experimental
2.1 Materials and chemicals
p-Cresol (≥98.0%), Ni(NO3)2·6H2O (≥98.0%), and SnC2O4 (≥98.5%) were obtained from Aladdin Chemical Reagent Co. Ltd., while Ce(NO3)3·6H2O (≥99.0%), Fe(NO3)3·9H2O (≥98.5%), and Co(NO3)2·6H2O (≥98.5%) were supplied by Sinopharm Chemical Reagent Co. Ltd. SiO2 was purchased from a local manufactory.2.2 Preparation of catalysts
Five catalysts, Ni/SiO2, Ni–Ce/SiO2, Ni–Ce/SiO2, Ni–Sn/SiO2, and Ni–Fe/SiO2, were prepared through an incipient wetness impregnation method in which the Ni loading was kept constant at 20 wt% (calculated by carrier quality) and the Ni/M molar ratio was maintained at 10. First, 15 g SiO2 were dried at 120 °C for 12 h. Second, 14.86 g Ni(NO3)2·6H2O with different bimetallic nitrates (2.21 g Ce(NO3)3, 1.48 g Co(NO3)2·6H2O, 1.05 g SnC2O4, or 1.23 g Fe(NO3)3·9H2O; in addition, SnC2O4 should be hydrolyzed first with hydrochloric acid) were dissolved in 15 mL deionized water (the volume of dissolved water was calculated by measuring the water absorption of the carrier) in a beaker and treated by ultrasonic dissolution for 20 min. Then, the metal salt solution was slowly added dropwise to the carrier with magnetic stirring for 12 h at 45 °C. Third, the precursors were removed from the immersion solution by drying at 120 °C for 12 h and finally calcined at 550 °C for 4 h. The catalyst precursors were subjected to reduction treatment before being used in the reaction. The reduction conditions are as follows: H2/N2 (10% H2) mixed gas with a flow rate of 200 mL min−1; increase temperature from 30 °C to 350 °C at 5 °C min−1, maintain for 60 min at 350 °C, then increase again from 350 °C to 550 °C at 2 °C min−1 and maintain for another 4 h.2.3 Characterization of catalysts
N2 adsorption–desorption isotherms of the catalysts were measured by a Quantachrome SI instrument at −196 °C. Before these measurements, the catalyst samples were degassed under vacuum at 350 °C for 3 h. The Brunauer–Emmett–Teller (BET) equation was used to calculate the specific surface area (SBET). The average pore diameter was calculated using the desorption branch of the isotherm according to the BJH method.Inductively coupled plasma-atomic emission spectroscopy (ICP-AES) measurements were carried out on an Agilent 725 instrument. Prior to these measurements, the samples were first dissolved in mixed acid with concentrated sulfuric acid and concentrated nitric acid in a certain proportion.
CO pulse chemisorption studies were carried out in an AutoChem II 2920 instrument (Micromeritics). 80 mg sample was first reduced in a H2 flow (50 mL min−1) at 550 °C for 1 h, then cooled with a He flow to 25 °C. After the TCD signal was stable, CO (50 μL) was pulsed in until the signal no longer changed; subsequently, the CO uptake was calculated.
Powder X-ray diffraction (XRD) was performed on a Bruker D8 Avance instrument with Cu-Kα radiation (λ = 1.5409 Å). The X-ray tube was operated at 40 kV and 40 mA. The XRD patterns of the samples were recorded in the range of 10° ≤ 2θ ≤ 80° with a scanning step size of 5°.
H2 temperature-programmed reduction (H2-TPR) was measured on a self-built test instrument containing a U-tube reactor, a heating jacket and a thermal conductivity detector (TCD). The catalyst sample (80 mg) was maintained in the reactor, then dried and degassed under N2 flow (50 mL min−1) at 200 °C for 2 h. Reduction was conducted at a heating rate of 10 °C min−1 from 200 °C to 800 °C in a 10 vol% H2/N2 flow (50 mL min−1).
Temperature-programmed desorption of ammonia (NH3-TPD) was carried out on the AutoChem II 2920 (Micromeritics) to evaluate the total acidities of the catalysts. The catalysts were reduced under flowing H2 (50 mL min−1) at 450 °C for 2 h. After cooling and cleaning with He (50 mL min−1) and adsorption of NH3 at 100 °C, the NH3-TPD was performed between 100 °C and 800 °C with a heating rate of 10 °C min−1 using a He flow.
X-ray photoelectron spectroscopy (XPS) was carried out using a ThermoFisher ESCALAB 250Xi with Al Kα radiation (1486.6 eV). The binding energies were referenced to C 1s at 284.8 eV.
2.4 HDO tests
HDO reactions were performed on a fixed-bed reactor (ID 17 mm, length 1200 mm) with separate gas and liquid feed systems and a thermocouple to monitor the reaction temperature. Before the hydrogenation reaction, 30 mL catalyst was placed in the reactor and reduced at 550 °C for 2 h in a H2 flow (100 mL min−1); then, the reactor was cooled to reaction temperature in H2 atmosphere. The liquid products were analyzed by an Agilent gas chromatograph (9790 A) equipped with an HP-INNOWax polar column (30 m × 0.32 mm × 0.5 mm) and a flame ionization detector.During the experiment, the main hydrogenation products were quantitatively analyzed by the external standard method; products with very low contents, such as biphenyls and olefin, were not calculated.
The conversion of p-cresol and the product selectivity were defined as follows:
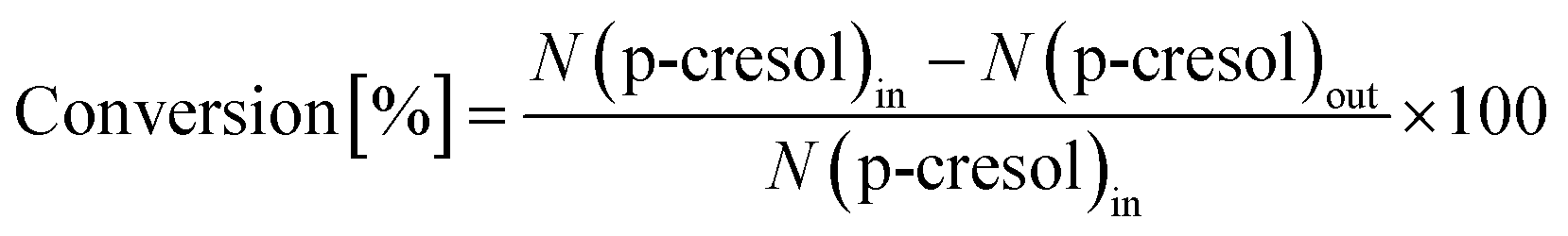
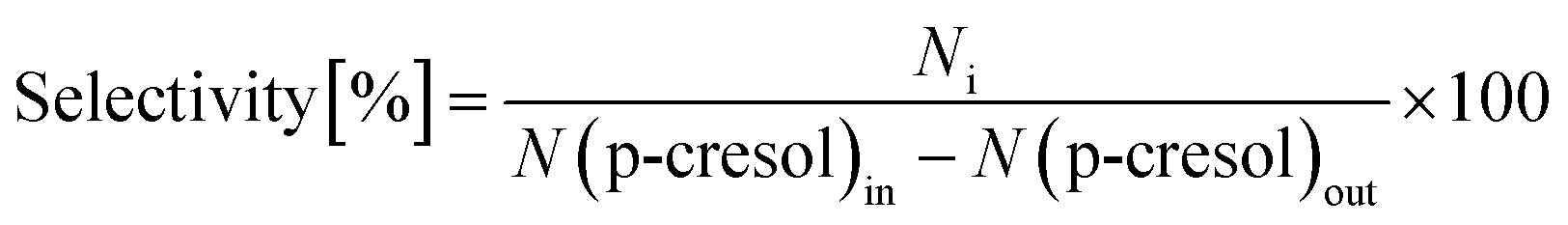
BTX products are the target products for this research. The main BTX products obtained by hydrogenation of p-cresol are benzene and cyclohexane. The content of xylene in the product is extremely low; thus, it was neglected when calculating selectivityBTX.
