
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePerovskite lattice oxygen contributes to low-temperature catalysis for exhaust gas cleaning†
Takuma Higo *a,
Kohei Uenoa,
Yuki Omoria,
Hiroto Tsuchiyaa,
Shuhei Ogoa,
Satoshi Hiroseb,
Hitoshi Mikamib and
Yasushi Sekinea
*a,
Kohei Uenoa,
Yuki Omoria,
Hiroto Tsuchiyaa,
Shuhei Ogoa,
Satoshi Hiroseb,
Hitoshi Mikamib and
Yasushi Sekinea
aDepartment of Applied Chemistry, Waseda University, 3-4-1, Okubo, Shinjuku, Tokyo 169-8555, Japan. E-mail: t-higo@aoni.waseda.jp
bHonda R&D, 4630, Shimo-Takanezawa, Tochigi 321-3393, Japan
First published on 23rd July 2019
Abstract
A Pd catalyst supported on Ba-substituted LaAlO3 perovskite (Pd/La0.9Ba0.1AlO3−δ) was investigated for NO reduction at low temperature by propylene, which revealed that Pd/La0.9Ba0.1AlO3−δ has remarkably higher activity than other Pd catalysts at low temperatures (≤573 K) for NO reduction by propylene. To elucidate the surface reaction pathway, transient response tests were conducted using 18O2. Also, X-ray photoelectron spectroscopy (XPS) and diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) measurements were conducted. Comparison with a Ba-impregnated catalyst (Pd/Ba/LaAlO3) demonstrated that Pd/La0.9Ba0.1AlO3−δ shows higher activity for the formation of oxygenated species (CxHyOz) as an intermediate for NO reduction because the surface lattice oxygen has improved mobility via Ba2+ substitution in LaAlO3. Therefore, Pd/La0.9Ba0.1AlO3−δ have high activity for NO reduction, even at low temperatures in a humid condition.
1. Introduction
Additional and increasingly severe emissions and fuel efficiency regulations are expected to be imposed on automobiles along with the global progress of motorization. In spite of the development for zero-emission vehicles (ZEVs) such as battery electric vehicles (BEVs) and fuel cell vehicles (FCVs), it is considered that internal combustion engine vehicles including plug-in hybrid vehicles (PHVs) will maintain a certain share of future vehicle markets. Therefore, purification of emission gases from gasoline combustion vehicles will remain an important technology in a future sustainable society. Currently, the most general technology for purifying the emission gases of gasoline combustion vehicles is conversion of pollutants using a three-way catalyst (TWC). On TWCs, reduction of nitrogen oxides (NOx), oxidation of unburned hydrocarbons (HC) and carbon monoxide (CO) proceed simultaneously through conversion respectively to N2, CO2, and H2O.1,2 Such catalysts can convert the pollutants in emissions by nearly 100% at the stoichiometric air/fuel ratio and at high temperatures (>673 K).3 One challenge for future TWCs is high catalytic performance at lower temperatures than those of the current standard operation conditions.Pd-based catalysts show higher activity for HC oxidation than those of either Pt and Rh, but the NOx reduction activity is poor.4,5 Many studies have been conducted to enhance the catalytic performance for Pd-based catalysts, with the addition of a promoter (alkali,6,7 alkaline earth,8 rare-earth8,9 etc.) or using bimetallic catalysts,10,11 and the development of support materials.12–22 Further increase in achieving catalytic activity at lower temperatures is anticipated.
The combination of noble metals and perovskite-type oxide has been studied as offering potential for effective de-NOx catalysts.15,18–22 A typical formula of the perovskite structure is ABO3. Perovskite oxides have been regarded as interesting catalyst supports because of their features, which include redox property and high thermal stability.15,20,23,24 Results have shown that the lattice oxygen reacts with various hydrocarbons to form partially oxidized hydrocarbon species (CxHyOz: oxygenated species) on Sr-substituted LaAlO3.25–27 Oxygenated species reacted with H2O as reductants to form H2. Then the lattice oxygen consumed is restored by H2O in atmosphere. Such a redox cycle of substituted LaAlO3 support is regarded as effective for NO reduction with hydrocarbon.
This study investigated Ba-substituted LaAlO3 perovskite as a support material for Pd catalyst for low-temperature NOx reduction by propylene. We confirmed the superiority of Pd catalyst supported on Ba-substituted perovskite and discussed factors related to its high catalytic performance.
2. Experimental
2.1 Catalyst preparation
Perovskite-type oxides (LaAlO3, La1−xBaxAlO3−δ; x = 0.03, 0.05, 0.1 and 0.2) were prepared using a citric acid complex method.25 Metal nitrate precursors (Kanto Chemical Co. Inc.) were dissolved in water. Then, excess citric acid and ethylene glycol (Kanto Chemical Co. Inc.) were added to the solution. The molar ratio of metal![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :citric acid:ethylene glycol was 1:3:3. After the obtained solution was evaporated in a water bath at ca. 353 K for 16 h, the solution was dried on a hot plate with stirring. The obtained powder was pre-calcined at 673 K for 2 h and was calcined at 1123 K for 10 h.
:citric acid:ethylene glycol was 1:3:3. After the obtained solution was evaporated in a water bath at ca. 353 K for 16 h, the solution was dried on a hot plate with stirring. The obtained powder was pre-calcined at 673 K for 2 h and was calcined at 1123 K for 10 h.
We prepared Pd-loaded catalysts using an impregnation method. For loading Pd, palladium acetate (Kanto Chemical Co. Inc.) was used as a precursor. Distilled acetone was used as a solvent. The Pd loading amount was 0.5 wt%. The prepared catalysts were calcined at 823 K for 3 h in air.
We prepared Pd/Ba/LaAlO3 using sequential impregnation of Ba and Pd. The Ba loading amount was 6.5 wt% (which is almost equal to the molar ratio of Ba in Pd/La0.9Ba0.1AlO3−δ). As a control catalyst, 0.53 wt% Pd/Al2O3 (Honda R&D Co. Ltd.) was used. γ-Al2O3 was prepared as support by calcination of boehmite (Sasol Ltd.) in air at 873 K. Loading of Pd was conducted using an ion-exchange method. Palladium nitrate (Kojima Chemicals Co. Ltd.) was used as a precursor. Calcination was conducted at 773 K in air for 2 h.
2.2 Activity tests
Catalytic activity tests for NO reduction were conducted in a fixed bed quartz reactor at atmospheric pressure. Catalysts (50 mg, 0.25–0.5 mm particle size) were mixed with SiC to reach a catalyst bed height of 1 cm. Activity tests were conducted in reaction gases of three compositions balanced by Ar gas: NO–C3H6 (2250 ppm NO, 250 ppm C3H6, λ = 1), NO–C3H6–O2 (1000 ppm NO, 500 ppm C3H6, 2000 ppm O2, λ = 1.11), and NO–C3H6–O2–H2O (1000 ppm NO, 500 ppm C3H6, 2000 ppm O2, 7 vol% H2O, λ = 1.11). The λ value is defined as eqn (1).
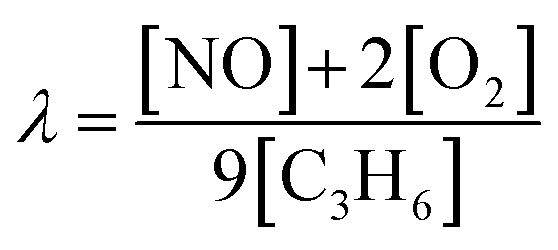 | (1) |
The total gas flow rate for the reaction was 200 mL min−1 (W/F = 0.1 g h mol−1). Pre-treatment of the catalyst was conducted sequentially for oxidation (5% O2, Ar balanced at 773 K for 15 min) and reduction (5% H2, Ar balanced at 773 K for 15 min). Then catalytic activities were measured at 673, 623, 573, 523, and 473 K. The outlet gas was analyzed using online GC-TCD (GC-8A; Shimadzu Corp.), GC-FID (GC-8A; Shimadzu Corp.) and a chemiluminescent method NOx analyser (NOA-7000; Shimadzu Corp.). The NO conversion and N2 yield are defined as shown below (eqn (2) and (3)).
