
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An unknown component of a selective and mild oxidant: structure and oxidative ability of a double salt-type complex having κ1O-coordinated permanganate anions and three- and four-fold coordinated silver cations†
Gréta Bettina Kovácsab,
Nóra V. May a,
Petra Alexandra Bombicza,
Szilvia Kléberta,
Péter Németha,
Alfréd Menyhárdc,
Gyula Novodárszkia,
Vladimir Petrusevskid,
Fernanda Paiva Franguellia,
József Magyarie,
Kende Béresa,
Imre Miklós Szilágyib and
László Kótai*af
a,
Petra Alexandra Bombicza,
Szilvia Kléberta,
Péter Németha,
Alfréd Menyhárdc,
Gyula Novodárszkia,
Vladimir Petrusevskid,
Fernanda Paiva Franguellia,
József Magyarie,
Kende Béresa,
Imre Miklós Szilágyib and
László Kótai*af
aResearch Centre for Natural Sciences, Hungarian Academy of Sciences, Magyar Tudósok krt. 2, Budapest, H-1117 Hungary. E-mail: kotai.laszlo@ttk.mta.hu
bDepartment of Inorganic and Analytical Chemistry, Budapest University of Technology and Economics, Műegyetem Rakpart 3, Budapest, H-1111 Hungary
cDepartment of Physical Chemistry and Materials Science, Budapest University of Technology and Economics, Budapest, Hungary
dFaculty of Natural Sciences and Mathematics, Ss. Cyril and Methodius University, Skopje, Republic of Macedonia
eDepartment of Chemistry, Biochemistry and Environmental Protection, Faculty of Sciences, University of Novi Sad, Trg Dositeja Obradovića 3, Novi Sad, 21000, Serbia
fDeuton-X Ltd., Selmeci u. 89, Érd, H-2030, Hungary
First published on 9th September 2019
Abstract
Compounds containing redox active permanganate anions and complexed silver cations with reducing pyridine ligands are used not only as selective and mild oxidants in organic chemistry but as precursors for nanocatalyst synthesis in low-temperature solid-phase quasi-intramolecular redox reactions. Here we show a novel compound (4Agpy2MnO4·Agpy4MnO4) that has unique structural features including (1) four coordinated and one non-coordinated permanganate anion, (2) κ1O-permanganate coordinated Ag, (3) chain-like [Ag(py)2]+ units, (4) non-coordinated ionic permanganate ions and an [Ag(py)4]+ tetrahedra as well as (5) unsymmetrical hydrogen bonds between pyridine α-CHs and a permanganate oxygen. As a result of the oxidizing permanganate anion and reducing pyridine ligand, a highly exothermic reaction occurs at 85 °C. If the decomposition heat is absorbed by alumina or oxidation-resistant organic solvents (the solvent absorbs the heat to evaporate), the decomposition reaction proceeds smoothly and safely. During heating of the solid material, pyridine is partly oxidized into carbon dioxide and water; the solid phase decomposition end product contains mainly metallic Ag, Mn3O4 and some encapsulated carbon dioxide. Surprisingly, the enigmatic carbon-dioxide is an intercalated gas instead of the expected chemisorbed carbonate form. The title compound is proved to be a mild and efficient oxidant toward benzyl alcohols with an almost quantitative yield of benzaldehydes.
Introduction
As a useful mild oxidant in organic chemistry, a ‘compound’ prepared by Firouzabadi and reported as ‘[Ag(py)2]MnO4’ (Firouzabadi's compound, FC) has been known for a long time.1 According to literature data, it is an important reagent in a range of oxidation reactions including the preparation of various oxocompounds and sulfones.1–5 It can be used as a catalyst for the CN coupling reactions of aromatic hydrocarbons and primary amines including the synthesis of the pharmaceutically important quinazoline heterocycles3 such as gefitinib, the drug used for treating breast and lung cancers.5 Furthermore, FC decomposes into Ag/manganese oxide particles, which are candidates for catalyzing the selective oxidation of 3-picoline into niacin, the key compound of vitamin B3 synthesis.6 The successful application of FC in organic oxidation reactions requires detailed knowledge about the structure, properties as well as the catalytically active chemical sites and the reactivity of the compound. However, the chemical identity of FC is questionable7 as its structural data are missing. According to Sajó et al.8 it is indeed a ∼1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of the 4[Ag(py)2MnO4]·[Ag(py)4]MnO4 double salt (compound 1) and the [Ag(py)2]MnO4 (compound 2).9 Therefore, the structural characterization and properties identification are imperative for both compound 1 and 2 in order to appreciate the mild and selective oxidation capacity of FC. In this work we study the structural and vibrational spectroscopic characteristics, as well as thermal properties and reactivity with selected organic materials of compound 1 and compare its structure and properties with the known perchlorate analogues (listed in Table 1) and selected pyridinesilver(I) permanganate compounds found during the syntheses of FC.8
:1 mixture of the 4[Ag(py)2MnO4]·[Ag(py)4]MnO4 double salt (compound 1) and the [Ag(py)2]MnO4 (compound 2).9 Therefore, the structural characterization and properties identification are imperative for both compound 1 and 2 in order to appreciate the mild and selective oxidation capacity of FC. In this work we study the structural and vibrational spectroscopic characteristics, as well as thermal properties and reactivity with selected organic materials of compound 1 and compare its structure and properties with the known perchlorate analogues (listed in Table 1) and selected pyridinesilver(I) permanganate compounds found during the syntheses of FC.8
| Compound | X = Mn | X = Cl |
|---|---|---|
| 4[Ag(py)2XO4]·[Ag(py)4]XO4 | 1 | 1-ClO4 |
| [Ag(py)2]XO4 | 2 | 2-ClO4 |
| [Ag(py)4]XO4 | 3 | 3-ClO4 |
| [Ag(py)2]XO4·0.5py | 4 | — |
| ∼1:1 mixture of 1 and 2 |
FC | — |
| Decomposition intermediate from compound 1 at 300 °C | I-300 | — |
Results and discussion
Syntheses
Compound 1 is prepared from the pyridine solution of AgMnO4 by dilution with water until reaching 10% pyridine concentration following the method described in the literature.8 The blackish purple block-shaped crystals are stable for a month at room temperature in the dark, but after 1–2 weeks a shiny silvery colour appears on the surface of the crystals, which is not accompanied by bulk compositional change (ESI1†).Crystal structure of compound 1
The double salt 1 (Fig. 1) crystallizes in the tetragonal crystal system in the space group I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) (a = 21.982(3) and c = 7.5974(15) Å, T = 293(2) K, Z = 2, Dcalcd = 1.885 g cm−3). There is one [Ag(py)2MnO4] and a quarter of [Ag(py)4]MnO4 in the asymmetric unit. The ratio of [Ag(py)2MnO4] and [Ag(py)4]MnO4 is 1:4. The Kitaigorodskii packing coefficient is 69.6%,10 and there is no residual solvent accessible void in the crystal lattice.
(a = 21.982(3) and c = 7.5974(15) Å, T = 293(2) K, Z = 2, Dcalcd = 1.885 g cm−3). There is one [Ag(py)2MnO4] and a quarter of [Ag(py)4]MnO4 in the asymmetric unit. The ratio of [Ag(py)2MnO4] and [Ag(py)4]MnO4 is 1:4. The Kitaigorodskii packing coefficient is 69.6%,10 and there is no residual solvent accessible void in the crystal lattice.
