
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMethanol promoted naphtha catalytic pyrolysis to light olefins on Zn-modified high-silicon HZSM-5 zeolite catalysts†
Qi-tong Cheng ,
Ben-xian Shen*,
Hui Sun*,
Ji-gang Zhao* and
Ji-chang Liu
,
Ben-xian Shen*,
Hui Sun*,
Ji-gang Zhao* and
Ji-chang Liu
Research Institute of Petroleum Processing, East China University of Science and Technology, Shanghai 200237, China. E-mail: sbx@ecust.edu.cn; sunhui@ecust.edu.cn; zjg@ecust.edu.cn
First published on 3rd July 2019
Abstract
The methanol promoted naphtha catalytic pyrolysis system to obtain light olefins (ethylene and propylene) was studied over Zn-modified high-silicon HZSM-5 (Zn/HZSM-5) catalysts. Compared with the individual naphtha catalytic pyrolysis to light olefins, the addition of methanol remarkably improved the naphtha conversion and the yield of the light olefins. All Zn/HZSM-5 samples were characterized by using a variety of techniques including inductively coupled plasma-optical emission spectrometry (ICP-OES), X-ray diffraction (XRD), scanning electron microscopy (SEM), N2 adsorption, NH3-temperature programmed desorption (NH3-TPD), X-ray photoelectron spectroscopy (XPS), pyridine adsorption infrared spectroscopy (Py-IR), and Fourier transform infrared spectroscopy (FT-IR). The performances of the catalysts for methanol promoted naphtha catalytic pyrolysis were evaluated in a fixed-bed reactor. In the methanol promoted naphtha catalytic pyrolysis reaction, the yield of the light olefins was strongly dependent on the reaction conditions and the degree of Zn ion-exchange. Due to the heterogeneous distribution of the protons of high silicon ZSM-5, two types of Lewis acid sites were formed by the interaction of Zn with hydroxyl groups (OH) adsorbed on HZSM-5. The 0.3-Zn/HZSM-5 (0.3 mol L−1 Zn ion-exchange HZSM-5) sample holds a comparatively high light olefin yield of 51.7 wt% possibly because of the moderate density and distribution of the acid sites on the catalyst.
1 Introduction
Light olefins (ethylene and propene), the key building blocks in the modern chemical industry, are in huge demand in the production of polymers, oxygenates, and many other important intermediate chemicals.Traditionally, ethylene is produced via naphtha steam cracking, which suffers from high reaction temperature (over 800 °C, leading to high energy consumption), low light olefin selectivity, and product slate inflexibility (particularly low propene/ethylene (P/E) ratio). Propene is primarily produced as a by-product in steam cracking plants (accounting for around 70% of the total propene production) and fluid catalytic cracking plants for gasoline production (accounting for the remaining 30%).1,2 The extended gap between the conventional light olefin supply and their increasing demand has stimulated intense efforts in the development of cost-effective naphtha catalytic cracking route, which can achieve highly flexible P/E ratio with respect to the fluctuant market demand and give high yields of propylene and ethylene at relatively low temperature. Although the use of alternative feed stocks such as methane (methane oxidation coupling and methane chloride process to olefin),3,4 methanol (methanol to olefin),5,6 ethanol (ethanol dehydration to ethylene),7 ethane and propane (dehydrogenation of ethane and propane to olefins)8–10 have been pursued, naphtha is still the most attractive feedstock in Europe and Asia due to a variety of practical and economic considerations.11–15
In order to achieve the efficient utilization of naphtha, recently, a number of studies on the coupling conversion of methanol and naphtha to light olefins have been published.16–18 Chang et al.16 reported the role of methanol addition in n-hexane catalytic cracking. An improved conversion of n-hexane in the temperature range of 400–500 °C was obtained because the addition of methanol increased the contribution of the faster bimolecular mechanism. Lücke et al.17 tested the coupled methanol–hydrocarbon cracking reaction with various liquid hydrocarbon feedstocks (n-hexane, cyclohexane, and naphtha). A relatively high ethylene yield of 24.0 wt% and propene yield of 23.7 wt% were attained under 670 °C, WHSV of 2.5 h−1, and methanol/naphtha molar ratio of 6 on the HZSM-5 zeolite. Yan et al.18 investigated the coupling transformation of light naphtha with methanol participation over the hybrid catalysts (HZSM-5 with Zn–Pd/Y2O3–Al2O3). The comparatively high light olefin (ethylene + propene) yield of 44.4 wt% was obtained under 670 °C, WHSV of 0.75 h−1, and 20 wt% methanol in the naphtha feed, and the effect of methanol content in the naphtha feed on the light olefin yield was also discussed.
Some ZSM-5 based catalysts including Fe-modified HZSM-5,19 La-modified HZSM-5,20 and Ni-modified HZSM-5,21 have been developed for the coupled reaction. Meanwhile, the relationship between the properties and the catalytic reactivity of the catalyst in the coupling conversion of methanol and naphtha were also investigated. Mier et al.21 prepared Ni/HZSM-5 by the impregnation method for the coupled reaction of methanol with n-butane. Compared with Ni/HZSM-5, the unmodified HZSM-5 gave a higher C2–C4 olefin yield of 24.4 wt% (11.5 wt% of propene) with a selectivity of 43 wt% at 575 °C and WHSV of 12.7 h−1 because of the high levels of acidity and acid strength (≥120 kJ mol NH3−1). Gong et al.20 prepared La/HZSM-5 by the wetness impregnation method for the coupling conversion of methanol with C4 hydrocarbon. The comparatively high propylene yield of 46.0 wt% was obtained on 1.5 wt% La/HZSM-5, and the effect of the density and distribution of the acid sites on the catalyst was emphasized. Martin et al.19 prepared H(Fe)-ZSM-5, H(Al)-ZSM-5, and H(Fe, Al)-ZSM-5 zeolites by the hydrothermal synthesis method for the coupled methanol-n-butane cracking process and found that the catalytic lifetime of the zeolites is prolonged by reducing the Brønsted acidity. Moreover, a lower decrease in the cracking activity was obtained together with the prolongation of the catalyst lifetime. Based on this, in order to develop effective catalysts, it is vital to fully understand the relationship between the properties and the catalytic reactivity of a catalyst in the coupling conversion of methanol and naphtha.
Although some significant achievements have been made in the coupled methanol–hydrocarbon catalytic system by the abovementioned researchers, it is still highly desirable to further explore the properties–reactivity relationship and develop an effective catalyst. Yan et al.18 reported that the (Zn–Pd) co-catalyst of the hybrid catalyst exerts its coke cleaning effect noticeably on the zeolite acid sites in the mixed naphtha-methanol feed. Biscardi et al.22 suggested that the Zn-modified HZSM-5 promotes the dehydrogenation of hydrocarbons due to the dissociative hydrogen adsorption of Zn species. In addition, the high-silicon HZSM-5 has been proven to exhibit extremely high hydrothermal stability, according to a previous investigation. Also, the heterogeneous distribution of the protons and the presence of the ‘hydroxyl nest’ defect sites (silanol groups) make it possible for high-silicon ZSM-5 to form a number of specific active sites with different metal ions.23–25 These results encourage us to attempt the use of Zn-modified high-silicon HZSM-5 catalysts (Zn/HZSM-5) in the coupled methanol–naphtha catalytic pyrolysis to light olefins. To the best of our knowledge, few research works concerning methanol promoted naphtha catalytic pyrolysis over Zn-modified high-silicon HZSM-5 have been reported so far.
In this work, a series of Zn-modified high-silicon HZSM-5 zeolites (Zn/HZSM-5) were prepared by the ion exchange method and used in the methanol promoted naphtha catalytic pyrolysis system. The effects of reaction conditions and the Zn ion-exchange degree on the catalytic performance of Zn/HZSM-5 in the methanol promoted naphtha catalytic pyrolysis system were investigated in detail. The properties of the catalysts were characterized by using a variety of techniques including ICP-OES, SEM, XRD, NH3-TPD, XPS, FT-IR, and Py-IR. Based on the obtained results, the relationship of the physiochemical properties with the catalytic performance was correlated to provide an insight into the improvement of light olefin yield by modifying the zeolite catalyst.