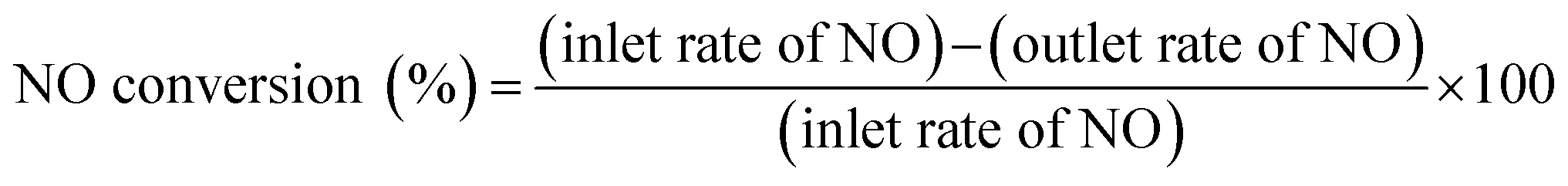 | (2) |
 | (3) |
Tests for dependence on NO and C3H6 concentration were conducted in NO–C3H6–O2–H2O condition at 523 K. The loaded amount of Pd/La0.9Ba0.1AlO3−δ was decreased to 25 mg to obtain kinetic values. The reaction gas composition for investigation of NO was NO (500, 750, 1000, 1250, or 1500 ppm): C3H6 500 ppm: O2 2000 ppm. The reaction gas composition for investigation of C3H6 was NO 1000 ppm: C3H6 (375, 425, 500, 575, 625, or 750 ppm): O2 2000 ppm. 7 vol% H2O was fed in both reaction conditions.
2.3 Characterization of catalysts
X-ray diffraction patterns were measured to confirm the crystalline structure of perovskite-type oxides (SmartLab 3; Rigaku Corp.). The Cu Kα radiation condition was at 40 kV and 40 mA. The respective BET specific surface areas of the catalysts were measured at 77 K using N2 adsorption (GeminiVII; Micromeritics Instrument Corp.). Using a field emission transmission electron microscope equipped with an energy dispersive X-ray spectrometer (TEM-EDX, JEM-2100F; JEOL Ltd.), we evaluated the Pd particle sizes of Pd/Al2O3, Pd/LaAlO3, Pd/La0.9Ba0.1AlO3−δ and Pd/Ba/LaAlO3 after oxidation (5% O2, Ar balance at 773 K for 15 min) and reduction (5% H2, Ar balance at 773 K for 15 min) as pre-treatments.2.4 In situ DRIFTS measurements
In situ DRIFTS measurements were taken using a Fourier transform infrared spectrometer (FT/IR 6200; Jasco Corp.) with an MCT detector and a ZnSe window. Catalyst sample powder (ca. 40 mg) was filled into a sample cell. After background (denoted as BKG) measurements were taken under inert Ar gas (100 mL min−1 at each temperature) after oxidation (5% O2 for 15 min) and reduction (5% H2 for 15 min) at 773 K, the following two measurements were taken.(1) Steady state measurement: reactant gases (NO 1000 ppm, O2 2000 ppm, C3H6 500 ppm, H2O 7 vol% and Ar balanced) were supplied to the sample cell for 10 min at each temperature (473–623 K). Each spectrum was recorded from 1000 to 4000 cm−1 with resolution of 4.0 cm−1 and accumulation of 10 scans.
(2) Transient measurement: C3H6-containing gas (C3H6 500 ppm and Ar) is supplied to the sample cell for 10 min at 523 K. Then, oxidant gases (O2 2000 ppm or NO 1000 ppm and Ar) were supplied for 10 min after Ar purging for 10 min. This procedure was regarded as one operation. Two operations were conducted continuously. The change of spectrum was recorded from 1000 to 4000 cm−1 with resolution of 4.0 cm−1 and accumulation of 10 scans.
2.5 Transient response tests using 18O2
Transient response tests using 18O2 were done to observe the contribution of lattice oxygen to the reaction. Catalyst of 200 mg was used for this test. Pretreatment was conducted similarly to the activity test. The feed gas was supplied for 15 min at 523 K after Ar purging for 5 min. The gas composition was 500 ppm C3H6: 2000 ppm 18O2 (balance gas was Ar; 200 mL min−1 total flow rate). The outlet gas was analysed using a quadrupole mass spectrometer (QGA; Hiden Analytical Ltd.). The analysis references of Q-mass for carbon dioxide species were m/z = 44(C16O2), 46(C16O18O), and 48(C18O2). Here, 16O detected in carbon dioxide was regarded as the amount of lattice oxygen used in the reaction.2.6 X-ray photoelectron spectroscopy (XPS)
To obtain data for the surface atomic ratio and the spectra of O1s region of the catalysts, XPS measurements were performed on Versa Probe II (Ulvac Phi Inc.) with Al Kα X-rays. Binding energies for each orbital were calibrated using C1s (284.8 eV). Catalysts were treated using the following procedure before XPS measurements. After heating treatment in Ar gas flow at 773 K for 30 min, samples were exposed sequentially in the order of 500 ppm C3H6 (step 1), 2000 ppm O2 (step 2), and 500 ppm C3H6 (step 3) flow at 523 K. To remove C3H6 or O2 from the sample cell, purging with Ar gas was performed for 10 min between the respective steps. Samples after each step were cooled to room temperature in Ar flow and were moved to the sample chamber using a transfer vessel to avoid exposure to air.3. Results and discussion
3.1 Catalytic performance and structure of substituted perovskite supported Pd catalyst