The BTX selectivity was defined as follows:
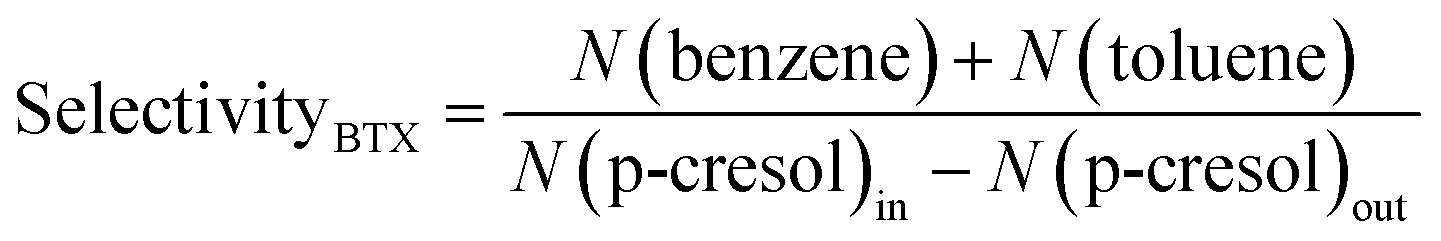
3 Results and discussion
3.1 Characterization of the catalysts
| Nia wt% | Ma wt% | BET surface areab (m2 g−1) | Pore volume | Pore size | Particle sizec nm | Dispersiond (%) | CO uptake mmol g−1 | Particle sizee nm | |
|---|---|---|---|---|---|---|---|---|---|
| a Determined by ICP-AES.b BET surface areas, pore volumes, and pore sizes determined by N2 physisorption.c Particle size calculated by the Scherrer formula.d Dispersion (D) calculated with particle size (d) by the equation D ≈ 0.9/d.e Calculated by CO pulse adsorption. | |||||||||
| SiO2 | 438 | 0.85 | 6.88 | ||||||
| Ni | 15.7 | — | 402 | 0.77 | 6.28 | 19.8 | 4.5 | 0.12 | 18.3 |
| Ni–Ce | 14.8 | 3.75 | 384 | 0.70 | 6.12 | 11.4 | 7.9 | 0.17 | 10.5 |
| Ni–Co | 14.6 | 1.48 | 371 | 0.69 | 6.01 | 12.2 | 7.4 | 0.16 | 11.5 |
| Ni–Sn | 14.3 | 3.10 | 389 | 0.72 | 6.04 | 14.6 | 5.5 | 0.18 | 9.9 |
| Ni–Fe | 15.0 | 1.42 | 360 | 0.71 | 6.14 | 13.7 | 6.6 | 0.13 | 15.1 |
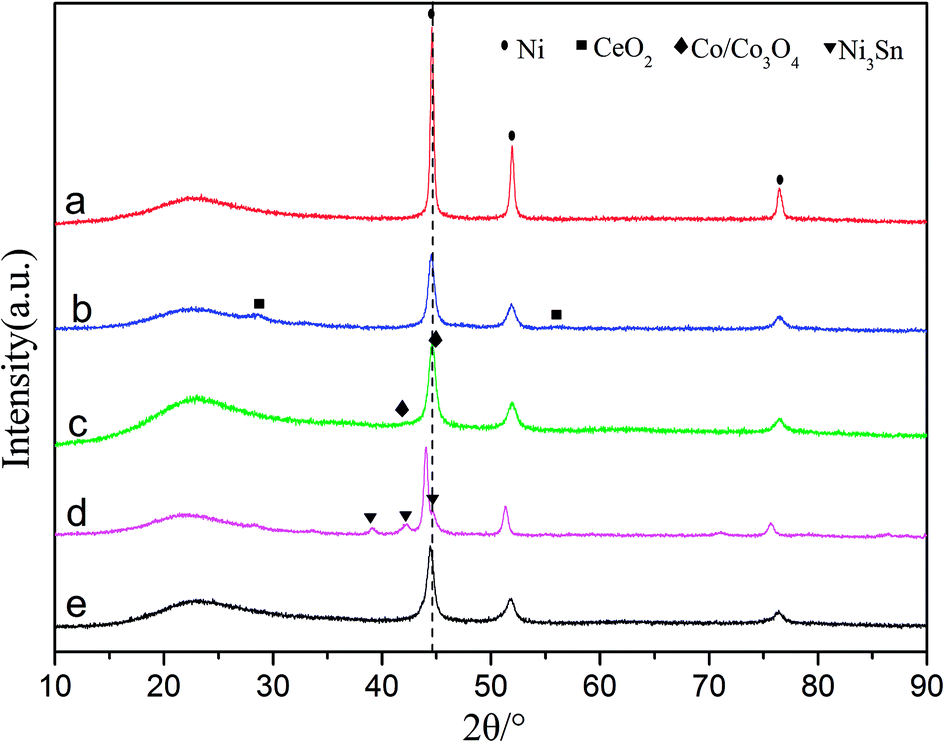 | ||
| Fig. 1 X-ray diffraction patterns for (a) Ni/SiO2, (b) Ni–Ce/SiO2, (c) Ni–Co/SiO2, (d) Ni–Sn/SiO2 and (e) Ni–Fe/SiO2. | ||
The average particle diameters of the Ni–M/SiO2 catalysts, as shown in Table 1, were calculated by Scherrer's formula through selecting the diffraction peak at 2θ = 51.8°; meanwhile, the dispersions of the active metal were calculated from the relationship between the particle diameter d and the degree of dispersion D (d ≈ 0.9/D). Simultaneously, CO chemisorption was used to characterize the particle sizes and dispersions, which are listed in Table 1. The particle sizes of the catalysts, measured by both methods, are distributed in the range of 10 to 18 nm; this indicates good consistency.37 After the addition of the binary metal, compared with Ni/SiO2, the particle size of Ni–M decreased, indicating that the dispersion of the active metal improved.27 Based on the premise that each active site adsorbs a CO molecule, the order of dispersion was Ni–Ce > Ni–Co > Ni–Fe > Ni–Sn > Ni, suggesting that the addition of Ce results in higher reactivity due to its effects on the dispersibility.
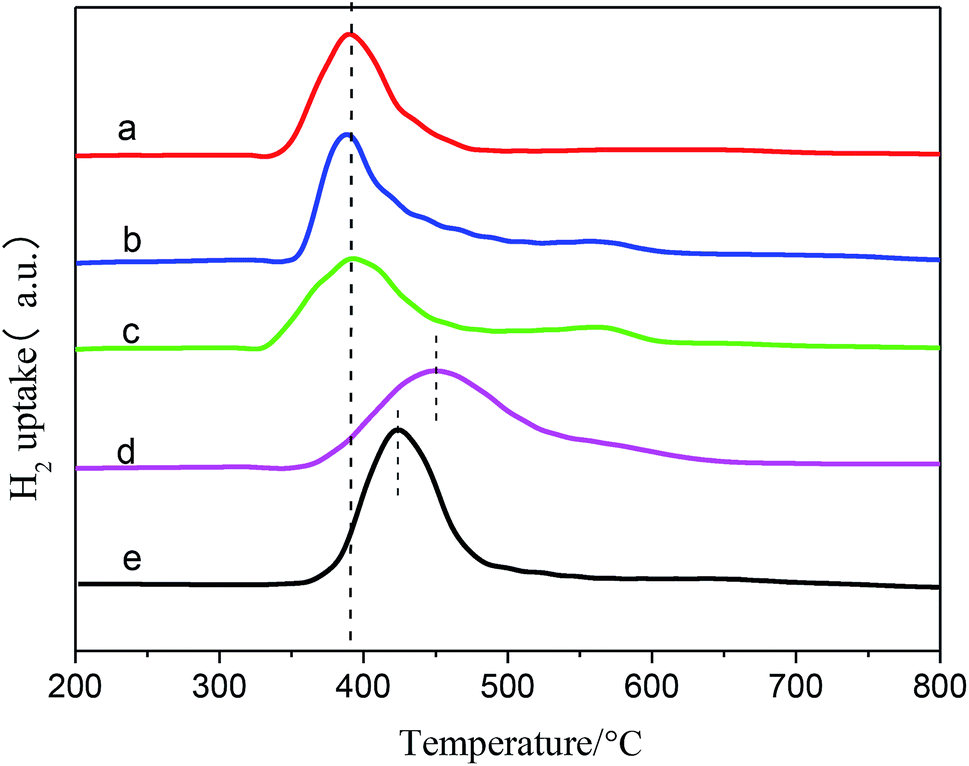 | ||
| Fig. 2 H2-TPR profiles of (a) Ni/SiO2, (b) Ni–Ce/SiO2, (c) Ni–Co/SiO2, (d) Ni–Sn/SiO2, (e) Ni–Fe/SiO2. | ||
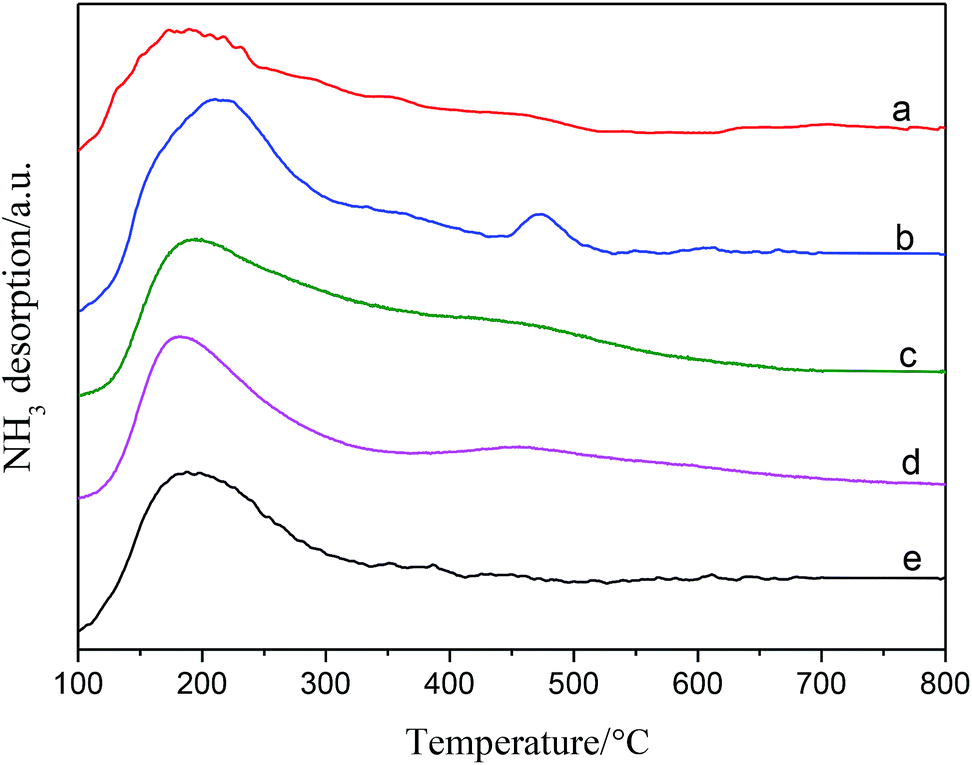 | ||
| Fig. 3 NH3-TPD profiles of (a) Ni/SiO2, (b) Ni–Ce/SiO2, (c) Ni–Co/SiO2, (d) Ni–Sn/SiO2, (e) Ni–Fe/SiO2. | ||
On the one hand, the acidity of the Ni–M catalyst increased in comparison with that of Ni/SiO2 with the addition of the binary metal, as can be seen from the acid quantitative results in Table 2.