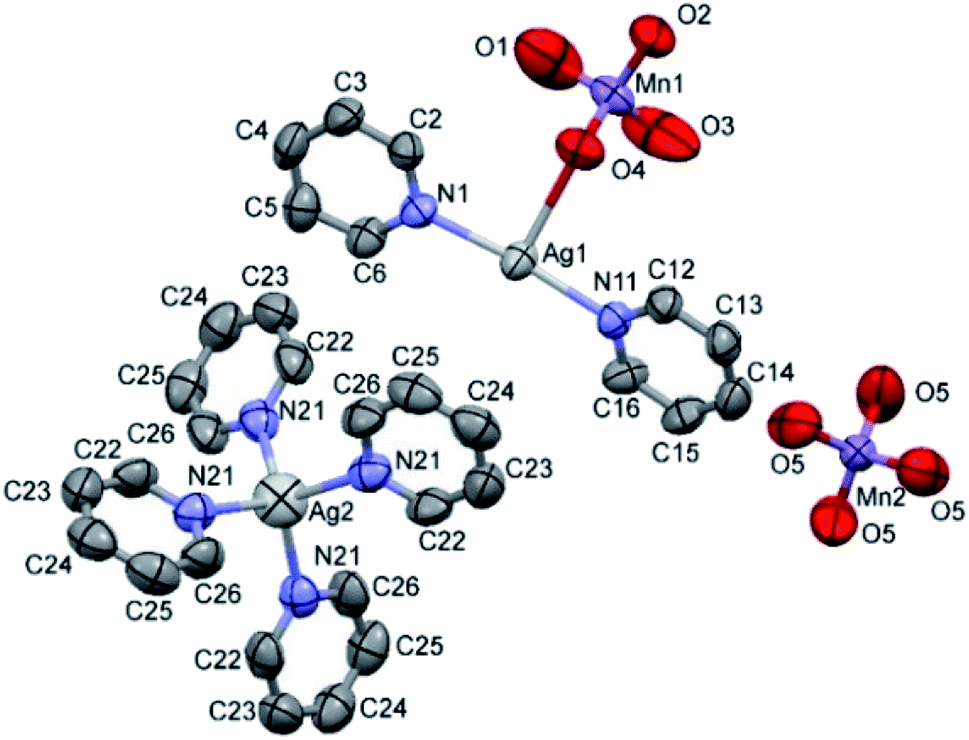 | ||
| Fig. 1 ORTEP presentation of the structure and atomic numbering scheme of compound 1. | ||
The asymmetric unit contains [Ag(py)2MnO4] and [Ag(py)4]MnO4 in the stoichiometric ratio of 1:1/4. The displacement ellipsoids are drawn at the 50% probability level.
The conformation of the [Ag(py)2MnO4] moiety is shown in Fig. 2. The pyridine rings are rotated by an angle of 12.03° respect to each other. The Cα–H⋯Opermanganate interactions are although weak, they contribute to the complex stability. The salt forms chains along the ‘c’ crystallographic axis (Fig. 3). These chains are arranged in a framework structure presented in Fig. 4.
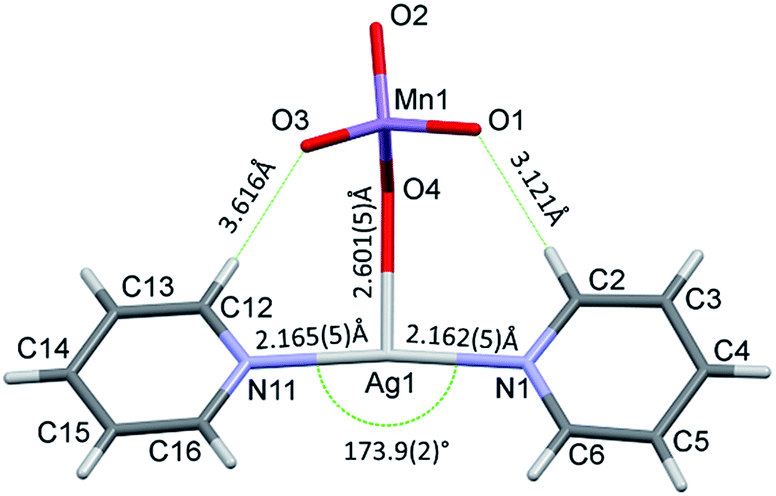 | ||
| Fig. 2 The [Ag(py)2MnO4] moiety in 1 shows the bond distances and angle around the Ag+. | ||
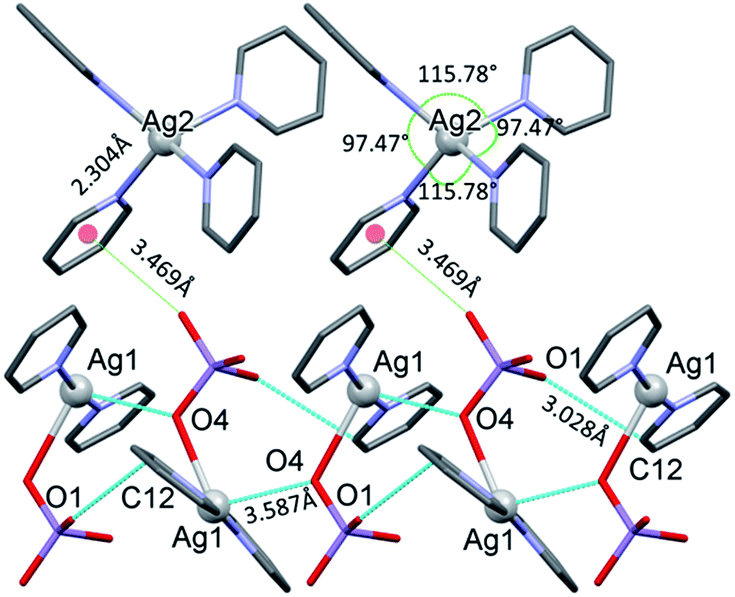 | ||
| Fig. 3 The [Ag(py)2MnO4] chains along the ‘c’ crystallographic axis and their Mn1–O2⋯π interactions (cyan dotted lines) with [Ag(py)4] cations in 1. | ||
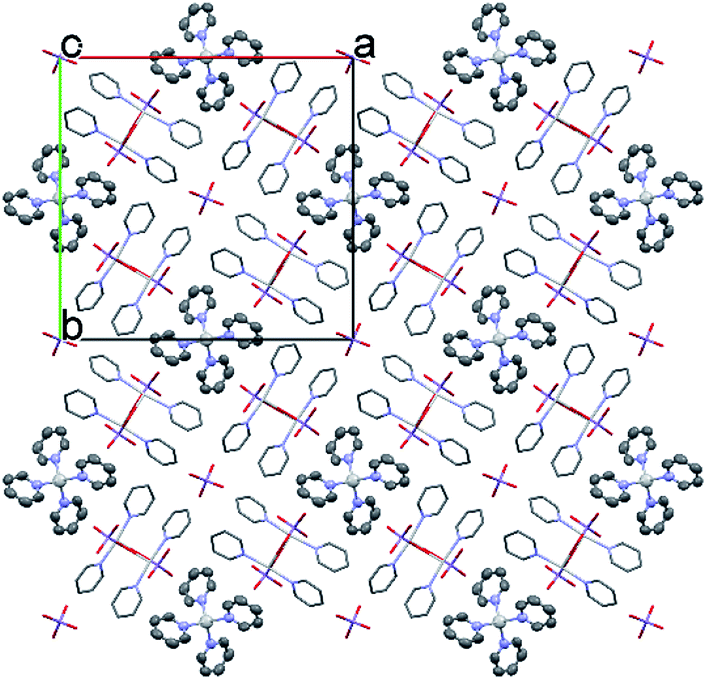 | ||
| Fig. 4 The packing arrangement of 1 [Ag(py)2]MnO4·[Ag(py)4]MnO4 viewing from the c crystallographic axis. | ||
The conformation of the [Ag(py)4]+ is shown in Fig. 3. The Ag+ cation is placed on a inversion axis. The [Ag(py)2MnO4] chain and the [Ag(py)4]+ cations are connected by Mn1–O2⋯π interactions (Fig. 3, where the Mn1–O2⋯Cg(py) distance is 3.469(7) Å and its angle is 161.0(3)°). The framework constructed by the interacting pyridine complexes contains neither classical hydrogen bonds nor π⋯π interaction (Fig. 4).