We investigated NO reduction by propylene as a model of unburned hydrocarbon in an exhaust gas on various Pd catalysts supported on perovskites (Pd/LaAlO3, Pd/Ba/LaAlO3, Pd/La0.9Ba0.1AlO3−δ) and Pd/Al2O3 as a control catalyst for comparison. The respective catalytic activities on these catalysts are presented in Fig. 1, S1 and Table S1 of ESI.† At low temperatures of 523–573 K, Pd/La0.9Ba0.1AlO3−δ catalyst showed higher NO conversion and N2 yield than the other catalysts. Table S2† shows results for the BET specific surface area, mean particle diameter and turnover frequency (TOF) for NO conversion at 523 K on the catalysts. The mean particle diameter was estimated from the Pd particle size histogram of TEM measurements (Fig. S2†). The TOF was calculated on the assumption that Pd particles are supported hemispherically. The value of TOF for Pd/La0.9Ba0.1AlO3−δ was the highest among these catalysts under this condition. To investigate the appropriate amount of Ba substitution in the LaAlO3 support, we evaluated the catalytic performances of various Ba-substituted catalysts (Pd/La1−xBaxAlO3−δ; x = 0, 0.03, 0.05, 0.1 and 0.2). Fig. S3† presents the results. Among these catalysts, Pd/La0.9Ba0.1AlO3−δ showed the best NO conversion. Therefore, we determined the appropriate substitution ratio of La with Ba as 10%. The XRD pattern and the lattice constant for each Ba-substituted LaAlO3 are presented in Fig. S4 and S5†.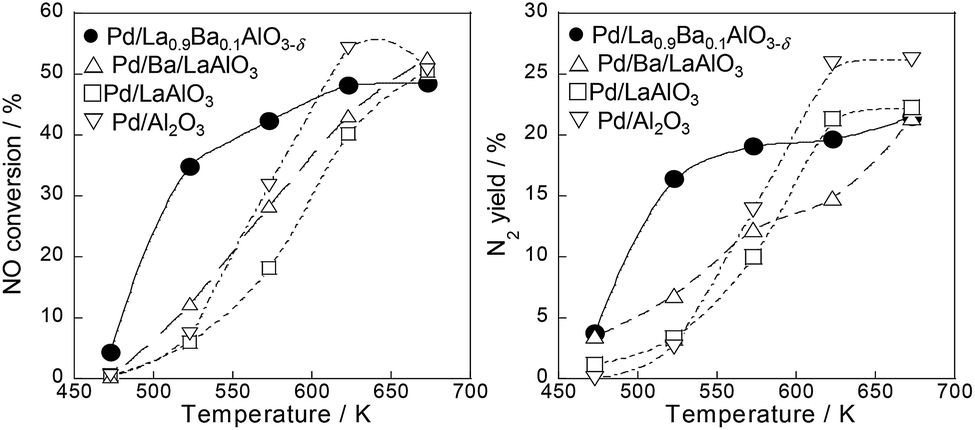 | ||
| Fig. 1 Comparison of NO conversions and N2 yields over Pd/La0.9Ba0.1AlO3−δ, Pd/Ba/LaAlO3, Pd/LaAlO3 and Pd/Al2O3. Reaction conditions: total flow rate = 200 mL min−1, NO 1000 ppm, C3H6 500 ppm, O2 2000 ppm, H2O 7 vol%. | ||
In other conditions, we compared the catalytic activities on NO–C3H6 reaction in a stoichiometric condition and a slightly lean condition without steam. Tables S3 and S4† present NO conversions on various catalysts at each temperature. Regarding NO–C3H6 reaction without O2 and H2O (NO, 2250 ppm; C3H6, 250 ppm), NO conversion over the catalysts at 523 K were 23.9% for Pd/Al2O3, 29.5% for Pd/LaAlO3, 44.3% for Pd/Ba/LaAlO3 and 46.5% for Pd/La0.9Ba0.1AlO3−δ. Results show that Pd/La0.9Ba0.1AlO3−δ and Pd/Ba/LaAlO3 have similarly high activities at 523 K. From earlier studies, the effects of promoter co-impregnated or sequentially impregnated on the catalytic activity were reported for PGM catalysts for NOx reduction.6–9,28–31 Reportedly, basic additives such as Na and Ba showed promotive effects on NO–C3H6 reaction.6,7 These results demonstrate that Ba species promote NO reduction by C3H6, irrespective of substitution or impregnation. However, the catalytic activity of Pd/La0.9Ba0.1AlO3−δ (i.e. Ba substituted catalyst) under a slightly lean condition was much higher than Pd/Ba/LaAlO3, as shown in Tables S1 (with H2O) and S4 (without H2O).† Under these conditions, Pd/La0.9Ba0.1AlO3−δ is a promising catalyst for NOx reduction.
3.2 In situ DRIFTS studies for steady-state surface species
To elucidate the reasons for high activity at low temperature with steam on Pd/La0.9Ba0.1AlO3−δ (i.e. Ba-substituted perovskite support) catalyst in terms of a reaction mechanism, in situ DRIFTS measurements were conducted for various catalysts. Fig. 2 presents DRIFT spectra of surface species on Pd/La0.9Ba0.1AlO3−δ. These spectra had two notable regions at 1100–1800 cm−1 and 2100–3000 cm−1. The notable band at 1227 cm−1 assignable to nitrite species32 was observed at all temperatures. The relative intensity of the nitrite band decreased concomitantly with increasing temperature. The other strong bands at 1307 and 1500–1800 cm−1 are overlapping bands derived from various surface species. These bands in this region are assigned to partially oxidized hydrocarbons, carbonate and nitrate species.32–37 The presence of weak bands near 2800–3000 cm−1 assignable to C–H stretching32,37 revealed the existence of hydrocarbon fragments on the catalyst surface. The relative intensity of these bands increased at temperatures up to 523 K and decreased at temperatures higher than 573 K. In addition to the bands described above, notable bands at 2168 cm−1 and 2198 cm−1 were observed and assigned to isocyanate (-NCO) species.32–35,37,38 These bands of isocyanate appeared clearly from 473 K to 573 K, and disappeared at 623 K. Earlier reports describe the surface isocyanate species as the intermediate species on various NOx reduction pathways.33,34,37,38 However, Burch et al. reported that isocyanate species is not a key intermediate but is instead a spectator.39 We infer that isocyanate, which contains N and C, is an indicator of the surface reaction of NO and hydrocarbon reductant. From observation of the spectra for Pd/La0.9Ba0.1AlO3−δ, the reaction pathway under this condition can be inferred as follows: NO adsorbs on the Ba species near Pd particles as bridging nitrite and nitrate species. Also, C3H6 is oxidized to form CxHyOz. The nitrite/nitrate and CxHyOz are the active intermediate species, which mutually react to form isocyanate. The isocyanate reacts with NO to convert to N2, CO2 and H2O as final products. The DRIFT spectra for Pd/Ba/LaAlO3 are depicted in Fig. S6†. The assignment and behaviour for the bands on spectra closely resemble those for Pd/La0.9Ba0.1AlO3−δ. Therefore, the results suggest that the surface intermediate species during steady state reactions on Pd/La0.9Ba0.1AlO3−δ and Pd/Ba/LaAlO3 are almost identical under this condition.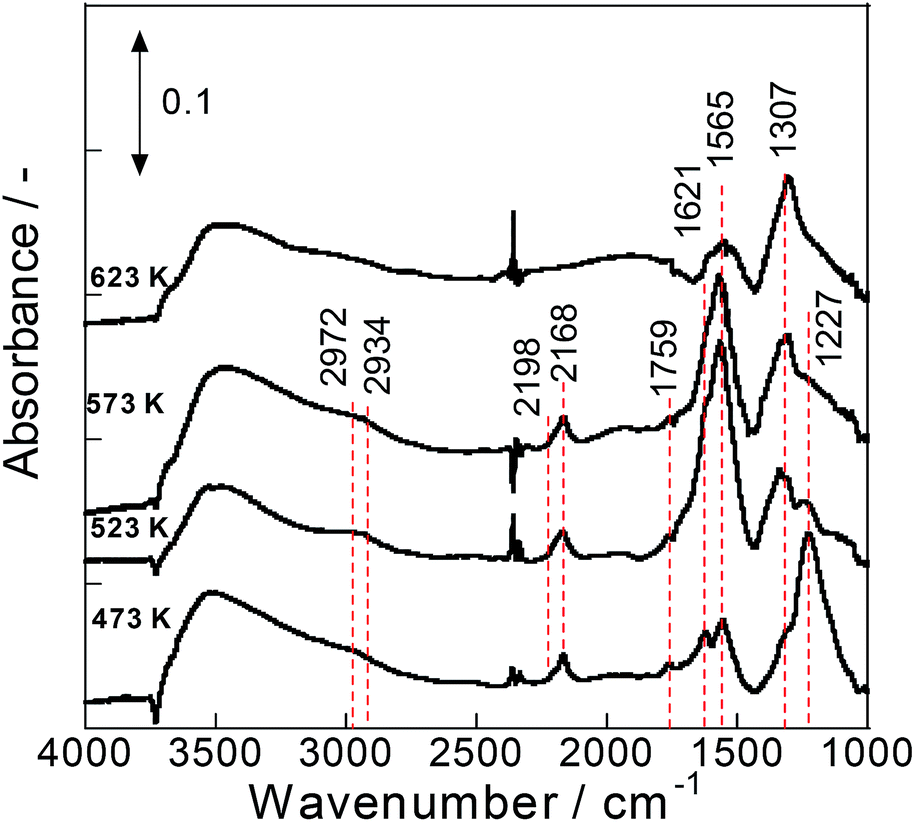 | ||