This indicates that the reduced or partially oxidized binary metal M increased the number of new acid sites, especially cerium ions and cobalt ions, which were not completely reduced. On the other hand, the addition of a binary metal resulted in an improvement in the dispersion of the active metal. The aggregation and mutual coverage of the active metal Ni were prevented; therefore, more Lewis acid sites could be formed.46
Therefore, the total acid sites of the catalysts increased in the order of Ni–Ce/SiO2 > Ni–Co/SiO2 > Ni–Sn/SiO2 > Ni–Fe/SiO2 > monometallic Ni/SiO2, which is similar to the order of metal dispersion.
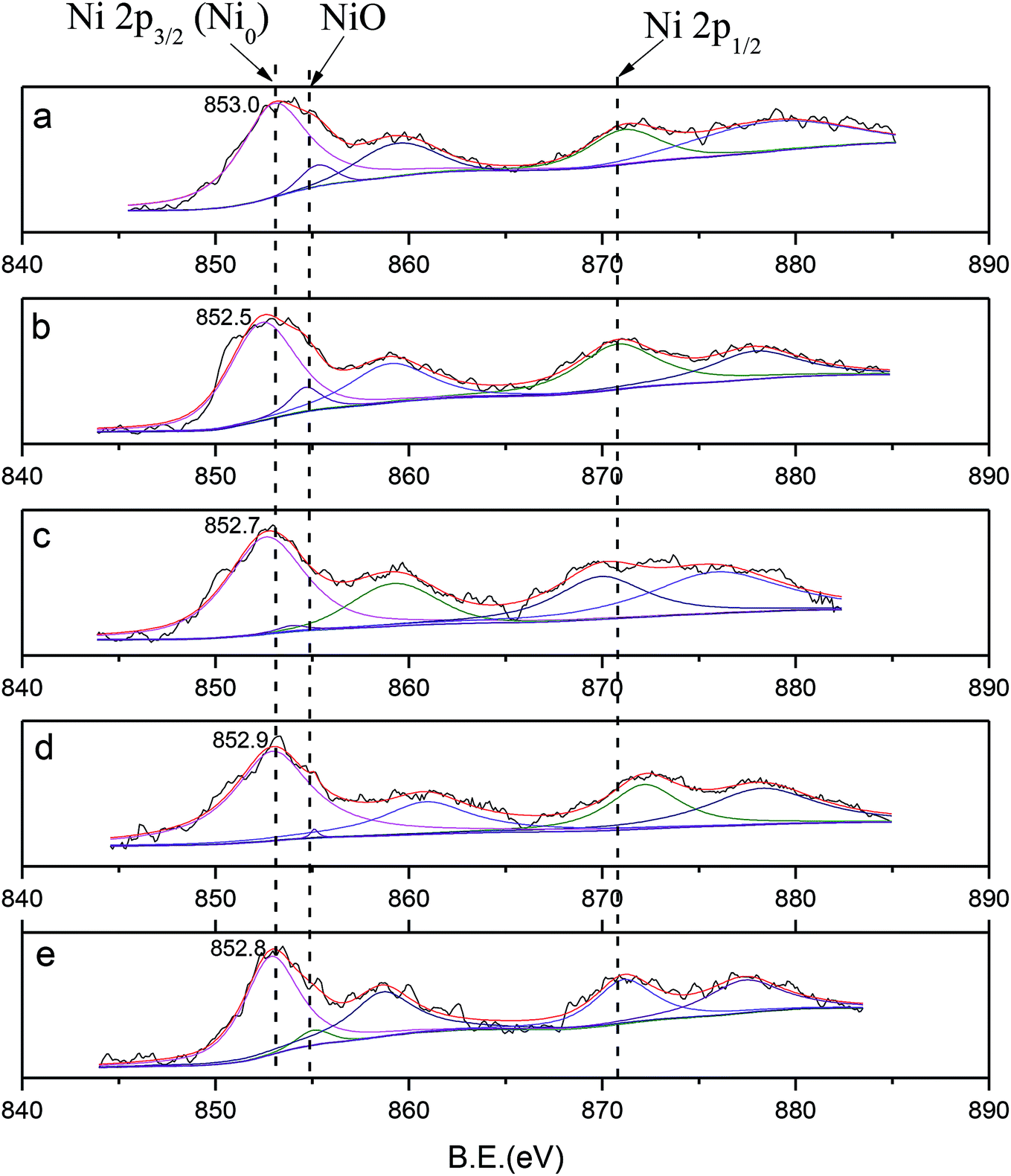 | ||
| Fig. 4 XPS profiles of (a) Ni/SiO2, (b) Ni–Ce/SiO2, (c) Ni–Co/SiO2, (d) Ni–Sn/SiO2, (e) Ni–Fe/SiO2. | ||
In comparison with monometallic Ni, shifts in BE towards lower values of about 0.1 eV to 0.5 eV were observed, from 853.0 eV (mono Ni) to 852.5 eV (Ce), 852.7 eV (Co), 852.9 eV (Sn), and 852.8 eV (Fe); this was in accordance with the electronegativity values of Ni (1.9), Ce (1.1), Co (1.8), Sn (1.7) and Fe (1.9). These shifts were mainly attributed to electron transfer from the binary metal M to nickel, which caused an increase in the electron density of the nickel atoms. In addition, the occurrence of electron transfer confirmed the interaction between Ni and the binary metal M.