Analogue perchlorate compounds
The perchlorate analogue of compound 1 (1-ClO4, DITCEO) is isostructural with compound 1, the crystallographic positions of the [Ag(py)4]+ cations are almost identical in the two crystal lattices (ESI9†). The unit cell volume of 1-ClO4 is 0.85% larger than that of compound 1; their cell similarity index,11 is 0.00044.The co-crystal 1 has lower stability than compound 1-ClO4 (Tdec = 86 and 95.6 °C,12–14 respectively). Compound 3 decomposes fast even at room temperature.8 Presence of [Ag(py)2]MnO4 (2) in its co-crystal (1) with compound 3 stabilizes the tetrapyridinesilver(I) cation in its crystal similarly to the perchlorate analogues of compound 3-ClO4 and 2-ClO4 in 1-ClO4 (Tdec = 68, 158 and 95.6 °C, respectively8,14,15). The conformational arrangement of the coordinated permanganate ([Ag(py)2MnO4]) containing unit in 1 and 4 (ref. 8) is presented in Fig. 5. Depending on the Ag:py:MnO4 stoichiometries of 1 and 4, the [Ag(py)2MnO4] units have distinct geometries. Superimposing the complexes17 the rmsd value is 0.7805 and the maxD value is 1.3312 Å.
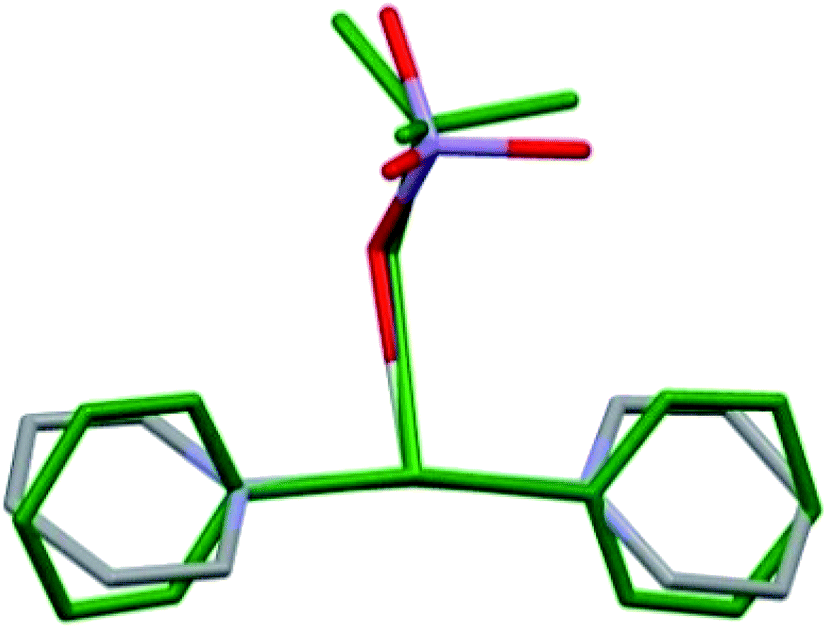 | ||
| Fig. 5 Comparison of the [Ag(py)2MnO4] moieties from the crystal structures of 1 (coloured by elements) and 4 (green). | ||
Vibrational modes in compound 1
| Wavenumber, cm−1 | Assignations | |
|---|---|---|
| IR | Raman | |
| a vs-very strong, m-medium, w-weak, vw-very weak. | ||
| 826 (w) | 826 (vs) | ν1(MnO), νs (A) |
| 339 (vw) | 345 (m) | ν2(MnO), δs (E) |
| 909, 917 (vs) | 887 (w), 902 (w), 913 (w) | ν3(MnO), νas (F2) |
| 382 (w) | 384 (vw) | ν4(MnO), δas (F2) |
There are 9 internal modes of the MnO4− anions at S4 sites. The B and E modes of the factor group (f-g) are IR active, whereas all f-g modes (of A, B and E symmetry) are Raman active. E modes are doubly and F modes are triply degenerated. There are 9 internal vibrational modes of the MnO4− anions at C1 sites (permanganates are coordinated to [Ag(py)2]+ cations). Each mode from the local (site) group splits into 3 components in the factor group. Thus, there are 9 A modes, 9 B modes and 9 E modes (the latter are doubly degenerated) giving rise to 36 vibrational degrees of freedom due to the 4 MnO4− anions on general positions (i.e. positions with C1 symmetry). There are 6 external vibrations of the MnO4− anions at S4 sites; 3 and 4 bands are expected in the far IR region, i.e. the low-frequency part of the Raman spectra, respectively. The external vibrations of the MnO4− anions at C1 sites result in a total of 24 vibrational degrees of freedom, which is in accordance with the 4 tetrahedral anions on general symmetry positions. The external vibration bands are also expected in the far IR and the low-frequency region of the Raman spectra. No predictions of the band intensities can a priory be given.
According to the factor group analysis (unit-cell group analysis), the two types of permanganate ions result in 4 vibrational modes (Table 2). However, the distinctions of permanganate bands belonging to S4 and C1 sites are challenging. One would expect a strong doublet due to the symmetric stretch (the breathing mode) in Raman, however, the observed spectrum does not show this expected feature (ESI3†). Three bands appear as a result of splitting of the ν3(MnO4) modes instead of the expected twice three components due to the pronounced distortion of the permanganate moieties at C1 symmetry.18a,b
Assigned cation modes of compound 1
Cation vibrations can arise from the hindered Ag translations within the [Ag(py)2]+ and [Ag(py)4]+ units. The correlation diagrams for the hindered translations of Ag+ cations at S4 sites and those at C1 sites are given in ESI2.† A total of 3 vibrational degrees of freedom exist for S4 and 12 for C1 sites. All f-g modes are active in Raman scattering, but only B and E modes are IR active. There are 27 internal vibrational modes of the pyridine rings at C1 sites. Each mode from the local (site) group splits into 3 components in the factor group. There are 27 A, 27 B and 27 E modes (the latter are double degenerated) giving rise to 108 vibrational degrees of freedom. For the three types of pyridine rings, there are 324 vibrational modes of freedom corresponding to internal vibrations of the pyridine molecules. Each mode of external vibrations from the local (site) group splits into 3 components in the factor group. There are 6 A, 6 B and 6 E modes (the latter are double degenerated) giving rise to 24 vibrational degrees of freedom. For the three types of pyridine rings, there are 72 vibrational modes of freedom corresponding to external vibrations of the pyridine molecules. The assignment of the heavily overlapped bands belonging to the pyridine ring is given in ESI2.† The assignments of Ag–N vibrations, arising from the [Ag(py)2]+ and [Ag(py)4]+ cations, in comparison with the analogous perchlorate complex (1-ClO4)13 are given in Table 3. The far-IR spectrum of compound 1 shows two bands at ∼246 and 166 cm−1, which are assigned to the symmetric and the antisymmetric ν(AgN) modes of the [Ag(py)2]+ ion. The proposed νas(AgN) assignments are in good agreement with the 185 cm−1 wavenumber obtained recently from density functional calculations for the species [Ag(py)]+.18c The νs(AgN) mode appearance in the IR spectrum may result from the deviation of the ideal (linear) N–Ag–N angle of [Ag(py)2]+ ion. Although the N–Ag–N unit slightly deviates from linear (173.9(2)° instead of 180°), this deviation may be sufficient to activate the νs(AgN) mode in the IR spectrum.13 Since the Ag–N distances in compounds 1 (ESI9†) and 1-ClO4 (ref. 12) are practically the same, the difference between the νs(Ag–N) band positions in 1 and 1-ClO4 suggests that the νs(AgN) mode of compound 1 is coupled with ν(AgO) modes.| Compound | IR | Raman | ||
|---|---|---|---|---|
| a w-weak, vw-very weak. | ||||
| [Ag(py)2]+ in compound 1 | 246 (νs(AgN)) | 166 (νas(AgN)) | 247 (νs(AgN)) | 150 (νas(AgN)) |
| [Ag(py)4]+ in compound 1 | — | 117 (νas(AgN)) | — | 124 (νas)AgN |
| [Ag(py)2]+ in compound 1-ClO4 | 254 (νs(AgN)) | 164 (νas(AgN)) | No data | No data |
| [Ag(py)4]+ in compound 3 | — | 122 (νas(AgN))13 | 88 (νs(AgN))13 | — |
A non-separable band system can be seen at frequencies higher than 100 cm−1 in the far-IR spectrum of compound 1 (ESI3†), which probably contains the combined bands of the coupled νas(AgN) mode of the tetrahedral [Ag(py)4]+ ion and ν(AgO) modes of the O3MnO–Ag(py)2 units. Although the Td symmetry of [Ag(py)4]+ ion is imperfect, the local symmetry of the AgN4 group is sufficiently close to the Td symmetry and thus one IR active νas(AgN) mode of tetrahedral symmetry is expected for this structure. In compound 1 the fully symmetric νs(AgN) mode of A1 symmetry is expected to occur at a lower frequency than our measured Raman range (4000–100 cm−1) (the νs(AgN) value for 1-ClO4 was found at 88 cm−1).13 The IR frequencies of the tetrahedrally coordinated [Ag(py)4]+ cations differ from those of the [Ag(py)2]+, which are consistent with the variations of the Ag–pyridine bond strength and the increasing coordination number in the complexes. The bands at 412 and 418 cm−1 for compound 1 (412 and 419 cm−1 for compound 1-ClO4)13 belong to coordinated pyridine modes of the [Ag(py)2]+ and the [Ag(py)4]+ cations, respectively. The appearance of single bands for [Ag(py)2]XO4 (X = Mn (2) and Cl (2-ClO4)) at 412 cm−1 and for [Ag(py)4](XO4) (X = Mn (3) and Cl (3-ClO4)) at 416 and 419 cm−1 confirm these assignations.8,13