| Fig. 2 IR spectra of surface species on Pd/La0.9Ba0.1AlO3−δ during NO + C3H6 + O2 + H2O reaction at 473 K, 523 K, 573 K and 623 K. | ||
Next, we investigated the DRIFT spectra on Pd/Al2O3 as a control catalyst support at various temperatures in the NO–C3H6–O2–H2O condition (1000 ppm NO + 500 ppm C3H6 + 2000 ppm O2 + 7 vol% H2O). Results are shown in Fig. S7†. The bands assigned to acetate species (1578 and 1455 cm−1) were observed at widely various temperatures. The results for this species on Al2O3 surface show good accordance with those explained in earlier reports.32–34 The band at 1647 cm−1 is an overlapped band of two-fold NO35 on Pd surface and bending mode of H2O. Moreover some weak bands assignable to formate (1375 and 2900–3000 cm−1) and isocyanate (–NCO; 2217 cm−1) were observed. Nitrite (NO2−) and nitrate (NO3−) species were not observed. At 623 K, the acetate band intensity became very weak. A strong band assigned to bidentate nitrate (1548 cm−1) appeared by accumulation on Al2O3.34,36 This behaviour suggests that acetate species reacted with NO on the Pd surface. Many research groups have reported the role of acetate as an essential reductant.33,34 Our DRIFTS results agreed well with earlier reports for Al2O3-based catalysts on NO reduction by C3H6 in the presence of O2.32–36 Fig. S8† presents DRIFT spectra for Pd/LaAlO3, i.e. no Ba on the catalyst surface. On the Pd/LaAlO3, the spectra were unclear. Moreover, the band intensity was low. The evident adsorbed species were NO adsorbed onto Pd (1667, 1731 cm−1) and isocyanate (2169, 2194 cm−1). On this catalyst, nitrite (1286 cm−1) is a spectator because its intensity was not changed at high temperatures. Therefore, results suggest that the reaction in steady state on Pd/LaAlO3 is similar to that on Pd/Al2O3.
3.3 Conversion rate dependence on NO and C3H6 concentration
To clarify details of the reaction mechanism, the dependence of the reaction rate at 523 K on the concentration of NO or C3H6 was investigated for Pd/La0.9Ba0.1AlO3−δ and Pd/Ba/LaAlO3. The dependence of the conversion rates on NO concentration are shown in Fig. S9†. No clear dependence of the conversion rates was found for any component on NO concentration, as shown in Fig. S9†. It was expected that the adsorption or activation of NO was sufficiently fast on these catalysts because the Ba species on surface promote the adsorption of NO as NO2− and NO3−. Results of the dependence on C3H6 concentration are presented in Fig. 3. In the lean region, the conversion rate of NO on both catalysts positively depended on C3H6 concentration. Apparently, the conversion rate dependences of C3H6 and O2 were low. However, the dependence on C3H6 concentration presented remarkably different trends on Pd/La0.9Ba0.1AlO3−δ and Pd/Ba/LaAlO3 in a rich region. On Pd/Ba/LaAlO3, the conversion rate of all feeds clearly showed negative dependence on the C3H6 concentration. However, in the case of Pd/La0.9Ba0.1AlO3−δ, positive dependence was shown. Comparison of these results with those for Pd/LaAlO3 in Fig. S10† reveals that the effect of surface Ba species can be regarded as described below. On Pd/LaAlO3, the reaction of NO and C3H6 is affected by the competitive adsorption of these reactants because there is no adsorption site other than Pd. The dependence on C3H6 concentration for Pd/LaAlO3 showed negative dependence under the same experimental conditions. C3H6 strongly adsorbing on Pd inhibits the adsorption of NO. This assumption agrees with an earlier report of the literature.6 The surface Ba species on Pd/Ba/LaAlO3 and Pd/La0.9Ba0.1AlO3−δ play an important role as alternative adsorption sites for NO in avoiding the competitive adsorption of NO and C3H6. Furthermore, nitrite on Ba species is an effective intermediate for NO reduction by C3H6. On Ba-containing catalysts, it is considered that the reaction pathway changes to the route via nitrite and CxHyOz, which can proceed efficiently at lower temperatures. However, at high C3H6 concentrations, NO conversion on Pd/Ba/LaAlO3 was inhibited by the strong adsorption of C3H6. This result suggests that, because C3H6 occupied Pd surface of Pd/Ba/LaAlO3 and C3H6 oxidation by O2 is inhibited, the CxHyOz formation does not occur so rapidly. Consequently, the rate of NO reduction by CxHyOz also decreases because the formation rate of CxHyOz decreases. In contrast, no inhibition phenomenon occurs on Pd/La0.9Ba0.1AlO3−δ because the adsorbed species derived from C3H6 was oxidized rapidly. Comparison of both catalysts suggests that the oxidation of C3H6 was promoted on Pd/La0.9Ba0.1AlO3−δ compared with Pd/Ba/LaAlO3.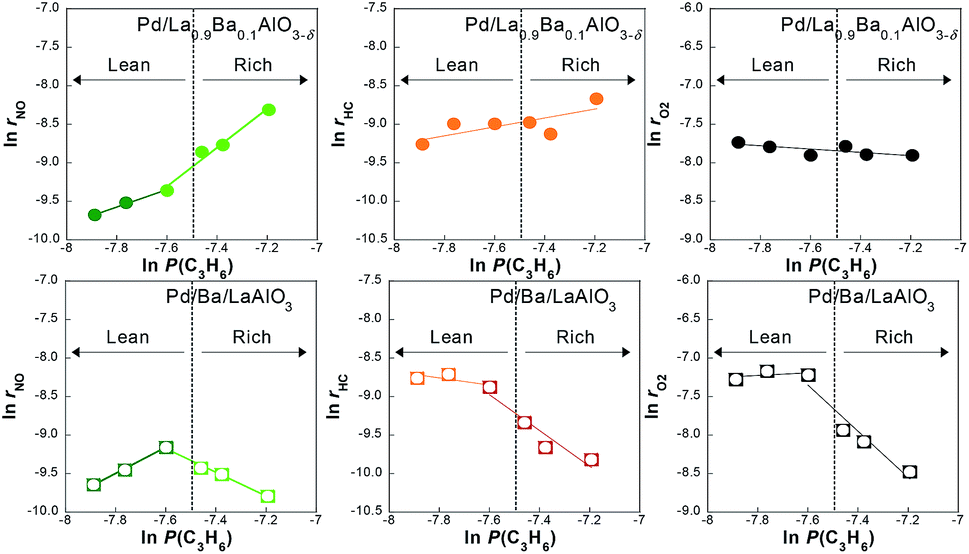 | ||