In addition, the distribution of the valence states and atomic ratios on the surface are listed in Table 3. Ni2+ species with a small intensity peak observed at 854.8 eV (Ni/SiO2) showed that the samples may be partially oxidized when exposed to air.49 However, the distributions of Ni2+ in Ni–Ce (3.8%), Ni–Co (1.1%), Ni–Sn (0.7%) and Ni–Fe (2.1%) are lower than that observed for monometallic Ni (5.8%); this indicates that due to the interaction of Ni and the binary metal M, the phenomenon of Ni oxidation was prevented. The effect of Sn is especially obvious. In addition, compared with monometallic Ni, the Ni/Si ratios decreased after addition of M; among the metals, Sn and Ce showed the most obvious decreases. The decrease in the surface content of Ni reflects the increase in dispersion. This is in agreement with the CO uptake results and also confirms the superiority of the catalytic activity of Ni–Sn. In addition, Ni/M decreased from the theoretical 10 to 1 to 3.5, indicating that the dispersibility of Ni is better than that of M and that M atom and Ni are co-distributed on the surface.
| Catalyst | Distribution of valence states (%) | Atomic ratio | ||||||
|---|---|---|---|---|---|---|---|---|
| Ni0 2p3/2 | Ni0 2p1/2 | Ni2+ | Ni/Si | M/Si | Ni/M | |||
| Main | Sat. | Main | Sat. | |||||
| Ni | 43.5 | 13.4 | 13.8 | 23.5 | 5.8 | 0.15 | ||
| Ni–Ce | 37.7 | 18.7 | 23.5 | 16.3 | 3.8 | 0.06 | 0.04 | 1.5 |
| Ni–Co | 36.5 | 14.4 | 19.9 | 28.0 | 1.1 | 0.07 | 0.06 | 1.2 |
| Ni–Sn | 41.9 | 17.4 | 19.7 | 20.4 | 0.7 | 0.04 | 0.03 | 2 |
| Ni–Fe | 37.1 | 22.1 | 19.3 | 18.3 | 2.1 | 0.10 | 0.03 | 3.3 |
3.2 HDO tests
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, respectively. The distribution of the main products is shown in Fig. 5. The conversion of p-cresol, the selectivity of each product and the selectivity of the target products BTX are listed in Table 4. Consistently, DDO and HYD are basically parallel processes, and p-cresol is hydroconverted to toluene and methylcyclohexane through these two paths, respectively.34 However, based on the detection of the products, it is believed that there is a third reaction path in HDO for the hydrogenation of p-cresol, namely a demethylation reaction, which ultimately results in the production of cyclohexane and benzene after the HDO of phenol. However, the xylene in the product is presumably derived from the transalkylation reaction of toluene and benzene. The main cause of the weak dealkylation and alkylation phenomena is presumed to be the Lewis acid sites provided by the oxide of the unreduced binary metal M, as characterized by NH3-TPD and TPR.50
:1, respectively. The distribution of the main products is shown in Fig. 5. The conversion of p-cresol, the selectivity of each product and the selectivity of the target products BTX are listed in Table 4. Consistently, DDO and HYD are basically parallel processes, and p-cresol is hydroconverted to toluene and methylcyclohexane through these two paths, respectively.34 However, based on the detection of the products, it is believed that there is a third reaction path in HDO for the hydrogenation of p-cresol, namely a demethylation reaction, which ultimately results in the production of cyclohexane and benzene after the HDO of phenol. However, the xylene in the product is presumably derived from the transalkylation reaction of toluene and benzene. The main cause of the weak dealkylation and alkylation phenomena is presumed to be the Lewis acid sites provided by the oxide of the unreduced binary metal M, as characterized by NH3-TPD and TPR.50
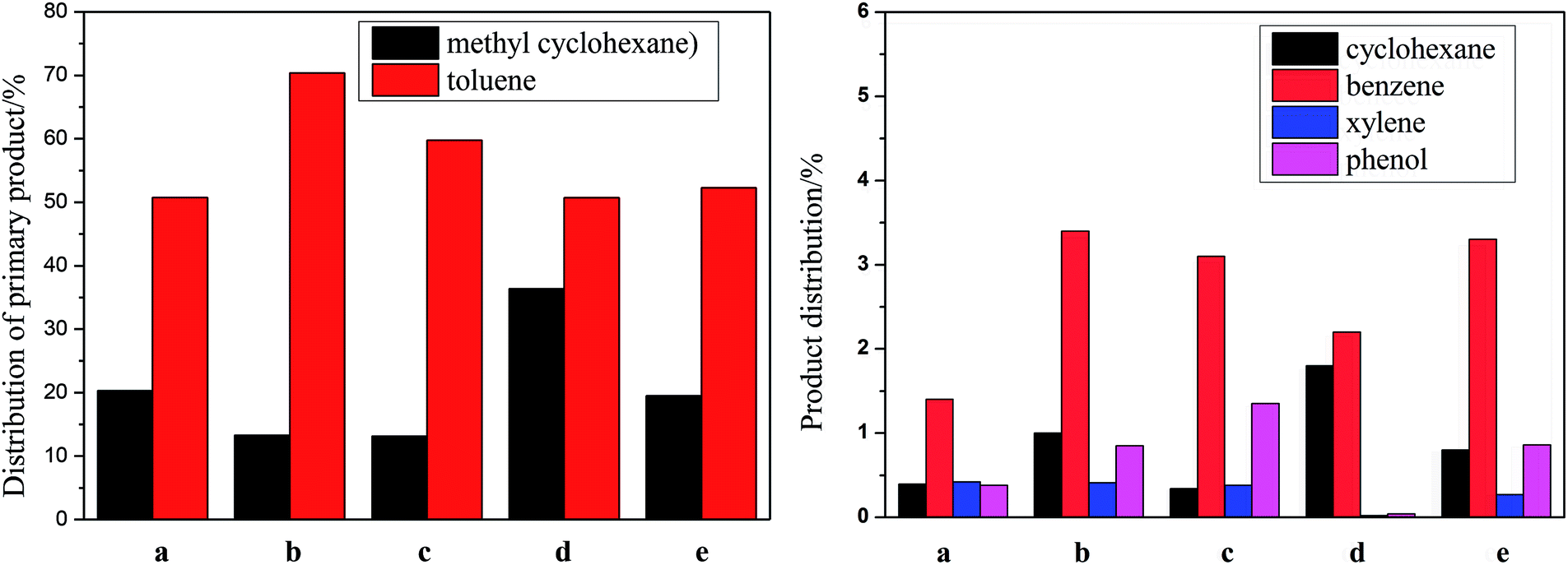 | ||
| Fig. 5 Product distributions of p-cresol over different Ni–M/SiO2 catalysts. (a) Ni/SiO2; (b) Ni–Ce/SiO2; (c) Ni–Co/SiO2; (d) Ni–Sn/SiO2; (e) Ni–Fe/SiO2. | ||
| Ni/SiO2 | Ni–Ce/SiO2 | Ni–Co/SiO2 | Ni–Sn/SiO2 | Ni–Fe/SiO2 | ||
|---|---|---|---|---|---|---|
| a Each catalyst was tested in three parallel experiments with an error less than 2%. | ||||||
| Conversiona/% | 80.5 | 95.4 | 88.5 | 94.1 | 82.5 | |
| TOF × 10−3/S−1 | 8.8 | 6.0 | 6.3 | 7.7 | 7.0 | |
| Selectivity/% | Cyclohexane | 0.7 | 1.0 | 0.4 | 1.9 | 1.0 |
| Benzene | 3.0 | 3.6 | 3.5 | 2.3 | 4.0 | |
| Methyl cyclohexane | 25.2 | 13.9 | 17.1 | 38.7 | 23.6 | |
| Toluene | 63.1 | 73.8 | 67.6 | 53.9 | 63.4 | |
| SBTX | 66.2 | 77.8 | 71.5 | 56.2 | 67.7 | |
| SBTX/Scycloparaffin | 2.5 | 5.2 | 4.1 | 1.4 | 2.7 | |
The conversion over these catalysts of HDO follows the order: Ni–Ce > Ni–Sn > Ni–Co > Ni–Fe > monometallic Ni. However, the TOFs calculated and listed in Table 4 are inconsistent with the conversion.
According to the characterization analysis of XPS, the addition of binary metal M, especially Ce and Co, resulted in a decrease in the number of d-bands of Ni atom due to the electron transfer from M to Ni; this eventually led to decreases in the intrinsic reaction rates of the catalysts. However, the addition of M improved the dispersion of the active metal; therefore, the specific activity of Ni–M/SiO2 reflected in the conversion was still higher than that of Ni/SiO2.51
In order to evaluate the selectivity of the different reaction pathways in the HDO of p-cresol, SBTX/Scycloparaffin was defined. The product distributions on Ni–Ce and Ni–Co were very different from that on the monometal Ni catalyst. In Ni–Ce and Ni–Co, the content of toluene as the main product of the DDO path increased observably; also, the selectivity of toluene, compared to that of the monometal Ni catalyst, increased from 63.1% to 77.8 and 71.5%, respectively. Moreover, SBTX/Scycloparaffin increased from 2.5 to 5.2 and 4.1, respectively. Interestingly, the DDO path had a dominant advantage on Ni–Co, although the activity of the catalyst did not improve greatly. The performance of Ni–Sn differs from the above two. Although the activity of Ni–Sn was significantly improved, SBTX/Scycloparaffin decreased sharply from 2.5 to 1.4; meanwhile, the selectivity of BTX product decreased from 66.2% (Ni) to 56.2%. This indicates that Ni–Sn promotes the aromatic hydrogenation of p-cresol; that is, the HYD path is enhanced, which ultimately leads to difficulties in obtaining BTX products. Unfortunately, the addition of Fe did not show good performance; the selectivity of the BTX product increased only slightly.