2 Experimental
2.1. Materials and reagents
HZSM-5 zeolite (Si/Al molar ratio of 350, particle size of 120 mesh) was purchased from the XFNANO company, Zn(NO3)2·6H2O (with purity higher than 99.0%) was supplied by Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Naphtha (the composition is shown in Fig. S1†) was supplied by Shanghai Gaoqiao Petrochemical Company (Shanghai, China). Methanol (with purity higher than 99.0%) was supplied by Sinopharm Chemical Reagent Co., Ltd (Shanghai, China).2.2. Catalyst preparation
The Zn-exchange was conducted at 110 °C using an aqueous solution of Zn(NO3)2·6H2O with the concentration of 0.1 mol L−1, 0.3 mol L−1, and 0.5 mol L−1. The same procedure was repeated thrice. The solid sample was washed with deionized water, dried at 120 °C for 8 h, and finally calcined at 700 °C for 5 h. The resulting zeolite samples were labelled according to the solution concentration. For example, 0.3-Zn/HZSM-5 represents the modified Zn/HZSM-5 zeolite sample exchanged using 0.3 mol L−1 Zn(NO3)2·6H2O. All HZSM-5 samples used for characterization and catalytic performance evaluation underwent the same thermal treatment of calcination at 700 °C for 5 h.2.3. Catalytic activity measurement
The measurement of catalytic activity was carried out on a customer-made fixed-bed reactor (Fig. 1). Naphtha and methanol were pumped from the top of the reactors. N2 flow was fed as a diluent. The hydrocarbon components in the gas products were collected and analyzed using a GC-920 gas chromatograph (Haixin chromatograph instrument Co., LTD., Shanghai) equipped with a PLOT/Al2O3 capillary column (30 m × 0.53 mm × 2.0 μm) and a flame ionization detector while the non-hydrocarbon components were determined using a GC-2014 gas chromatograph (Shimadzu Group Co., LTD., Japan) equipped with a packed column filled with zeolite 13X, PQ, and a thermal conductivity detector. The liquid products were analyzed using a GC-2010 gas chromatograph (Shimadzu Group Co., LTD., Japan) equipped with a Rtx-1 PONA (100 m × 0.25 mm × 0.5 μm) capillary column and a flame ionization detector.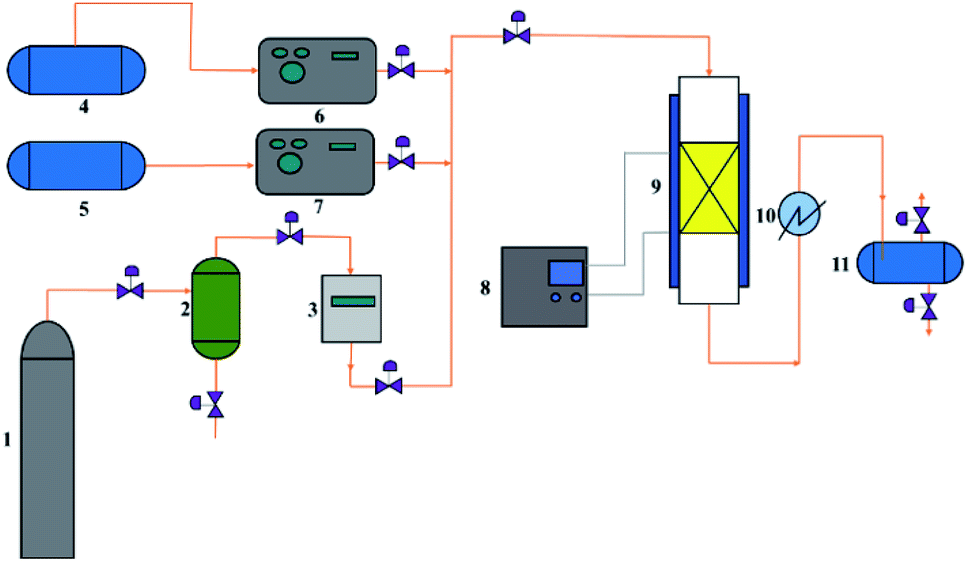 | ||
| Fig. 1 Experimental setup for the catalytic performance evaluation. 1-N2 cylinder, 2-desiccator, 3-mass flow controller, 4-naphtha container, 5-methanol container, 6-constant flux pump, 7-constant flux pump, 8-temperature controller, 9-fixed-bed reactor, 10-condenser, 11-liquid product container. | ||
Under the reaction conditions used, methanol almost completely converted on the investigated catalysts. The total conversion Ct is defined as the percentage of the weight of (all the components of) the organic feed (naphtha and eventually methanol) converted into the final products as follows:
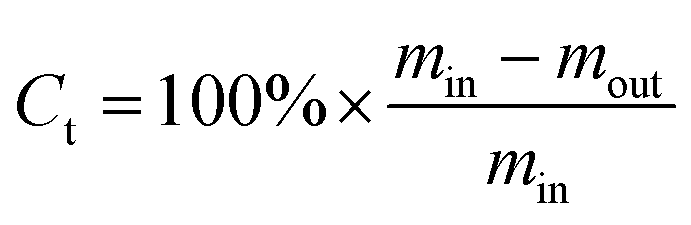
The yield of the product i (Yi, in wt%) was expressed as follows:
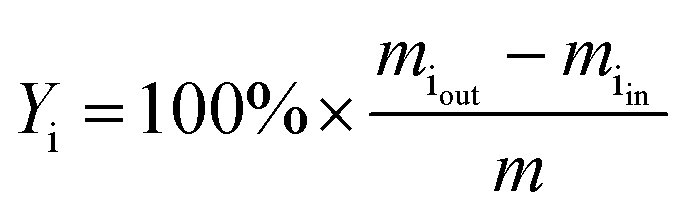
2.4. Catalyst characterization
3 Results and discussion
3.1. Catalysts' characterization
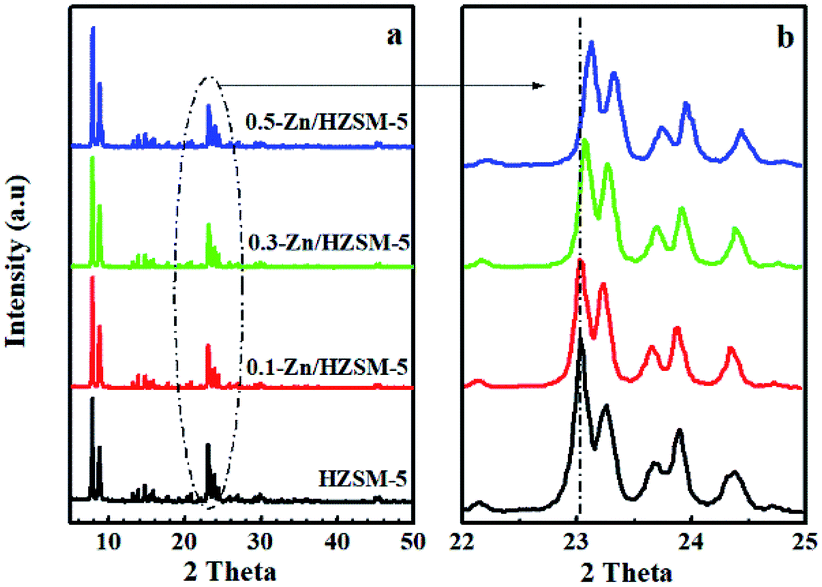 | ||
| Fig. 2 XRD patterns at 2θ in the range of 5–50° (a) and the scaled-up ones at 2θ in the range of 22–25° (b) of HZSM-5, 0.1-Zn/HZSM-5, 0.3-Zn/HZSM-5, and 0.5-Zn/HZSM-5. | ||
 | ||
| Fig. 3 SEM images of (a) and (a-1) HZSM-5, (b) and (b-1) 0.3-Zn/HZSM-5, and (c) and (c-1) 0.5-Zn/HZSM-5. | ||
The SEM images of the parent HZSM-5 and the modified Zn/HZSM-5 samples are shown in Fig. 3. The HZSM-5 sample exhibited a prism-like crystal morphology having a particle size of about 700 × 200 × 80 nm. It also can be seen that the modified Zn/HZSM-5 samples showed similar morphologies to that of the parent HZSM-5 sample.
The pore structure and the elemental content of the parent HZSM-5 and the modified zeolites were measured by N2 adsorption and ICP-OES. The results are shown in Table 1. Compared with HZSM-5, the Al content decreased and the Zn content increased in the Zn/HZSM-5 zeolites, which indicated that the dealumination of the skeleton occurred during the zinc ion-exchange process.
| Samples | Si content (wt%) | Al content (wt%) | Zn content (wt%) | Surface area (m2 g−1) | Pore volume (cm3 g−1) | ||||
|---|---|---|---|---|---|---|---|---|---|
| SBETa | Smicb | Smes | Vt c | Vmicb | Vmes | ||||
| a BET specific surface area.b Micropore specific surface area and micropore volume calculated using the t-plot method.c Total pore volume estimated based on the volume adsorbed at P/P0 = 0–0.99. | |||||||||
| HZSM-5 | 42 | 0.6 | 0.00 | 343 | 192 | 151 | 0.220 | 0.099 | 0.121 |
| 0.1-Zn/HZSM-5 | 43 | 0.44 | 0.15 | 344 | 190 | 154 | 0.224 | 0.097 | 0.127 |
| 0.3-Zn/HZSM-5 | 43 | 0.35 | 0.19 | 350 | 182 | 168 | 0.232 | 0.085 | 0.137 |
| 0.5-Zn/HZSM-5 | 43 | 0.30 | 0.21 | 383 | 128 | 254 | 0.237 | 0.066 | 0.171 |
Moreover, the Zn-exchanged samples showed less micropores and more mesopores as compared to HZSM-5, which were caused by the Zn incorporation and the skeleton dealumination, respectively. The N2 adsorption–desorption isotherm also supports the above view (see Fig. S2†). The abundant mesopores can effectively interconnect the micropores to offer additional diffusional paths, which are of great benefit in adsorption and catalysis. In addition, the degree of skeleton dealumination increased with the modified solution concentration increasing from 0.3 mol L−1 to 0.5 mol L−1. Combined with the XRD spectra (Fig. 2), the shift in the peaks at 2θ of 22–25° can be attributed to both the different Zn contents and the skeleton dealumination in the zeolite lattice.