| Fig. 3 Dependence of the reaction rate on the partial pressure of C3H6 on Pd/La0.9Ba0.1AlO3−δ and Pd/Ba/LaAlO3 at 523 K during NO–C3H6–O2–H2O reaction. | ||
3.4 Role of surface lattice oxygen of Pd/La0.9Ba0.1AlO3−δ
From investigation of the conversion rate dependence, results demonstrated that C3H6 oxidation is the important factor on Pd/La0.9Ba0.1AlO3−δ. It is inferred that pathways for the formation of the intermediates derived from C3H6 differ among Pd/La0.9Ba0.1AlO3−δ and Pd/Ba/LaAlO3. In our earlier studies of steam reforming of toluene over Ni catalyst supported on Sr-substituted LaAlO3 (La0.7Sr0.3AlO3−δ),26,27 we reported that hydrocarbons were oxidized by the lattice oxygen of La0.7Sr0.3AlO3−δ. The lattice oxygen of La0.7Sr0.3AlO3−δ, having high mobility, can contribute to the surface reaction via release and restoration. Therefore, we investigated the contribution of surface lattice oxygen to the formation of oxidized C3H6 species by transient response test using 18O2, XPS and DRIFTS measurements. To confirm the release of surface lattice oxygen, we conducted the transient response test using 18O2 according to the procedures described in Section 2. The test results are presented in Fig. 4 and Table 1. After gas switching, C16O2 and C16O18O were observed on Pd/La0.9Ba0.1AlO3−δ. Formation of these 16O-containing products indicates that C3H6 is oxidized by lattice oxygen of La0.9Ba0.1AlO3−δ. The amount of 16O detected during the test is 91 μmol per 200 mg catalyst, which has 2753 μmol of lattice oxygen. Particularly, 45.4 μmol of 16O was released as the C16O2 during this test. This value is comparable to the amount of surface lattice oxygen of about 1–2 layer(s). We estimated the amount of surface lattice oxygen on (100) or (110) to be 32 μmol or 22 μmol per 200 mg from BET surface area and lattice structure of La0.9Ba0.1AlO3−δ. These 16O-containing products were detected before C18O2. Therefore, the surface lattice oxygen of La0.9Ba0.1AlO3−δ is considered to react to C3H6 faster than 18O species derived from adsorbed 18O2 on surface of Pd. The results for Pd/Ba/LaAlO3 are presented in Fig. S11 and Table S5:† the C16O2 formation amount was only 6.5 μmol during this test. Therefore, only a small amount of the surface lattice oxygen of Pd/Ba/LaAlO3 contributed to C3H6 oxidation during this test. Additionally, because of high mobility of surface lattice oxygen of La0.9Ba0.1AlO3−δ, on Pd/La0.9Ba0.1AlO3−δ, the peak of MS signal for C16O2 was detected 90 s faster than on Pd/Ba/LaAlO3.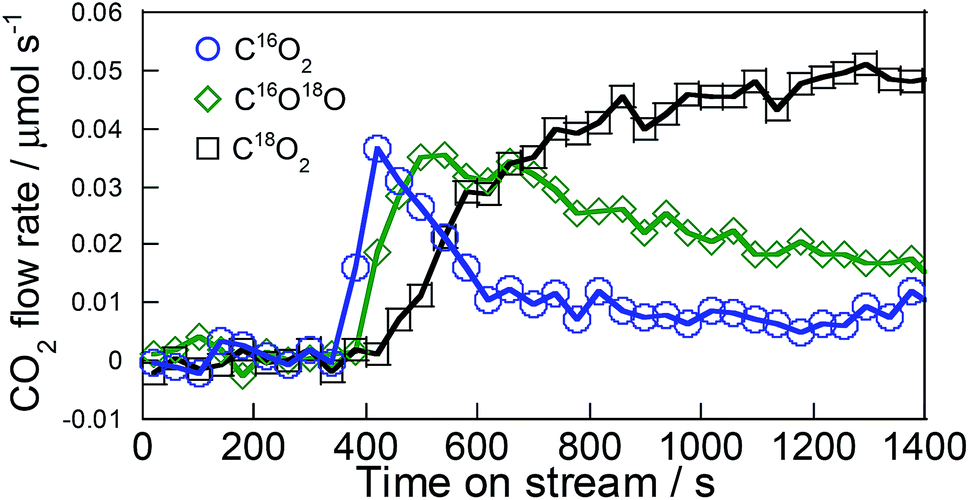 | ||
| Fig. 4 Transient response curves for typical products on Pd/La0.9Ba0.1AlO3−δ following Ar → C3H6 + 18O2 switch at 523 K. | ||
| C16O2/μmol | C16O18O/μmol | C18O2/μmol | |
|---|---|---|---|
| Pd/La0.9Ba0.1AlO3−δ | 22.7 | 45.6 | 51.8 |
We conducted XPS measurements of these catalysts with treatment according to the following procedure. First, the heat treatment in Ar flow for 30 min at 773 K. Then, at 523 K, the catalysts were alternately exposed to 500 ppm C3H6 flow or O2 flow diluted with Ar for 15 min each. The flow order is C3H6 (step 1) → O2 (step 2) → C3H6 (step 3). To remove C3H6 and O2 from sample cell, purging with Ar gas was performed for 10 min between the respective treatments. XP spectra of O1s region for Pd/La0.9Ba0.1AlO3−δ after each treatment are shown in Fig. 5. On the spectra, O1s region had four components corresponding to various oxygen species as OI (at 528.9–529.1 eV), OII (at 530.1–530.2 eV), OIII (at 531.1–531.5 eV) and OIV (at 532.3–532.7 eV). The binding energies of these peaks and the ratios of Ox/La are presented in Table 2. The OI is assigned to O2− as lattice oxygen of La0.9Ba0.1AlO3−δ.40–42 The OII assigned to O−, which is a superficial chemisorbed or lattice oxygen species.40–42 We infer that O− is the intermediate species for the release and restoration of lattice oxygen according to the following scheme.1,43
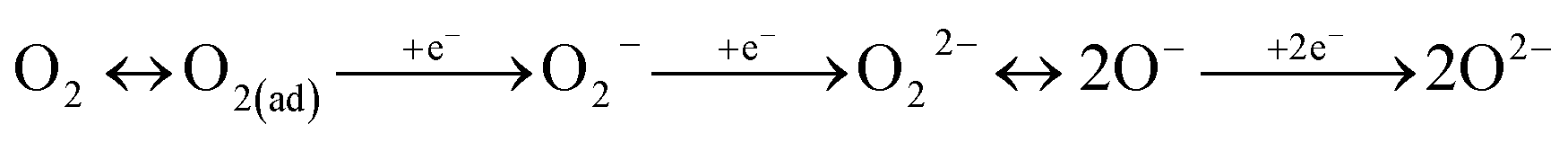
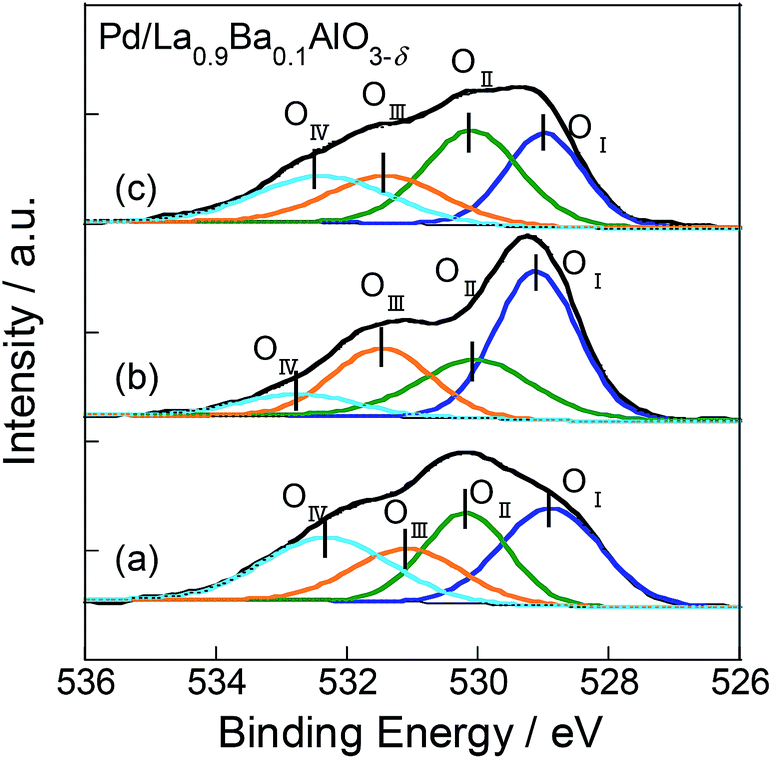 | ||
| Fig. 5 XP spectra of O1s for Pd/La0.9Ba0.1AlO3−δ (a) after exposure in C3H6 flow, (b) after exposure in O2 and (c) after exposure of C3H6 flow again. | ||
| Spectrum | Binding energy/eV | OI/La | OII/La | OIII/La | OIV/La | |||
|---|---|---|---|---|---|---|---|---|
| OI | OII | OIII | OIV | |||||
| (a) | 528.9 | 530.2 | 531.1 | 532.3 | 2.3 | 1.8 | 1.6 | 1.9 |
| (b) | 529.1 | 530.2 | 531.5 | 532.7 | 3.0 | 1.7 | 1.6 | 0.6 |
| (c) | 529.0 | 530.1 | 531.4 | 523.4 | 2.0 | 2.5 | 1.5 | 1.6 |
The OIII and OIV are attributed to adsorbed carbonate40,42,44 and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O(C