UV-spectral studies
The diffuse reflection UV-Vis spectrum of compound 1 contains a band system consisting of pyridine n–π*, Ag+-pyridine MLCT and permanganate t1–4t2, 3t2–2e transitions.18 The UV-silent counterion containing [Ag(py)2]NO3 and KMnO4 spectra confirm the assignations and multi-complex nature of the UV bands given for compound 1 in Table 4 and ESI4.† The band maxima and their assignations are shown in Table 4.| Compound/band | Compound 1 | [Ag(py)2]NO3 | KMnO4 |
|---|---|---|---|
| Ag–py CT | 219.9 mixed band | 219.1 | — |
| MnO4, 1A1–1T2 (t1–4t2) | — | 227.3 | |
| MnO4, 1A1–1T2 (3t2–2e) | 258.4 mixed band | −252.2 | 259.0 |
| Pyridine, 1A1–1B2 (n → π*) | 291.0 | — | |
| MnO4, (1A1–1T2) (t1–2e) | 521.9 | — | 499.8, 513.7, 533.4, 562.8 |
| MnO4, (1A1–1T1) (t1–2e) | 710.1 | 720.8 |
The number of bands belonging to each transition of compound 1 listed in Table 4 can be multiplied due to the presence of two kinds of pyridine containing cations ([Ag(py)2]+ and [Ag(py)4]+) and the coordinated/non-coordinated type of permanganate anions. The 219.9 nm band may be assigned as frequencies for combined bands consisting of Ag–py (CT) and MnO4 – (1A1–1T2 (t1–4t2)), whereas the 258.4 nm band contains the MnO4− (1A1–1T2 (3t2–2e)) transitions and the components of the pyridine (1A1–1B2 (n → π*)) transitions. 219.9 and 258.4 nm bands are close to those found for [Ag(py)2]NO3 (219.4 and 252.2 nm, respectively) and KMnO4 (227.3 and 259 nm, respectively) as well. The main text of the article should appear here with headings as appropriate.
The bands found at 521.9 and 710.1 nm belong to the components of permanganate (1A1–1T2)(t1–2e) and 1A1–1T1 (t1–2e) transitions, respectively. The hypsochromic band shift belonging to compound 1 toward 1A1–1T1 (t1–2e) transition at 521.9 nm of KMnO4 might arise from the coordinated nature of permanganates in the lattice of compound 1.
Thermal decomposition
To initiate the redox reaction between the redox active cationic and anionic parts of compound 1, we heated the material in inert and O2-containing atmospheres.16 The thermal decomposition process of compound 1 in an inert atmosphere is proved to be a strongly exothermic explosion reaction; a part of the decomposition products practically burns out from the crucible. Therefore, the sample has to be diluted with an inert heat absorbing material (70% alumina). The reaction proceeds with 48.0% (wt) mass loss at 85 °C peak (TG and DTG) temperature (ESI5† and Table 5). The same exothermic characteristic is observed in the experiment performed in air flow (Table 5 and ESI5†), thus the oxygen of the air oxygen is not essential for initiating the decomposition process. The second decomposition step is a slower process in comparison to the first one and occurs between 410 and 500 °C and results in 8.0% (wt) mass loss (DTG peak temperature corresponds to 428 °C).| Δm, % | Temperatures, °C | ||||
|---|---|---|---|---|---|
| TG range | DTG peak | DSC range | DSC peak | ||
| Inert atmosphere | |||||
| Step 1 | 48.0 | 70–90 | 85 | 92.8–101.2 | 86 |
| Step 2 | 8.0 | 410–500 | 428 | — | — |
|
|||||
| Under air flow | |||||
| Step 1 | 48.4 | 70–110 | 89 | 60–110 | 91 |
| Step 2 | 7.9 | 110–220 | 184 | 110–210 | 177 |
| Step 3 | 2.4 | 280–750 | 434 | 280–500 | — |
| Step 4 | 744 | 500–750 | 744 | ||
The autocatalytic character of the decomposition intermediates
The total mass loss in the decomposition reaction of compound 1 under He suggests the formation of elementary silver and Mn3O4 (or equivalent amount of Mn2O3 and MnO) (Δmtheor = 55.75%, Δmfound = 55.80%). The redox titration of the decomposition product formed at 700 °C with oxalic acid results in close to the 2:1 ratio of MnIII/MnII content, and according to the stoichiometry of the compound 1 (Ag:Mn ratio = 1:1), the decomposition product consists of ∼60% Ag and ∼40% manganese oxides (γ-Mn2O3, Mn3O4 and MnO). Compound 1 contains reducing pyridine ligands, which reduces the silver(I) ions into metallic silver and the permanganate (Mn(VII)) anion into lower valence manganese oxides (MnO2, γ-Mn2O3, Mn3O4 and MnO). The autocatalytic character of the explosive exothermic decomposition of compound 1 may be attributed to the in situ formation of Ag/Mn-oxides (Koerbl's catalyst19), which are expected to accelerate the oxidation of such N-heterocycles as pyridine. The complete lack of oxygen evolution (see TG-MS results) and the exothermic character of the decomposition process (ESI5†) even at the initial stage excludes that the decomposition of compound 1 would start with endothermic ligands loss and consecutive oxidation of the liberated pyridine.
The intermediate formed at 300 °C (I-300) consists of a homogeneous mass of manganese oxides, which is covered by 1–5 μm size crystallites of silver (light grains in Fig. 6). The powder XRD of the intermediate (I-300) confirms the presence of metallic silver, a small amount of MnO and a phase, which could be equally assigned to Mn3O4 (hausmannite) or γ-Mn2O3 (ESI6†). These oxides have pseudo-spinel structures with identical lattice constants,20 thus PXRD is inappropriate to distinguish between them. Similarly, far-IR studies of I-300 (ESI6†) could not help in these manganese-oxide identifications. The characteristic IR bands of Mn3O4 and γ-Mn2O3 are too close to each other;21 and IR band shift resulted from the small grain size22 and the distribution of MnII and MnIII cations between the tetrahedral and octahedral sites possibly relating to the ferro/ferrimagnetic ordering of crystals,23 makes the IR identification ambiguous.
 | ||
| Fig. 6 Backscattered SEM image of I-300 (Ag/manganese oxides). | ||
Since titrimetric chemical analysis of I-300 suggests that the MnIII/MnII ratio is ∼2:1, and PXRD does not show γ-Mn2O3 (bixbyite) contribution, we conclude the main manganese oxide component is Mn3O4, whereas MnO and γ-Mn2O3 (in ∼1:1 molar ratio) are present in minor amounts. The same PXRD peaks are observed for the product formed at 700 °C and the I-300 (ESI6†) thus the second decomposition step observed at ∼430 °C does not change the chemical identity of the main crystalline phases of I-300. PXRD does not indicate any MnO2 contribution, but a weak IR band at 732 cm−1 suggests22d that it is a minor component of I-300.