From the NH3-TPD profiles of HZSM-5 (Fig. 4), two desorbed peaks were found in the ranges of 100–170 °C and 300–420 °C, corresponding to the weak and medium strength acid sites, respectively. After the Zn-modification process, the desorption peak located at 125 °C widened and moved towards higher temperature, accompanied by the decreased intensity of the desorption peak at 380 °C. On the other hand, a new desorption peak at 520 °C appeared for desorption from Zn-modified HZSM-5. The results indicate that the B acid sites decrease and the strong L acid sites increase with the incorporation of Zn. On comparing the NH3-TPD profiles of the 0.3-Zn/HZSM-5 and 0.5-Zn/HZSM-5 catalysts, subtle differences can be found. The medium acid sites' desorption peak temperature of 0.5-Zn/HZSM-5 shifted to lower temperature. However, the variation of the curve cannot be realized due to the fewer active sites in high-silicon zeolites. Therefore, the concentration and distribution of the Brønsted and Lewis acid sites on the catalysts were detected by using Py-IR.
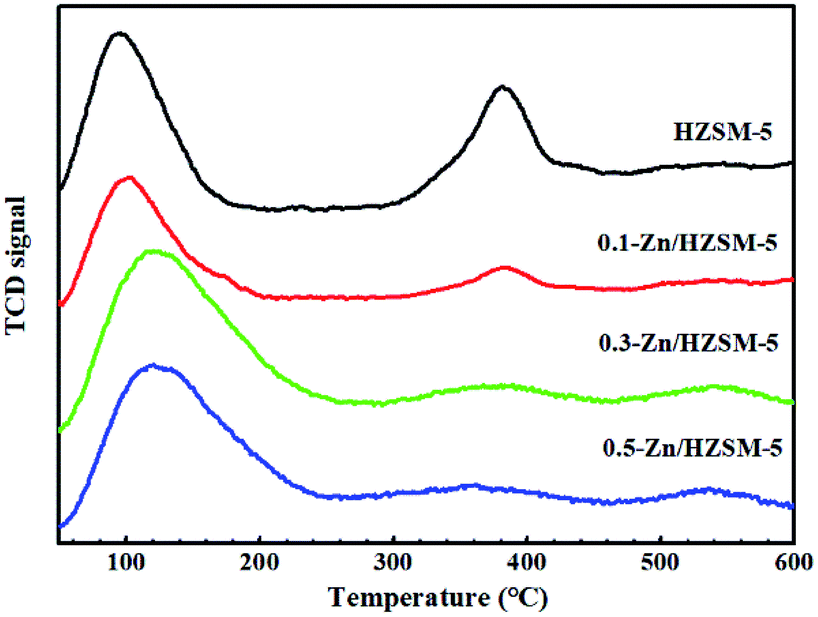 | ||
| Fig. 4 NH3-TPD profiles of the HZSM-5 and modified Zn/HZSM-5 zeolites with different Zn-exchange degree. | ||
The Py-IR spectra of the catalysts are displayed in Fig. 5. At 200 °C, the pyridine ring-mode bands indicative of the Brønsted sites (PyH+ at 1545 cm−1) and of Lewis sites (Lewis-bonded pyridine at 1451 cm−1) on HZSM-5 are visible. The desorption band of H-bonded pyridine emerged at 1446 cm−1. Upon modification by Zn exchange, the band at 1451 cm−1 evolved into more prominent ones. Combined with the broadened desorption peaks in the range of 50–170 °C and the damped proton acidic desorption peak at 380 °C in NH3-TPD curves, it can be deduced that some new Lewis acid sites are formed by the conversion of Brønsted acid sites with Zn species. On the other hand, even after desorption at 450 °C, the Zn/HZSM-5 catalyst holds more Lewis-bound pyridine than HZSM-5, corresponding to the appearance of the high temperature desorption peak in the NH3-TPD profiles of the Zn/HZSM-5 samples (Fig. 4). This may suggest that there is also a minority of strong Lewis sites. It is reasonable to speculate that at least two kinds of Lewis acid sites were formed over the Zn-modified HZSM-5 zeolites.
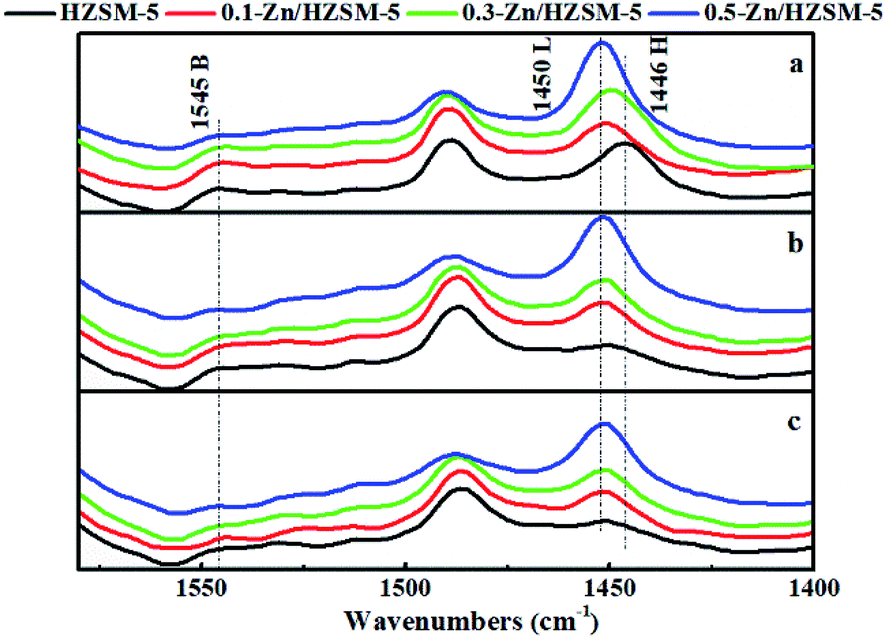 | ||
| Fig. 5 Py-IR spectra of HZSM-5 and Zn/HZSM-5 desorbed at (a) 200 °C, (b) 350 °C, and (c) 450 °C. | ||
The concentration and distribution of the Brønsted and Lewis acid sites on the catalysts are shown in Table 2. As seen from Table 2, the HZSM-5 zeolite has a higher total concentration of the B acid sites and less total concentration of the L acid sites than any other Zn-modified catalysts; moreover, it possessed the highest B/L ratio of 0.404 among these catalysts. For the Zn/HZSM-5 catalyst, the concentration of the B acid sites and the B/L ratio decreased with an increase in the Zn ion-exchange degree, whereas the concentration of the L acid sites increased. In addition, the discrepancies in the acid properties between the samples 0.3-Zn-ZSM-5 and 0.5-Zn-ZSM-5 increased. Combined with the data from ICP-OES, the increased discrepancies in the acid properties between 0.3-Zn/HZSM-5 and 0.5-Zn/HZSM-5 samples may be caused by both the amount of Zn incorporation and the degree of skeleton dealumination. This will be discussed further below.