O(C![[O with combining low line]](https://www.rsc.org/images/entities/char_004f_0332.gif) OR).45 The appearance of OIV suggests that a oxygenated hydrocarbon species adsorbed onto the surface. The spectra changed drastically for each step in the procedure of the experiment on Pd/La0.9Ba0.1AlO3−δ. In spectrum (b), increase of OI and decrease of OIV were observed. These behaviours indicated the restoration of O2− as lattice oxygen and the total oxidation of adsorbed hydrocarbon species by O2 treatment. Furthermore, in spectrum (c), the amounts of OI and OIV changed, and the values were close to those in spectrum (a). Partial oxidation of C3H6 probably occurred by lattice oxygen, which was restored by O2 treatment. However, as shown in the results for Pd/Ba/LaAlO3 in Fig. S12 and Table S6†, the respective intensities of OI and OIV were only slightly changed at each step. Only OII increased in step 2 because the adsorbed oxygen on Pd was increased by O2 treatment. These results indicate that the surface lattice oxygen on Pd/Ba/LaAlO3 does not contribute to C3H6 oxidation to any considerable degree. From the results described above, we confirmed the release and restoration of the surface lattice oxygen during the oxidation of C3H6 on Pd/La0.9Ba0.1AlO3−δ.
OR).45 The appearance of OIV suggests that a oxygenated hydrocarbon species adsorbed onto the surface. The spectra changed drastically for each step in the procedure of the experiment on Pd/La0.9Ba0.1AlO3−δ. In spectrum (b), increase of OI and decrease of OIV were observed. These behaviours indicated the restoration of O2− as lattice oxygen and the total oxidation of adsorbed hydrocarbon species by O2 treatment. Furthermore, in spectrum (c), the amounts of OI and OIV changed, and the values were close to those in spectrum (a). Partial oxidation of C3H6 probably occurred by lattice oxygen, which was restored by O2 treatment. However, as shown in the results for Pd/Ba/LaAlO3 in Fig. S12 and Table S6†, the respective intensities of OI and OIV were only slightly changed at each step. Only OII increased in step 2 because the adsorbed oxygen on Pd was increased by O2 treatment. These results indicate that the surface lattice oxygen on Pd/Ba/LaAlO3 does not contribute to C3H6 oxidation to any considerable degree. From the results described above, we confirmed the release and restoration of the surface lattice oxygen during the oxidation of C3H6 on Pd/La0.9Ba0.1AlO3−δ.
Then, to ascertain behaviours of surface adsorbed species on the oxidation of C3H6 using the surface lattice oxygen, we observed DRIFT spectra for Pd/La0.9Ba0.1AlO3−δ and Pd/Ba/LaAlO3 using the following procedure. First, 500 ppm C3H6 diluted by Ar flow was applied for 15 min at 523 K after pre-treatment as the same to activity test. Then, 2000 ppm O2 flowed for 15 min sequentially. This procedure was one operation; it was conducted twice. Results are shown in Fig. 6 (first operation) and S13† (second operation). In spectra for Pd/La0.9Ba0.1AlO3−δ, absorption bands were observed at 1300, 1598, 1717 and 1760 cm−1. These bands were assigned respectively to carbonate (1300 and 1598 cm−1)35 and a CO group of aldehyde or ketone species (1717 and 1760 cm−1).32,46,47 The oxygen source was only lattice oxygen of a support material in this experimental condition. Therefore, it is considered that C3H6 was oxidized by the surface lattice oxygen of La0.9Ba0.1AlO3−δ to form CO2 and oxygenated species having CO group. Furthermore, an increase in the intensity of bands for carbonate and a decrease of that for CO group were observed on Pd/La0.9Ba0.1AlO3−δ simultaneously by introducing O2 sequentially. These behaviours of the bands indicate the oxidation of oxygenated species and CO2 formation. Fig. S13† portrays spectra measured using the same procedure after introduction of O2 in the first operation. Even in the second operation, the same bands and behaviours as in the first operation were observed on Pd/La0.9Ba0.1AlO3−δ. Therefore, it is deduced that the release and restoration of lattice oxygen occur on the surface of Pd/La0.9Ba0.1AlO3−δ at 523 K. These results agree well with those for the transient test and XPS measurements. On Pd/Ba/LaAlO3, when C3H6 was introduced after pre-treatment, bands with very weak intensity were observed at 1341 cm−1 and 1540 cm−1. These bands were assigned to carbonate species.33–38 The different wavenumbers of carbonate species between the surface of La0.9Ba0.1AlO3−δ and Ba/LaAlO3 derive from different adsorption states of carbonate. In subsequent steps of this DRIFTS measurement, spectra only slightly changed on Pd/Ba/LaAlO3 until the end of the second operation (spectra (f)–(h) and Fig. S13†). Therefore, from transient tests, XPS and DRIFTS measurements, we inferred that Pd/Ba/LaAlO3 has a slight amount of reactive lattice oxygen species on its surface and that these oxygen species do not contribute to the reaction steadily at low temperatures such as 523 K because the mobility of surface lattice oxygen is very low on Pd/Ba/LaAlO3.