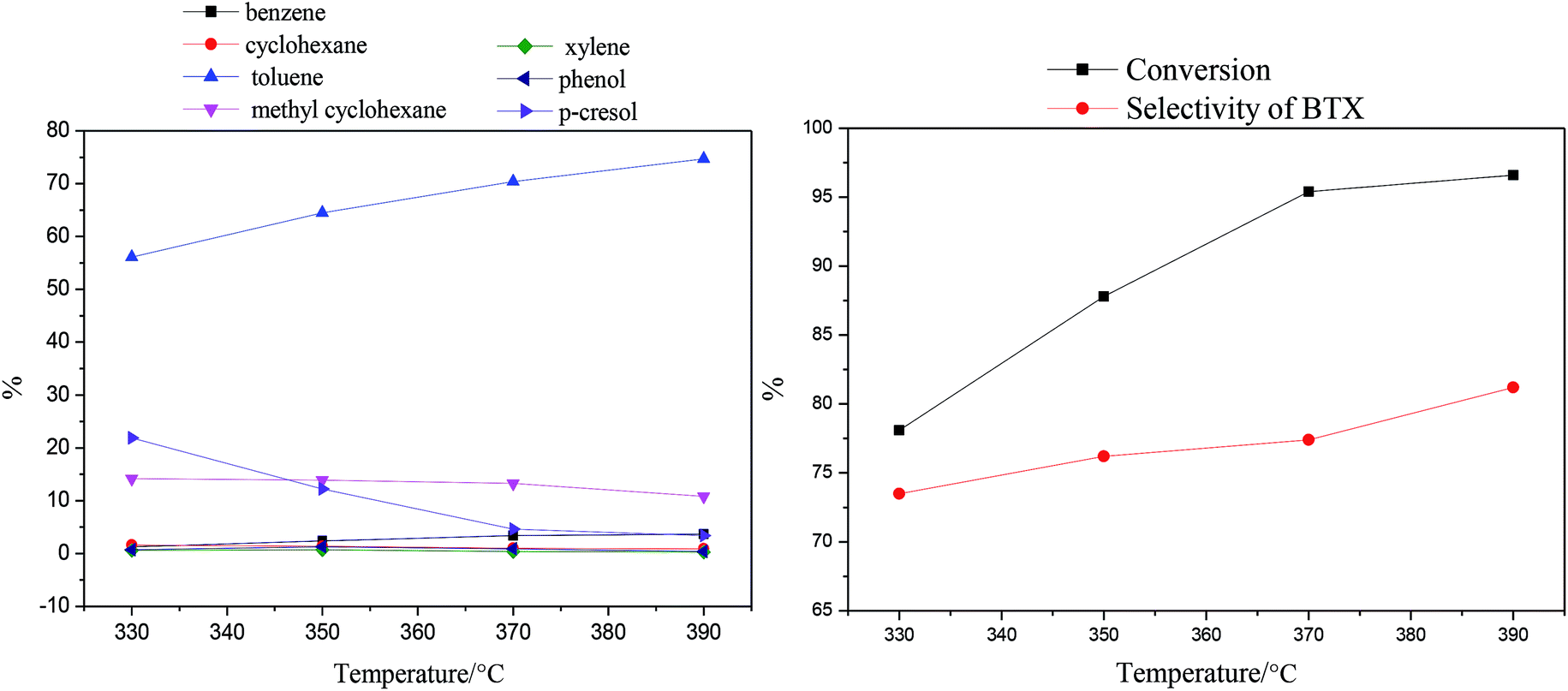 | ||
| Fig. 6 Effects of reaction temperature on the products and p-cresol content via HDO (LHSV = 0.5 h−1). | ||
As the temperature increases, the amount of p-cresol decreases from 21.9% to 3.4%; this indicates that the conversion increased from 78.1% to 96.6%. The amount of toluene obviously increased, reaching the maximum value of 74.7% at 390 °C. In contrast, the amount of methyl cyclohexane decreased gradually from 14.2% to 10.8% with increasing reaction temperature from 330 °C to 390 °C. In other words, the process of HDO of p-cresol, from which toluene is obtained by the cleavage of Caromatic–O bonds, was enhanced with increasing reaction temperature.52 Meanwhile, the process of HDO of p-cresol to the cycloalkane through the HYD path was prevented. Similarly, the amounts of benzene and cyclohexane were similar to those of toluene and methylcyclohexane, respectively. It is possible that the high reaction temperature not only promoted the cleavage of Caromatic–O bonds to improve the selectivity of the DDO pathway, but also enhanced the dealkylation reaction. In addition, the amounts of xylene and phenol, which were lower in the product distribution, both increased initially at temperatures below 350 °C and then decreased as the reaction temperature continued to rise. In summary, the selectivity of the BTX product eventually increased from 74.8% to 81.2% as the reaction temperature increased.
Fig. 7 shows the amounts of p-cresol and HDO products as well as the conversion and selectivity of BTX, with LHSV values of 0.25 h−1 to 1.0 h−1 at 370 °C.
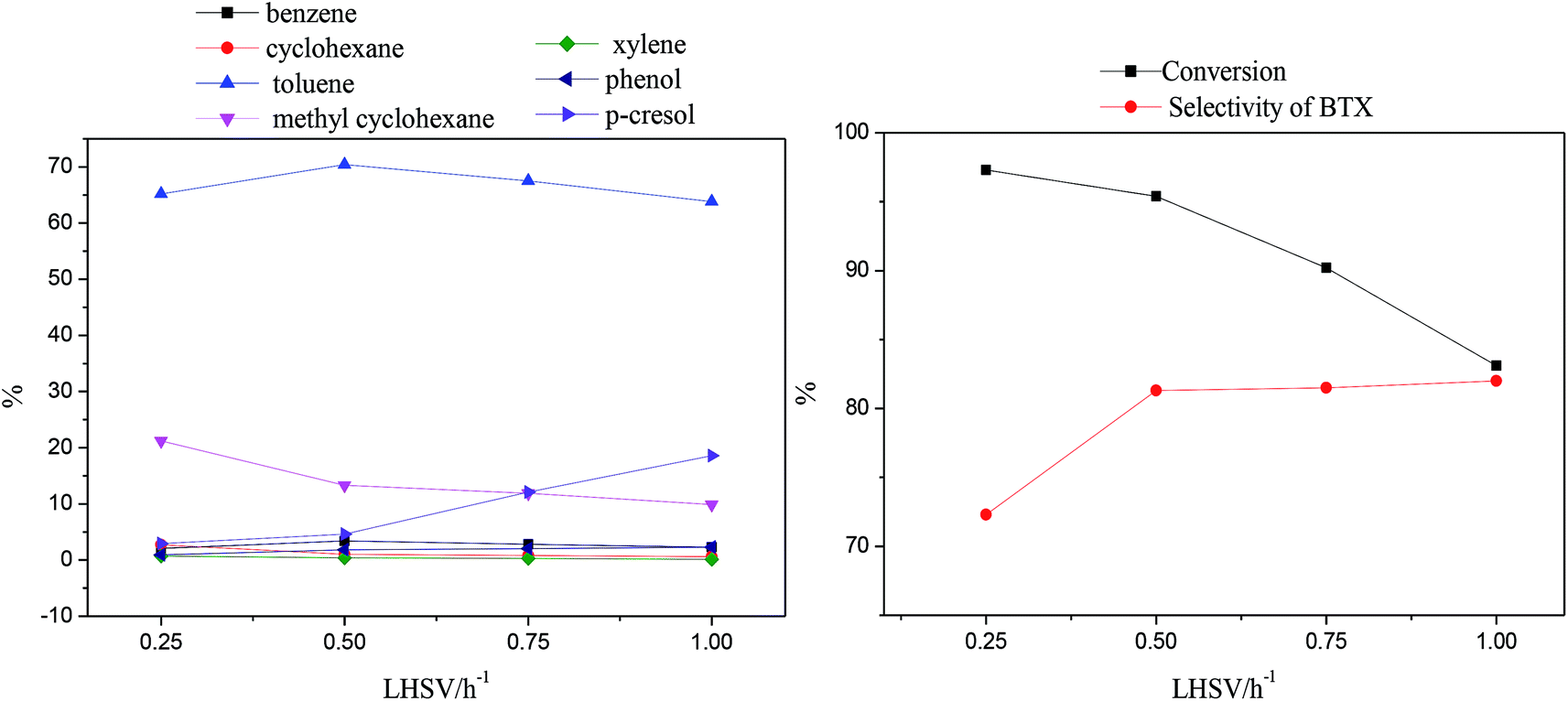 | ||
| Fig. 7 Effects of LHSV on the products and p-cresol content via HDO. | ||
As LHSV increased, the content of p-cresol increased from 2.7% to 16.9%, indicating that the conversion decreased from 97.3% to 83.1%; this is mainly because a higher LHSV results in shorter contact times of the reactants with the catalyst sites.