| Catalyst | Concn. of B (mmol g−1) | Concn. of L (mmol g−1) | B/L | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Weak 200 °C | Medium 350 °C | Strong 450 °C | Total | Weak 200 °C | Medium 350 °C | Strong 450 °C | Total | ||
| HZSM-5 | 0.157 | 0.093 | 0.057 | 0.307 | 0.549 | 0.124 | 0.088 | 0.760 | 0.404 |
| 0.1-Zn/HZSM-5 | 0.151 | 0.071 | 0.032 | 0.253 | 0.633 | 0.245 | 0.214 | 1.092 | 0.232 |
| 0.3-Zn/HZSM-5 | 0.138 | 0.049 | 0.023 | 0.210 | 0.754 | 0.312 | 0.266 | 1.331 | 0.158 |
| 0.5-Zn/HZSM-5 | 0.056 | 0.020 | 0.017 | 0.092 | 0.979 | 0.611 | 0.527 | 2.117 | 0.044 |
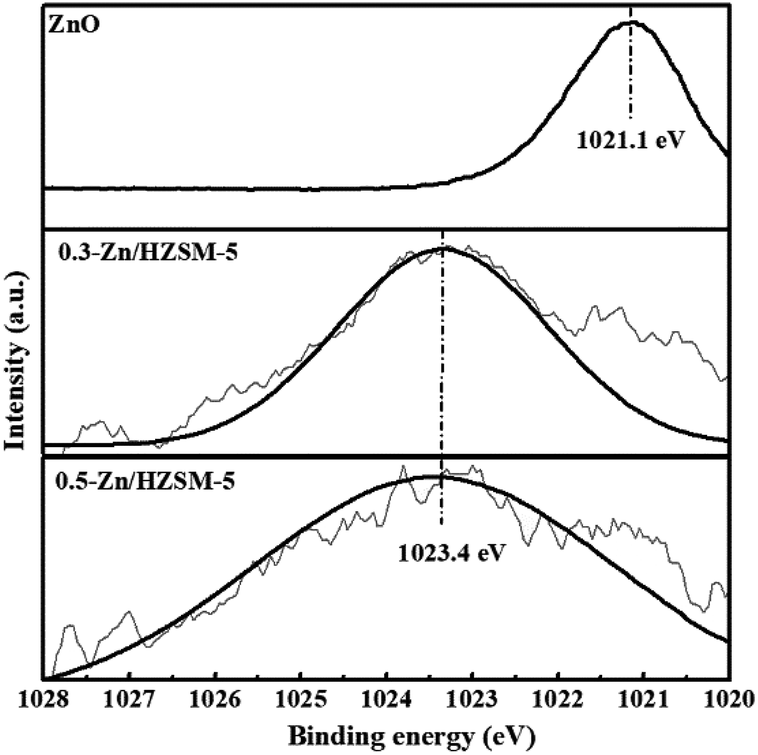 | ||
| Fig. 6 XPS spectra of Zn (2p3/2) for ZnO and Zn/HZSM-5. | ||
The IR spectra of the catalysts display the infrared characteristic bands of the ZSM-5 framework with stretching vibrations at 450, 546, and 798 cm−1, as shown in Fig. 7. The symmetrical stretching vibration of the T–O (νintsym(TO)) bond at 798 cm−1 and the T–O bond angular vibration at 450 cm−1 belong to the inner TO4 tetrahedral of the zeolite framework. The absorption event happening at 546 cm−1 is related to the unique symmetry five-membered ring structural unit of the ZSM-5 framework and is sensitive to the skeletal structure of ZSM-5 zeolite. With the improvement in the Zn-exchange degree, the new absorption band at 879 cm−1 appeared and is assigned to an out-of-plane deformation vibrational mode of a strongly H-bonded zeolitic OH group, γ(Z(O)–H⋯O).28–30
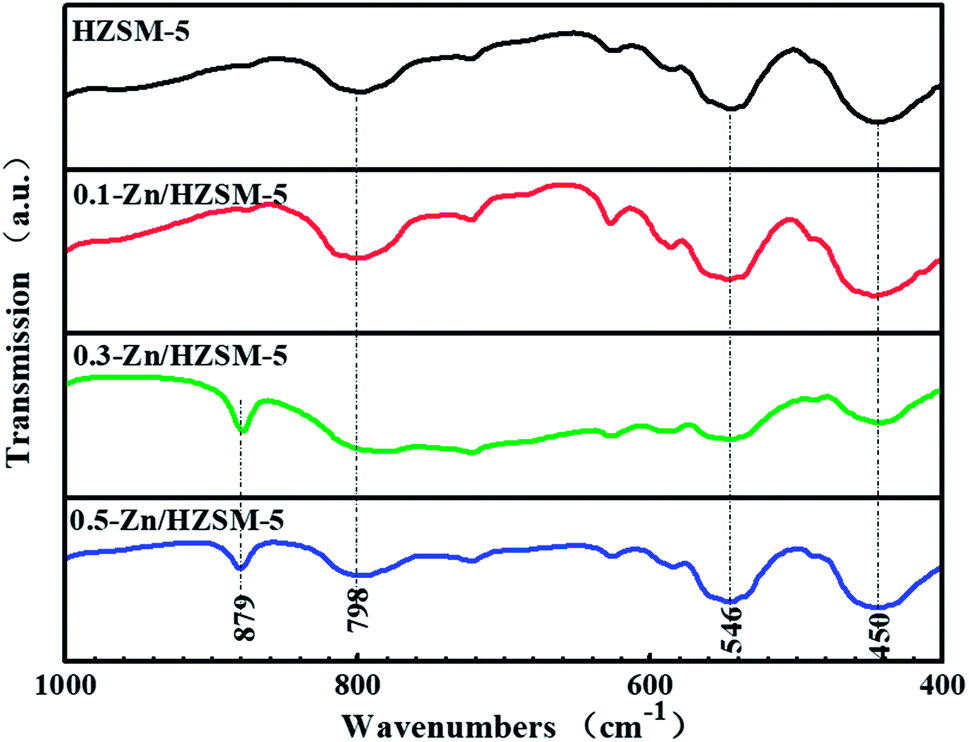 | ||
| Fig. 7 The IR spectra of HZSM-5, 0.1-Zn/HZSM-5, 0.3-Zn/HZSM-5, and 0.5-Zn/HZSM-5. | ||
The IR bands of these catalysts degassed at 200 °C were recorded in the range of wavenumbers from 3000 to 4000 cm−1 for detecting the surface hydroxyl (OH) groups. As seen from Fig. 8, the IR bands at 3610, 3740, and 3670 cm−1 of HZSM-5 are attributed to the bridged O–H stretching vibration, isolated silanol group stretching vibration of the ‘hydroxyl nests’, and AlOH vibrations, respectively.31,32 Instead of the bridged O–H bond, the broad H-bonded OH stretching vibration band centered at ∼3500 cm−1 was obtained and became prominent for the 0.3-Zn/HZSM-5 and 0.5-Zn/HZSM-5 samples.28,29 This corresponds to the γ(Z(O)–H⋯O) deformation vibrational band at 879 cm−1 shown in Fig. 7. In addition, the intensity of the SiOH band centered at 3740 cm−1 increased due to the skeleton dealumination of the Zn/HZSM-5 samples. However, the infrared absorption band intensity of SiOH in 0.5-Zn/HZSM-5 decreased compared to the other samples. It can be inferred that interactions between Zn and SiOH occurred in the 0.5-Zn/HZSM-5 sample, which led to significantly distinct Lewis acid sites in 0.3-Zn/HZSM-5 and 0.5-Zn/HZSM-5.
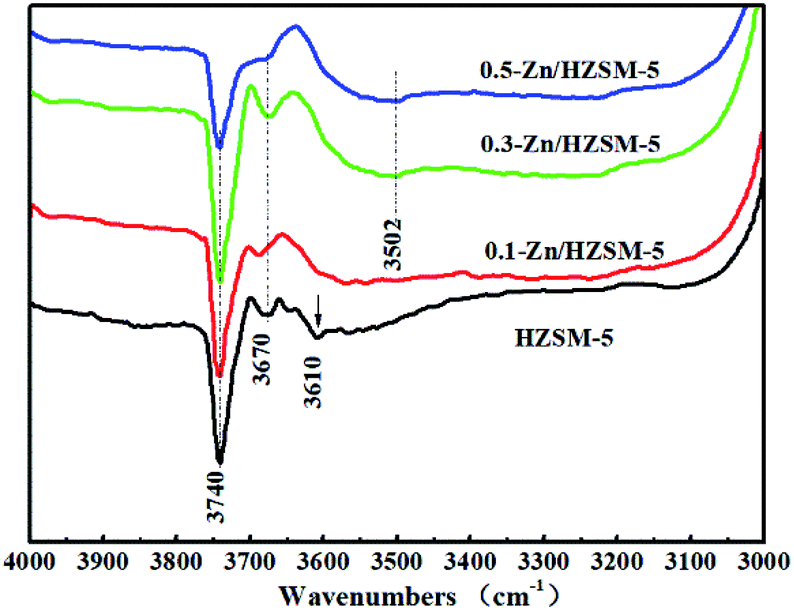 | ||
| Fig. 8 IR spectra of the catalysts degassed at 200 °C: (a) HZSM-5, (b) 0.1-Zn/HZSM-5, (c) 0.3-Zn/HZSM-5, and (d) 0.5-Zn/HZSM-5. | ||
In view of the high Si/Al ratio of HZSM-5, the Al distribution in the frameworks is heterogeneous and the probability of a five-membered ring containing only one Al ion is relatively high. With the incorporation of Zn, despite the presence of only one Al-centered tetrahedron ([AlO2]−) in the vicinity of Zn2+, it is also coordinated to four framework oxygen atoms. Then, the framework bonds distort to stabilize Zn2+ in this unusual situation.33 Meanwhile, the distortion in the framework bonds facilitated the formation of H-bonded acidic OH.29 Combined with the result of Zn (2p3/2) XPS spectrum, the possible location of Zn in high-silicon ZSM-5 is shown in Fig. 9.