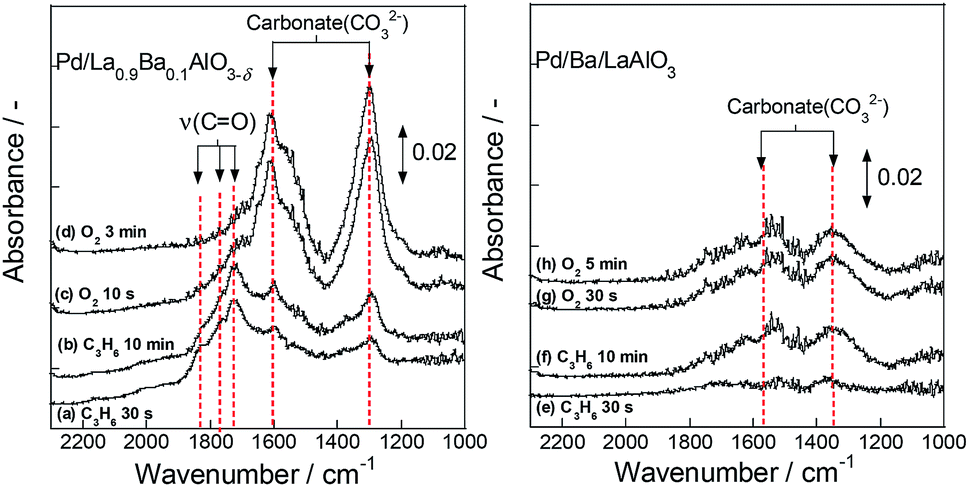 | ||
| Fig. 6 DRIFT spectra of Pd/La0.9Ba0.1AlO3−δ and Pd/Ba/LaAlO3 during transient test (switching from C3H6 to O2 flow). | ||
Subsequently, we took another DRIFTS measurement to elucidate whether the adsorbed species is an intermediate species for NO reduction or not. This test was conducted using the same procedure by flowing 1000 ppm NO instead of O2 in the second operation. Additionally, the atmosphere for this test contained 7 vol% H2O. Fig. 7(A) shows that the same oxygenated species was also formed in a wet atmosphere. Fig. 7(B) presents spectra obtained when NO was introduced to IR cell after C3H6 flow. During the introduction of NO, the absorption bands for oxygenated species disappeared. The band intensity for carbonate species increased. In addition, new bands were observed at 1230, 1542 and 2163 cm−1. The assignments for these bands were, respectively, nitrite,34,36 nitrate32–37 and isocyanate (–NCO) species.32–35,37,38 The formation of isocyanate species indicates that introduced NO reacts with the oxygenated species. Therefore, results clarified that the oxygenated species formed by the surface lattice oxygen is the intermediate species for NO reduction.
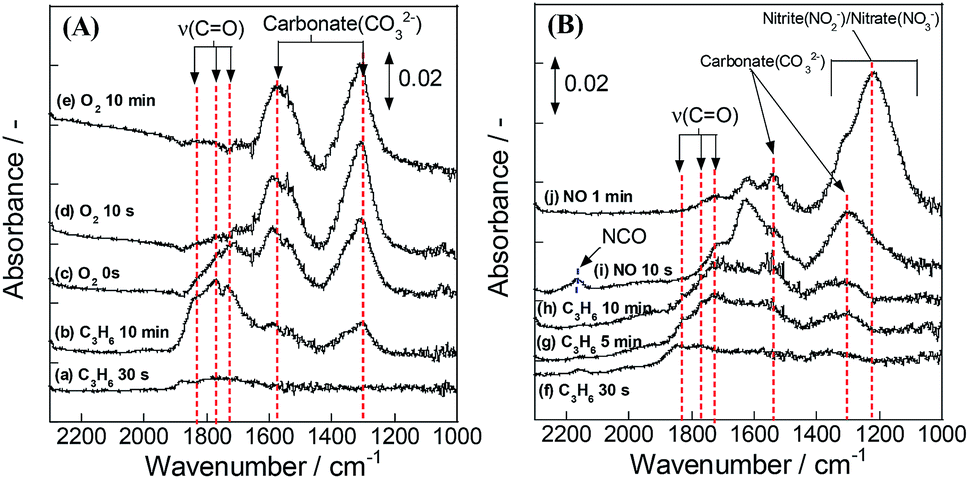 | ||
| Fig. 7 DRIFT spectra of Pd/La0.9Ba0.1AlO3−δ during transient test in humid atmosphere: (A) first operation – switching from C3H6 to O2 flow; (B) second operation – switching from C3H6 to NO flow. | ||
Fig. 8 portrays the presumed reaction scheme on Pd/La0.9Ba0.1AlO3−δ under this slightly lean condition. NO adsorbed as NO2−/NO3− on La0.9Ba0.1AlO3−δ. At the same time, the surface lattice oxygen oxidized C3H6 via the interface of Pd particles and La0.9Ba0.1AlO3−δ to form CxHyOz species. Finally, NO2−/NO3− and CxHyOz species react to form R–NCO as intermediate species. Isocyanate species react with NO or NO2−/NO3− to convert to N2, CO2 and H2O.
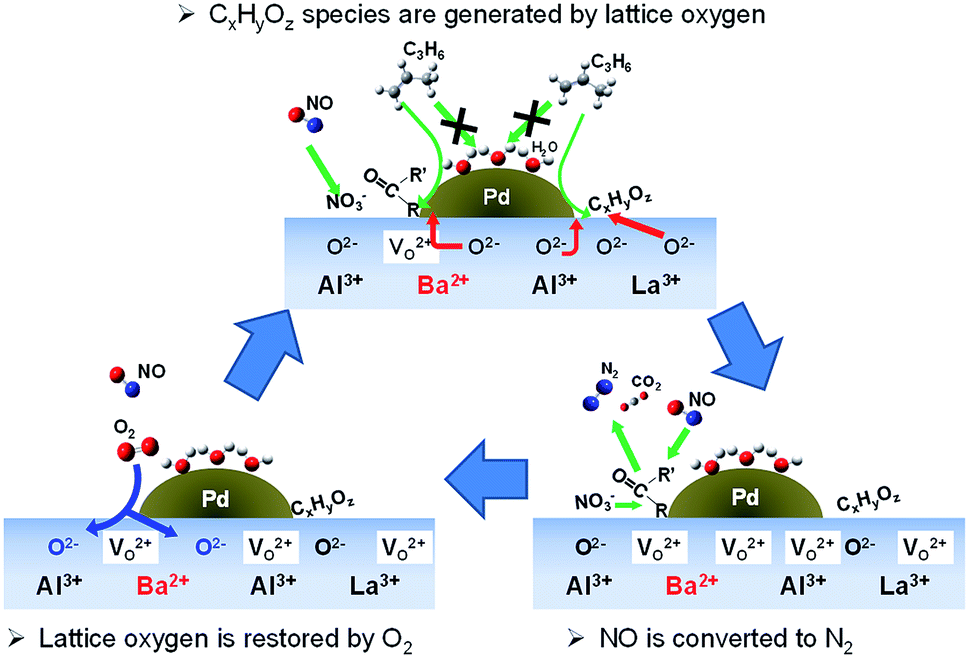 | ||
| Fig. 8 Presumed reaction scheme for NO reduction on Pd/La0.9Ba0.1AlO3−δ. | ||
4. Conclusions
We conducted reduction of NO by C3H6 under slightly lean conditions with the coexistence of steam. Pd/La0.9Ba0.1AlO3−δ catalyst showed higher catalytic activity than Pd/LaAlO3, Pd/Ba/LaAlO3 or Pd/Al2O3, especially at low temperatures (573 K or lower). For the steady-state surface reaction, NO2−/NO3− and oxygenated hydrocarbon species are active surface species on Pd/La0.9Ba0.1AlO3−δ catalyst. The cycle of release and restoration of the surface lattice oxygen was confirmed for Pd/La0.9Ba0.1AlO3−δ by transient tests and XPS measurements. The lattice oxygen reacts with C3H6 to oxygenated hydrocarbons very rapidly on Pd/La0.9Ba0.1AlO3−δ. The surface lattice oxygen contributes to the formation of intermediates and accelerates NO reduction on Pd/La0.9Ba0.1AlO3−δ, even in humid conditions at low temperatures.Conflicts of interest
The authors have no conflict to declare.Notes and references
- M. V. Twigg, Appl. Catal., B, 2007, 70, 2–15 CrossRef CAS.
- H. S. Gandhi, G. W. Graham and R. W. McCabe, J. Catal., 2003, 216, 433–442 CrossRef CAS.
- H. Y. Chen and H. L. Chang, Johnson Matthey Technol. Rev., 2015, 59, 64–67 CrossRef CAS.