The amounts of cyclohexane and methylcyclohexane decreased respectively from 2.7% and 21.2% to 0.6% and 9.9% with increasing LHSV; this also indicates that the decrease in residence time decreased the depth of hydrogenation, resulting in the suppression of aromatic saturation. The amounts of benzene and toluene, as the main BTX products, showed changes of an initial increase followed by a decrease with increasing LHSV, reaching maximum values of 3.4% and 70.4% at 0.5 h−1, respectively. It can be speculated that the decreased amounts of BTX products caused by the high LHSV corresponds to low conversion; however, at 0.3 h−1, a low LHSV led to a large amount of aromatic ring consumption through full hydrogenation, which resulted in the conversion of aromatic hydrocarbons to naphthenic products.53
Moreover, the amount of xylene slightly decreased but the content of phenol obviously increased from 0.9% to 2.3% with increasing LHSV. It is possible that the longer residence time corresponding to a low LHSV was beneficial to the transalkylation reaction but also facilitated the consumption of phenol by further hydrodeoxygenation.
Previous studies have been conducted on the path of methylphenol hydrodeoxygenation.11,33,54 Odebunmi55 obtained similar reaction paths for the hydrodeoxygenation of cresol with Co–Mo/γ-Al2O3 catalyst. They concluded that cresol first produced an intermediate and then continued to produce toluene (k1), methylcyclohexane (k2) and phenol (k4), respectively, and that there was interconversion between toluene and methylcyclohexane (k3), showing (k1, k3) ≫ k2, k4 on fresh catalyst. Kirby56 also speculated that the reaction pathway of dibutylmethyl phenol hydrodeoxygenation on a novel organometallic catalyst precursor included dealkylation, direct deoxygenation, hydrodeoxygenation, and alkyl isomerization. By analyzing the product distributions at different reaction temperatures and LHSVs, a reaction conversion pathway of p-cresol on Ni–Ce/SiO2 is proposed in Scheme 1.
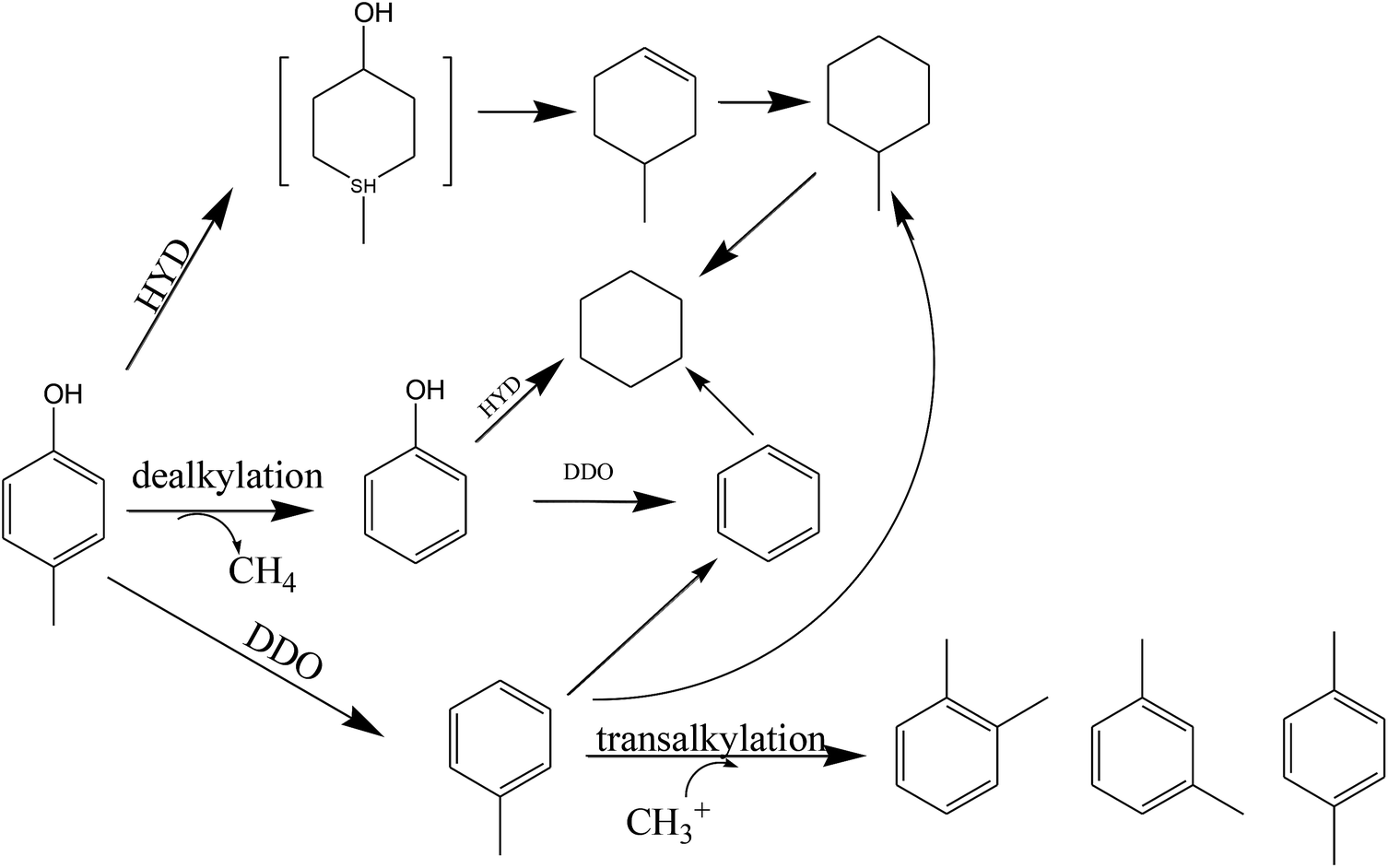 | ||
| Scheme 1 Reaction path of p-cresol on Ni–M/SiO2 catalysts. | ||
It can be seen from the HDO test that the addition of a small amount of different binary metal M results in distinct reaction path selectivity. Supported Ni, a group VIII metal, especially when used for HDO on phenolic compounds, can adsorb the aromatic ring at the bridge site and repel C–O bond adsorption simultaneously, resulting in relatively strong hydrogenation reactivity and saturation ability for aromatic rings or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds.
C bonds.
After the addition of the binary metal M, the electron density of the nickel atoms changes significantly, which may cause a change in the adsorption performance of the catalyst surface for the groups of p-cresol and activate cleavage of the C–O bonds; that is, the priority of the hydrogenation reactions changes between the aromatic ring and C–O bond.57 The increased electron density of the nickel atoms, which is related to the transfer of electrons from the binary metal M to Ni, tends to attack the C–O bonds and repel the electron-rich aromatic rings. This results in a preference for the hydrogenation process to involve direct deoxygenation after cleavage of the C–O bond rather than saturation of the aromatic ring.58,59 Moreover, changes in the acidity of the catalyst also affect the selectivity of HDO products on phenols. The low oxidation states of Co and Ce (observed from XRD and TPD) added to Ni/SiO2 at lower reduction temperatures leads to the formation of more Lewis acid sites, resulting in optimization of hydrogenation performance.29 In addition, the structural effects imparted by Ce and Co may cause changes in selectivity. Yang et al.57 observed that for monometallic Ni catalyst, the nickel atom arrangement is continuous, so that the aromatic ring of phenol can be adsorbed in a plane form but the C–O bond is rejected; therefore, the cycloalkane is more favorable as the saturated product. However, after the binary metal entered the nickel alloy, the above continuity was destroyed, resulting in surface roughening. This created a rough atomic plane to prevent the adsorption of the benzene ring; therefore, aromatic ring hydrogenation was less likely to occur, and the adsorption probability of the C–O bond greatly increased. Other factors may exist in the effects of Fe on the increased selectivity of the DDO path. We can speculate that the combination of Fe, an oxophilic metal, with nickel not only enhanced the attraction of the catalyst to Caromatic–O but also caused rejection of the π-electron cloud of the aromatic ring.44 This resulted in an increasing tendency of Caromatic–O bond cleavage and a weakened tendency of aromatic ring hydrogenation during the hydrogenation process.28,60 In contrast, Ni–Sn exhibited saturation of the phenolic ring. Due to the strong interactions between Sn and Ni, under the low load of Sn, an alloy state such as Ni3Sn is formed, which was detected by XRD. Some previous studies showed that the formation of Ni3Sn affects the hydrogenation priority of different groups. Rodiansono61 et al. found that the presence of Ni3Sn alloy in Ni–Sn alloy catalysts increased the hydrogenation selectivity of CO over that of CC groups in unsaturated compounds. Therefore, we can speculate that Ni3Sn was more favorable for promoting the hydrogenation of aromatic rings than the cleavage of C–O bonds in the process of hydrodeoxygenation of oxygen-containing phenolic compounds. Because the tin atoms are likely to be located in the vicinity of the steps, the influence on the continuity of the atomic surface is weak; thus, the surface readily adsorbs and is enriched by a large amount of hydrogen, and the phenol ring is thus more inclined to be saturated.50,57,61
4 Conclusions
A series of bimetallic catalysts, Ni–M/SiO2, were prepared and evaluated in a reduced state by HDO with p-cresol as a model. In the hydrodeoxygenation process, deoxygenation and transalkylation mainly occur, yielding saturated products such as methylcyclohexane and cyclohexane as well as unsaturated products such as toluene and benzene along with small amounts of phenol and xylene. The catalyst activity was as follows: Ni–Ce > Ni–Sn > Ni–Co > Ni–Fe > monometallic Ni. After the addition of the binary metal M, in comparison with the monometallic nickel, the dispersibility of the metal was observed to improve; meanwhile, the electron density also changed, which may be a factor contributing to the improvement of the activity.Different binary metals exhibit great differences in the distribution of reaction products; Ce, Co, and Fe effectively increased the BTX selectivity and SBTX/Scycloparaffin, while Sn had the opposite effect. The reason for the difference in selectivity may be the joint effect of Lewis acid sites, the electron-rich phenomenon of nickel and reconstruction of the surface atomic arrangements. These changes may have a congenerous effect which not only strengthens the adsorption and cleavage of the polar C–O bond but also weakens the adsorption and saturation of the aromatic ring.