Since the temperatures of the MnO2 → Mn2O3 → Mn3O4 → MnO decomposition reactions occur at 542, 918 and 1027 °C,24 taking into consideration the complete lack of oxygen evolution (Fig. 7), the I-300 phases could unambiguously be formed only from the reduction reactions of permanganate ions. The MnO2 IR band disappears on further heating (it is the strongest oxidant among the Mn-oxides present), and the intensity of IR bands belonging to carbon-rich residues in I-300 (aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN bonds at 1500–1600 and ∼2300 cm−1, respectively) decreases/disappears with the appearance of the oxidative cleavage products of aromatic CC bonds (C–O–C at ∼1083 cm−1) (ESI6†).
C and CN bonds at 1500–1600 and ∼2300 cm−1, respectively) decreases/disappears with the appearance of the oxidative cleavage products of aromatic CC bonds (C–O–C at ∼1083 cm−1) (ESI6†).
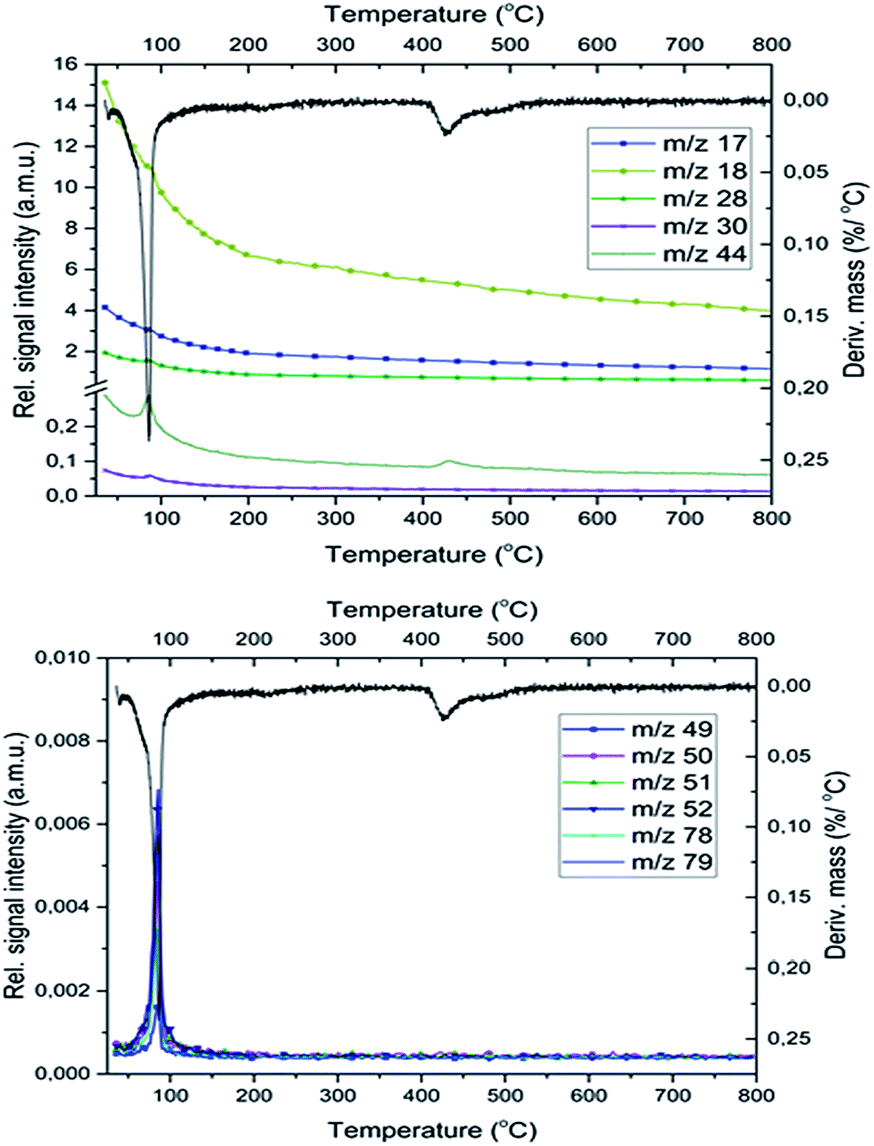 | ||
| Fig. 7 The gaseous products of the redox reaction between the pyridine ligands and permanganate anions in compound 1. The fragment ion intensities belonging to the liberated pyridine during the decomposition of compound 1. | ||
Evolved gas composition and the mechanism of the redox reactions
MS studies on the gas-phase formed during the decomposition of 1 indicate the presence of free pyridine (m/z = 79, 78, 52), which is liberated together with its oxidation products (CO2, H2O, NO, and N2, m/z = 44, 18, 30 and 28, respectively) both in the inert and oxygen-containing atmosphere. Based on previous studies25 in an inert atmosphere (He) the relative intensities of CO and CO2 peaks (m/z = 28 and 44, respectively) show that the m/z = 28 signal is a combined signal of CO and N2.The decomposition steps of [Ag(py)4]+ and [Ag(py)2]+ cations contemporarily occur in the first decomposition step of compound 1, the ligand losses/oxidations of both cations proceed during a multi-component simultaneous process.
The decomposition temperature of compound 1 (Tdec = 86 °C) is lower than those compounds that contain the same [Ag(py)2]+ and [Ag(py)4]+ cations such as compound 1-ClO4 (Tdec = 95.6 °C),12 compound 2-ClO4 (Tdec = 158 °C),26 or AgMnO4 (Tdec = 135).27 The decomposition of compound 1 is consistent with a solid phase quasi-intramolecular redox reaction and not with a ligand loss/permanganate decomposition followed by consecutive oxidation of the liberated pyridine. The solid phase quasi-intramolecular redox behaviour can be explained by the presence of the weak hydrogen bond interactions between the α-CH hydrogen of pyridine ring in the [Ag(py)2]+ units and the permanganate ion (Fig. 2) of compound 1. A similar structural feature was observed in compound 4 (hemipyridine solvate of compound 2);8 the reaction between the coordinated pyridine ligands and permanganate anions was attributed to the presence of the mentioned N–CH⋯O–Mn hydrogen-bond mediated redox reaction centre.7c The evolved heat of the redox reaction between the permanganate anion and pyridine ligands in compound 1 overcompensates the energy demand of the further endothermic ligand loss and initiates the completion of the decomposition process of compound 1 in one main step. In oxygen-containing air flow the decomposition process starts similarly, however, the organic residues of the solid phase are oxidized due to the catalytic activity of the formed Ag and Mn containing redox active intermediate (ESI5†) shown by the exothermic DSC peaks at 177 °C.
The role of the atmosphere in the decomposition process of compound 1
The pyridine permanganate = 2.4 molar ratio in compound 1 (CH:MnO4 = 12:1 → CH/O = 3) suggests that only a part of the pyridine can be oxidized in inert atmosphere by the oxygen content of compound 1, whereas in air, the oxygen content of the atmosphere plays a role in the pyridine oxidation process confirmed by the DSC experiments performed under pure N2 and O2 (Fig. 8 and ESI7†).