- M. Shelef and G. W. Graham, Catal. Rev.: Sci. Eng., 1994, 36(3), 433–457 CrossRef CAS.
- J. Wang, H. Chen, Z. Hu, M. Yao and Y. Li, Catal. Rev.: Sci. Eng., 2015, 57, 79–144 CrossRef CAS.
- I. V. Yentekakis, R. M. Lambert, M. S. Tikhov, M. Konsolakis and V. Kiousis, J. Catal., 1998, 176, 82–92 CrossRef CAS.
- M. Konsolakis and I. V. Yentekakis, J. Hazard. Mater., 2007, 149, 619–624 CrossRef CAS PubMed.
- T. Kobayashi, T. Yamada and K. Kayano, Appl. Catal., B, 2001, 30, 287–292 CrossRef CAS.
- D. H. Kim, S.-I. Woo, J.-M. Leeb and O.-B. Yang, Catal. Lett., 2000, 70, 35–41 CrossRef CAS.
- A. E. Hamdaoui, G. Bergeret, J. Massardier, M. Primet and A. Renouprez, J. Catal., 1994, 148, 47–55 CrossRef.
- A. A. Vedyagin, M. S. Gavrilov, A. M. Volodin, V. O. Stoyanovskii, E. M. Slavinskaya, I. V. Mishakov and Y. V. Shubin, Top. Catal., 2013, 56, 1008–1014 CrossRef CAS.
- B. Zhao, G. Li, C. Ge, Q. Wang and R. Zhou, Appl. Catal., B, 2010, 96, 338–349 CrossRef CAS.
- L. Lan, S. Chen, Y. Cao, S. Wang, Q. Wu, Y. Zhou and M. Huang, J. Mol. Catal. A: Chem., 2015, 410, 100–109 CrossRef CAS.
- V. G. Papadakis, C. A. Pliangos, I. V. Yentekakis, X. E. Verykios and C. G. Vayenas, Catal. Today, 1996, 29, 71–75 CrossRef CAS.
- A. Tou, H. Einaga and Y. Teraoka, Catal. Today, 2013, 201, 103–108 CrossRef CAS.
- J. Zhu and A. Thomas, Appl. Catal., B, 2009, 92, 225–233 CrossRef CAS.
- M. Valden, R. L. Keiski, N. Xiang, J. Pere, J. Aaltonen, M. Pessa, T. Maunula, A. Savimäkic, A. Lahti and M. Härkönen, J. Catal., 1996, 161, 614–625 CrossRef CAS.
- R. B. Biniwale, J. V. Pande, M. Dhakad, N. K. Labhsetwar and M. Ichikawa, Catal. Lett., 2008, 123, 164–171 CrossRef CAS.
- Y. Nishihata, J. Mizuki, T. Akao, H. Tanaka, M. Uenishi, M. Kimura, T. Okamoto and N. Hamada, Nature, 2002, 418, 164–166 CrossRef CAS PubMed.
- D. Y. Yoon, Y. J. Kim, J. H. Lim, B. K. Cho, S. B. Hong, I. S. Nam and J. W. Choung, J. Catal., 2015, 330, 71–83 CrossRef CAS.
- K. Zhou, H. Chen, Q. Tian, Z. Hao, D. Shen and X. Xu, J. Mol. Catal. A: Chem., 2002, 189, 225–232 CrossRef CAS.
- H. Tanaka, Catal. Surv. Asia, 2005, 9, 63–74 CrossRef CAS.
- H. Tanaka and M. Misono, Curr. Opin. Solid State Mater. Sci., 2001, 5, 381–387 CrossRef CAS.
- K. Beppu, S. Hosokawa, H. Asakura, K. Teramura and T. Tanaka, Catal. Sci. Technol., 2018, 8, 147–153 RSC.
- D. Mukai, S. Tochiya, Y. Murai, M. Imori, T. Hashimoto, Y. Sugiura and Y. Sekine, Appl. Catal., A, 2013, 464–465, 78–86 CrossRef CAS.
- D. Mukai, Y. Murai, T. Higo, S. Tochiya, T. Hashimoto, Y. Sugiura and Y. Sekine, Appl. Catal., A, 2013, 466, 190–197 CrossRef CAS.
- T. Higo, T. Hashimoto, D. Mukai, S. Nagatake, S. Ogo, Y. Sugiura and Y. Sekine, J. Jpn Petrol. Inst., 2015, 58, 86–96 CrossRef CAS.
- R. Burch and T. C. Watling, Appl. Catal., B, 1997, 11, 207–216 CrossRef CAS.
- I. V. Yentekakis, M. Konsolakis, R. M. Lambert, N. Macleod and L. Nalbantian, Appl. Catal., B, 1999, 22, 123–133 CrossRef CAS.
- M. Konsolakis and I. V. Yentekakis, J. Catal., 2001, 198, 142–150 CrossRef CAS.
- M. Konsolakis and I. V. Yentekakis, Appl. Catal., B, 2001, 29, 103–113 CrossRef CAS.
- M. Haneda, N. Bion, M. Daturi, J. Saussey, J. C. Lavalley, D. Duprez and H. Hamada, J. Catal., 2002, 206, 114–124 CrossRef CAS.
- M. Huuhtanen, T. Kolli, T. Maunula and R. L. Keiski, Catal. Today, 2002, 75, 379–384 CrossRef CAS.
- A. Satsuma and K. Shimizu, Prog. Energy Combust. Sci., 2003, 29, 71–84 CrossRef CAS.
- T. E. Hoost, K. Otto and K. A. Laframboise, J. Catal., 1995, 155, 303–311 CrossRef CAS.
- U. Bentrup, M. Richter and R. Fricke, Appl. Catal., B, 2005, 55, 213–220 CrossRef CAS.
- V. Matsouka, M. Konsolakis, R. M. Lambert and I. V. Yentekakis, Appl. Catal., B, 2008, 84, 715–722 CrossRef CAS.
- F. C. Meunier, J. P. Breen, V. Zuzaniuk, M. Olsson and J. R. H. Ross, J. Catal., 1999, 187, 493–505 CrossRef CAS.
- R. Burch, J. P. Breen and F. C. Meunier, Appl. Catal., B, 2002, 39, 283–303 CrossRef CAS.
- N. A. Merino, B. P. Barbero, P. Eloy and L. E. Cadús, Appl. Surf. Sci., 2006, 253, 1489–1493 CrossRef CAS.
- H. Wang, J. Liu, Z. Zhao, Y. Wei and C. Xu, Catal. Today, 2012, 184, 288–300 CrossRef CAS.
- B. Liua, Y. Zhang and L. Tang, Int. J. Hydrogen Energy, 2009, 34, 435–439 CrossRef.
- Q. Meng, W. Wang, X. Weng, Y. Liu, H. Wang and Z. Wu, J. Phys. Chem. C, 2016, 120, 3259–3266 CrossRef CAS.
- V. I. Bukhtiyarov, A. I. Nizovskii, H. Bluhm, M. Hävecker, E. Kleimenov, A. Knop-Gericke and R. Schlögl, J. Catal., 2006, 238, 260–269 CrossRef CAS.
- S. Hamoudi, F. Larachi and A. Sayari, J. Catal., 1998, 177, 247–258 CrossRef.
- A. M. Hernández Giménez, J. Ruiz Martínez, B. Puértolas, J. Pérez Ramírez, P. C. A. Bruijnincx and B. M. Weckhuysen, Top. Catal., 2017, 60, 1522–1536 CrossRef.
- L. Cheng and X. P. Ye, Catal. Lett., 2009, 130, 100–107 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03050f |
| This journal is © The Royal Society of Chemistry 2019 |