Conflicts of interest
There are no conflicts to declare.References
- T. Kan, X. Sun and H. Wang, et al., Production of Gasoline and Diesel from Coal Tar via Its Catalytic Hydrogenation in Serial Fixed Beds, Energy Fuels, 2012, 26, 3604–3611 CrossRef CAS.
- H. Clough, Gasoline Production from Coal Tar Oils, Ind. Eng. Chem., 1957, 49(4), 673–678 CrossRef CAS.
- D. Li, Z. Li and W. Li, et al., Hydrotreating of low temperature coal tar to produce clean liquid fuels, J. Anal. Appl. Pyrolysis, 2013, 100, 245–252 CrossRef CAS.
- S. Caplan, J. Ross and G. M. Sevag, et al., Examination of the phenols of a low-temperature coal tar, Ind. Eng. Chem., Anal. Ed., 2002, 6(1), 379–380 Search PubMed.
- C. Karr, P. M. Brown and P. A. Estep, et al., Identification and Determination of Low-Boiling Phenols in Low Temperature Coal Tar, Anal. Chem., 1958, 30(8), 1413–1415 CrossRef CAS.
- R. Wang, M. Sun and Q. Liu, et al., Extraction and GC/MS analysis of phenolic compounds in low temperature coal tar from Northern Shaanxi, J. China Coal Soc., 2011, 36(4), 664–669 CAS.
- M. Niu, X. Sun and R. Gao, et al., Effect of Dephenolization on Low-Temperature Coal Tar Hydrogenation To Produce Fuel Oil, Energy Fuels, 2016, 30(12), 10215–10221 CrossRef CAS.
- L. Wang, D. Li and F. Han, et al., Experimental optimization and reactor simulation of coal-derived naphtha reforming over Pt–Re/γ-Al2O3 using design of experiment and response surface methodology, React. Kinet., Mech. Catal., 2018, 125(1), 245–269 CrossRef CAS.
- X. Wei Xu and E. Chen Jiang, “BTX” from guaiacol HDO under atmospheric pressure: the effect of support and the carbon deposition, Energy Fuels, 2017, 31(3), 2855–2864 CrossRef.
- X. Xu, E. Jiang and Y. Du, et al., BTX from the gas-phase hydrodeoxygenation and transmethylation of guaiacol at room pressure, Renewable Energy, 2016, 96, 458–468 CrossRef CAS.
- Y. Romero, F. Richard and S. Brunet, Hydrodeoxygenation of 2-ethylphenol as a model compound of bio-crude over sulfided Mo-based catalysts: promoting effect and reaction mechanism, Appl. Catal., B, 2010, 98(3–4), 213–223 CrossRef CAS.
- S. B. Gevert, M. Eriksson and P. Eriksson, et al., Direct hydrodeoxygenation and hydrogenation of 2,6- and 3,5-dimethylphenol over sulphided CoMo catalyst, Appl. Catal., A, 1994, 117(2), 151–162 CrossRef CAS.
- W. Tang, M. Fang and H. Wang, et al., Mild hydrotreatment of low temperature coal tar distillate: product composition, Chem. Eng. J., 2014, 236, 529–537 CrossRef CAS.
- T. Kan, H. Wang and H. He, et al., Experimental study on two-stage catalytic hydroprocessing of middle-temperature coal tar to clean liquid fuels, Fuel, 2011, 90(11), 3404–3409 CrossRef CAS.
- W. Cui, W. Li and R. Gao, et al., Hydroprocessing of Low-Temperature Coal Tar for the Production of Clean Fuel over Fluorinated NiW/Al2O3–SiO2 Catalyst, Energy Fuels, 2017, 31(4), 3768–3783 CrossRef CAS.
- E. Laurent, A. Centeno, and B. Delmon, Coke Formation during the Hydrotreating of Biomass Pyrolysis Oils: Influence of Guaiacol Type Compounds, in Studies in Surface Science and Catalysis, ed. B. Delmon and G. F. Froment, Elsevier, 1994, pp. 573–578 Search PubMed.
- Y. Yoshimura, T. Sato and H. Shimada, et al., Influences of oxygen-containing substances on deactivation of sulfided molybdate catalysts, Appl. Catal., 1991, 73(1), 55–63 CrossRef CAS.
- O. İ. Şenol, E. M. Ryymin and T. R. Viljava, et al., Effect of hydrogen sulphide on the hydrodeoxygenation of aromatic and aliphatic oxygenates on sulphided catalysts, J. Mol. Catal. A: Chem., 2007, 277(1), 107–112 CrossRef.
- T. R. Viljava, R. S. Komulainen and A. O. I. Krause, Effect of H2S on the stability of CoMo/Al2O3 catalysts during hydrodeoxygenation, Catal. Today, 2000, 60(1), 83–92 CrossRef CAS.
- P. M. de Souza, L. Nie and L. E. P. Borges, et al., Role of Oxophilic Supports in the Selective Hydrodeoxygenation of m-Cresol on Pd Catalysts, Catal. Lett., 2014, 144(12), 2005–2011 CrossRef CAS.
- C. A. Fisk, T. Morgan and Y. Ji, et al., Bio-oil upgrading over platinum catalysts using in situ generated hydrogen, Appl. Catal., A, 2009, 358(2), 150–156 CrossRef CAS.
- A. Gutierrez, R. K. Kaila and M. L. Honkela, et al., Hydrodeoxygenation of guaiacol on noble metal catalysts, Catal. Today, 2009, 147(3–4), 239–246 CrossRef CAS.
- K. Li, R. Wang and J. Chen, Hydrodeoxygenation of Anisole over Silica-Supported Ni2P, MoP, and NiMoP Catalysts, Energy Fuels, 2011, 25(3), 854–863 CrossRef CAS.
- M. D. Romero, J. A. Calles and A. Rodríguez, Influence of the Preparation Method and Metal Precursor Compound on the Bifunctional Ni/HZSM-5 Catalysts, Ind. Eng. Chem. Res., 1997, 36(9), 3533–3540 CrossRef CAS.
- J.-S. Moon, E.-G. Kim and Y.-K. Lee, Active sites of Ni2P/SiO2 catalyst for hydrodeoxygenation of guaiacol: a joint XAFS and DFT study, J. Catal., 2014, 311, 144–152 CrossRef CAS.
- Y. Yang, A. Gsilbert and C. Xu, Hydrodeoxygenation of bio-crude in supercritical hexane with sulfided CoMo and CoMoP catalysts supported on MgO: a model compound study using phenol, Appl. Catal., A, 2009, 360(2), 242–249 CrossRef CAS.
- H. Y. Zhao, D. Li and P. Bui, et al., Hydrodeoxygenation of guaiacol as model compound for pyrolysis oil on transition metal phosphide hydroprocessing catalysts, Appl. Catal., A, 2011, 391(1), 305–310 CrossRef CAS.