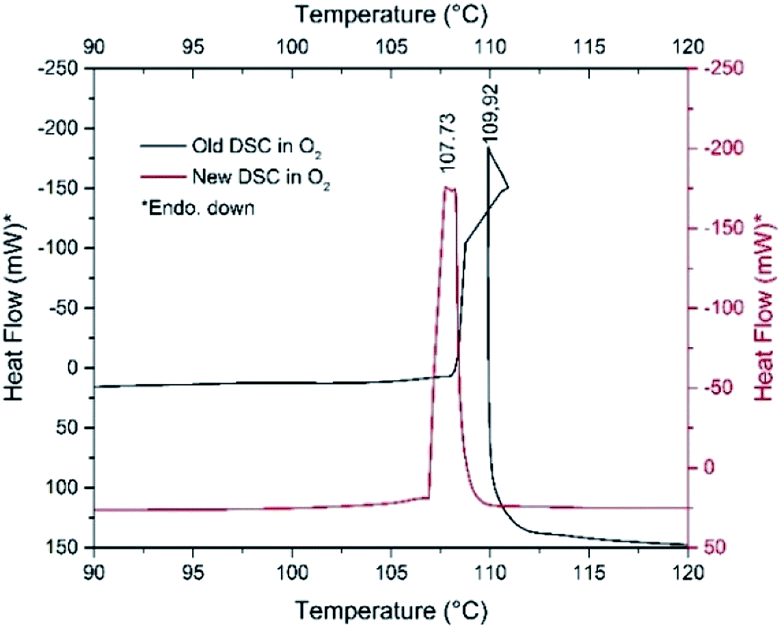 | ||
| Fig. 8 DSC curves of fresh (red line) and one-month-old (black line) samples of compound 1 under O2 atmospheres. | ||
The decomposition heats are −756.94 and −895.02 kJ mol−1 under N2 and O2, respectively. However, sample aging fundamentally influences the decomposition process. The decomposition starting temperature of the fresh material under N2 is 107 °C, whereas it is 101 °C for the one-month-old, Koerbl-catalysts containing sample.
The decomposition heat of one-month-old sample under N2 slightly increases (−850.35 kJ mol−1), which may be attributed to either different reaction products or different distribution of the same products as for the fresh sample. A secondary process may also be found at 129 °C which we explain as the reaction of residual organic content. Under O2, the catalyzing effect of decomposition products becomes obvious, the shape of DSC curve and the decomposition heat are strongly altered (Fig. 8). The unusual sigmoid character of the DSC curve is the consequence of the asymmetric heating of the crucible resulted from the non-symmetrical ignition profile of the sample. The decomposition (including the ignition) heat increases significantly, it is −1651.23 kJ mol−1 respect to −756.94 kJ mol−1 of fresh sample. This increase unambiguously confirms both the role of outer oxygen in the decomposition process and the catalytic effect of the intermediates formed under storage. The appearance of NO and C4H4 fragments in the TG-MS of compound 1 also suggests that redox reactions of the pyridine rings should proceed similarly to that of compound 2.8 Pyridine reduces the permanganate into manganese oxides, which is not accompanied by O2 evolution; the permanganate disappears even before its expected decomposition temperature.7a
The appearance of NO and C4H4 fragments in the TG-MS of compound 1 also suggests that redox reactions of the pyridine rings should proceed similarly to that of compound 2.8 Pyridine reduces the permanganate into manganese oxides, which is not accompanied by O2 evolution; the permanganate disappears even before its expected decomposition temperature.7a
Enigmatic carbon dioxide in the Ag/manganese oxide matrix
Not only the lack of O2 evolution but also the appearance of CO2 evolution at 400–430 °C during the decomposition of compound 1 deserves recognition.This temperature range coincides with the thermal decomposition temperature of MnCO3 into MnO and CO2, however, there is no indication of MnCO3 by PXRD in I-300 and there is no observed reduction of CO2 by MnO into CO as it should occur in an inert gas stream:28
| MnCO3 = MnO + CO2 |
| 3MnO + CO2 = Mn3O4 + CO |
The basic silver carbonate would similarly decompose with CO2 evolution in this temperature range,29 however, neither PXRD nor IR studies show any silver carbonate30 compounds in I-300. Therefore, the in situ formed carbonates as sources of CO2 evolution could completely be excluded.
The oxidation of carbonaceous materials with MnO2 (or partly with Mn2O3) starts above 300 °C. However, the IR spectrum of I-300 unambiguously shows the presence of intercalated gaseous CO2 (2350 cm−1)31 and its amount decreases significantly during heating up to 700 °C (ESI6†). The formation of this gas can be attributed to the solid-phase redox reaction, during which the formed manganese oxide matrix encloses the locally evolved gas. The high-temperature CO2 formation would suggest a strong bond between CO2 and the porous manganese oxide sorbents,32a which could occur via the MnO + CO2 = Mn(CO3) chemisorption reaction. However, the BET surface area does not support strong interaction between the CO2 and the Ag/Mn-oxide matrix as the N2 and CO2 adsorption measurement shows only 6 and 3 m2 g−1, respectively. The CO2 sorption–desorption isotherms of I-300 (ESI8†) also suggest that the sample contains a simple gas inclusion, which is consistent with the gas-impermeable character of the formed mixture.
In the O2 atmosphere, three small peaks of CO2 formation could be detected during decomposition of compound 1. In addition to the peak of CO2 formed at 430 °C in an inert atmosphere, two additional peaks appear at 200 °C and 750 °C due to the formation of carbon dioxide in the aerial oxidation of organic residues.
The role of the oxygen gas flow on the thermal decomposition process of compound 1
DSC study of compound 1 at 10 °C min−1 heating rate under Ar shows that compound 1 has no polymorphic phase transition between −150 °C and the decomposition temperature. The decomposition peak temperatures under Ar or O2 atmosphere are found to be almost identical, 101 and 108 °C, respectively (Fig. 8, ESI7†). Under O2 gas almost twice more heat is evolved than under Ar, and two very small exothermic peaks (93 and 98 °C) also appear as a result of aerial oxygen during decomposition.Organic oxidation reactions
Firouzabadi synthesis results in a mixture of compounds 1 and 2, and the recrystallization of the mixed raw product from benzene gives rise to the formation of pure [Ag(py)2]MnO4·0.5py (compound 4).8 In order to clarify which compound or which component of the reaction mixture prepared and reported by Firouzabadi1a is responsible for the mild and useful oxidation effect, the oxidation abilities of compounds 1, 2 and 4 have to be studied on well-selected test materials systematically. As a part of this systematic study, the oxidation ability of compound 1 has been tested in the oxidation reaction of benzyl alcohols (BzOH, 2- and 4-nitro and 2-methoxy-substituted benzyl alcohol) in various organic solvents, at room and reflux temperatures (CHCl3-61 °C, CCl4-76 °C and benzene-80 °C). The results of oxidation reactions with selected solvents are given in Tables 6 and 7.| Solvents | T, °C | t, min | Reaction products, in %a | BzOH conversion | ||
|---|---|---|---|---|---|---|
| PhCHO | PhCOOH | PhPh | ||||
| a From GC-MS ion chromatograms. The relative error of measurements was below ±0.4%.b Using freshly prepared compound 1 without silvery colour.c In the presence of an artificial silver mirror prepared from diamminesilver(I) chloride and glucose.33 | ||||||
| CCl4 | Reflux | 30 | 77.7 | 20.1 | 0 | Incomplete |
| CCl4 | Reflux | 120 | 71.1 | 28.9 | 0 | Complete |
| C6H6 | Ambient | 180 | 19.6 | 3.4 | 0 | Incomplete |
| C6H6 | Reflux | 30 | 41.9 | 51.2 | 4.9 | Incomplete |
| C6H6 | Reflux | 120 | 34.3 | 60.4 | 5.3 | Complete |
| C6H6b | Reflux | 240 | 100 | 0 | 0 | Complete |
| C6H6c | Reflux | 240 | 61.7 | 34.2 | 4.1 | Complete |
| Compounds | Solvent | T, °C | t, min | Reaction products, in %a, PhCHO | |
|---|---|---|---|---|---|
| RC6H4CH2OH | RC6H4CHO | RC6H4COOH | |||
| a The relative error of measurements was below ±0.4%. | |||||
| R = H | CHCl3 | Reflux | 30 | 89.0 | 0 |
| R = H | CHCl3 | Ambient | 180 | 97.3 | 0 |
| R = 2-MeO | CHCl3 | Ambient | 30 | 98.9 | 0 |
| R = 2-NO2 | CHCl3 | Ambient | 30 | 100 | 0 |
| R = 4-NO2 | CHCl3 | Ambient | 30 | 100 | 0 |
| R = 4-NO2 | CHCl3 | Reflux | 30 | 100 | 0 |
Oxidation of benzyl alcohol by 1
Under analogous conditions used by Firouzabadi,1a benzaldehyde (PhCHO) and benzoic acid (PhCOOH) are formed together in a PhCHO/PhCOOH = 0.5 ratio. Firouzabadi reported 96% isolated PhCHO yield without by-products,1a in contrast, our experiment unambiguously showed that compound 1 could oxidize the PhCHO formed from BzOH further to PhCOOH. A further difference is the appearance of a small amount of diphenyl (PhPh).Increasing reaction time (from 30 to 120 min in benzene as solvent) under reflux slightly influences the distribution of the oxidation products (Table 6). In contrast, the reaction temperature greatly influences the distribution of the products. At room temperature the PhCHO/PhCOOH acid ratio is ca. 6 and the BzOH conversion is ca. 20%. The appearance of PhPh is observed only at reflux temperature (80 °C) (Schemes 1 and 2).