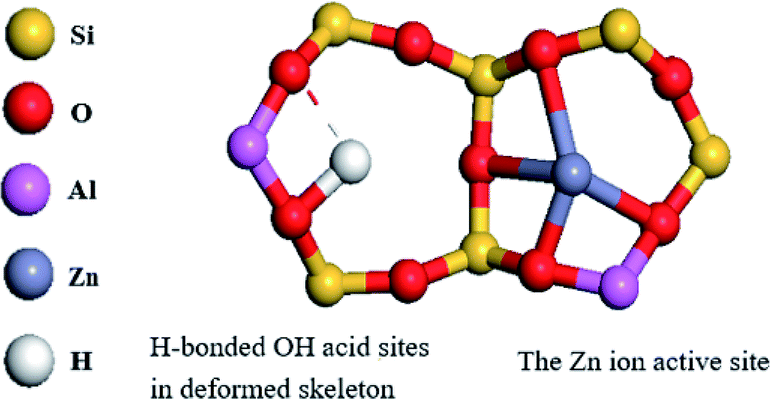 | ||
| Fig. 9 Active sites of Zn in the high-silicon HZSM-5 framework. | ||
3.2. Catalytic pyrolysis performance of Zn/HZSM-5 zeolites
Table 3 shows the effect of Zn ion-exchange degree on the reactivity of Zn/HZSM-5 in the methanol promoted naphtha catalytic pyrolysis system. Under the reaction conditions used, the naphtha conversion and light olefin yield were obtained for these catalysts. Compared with the parent sample, the naphtha conversion and propylene yield first increased and then gradually decreased with increasing Zn-exchange degree. The propylene yield initially increased to the maximum value of 28.3% on 0.3-Zn/HZSM-5 and then dropped with the increase in Zn ion-exchange degree. On the contrary, the ethylene yield improved with the increase in Zn ion-exchange degree. Thus, Zn loading has an influence on the catalytic reactivity of Zn/HZSM-5 and the 0.3-Zn/HZSM-5 sample is the optimal Zn-modified HZSM-5 catalyst under the reaction conditions used.| Sample | Methanol conversion (wt%) | Naphtha conversion (wt%) | Yield (wt%) | |||
|---|---|---|---|---|---|---|
| CH4 | C2H4 | C3H6 | C4H8a | |||
| a Butene (C4H8) was mainly 1-butene and 2-butene (including trans-2-butene and cis-2-butene). Reaction conditions: reaction temperature = 650 °C, WHSV = 3.83 h−1, N2 = 60 mL min−1, methanol/naphtha = 15 wt%, time on stream = 3 h. | ||||||
| HZSM-5 | >99 | 76.8 | 5.2 | 15.5 | 23.6 | 9.4 |
| 0.1-Zn/HZSM-5 | >99 | 80.4 | 6.6 | 18.1 | 26.1 | 8.2 |
| 0.3-Zn/HZSM-5 | >99 | 92.1 | 7.9 | 23.4 | 28.3 | 7.4 |
| 0.5-Zn/HZSM-5 | >99 | 91.3 | 7.1 | 24.1 | 26.3 | 7.8 |
On combining the acid site concentration (Table 2) with the light olefin yield (Table 3), the effect of the acidity on the catalytic reactivity of Zn/HZSM-5 is revealed. It can be seen from Fig. 10 that the acidity has complex influences on the catalytic reactivity of Zn/HZSM-5. The moderate density of the acid sites and B/L ratio on Zn/HZSM-5 favoured the naphtha conversion and propylene formation, while ethylene yield increased with the increase in the density of the acid sites in the methanol promoted naphtha catalytic pyrolysis reaction.
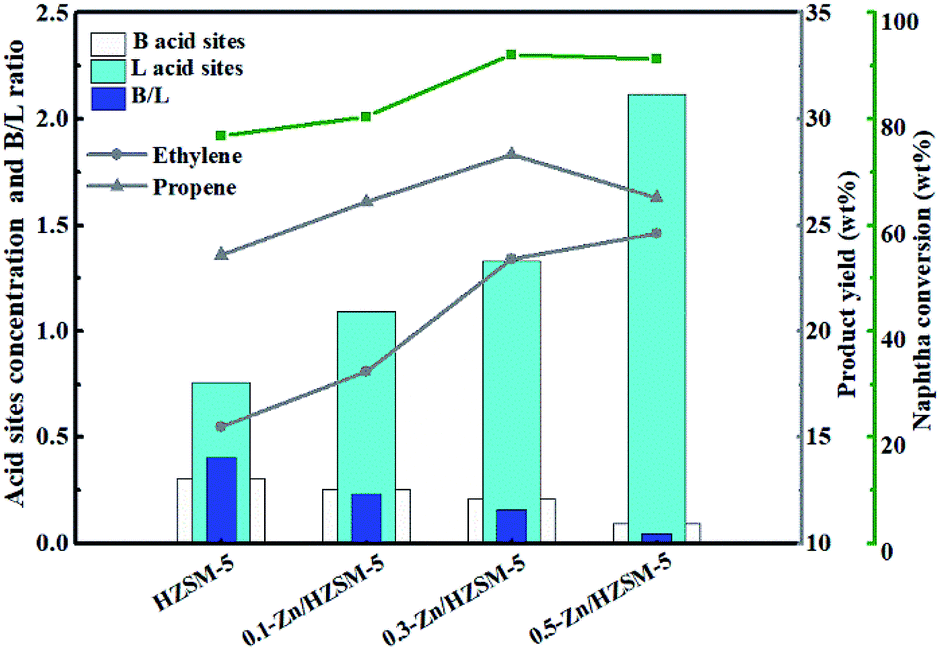 | ||
| Fig. 10 Effect of acidity on the catalytic reactivity of Zn/HZSM-5 in the methanol promoted naphtha catalytic pyrolysis system. Reaction conditions: reaction temperature = 650 °C, WHSV = 3.83 h−1, N2 = 60 mL min−1, methanol/naphtha = 15 wt%, time on stream = 3 h. | ||
It is widely known that naphtha catalytic pyrolysis follows the carbonium ion mechanism including monomolecular reaction pathway and bimolecular reaction pathway (i.e., hydride transfer, isomerization, alkylation, and β scission). As for MTO on HZSM-5, the dual cycle mechanism is followed, in which the aromatic- and olefin-based routes are accepted and ethylene is mainly produced through the aromatic-based route on HZSM-5.34
When the mixed feed of naphtha and methanol is fed to the zeolite, the aromatics are adsorbed on the B acid sites prior to methanol and methanol is adsorbed on the B acid sites prior to the alkanes in the feed due to the difference in their proton affinities (Table S1†).35 The Zn sites facilitate the dehydrogenation of alkanes to alkenes, thus improving the naphtha conversion. Then, the product alkenes are protonated and undergo the β-scission to light olefins. However, for the 0.5-Zn/HZSM-5 with fewer B acid sites and more Zn sites, the competitive adsorption of the feed component on the B acid sites decreases the protolytic cracking reaction of alkenes and enhances the methylation/cracking reaction between methanol and polymethylbenzenes to ethylene. Accordingly, the excess alkenes are further dehydrocycled to the aromatics on the Zn sites, leading to decreased propene yield.34
Fig. 11 shows the effect of methanol content on the catalytic reactivity of the 0.3-Zn/HZSM-5 catalyst in the methanol promoted naphtha catalytic pyrolysis system. Compared with only naphtha as the feed, the addition of methanol improves the conversion of naphtha. In addition, the naphtha conversion initially increased to the maximum value and then decreased with the increase in methanol content. It can be deduced that the coupling conversion of methanol–naphtha occurred on the 0.3-Zn/HZSM-5 catalyst. The methanol/naphtha = 15 wt% gave the highest conversion under the reaction conditions used.
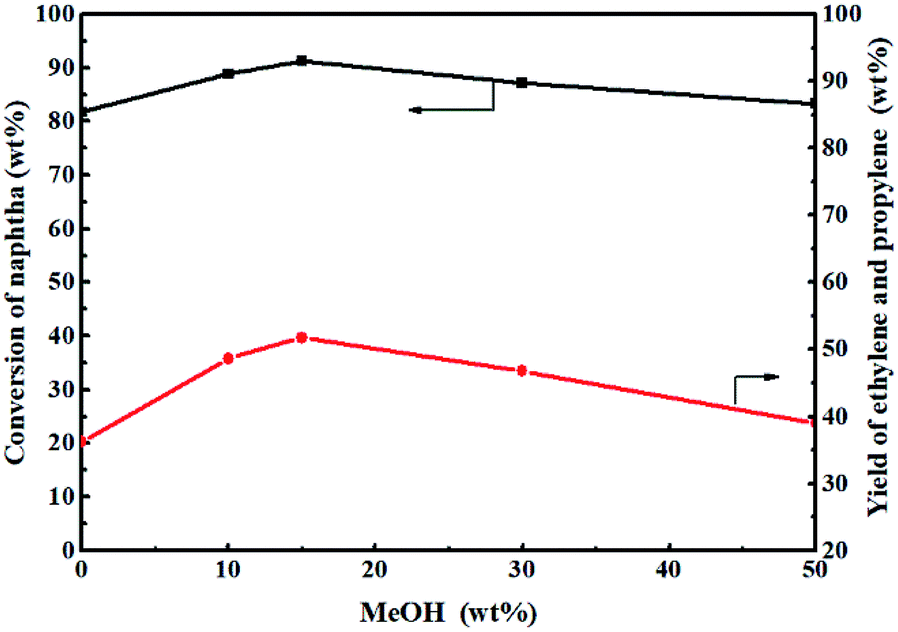 | ||
| Fig. 11 Effect of methanol content in the feed (wt%) on the reactivity of 0.3-Zn/HZSM-5 in the methanol promoted naphtha catalytic pyrolysis system. Reaction conditions: reaction temperature = 650 °C, WHSV = 3.83 h−1, time on stream = 3 h. | ||
Fig. 12 shows the effect of reaction temperature on the reactivity of 0.3-Zn/HZSM-5 in the methanol promoted naphtha catalytic pyrolysis system. Methanol was almost completely converted in the investigated reaction temperature range. As expected, the conversion of naphtha increased with the increase in reaction temperature. The yield of ethylene constantly increased with increasing temperature and reached a maximum value of 31.4 wt% at 700 °C. The propylene yield initially increased to a maximum value of 28.3 wt% at 650 °C and subsequently decreased to 26.8 wt% at 700 °C. Tang et al.36 carried out the thermodynamic analysis of naphtha cracking to olefins. The analysis illustrated that the propylene yield was limited by the thermodynamic equilibrium, while the feed conversion was not limited. When the reaction temperature exceeds a certain value, the dominant reaction of hydrocarbon cracking gradually changes from catalytic cracking to thermal cracking. Therefore, it can be inferred that for the methanol promoting naphtha catalytic pyrolysis system, the thermal cracking began to become prominent at reaction temperatures above 650 °C. The maximum light olefin (ethylene + propylene) yield of 58.2 wt% was attained at 700 °C. However, this is not the true performance of the modified catalysts under these reaction conditions. Moreover, a much higher reaction temperature will result in the rapid deactivation of the catalyst. Thus, the optimal reaction temperature for methanol promoted naphtha catalytic pyrolysis process is 650 °C.