- S. Leng, X. Wang and X. He, et al., NiFe/γ-Al2O3: a universal catalyst for the hydrodeoxygenation of bio-oil and its model compounds, Catal. Commun., 2013, 41, 34–37 CrossRef CAS.
- P. M. Mortensen, J.-D. Grunwaldt and P. A. Jensen, et al., Screening of Catalysts for Hydrodeoxygenation of Phenol as a Model Compound for Bio-oil, ACS Catal., 2013, 3(8), 1774–1785 CrossRef CAS.
- G. Bai, L. Niu and Z. Zhao, et al., Ni–La–B amorphous alloys supported on SiO2 and γ-Al2O3 for selective hydrogenation of benzophenone, J. Mol. Catal. A: Chem., 2012, 363–364, 411–416 CrossRef CAS.
- A. Y. Bunch, X. Wang and U. S. Ozkan, Adsorption characteristics of reduced Mo and Ni–Mo catalysts in the hydrodeoxygenation of benzofuran, Appl. Catal., A, 2008, 346(1), 96–103 CrossRef CAS.
- S. A. Khromova, A. A. Smirnov and O. A. Bulavchenko, et al., Anisole hydrodeoxygenation over Ni–Cu bimetallic catalysts: the effect of Ni/Cu ratio on selectivity, Appl. Catal., A, 2014, 470, 261–270 CrossRef CAS.
- L. Nie, P. M. de Souza and F. B. Noronha, et al., Selective conversion of m-cresol to toluene over bimetallic Ni–Fe catalysts, J. Mol. Catal. A: Chem., 2014, 388–389, 47–55 CrossRef CAS.
- L. li, W. Yi and T.-W. Liu, et al., Hydrogenation of 3-Hydroxypropanal to 1,3-Propanediol over Cu-V/Ni/SiO2 Catalyst, New J. Chem., 2017, 41, 8965–8976 RSC.
- Z. Wang, Q. Liu and J. Yu, et al., Surface structure and catalytic behavior of silica-supported copper catalysts prepared by impregnation and sol–gel methods, Appl. Catal., A, 2003, 239(1), 87–94 CrossRef CAS.
- F. Yang, D. Liu and Y. Zhao, et al., Size Dependence of Vapor Phase Hydrodeoxygenation of m-Cresol on Ni/SiO2 Catalysts, ACS Catal., 2018, 8(3), 1672–1682 CrossRef CAS.
- M. G. Prakash, R. Mahalakshmy and K. R. Krishnamurthy, et al., Studies on Ni–M (M = Cu, Ag, Au) bimetallic catalysts for selective hydrogenation of cinnamaldehyde, Catal. Today, 2016, 263, 105–111 CrossRef CAS.
- M. V. Bykova, D. Y. Ermakov and V. V. Kaichev, et al., Ni-based sol–gel catalysts as promising systems for crude bio-oil upgrading: guaiacol hydrodeoxygenation study, Appl. Catal., B, 2012, 113–114, 296–307 CrossRef CAS.
- O. Clause, L. Bonneviot and M. Che, Effect of the preparation method on the thermal stability of silica-supported nickel oxide as studied by EXAFS and TPR techniques, J. Catal., 1992, 138(1), 195–205 CrossRef CAS.
- S. Zhang, S. Muratsugu and N. Ishiguro, et al., Ceria-Doped Ni/SBA-16 Catalysts for Dry Reforming of Methane, ACS Catal., 2013, 3(8), 1855–1864 CrossRef CAS.
- S. Hwang, J. Lee and U. G. Hong, et al., Hydrogenation of carbon monoxide to methane over mesoporous nickel-M-alumina (M=Fe, Ni, Co, Ce, and La) xerogel catalysts, J. Ind. Eng. Chem., 2012, 18(1), 243–248 CrossRef CAS.
- T. Ishihara, K. Eguchi and H. Arai, Hydrogenation of carbon monoxide over SiO2-supported Fe-Co, Co-Ni and Ni-Fe bimetallic catalysts, Appl. Catal., 1987, 30(2), 225–238 CrossRef CAS.
- A. Onda, T. Komatsu and T. Yashima, Characterizations and catalytic properties of fine particles of Ni–Sn intermetallic compounds supported on SiO2, J. Catal., 2004, 221(2), 378–385 CrossRef CAS.
- S. Sitthisa, W. An and D. E. Resasco, Selective conversion of furfural to methylfuran over silica-supported NiFe bimetallic catalysts, J. Catal., 2011, 284(1), 90–101 CrossRef CAS.
- C. Minchev, S. A. Zubkov and V. Valtchev, et al., Nature of the active sites and catalytic activity of SAPO-5 synthesized in the presence of nickel cations, Appl. Catal., A, 1994, 119(2), 195–204 CrossRef CAS.
- K. Fang, J. Ren and Y. Sun, Effect of nickel precursors on the performance of Ni/AlMCM-41 catalysts for n-dodecane hydroconversion, J. Mol. Catal. A: Chem., 2005, 229(1), 51–58 CrossRef CAS.
- S. G. Zhang, K. Yorimitsu and S. Nohara, et al., Surface analysis of an amorphous MgNi alloy prepared by mechanical alloying for use in nickel–metal hydride batteries, J. Alloys Compd., 1998, 270(1), 123–126 CrossRef CAS.
- H. W. Nesbitt, D. Legrand and G. M. Bancroft, Interpretation of Ni 2p XPS spectra of Ni conductors and Ni insulators, Phys. Chem. Miner., 2000, 27(5), 357–366 CrossRef CAS.
- S. J. Sawhill, D. C. Phillips and M. E. Bussell, Thiophene hydrodesulfurization over supported nickel phosphide catalysts, J. Catal., 2003, 215(2), 208–219 CrossRef CAS.
- A. Borgna, B. Anderson and A. Saib, et al., Pt−Co/SiO2 Bimetallic Planar Model Catalysts for Selective Hydrogenation of Crotonaldehyde, J. Phys. Chem. B, 2004, 108(46), 17905–17914 CrossRef CAS.
- M. H. Peyrovi, T. Rostamikia and N. Parsafard, Competitive Hydrogenation of Benzene in Reformate Gasoline over Ni Supported on SiO2, SiO2–Al2O3, and Al2O3 Catalysts: Influence of Support Nature, Energy Fuels, 2018, 32(11), 11432–11439 CrossRef CAS.
- Y. Li, J. Fu and B. Chen, Highly selective hydrodeoxygenation of anisole, phenol and guaiacol to benzene over nickel phosphide, RSC Adv., 2017, 7(25), 15272–15277 RSC.
- X. Xu, Z. Li and Y. Sun, et al., High quality fuel from the upgrading of heavy bio-oil by the combination ultrasonic with mutual solvent, Energy Fuels, 2018, 32, 3477–3487 CrossRef CAS.
- E. Furimsky, Catalytic hydrodeoxygenation, Appl. Catal., A, 2000, 199(2), 147–190 CrossRef CAS.
- E. O. Odebunmi and D. F. Ollis, Catalytic hydrodeoxygenation: I Conversions of o-, p-, and m-cresols, J. Catal., 1983, 80(1), 56–64 CrossRef CAS.
- S. R. Kirby, C. Song and H. H. Schobert, Hydrodeoxygenation of O-containing polycyclic model compounds using a novel organometallic catalyst precursor, Catal. Today, 1996, 31(1), 121–135 CrossRef CAS.
- F. Yang, D. Liu and H. Wang, et al., Geometric and electronic effects of bimetallic Ni–Re catalysts for selective deoxygenation of m-cresol to toluene, J. Catal., 2017, 349, 84–97 CrossRef CAS.
- X. Wang and J. Chen, Effects of indium on Ni/SiO2 catalytic performance in hydrodeoxygenation of anisole as model bio-oil compound: suppression of benzene ring hydrogenation and C–C bond hydrogenolysis, Chin. J. Catal., 2017, 38(11), 1818–1830 CrossRef CAS.
- C. Li, Y. Chen and S. Zhang, et al., Ni–In Intermetallic Nanocrystals as Efficient Catalysts toward Unsaturated Aldehydes Hydrogenation, Chem. Mater., 2013, 25(19), 3888–3896 CrossRef CAS.
- A. N. Mansour, D. E. Sayers and J. W. Cook, et al., X-ray absorption studies of some platinum oxides, J. Phys. Chem., 1984, 88(9), 1778–1781 CrossRef CAS.
- Rodiansono, S. Khairi and T. Hara, et al., Highly efficient and selective hydrogenation of unsaturated carbonyl compounds using Ni–Sn alloy catalysts, Catal. Sci. Technol., 2012, 2(10), 2139 RSC.
| This journal is © The Royal Society of Chemistry 2019 |