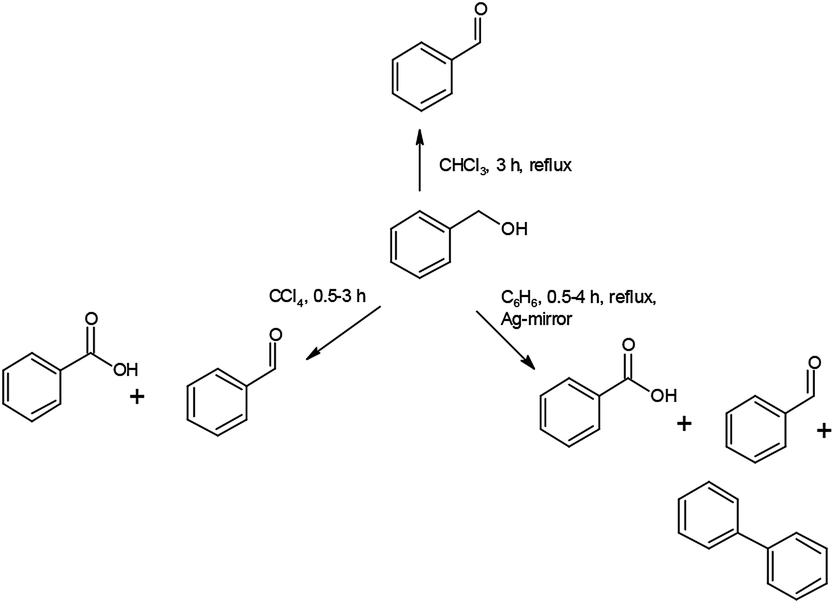 | ||
| Scheme 1 Oxidation reactions of 1 towards benzyl alcohol. | ||
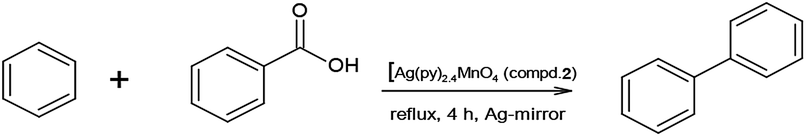 | ||
| Scheme 2 The reaction route to form PhPh. | ||
Since carbon tetrachloride does not result in PhPh formation even at reflux temperature (76 °C) in either 30 or 120 min reaction time, thus the influence of solvents also has to be tested on the formation of PhCOOH and PhPh (Table 6).
Compound 1 is purple when freshly prepared but gets a shiny silvery colour in due time, which is attributed to the formation of a thin surface film of metallic silver, similarly to the AgMnO4-sourced silver particles formation.7a Silver nucleating centers result in silver mirror formation on the wall of the vessel (both in CCl4 and benzene). Since the PhPh formation is observed only in benzene solvent, the decarboxylation of PhCOOH and dimerization of phenyl radicals are improbable, thus benzene is the key factor in the PhPh formation.
The silver catalyzed reaction of benzene with PhCOOH in the presence of a strong oxidant (e.g., persulphate) has already been investigated,34 thus the PhPh formation is supposed as a result of the reaction of benzene and PhCOOH with permanganate as oxidant and silver as a catalyst. To confirm this hypothesis, fresh compound 1 (without silvery lustre) was prepared and used it immediately. During 240 min reflux in benzene, it quantitatively yields PhCHO, without diphenyl or benzoic acid formation. Preparing a silver mirror from diamminesilver(I) chloride and glucose as reducing agent,35 and using the silver-coated vessel to perform the reaction of BzOH and freshly prepared compound 1 (without silvery lustre), under analogous conditions, a complete BzOH conversion with PhCHO/PhCOOH = ∼2:1 ratio and several percent of PhPh formation could be observed. This experiment unambiguously shows the catalytic effect of silver in the PhPh and PhCOOH formation reaction.
Testing of PhCOOH decarboxylation in benzene under reflux for 240 min without any oxidizing agent (e.g., compound 1) fails to form PhPh. Thus, not only the benzene as solvent and metallic silver as a catalyst play key role in the formation of PhPh but also the presence of oxidant is essential to form PhPh.
As it was revealed that the polarity of the solvent and the reaction temperature play an essential role in the conversion of benzyl alcohol and the distribution of the oxidation products, a low-boiling and oxidation-resistant but polar-solvent (CHCl3) was also tested in the reaction between compound 1 and various benzyl alcohols (Table 7). Roughly 90% conversion was found in 30 min at reflux temperature, notwithstanding at room temperature, 180 min reaction time gave an almost complete conversion of BzOH into PhCHO without PhCOOH formation.
Substituted benzyl alcohols oxidation with compound 1
Since the chloroform was found to be the best choice among the tested solvents, with the aim of preparation of the appropriately substituted benzaldehydes, a couple of oxidation reactions were examined. In particular, the 2- and 4-nitrobenzyl alcohols (electron-withdrawing substituents) as well as 2-methoxybenzyl alcohol (electron-donating substituent) substrates were tested. The oxidation reactions were performed in chloroform at room and reflux temperatures (Table 7). The phenyl ring substituents in benzyl alcohol increased the reactivity towards oxidation with compound 1. Independently from the nature and position of the substituents, practically complete conversion of the benzyl alcohols into the appropriate benzaldehyde occurred even at room temperature in 30 min. To examine the effect of temperature, the oxidation reaction of 4-nitrobenzyl alcohol was tested at reflux temperature as well, but the oxidation of the formed 4-nitrobenzaldehyde did not occur at all. These preliminary results confirmed the oxidation ability of compound 1. However, similar structural and reactivity studies of compound 2 are necessary in order to use FC for preparing commercial pharmaceuticals.Experimental
In the synthesis and analytical experiments analytical grade pyridine, silver(I) nitrate, 40% aq. NaMnO4 and solid KMnO4, twice distilled water, hydrochloric acid (37%) and nitric acid (67%) (Deuton-X Ltd) were used. All experiments with pyridine ligand containing silver permanganate and perchlorate complexes have to be performed very carefully due to the existing a possible hazard of explosion. The procedure to prepare the compound 1 can be safely, without risk of explosion, performed in the following way: freshly prepared wet silver(I) permanganate (2.27 g, precipitated in the reaction of saturated aq. AgNO3 and 40% aq. NaMnO4 solutions, at 1:1 Ag:MnO4 ratio, with washing the precipitate with copious amounts of cold water) was dissolved in pure pyridine, then the saturated purple solution formed was immediately diluted with water to reach a pyridine content of 10%. A precipitate was immediately formed which proved to be pure 1.8 Using wet AgMnO4 is essential, because old-samples of AgMnO4 can ignite the pure pyridine due to the catalytic effect of the silver manganese oxides formed on the surface of the old and dry samples.