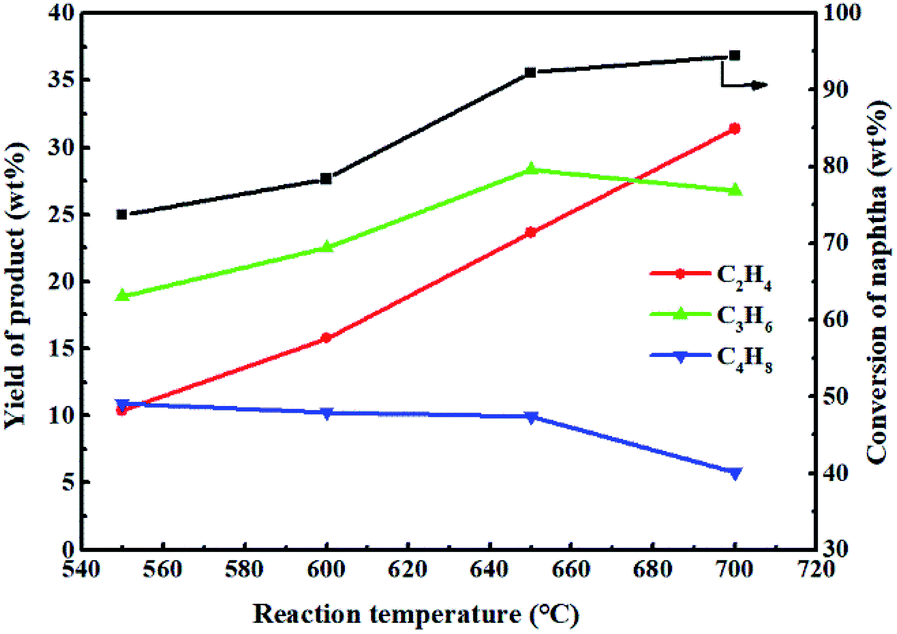 | ||
| Fig. 12 Effect of reaction temperature on the reactivity of 0.3-ZnZSM-5 in the methanol promoted naphtha catalytic pyrolysis system. Reaction conditions: methanol/naphtha = 15 wt%, WHSV = 3.83 h−1, N2 = 60 mL min−1, time on stream = 3 h. | ||
Fig. 13 shows the effect of weight hourly space velocity (WHSV) on the reactivity of 0.3-Zn/HZSM-5 in methanol promoted naphtha catalytic pyrolysis system at 650 °C. The conversion of naphtha gradually decreased with the increase in WHSV because high WHSV diminished the contact of the reactants with the active sites. The ethylene and propene yield first increased and then decreased with the increase in WHSV. However, the BTX (benzene, toluene, and xylene) yield decreased with the increase in WHSV except for a slight increase in the WHSV of 7. This indicates that the product distribution is closely related to the WHSV. With the initial increase in WHSV, the secondary conversion of light olefins was hindered, which improved the light olefin yield. However, the further improvement of WHSV was not conducive for high naphtha conversion and so, the light olefin yield diminished. Moreover, when WHSV was less than 8, the propene yield decreased with the decrease in WHSV. When WHSV was less than 3.8, the ethylene yield decreased with the decrease in WHSV. The result implies that propylene is more likely to have a secondary reaction than ethylene. To sum up, in order to achieve high light olefin yield, the optimal WHSV is 3.83 h−1 under the reaction conditions used.
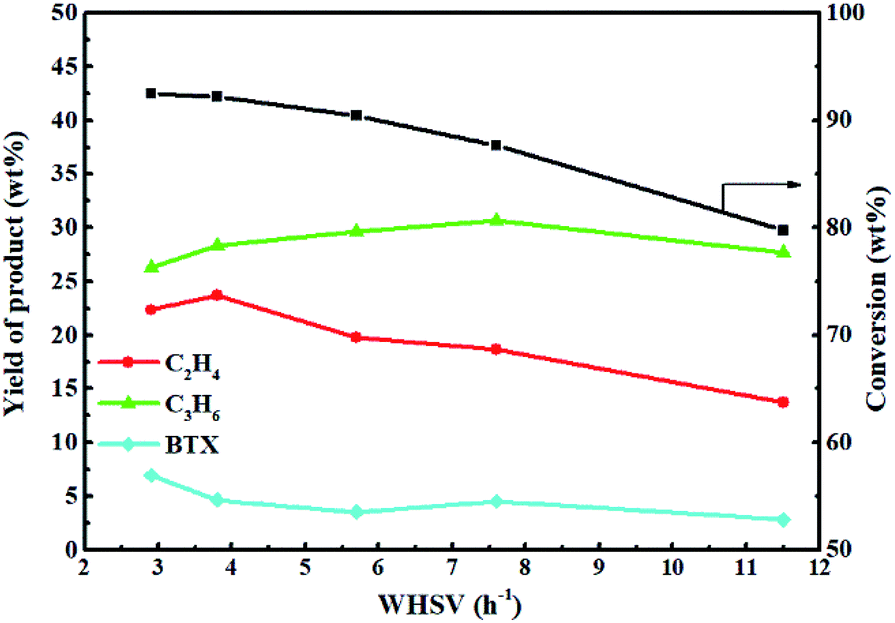 | ||
| Fig. 13 Effect of WHSV on the reactivity of 0.3-ZnZSM-5 in the methanol promoted naphtha catalytic pyrolysis system. Reaction conditions: reaction temperature = 650 °C, methanol/naphtha = 15 wt%, N2 = 60 mL min−1, time on stream = 3 h. | ||
Because the main objective of adding methanol to naphtha was to achieve the efficient utilization of naphtha and produce as much ethylene and propylene as possible, the reaction had to be performed at a reasonably higher temperature (650 °C) and at a relatively lower WHSV (3.83 h−1).
3.3. Coke deposition
Fig. 14 and 15 show the TG-DSC diagrams of HZSM-5 and 0.3-Zn/HZSM-5 that had been tested with mixed methanol–naphtha feed. According to the DSC curves, the exothermic peaks were obtained at 620 and 633 °C for the used HZSM-5 and the used 0.3-Zn/HZSM-5, respectively. We can reasonably assume that the peak corresponded to the combustion of coke. The weight loss between 300 and 800 °C corresponds to the coke amount. The weight loss due to coke combustion was 3.1 and 3.7 wt% for the used HZSM-5 and the used 0.3-Zn/HZSM-5, respectively. A subtle difference in the deposited amount of carbon was obtained between the used HZSM-5 and the used 0.3-Zn/HZSM-5. The result indicates that the incorporation of Zn into the HZSM-5 zeolite inevitably aggravated the carbon deposition due to the dehydrogenation of alkane on the Zn acid sites at the beginning of the reaction. However, the increase of mesopore and the enhancement of β-scission reaction alleviated the formation of coke on Zn/HZSM-5.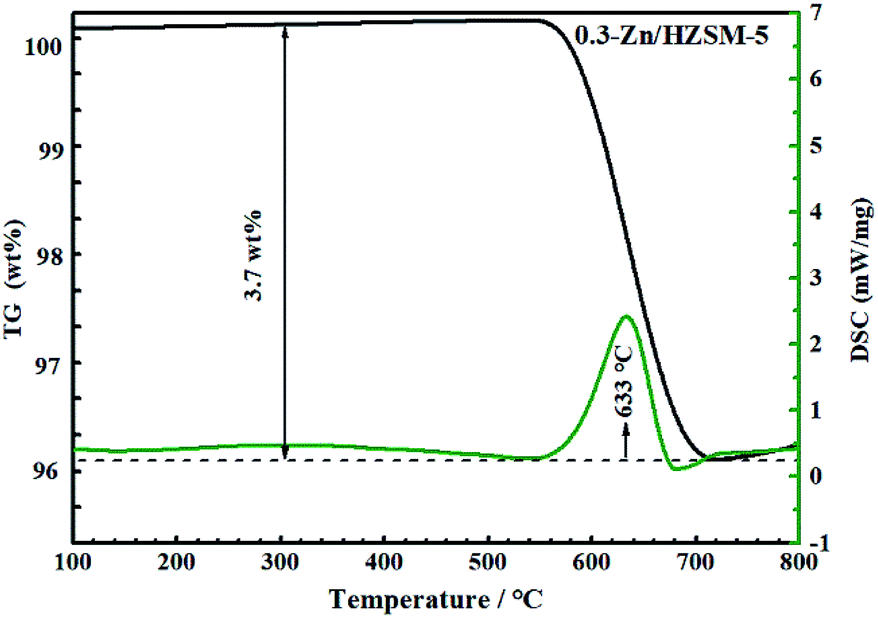 | ||
| Fig. 14 TG/DSC curves of used 0.3-Zn/HZSM-5 in flowing air. Reaction conditions: reaction temperature = 650 °C, methanol/naphtha = 15 wt%, WHSV = 3.83 h−1, N2 = 60 mL min−1, time on stream = 7 h. Feed: mixed feed of methanol–naphtha. | ||
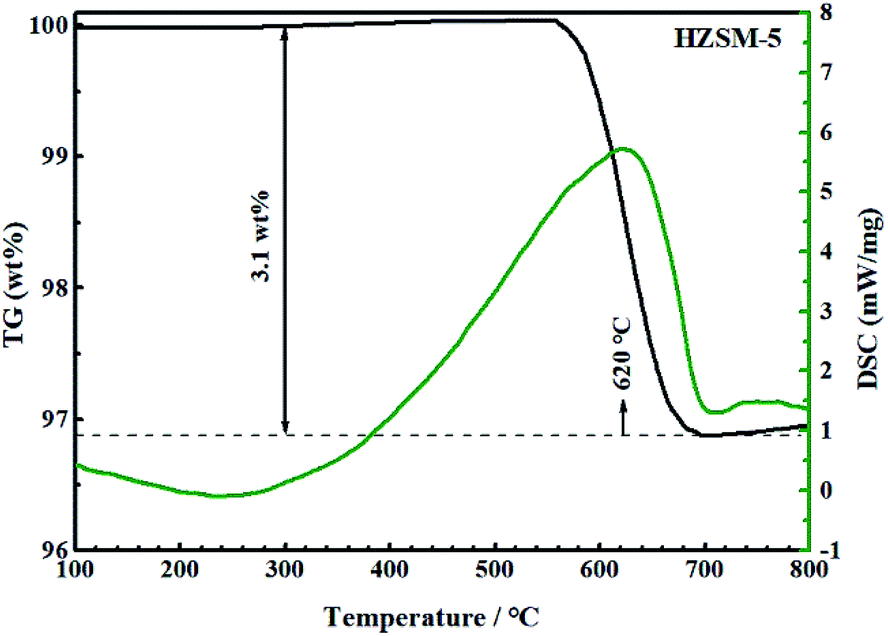 | ||
| Fig. 15 TG/DSC curves of used HZSM-5 in flowing air. Reaction conditions: reaction temperature = 650 °C, methanol/naphtha = 15 wt%, WHSV = 3.83 h−1, N2 = 60 mL min−1, time on stream = 7 h. Feed: mixed methanol–naphtha. | ||
Fig. 16 depicts the TG-DSC measurements of the 0.3-Zn/HZSM-5 catalyst that had been tested with pure naphtha. The weight loss between 300 and 800 °C was 7.5 wt% for 0.3-Zn/HZSM-5 run with pure naphtha, which is higher than that of the 0.3-Zn/HZSM-5 catalyst run with the mix feed. This result indicates that the addition of methanol in naphtha gave rise to a noticeable decrease in coke deposition. Yan et al.18 reported that the presence of methanol led to a higher turnover “reactant adsorption/product desorption” on the zeolite acid sites, thus lowering the formation of coke precursors and finally decreasing the coke deposition rate. In addition, the presence of water in the products may also prevent the deactivation of the catalysts.
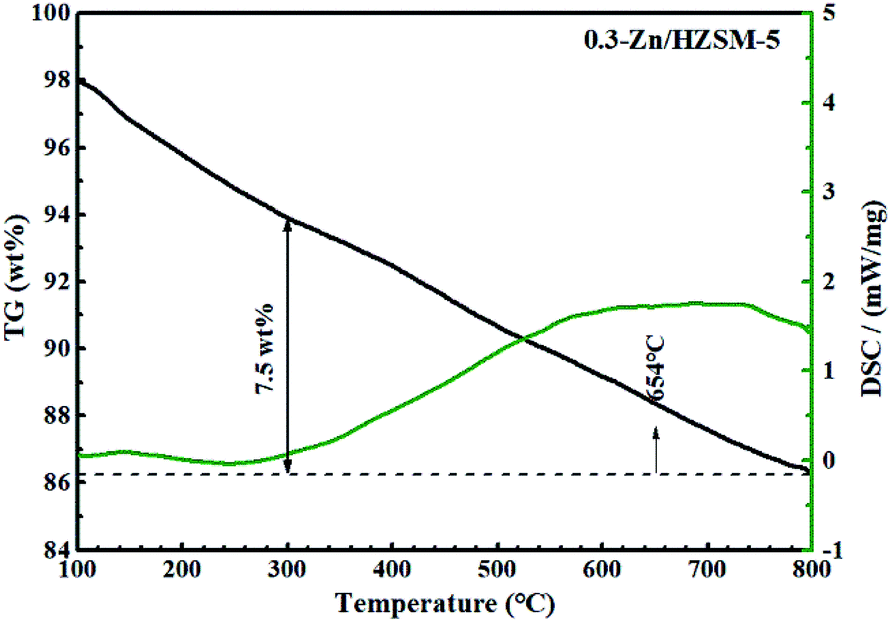 | ||
| Fig. 16 TG/DSC curves of used 0.3-Zn/HZSM-5 in flowing air. Reaction conditions: reaction temperature = 650 °C, WHSV = 3.83 h−1, N2 = 60 mL min−1, time on stream = 7 h. Feed: pure naphtha. | ||
3.4. Transformation pathways of alkanes over Zn/HZSM-5 in methanol promoted naphtha catalytic pyrolysis system
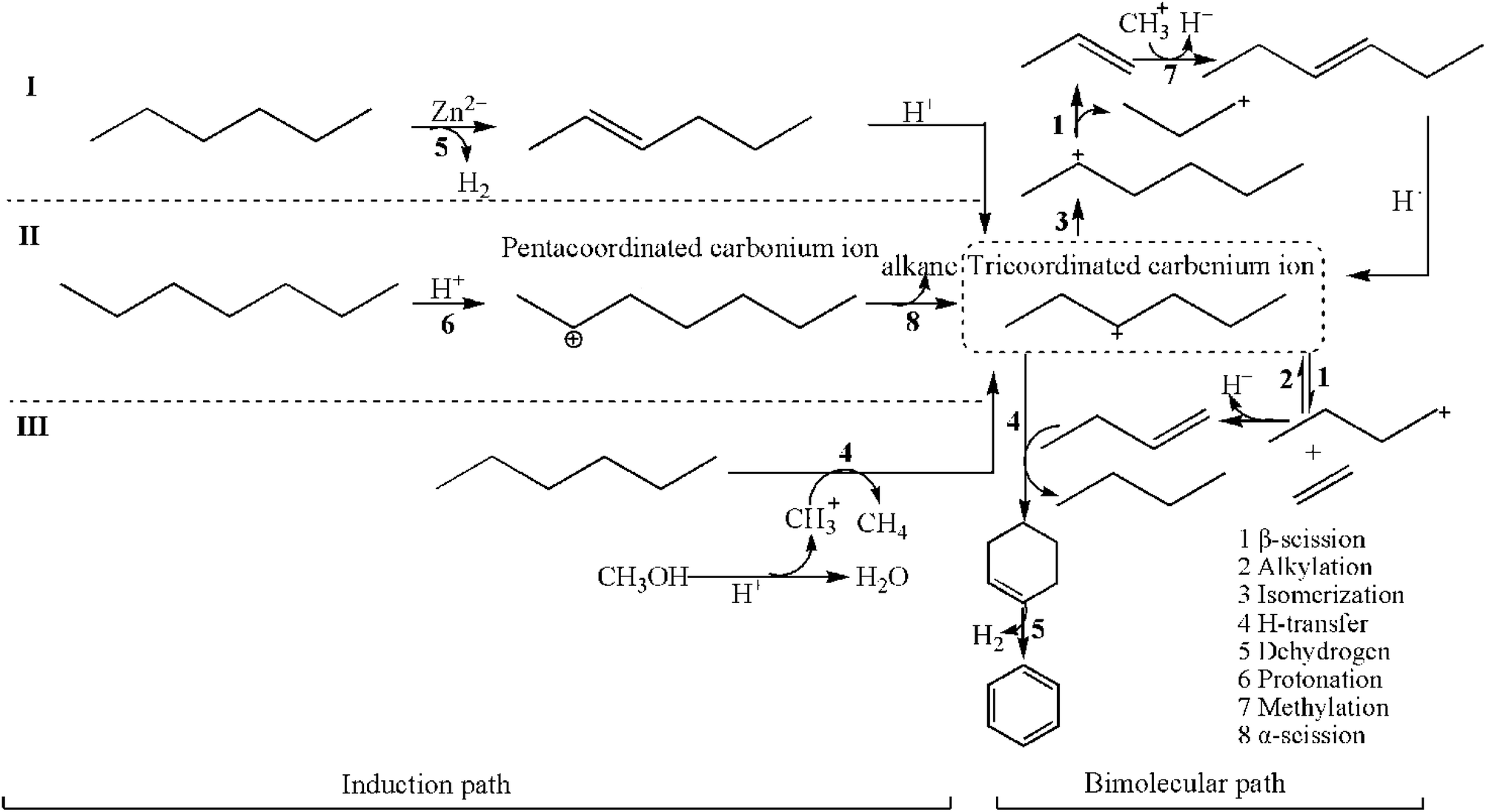 | ||
| Scheme 1 Transformation pathways of alkanes over Zn/HZSM-5 in the methanol promoted naphtha catalytic pyrolysis system. | ||
The consecutive isomerization and β-scission of the reactant-sized tricoordinated carbenium ion result in an alkene and a new tricoordinated carbenium ion. The H-transfer between the reactant alkane and a smaller tricoordinated carbenium ion produces a smaller alkane product and a reactant-sized tricoordinated carbenium ion. Apparently, these reactions do not involve the formation of carbonaceous materials in zeolites. The olefin products are determined by the priority of the β-scission reaction. Moreover, the larger tricoordinated carbenium ions are formed by alkylation between the tricoordinated carbenium ion and an alkene. The larger tricoordinated carbenium ions undergo sequential H-transfer and dehydrocyclization to form aromatics. It can be seen that the relative rates of alkylation and β-scission determine the olefins and aromatic product distribution in the zeolite.38
It can be seen that for the activation of alkanes, whether by immediate protonation (Scheme 1(II)) or H-transfer with CH3+ (Scheme 1(III)), the adsorption at the Brønsted acid sites is essential.
4 Conclusion
A series of Zn-modified high-silicon HZSM-5 zeolites were prepared and characterized. Their performances for the methanol promoted naphtha catalytic pyrolysis system were evaluated in a fixed-bed reactor. The comparatively high light olefin yield of 51.7 wt% was obtained on 0.3-Zn/HZSM-5 catalyst under the optimal reaction conditions of 650 °C, WHSV = 3.83 h−1, methanol/naphtha = 15 wt%, and flow rate of N2 = 60 mL min−1. Under the optimal coupling reaction conditions, the light olefin yield obtained from the 0.3-Zn/HZSM-5 catalyst was higher than that from the other HZSM-5 based catalysts, including H(Fe, Al)-ZSM-5 and the hybrid catalysts (HZSM-5 with Zn–Pd/Y2O3–Al2O3).17,18 The introduction of Zn species brought about new active sites and reaction pathways for the reaction system. The activation of alkane on the Brønsted acid sites was decreased. Moreover, with the increase of Zn species in the HZSM-5 framework, the density of the Brønsted acid sites reduced, which modified the density and distribution of the acid sites on Zn/HZSM-5. The results from the experiments indicate that the acidity of Zn/HZSM-5 has an important role on its catalytic reactivity in the methanol promoted naphtha catalytic pyrolysis reaction. The 0.3-Zn/HZSM-5 catalyst exhibited high light olefin yield mainly owing to its moderate density and distribution of the acid sites (involving the Zn and Brønsted acid sites).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (Grant 91634112, 21878097) and the Natural Science Foundation of Shanghai (Grant 16ZR1408100).References
- R. L. V. Mao, A. Muntasar, H. T. Yan and Q. Zhao, Catal. Lett., 2009, 130, 86–92 CrossRef.
- S. M. Sadrameli, Fuel, 2016, 173, 285–297 CrossRef CAS.
- G. E. Keller and M. M. Bhasin, J. Catal., 1982, 73, 9–19 CrossRef CAS.
- G. S. Kumar, L. J. Lau, M. D. Krcha and M. J. Janik, ACS Catal., 2016, 6, 1812–1821 CrossRef CAS.
- S. Svelle, F. Joensen, J. Nerlov, U. Olsbye, K. P. Lillerud, S. Kolboe and M. J. Bjørgen, J. Am. Chem. Soc., 2006, 128, 14770–14771 CrossRef CAS PubMed.
- M. Bjørgen, S. Svelle, F. Joensen, J. Nerlov, S. Kolboe, F. Bonino, L. Palumbo, S. Bordiga and U. Olsbye, J. Catal., 2007, 249, 195–207 CrossRef.
- J. Bedia, R. Barrionuevo, J. Rodríguez-Mirasol and T. Cordero, Appl. Catal., B, 2011, 103, 302–310 CrossRef CAS.
- F. Cabrera, D. Ardissone and O. F. Gorriz, Catal. Today, 2008, 133–135, 800–804 CrossRef CAS.
- F. Cavani and F. Trifirò, Catal. Today, 1995, 24, 307–313 CrossRef CAS.
- H. H. Kung, Adv. Catal., 1994, 40, 1–8 CAS.
- Y. Yoshimura, N. Kijima, T. Hayakawa, K. Murata, K. Suzuki, F. Mizukami, K. Matano, T. Konishi, T. Oikawa, M. Saito, T. Shiojima, K. Shiozawa, K. Wakui, G. Sawada, K. Sato, S. Matsuo and N. Yamaoka, Catal. Surv. Jpn., 2001, 4, 157–167 CrossRef.
- A. Usman, M. Siddiqui, A. Hussain, A. Aitani and S. Khattaf, Chem. Eng. Res. Des., 2017, 120, 121–137 CrossRef CAS.
- S. M. Alipour, Chin. J. Catal., 2016, 37, 671–680 CrossRef.
- H. Mochizuki, T. Yokoi, H. Imai, R. Watanabe, S. Namba, J. N. Kondo and T. Tatsumi, Microporous Mesoporous Mater., 2011, 145, 165–171 CrossRef CAS.
- N. Rahimi and R. Karimzadeh, Appl. Catal., A, 2011, 398, 1–17 CrossRef CAS.
- F. X. Chang, Y. X. Wei, X. B. Liu, Y. F. Zhao, L. Xu, Y. Sun, D. Z. Zhang, Y. L. He and Z. M. Liu, Appl. Catal., A, 2007, 328, 163–173 CrossRef CAS.
- B. Lücke, A. Martin, H. Günschel and S. Nowak, Microporous Mesoporous Mater., 1999, 29, 145–157 CrossRef.
- H. T. Yan and R. L. V. Mao, Catal. Lett., 2011, 141, 691–698 CrossRef CAS.
- A. Martin, S. Nowak, B. Lücke, W. Wieker and B. Fahlke, Appl. Catal., 1990, 57, 203–214 CrossRef CAS.
- T. Gong, X. Zhang, T. Bai, Q. Q. Zhang, L. Tao, M. Qi, C. Duan and L. Zhang, Ind. Eng. Chem. Res., 2012, 51, 13589–13598 CrossRef CAS.
- D. Mier, A. T. Aguayo, A. G. Gayubo, M. Olazar and J. Bilbao, Appl. Catal., A, 2010, 383, 202–210 CrossRef CAS.
- J. A. Biscardi and E. Iglesia, J. Catal., 1999, 182, 117–128 CrossRef CAS.
- V. B. Kazansky and A. I. Serykh, Phys. Chem. Chem. Phys., 2004, 6, 3760–3764 RSC.
- A. A. Sokol, C. R. A. Catlow, J. M. Garces and A. Kuperman, J. Phys.: Condens. Matter, 2004, 16, 2781–2794 CrossRef.
- N. Xue, N. Liu, L. Nie, Y. Yu, M. Gu, L. Peng, X. Guo and W. Ding, J. Mol. Catal. A: Chem., 2010, 327, 12–19 CrossRef CAS.
- X. Niu, J. Gao, Q. Miao, M. Dong, G. Wang, W. Fan, Z. Qin and J. Wang, Microporous Mesoporous Mater., 2014, 197, 252–261 CrossRef CAS.
- S. Tamiyakul, T. Sooknoi, L. L. Lobban and S. Jongpatiwut, Appl. Catal., A, 2016, 525, 190–196 CrossRef CAS.
- J. Sárkány, Appl. Catal., A, 1999, 188, 369–379 CrossRef.
- V. L. Zholobenko, L. M. Kustov, V. Y. Borovkov and V. B. Kazansky, Zeolites, 1988, 8, 175–178 CrossRef CAS.
- A. G. Pelmenschikov, J. H. M. C. V. Wolput, J. Jaenchen and R. A. V. Santen, J. Phys. Chem., 1995, 99, 3612–3617 CrossRef CAS.
- O. Awayssa, N. A. Yassir, A. Aitani and S. A. Khattaf, Appl. Catal., A, 2014, 477, 172–183 CrossRef CAS.
- El-M. El-Malki, R. A. van Santen and W. M. H. Sachtler, J. Phys. Chem. B, 1999, 103, 4611–4622 CrossRef CAS.
- A. L. Yakovlev, A. A. Shubin, G. M. Zhidomirovb and R. A. V. Santen, Catal. Lett., 2000, 70, 175–181 CrossRef CAS.
- S. Ilias, R. Khare, A. Malek and A. Bhan, J. Catal., 2013, 303, 135–140 CrossRef CAS.
- J. F. Haw, Phys. Chem. Chem. Phys., 2002, 4, 5431–5441 RSC.
- X. P. Tang, H. Q. Zhou, F. Wei and Y. Jin, Acta Pet. Sin., Pet. Process. Sect., 2008, 24, 22–27 CAS.
- F. X. Chang, Y. X. Wei, X. B. Liu, Y. Qi, D. Z. Zhang, Y. L. He and Z. M. Liu, Catal. Lett., 2005, 106, 171–176 CrossRef.
- B. A. Williams, W. Ji, J. T. Miller, R. Q. Snurr and H. H. Kung, Appl. Catal., A, 2000, 203, 179–190 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02793a |
| This journal is © The Royal Society of Chemistry 2019 |