The digestion of samples for ICP measurements was done in the 1:1 mixture of 67% nitric acid and 37% hydrochloric acid. The organic (benzyl alcohol, o- and p-nitrobenzyl alcohol and o-methoxybenzyl alcohol) were reagent grade chemicals (Deuton-X Ltd). [Ag(py)2]NO3 was prepared according to the method given by Jörgenson.35
The organic oxidation reactions have been performed with 0.01 mol of benzyl alcohol derivative dissolved in the appropriate organic solvent (CHCl3, CCl4 or benzene) and 1.5-fold molar excess of compound 1. The mixture was stirred at room temperature for 30 or 120 min, and another portion of the reaction mixture was refluxed (CHCl3-61 °C, CCl4-76 °C and C6H6-80 °C) for 30 or 120 min. The conversion was followed by GC-MS ion intensities until reaching the complete conversion. The partial conversion data was calculated from ion-chromatogram intensities which define a rough estimation of molar conversions. The calibration was performed using 2,4-dinitrophenylhydrazones.
Manganese(III) (Mn2O3 and Mn3O4) content of the thermal decomposition products were determined by reacting the mixtures containing these materials with oxalic acid in the presence of 20% sulfuric acid, with 2 h boiling then the oxalic acid excess was titrated back with 0.002 M potassium permanganate solution.
The Ag and Mn content of compound 1 were determined by atomic emission spectroscopy using a Spectro Genesis ICP-OES (SPECTRO Analytical Instruments GmbH, Kleve, Germany) simultaneous spectrometer with axial plasma observation. The multielement standard solution for ICP (Merck Chemicals GmbH, Darmstadt, Germany) was used for calibration. The carbon, hydrogen and nitrogen content were measured by elemental analysis (Fisons model CHN 1018S). The phase purity of compound 1 was checked by powder X-ray diffractometry. X-ray powder diffraction measurements were performed using a Philips PW-1050 Bragg–Brentano parafocusing goniometer equipped with a Cu tube operated at 40 kV and 35 mA power. A secondary beam graphite monochromator and a proportional counter were also equipped. Scans were recorded in step mode. Full profile fitting techniques were used for the evaluation of the diffraction patterns.
The FT-IR spectrum of compound 1 was recorded in the attenuated total reflection (ATR) mode on a Bruker Tensor 27 Platinum ATR FT-IR spectrometer (2 cm−1 resolution) between 4000 and 400 cm−1. The far-IR measurement was performed on a BioRad-Digilab FTS-30-FIR spectrometer for the 400–40 cm−1 range in polyethylene pellet.
The Raman measurement was performed using Horiba Jobin-Yvon LabRAM-type microspectrometer with external 532 nm Nd-YAG laser source (∼40 mW) and Olympus BX-40 optical microscope. The laser beam was focused by an objective (10×) and a D1 intensity filter decreased the laser power to 10% to avoid thermal degradation. The confocal hole of 1000 μm and 1800 groove mm−1 grating monochromator were also used in a confocal system and for light dispersion, respectively. The spectral range of 100–4000 cm−1 was detected with 3 cm−1 resolution. Each spectrum was collected at 240 s per point.
Diffuse reflectance spectrum in the UV-Vis region (200–800 nm) was detected at ambient conditions by a Jasco V-670 UV-Vis spectrophotometer equipped with NV-470 type integrating sphere using the official BaSO4 standard as a reference.
Thermal data were collected using a TA Instruments SDT Q600 thermal analyzer coupled to a Hiden Analytical HPR-20/QIC mass spectrometer. The decomposition was followed between room temperature and 800 °C at 2 °C min−1 heating rate in He and air as carrier gas (flow rate = 50 cm3 min−1). Alumina crucible and an empty alumina crucible were used as a sample holder and as a reference, respectively. Sample mass was ∼14 mg, 30% compound 1 with 70% (wt) calcined alumina. Selected ions between m/z = 1–120 were monitored in Multiple Ion Detection Mode (MID).
The non-isothermal DSC curve between −150 and 170 °C was recorded using a PerkinElmer DSC 7 apparatus. Sample mass was 4 mg and measured at 10 °C min−1 heating rate under continuous nitrogen flow (20 cm3 min−1) in an unsealed aluminum pan. The measurement was also performed under O2 flow between −50 and 170 °C.
Scanning Electron Microscopy (SEM) measurements were performed using a Zeiss EVO40 microscope operating at 20 kV. The single crystals of 4[Ag(py)2MnO4]·[Ag(py)4]MnO4 (1) were grown from the pyridine solution of AgMnO4 by adding 10-fold amount excess of water and leaving the solution to crystallize at room temperature. The diffraction pattern of the blackish purple, block type single crystal with the size of 0.25 × 0.25 × 0.20 mm was measured. Cell parameters were determined by least-squares using 30439 (6.78 ≤ θ ≤ 25.28°) reflections. Intensity data were collected on a Rigaku RAxis Rapid II diffractometer (monochromator; Mo-Kα radiation, λ = 0.71073 Å) at 293(2) K in the range 3.389 ≤ θ ≤ 25.244.36 A total of 21264 reflections were collected of which 1912 were unique [R(int) = 0.0301, R(σ) = 0.0188]; intensities of 1805 reflections were greater than 2σ(I). Completeness to θ = 0.994. The crystal contains two Ag complex cations with pyridine molecules as ligands and two permanganate anions. The ratio of the two complexes is 1:4 in the double salt. The lattice has high I symmetry. It results in low data to parameter ratio. In case of one tetrahedral cation and one tetrahedral anion there is only one-fourth of the molecule in the asymmetric unit.
Numerical absorption correction was applied to the data (the minimum and maximum transmission factors were 0.8567 and 0.9965).
The structure was solved by direct methods.36 Anisotropic full-matrix least-squares refinement36 on F2 for all non-hydrogen atoms yielded R1 = 0.0208 and wR2 = 0.0440 for 1332 [I > 2σ(I)] and R1 = 0.0235 and wR2 = 0.0445 for all (1912) intensity data, (number of parameters = 230, goodness-of-fit = 1.032, the maximum and mean shift/esd is 0.000 and 0.000). The absolute structure parameter is 0.015(9) (Friedel coverage: 0.821, Friedel fraction max.: 0.998, Friedel fraction full: 0.998). The maximum and minimum residual electron density in the final difference map was 0.386 and −0.274 e Å−3. The weighting scheme applied was w = 1/[σ2(Fo2) + (0.02940.0000P)2 + 0.0000P], where P = (Fo2 + 2Fc2)/3.
Hydrogen atomic positions were calculated from assumed geometries. Hydrogen atoms were included in structure factor calculations, but they were not refined. The isotropic displacement parameters of the hydrogen atoms were approximated from the U(eq.) value of the atom they were bonded to.
The liquid products were analyzed using a GC-MS (Shimadzu QP2010, He as the carrier gas) equipped with an RXi-5SIL MS capillary column (30 m × 0.2 mm × 0.25 μm). The column temperature was raised from 50 to 340 °C with a heating rate of 10 °C min−1.
CCDC-1879263 (1) contains the supplementary crystallographic data for this paper (ESI9†).
Conclusions
The compound 1 (4[Ag(py)2MnO4]·[Ag(py)4]MnO4) with four [Ag(py)2]+ and one [Ag(py)4]+ cations and having four coordinated and one non-coordinated permanganate anion has been synthesized. The compound 1 has several unique structural features including κ1O-coordinated permanganates to the silver cations of dimerized [Ag(py)2]+ units, non-coordinating ionic MnO4− and [Ag(py)4]+ tetrahedra as well as non-linear py–Ag–py moieties resulted from non-equivalent hydrogen bonds between the α-CH of pyridine rings in an [Ag(py)2] unit and neighbouring O atoms of the unprecedented κ1-O-coordinated MnO4−. The hydrogen bonds act as reactive centres in the thermally initiated, solid-phase redox reactions between the oxidizing MnO4− anion and reducing pyridine ligand. As a result a highly exothermic and explosive reaction, autocatalyzed by the intermediate silver manganese oxides (Körbl's catalysts) the pyridine content is partly oxidized into CO2 and H2O; the solid phase decomposition end products contain mainly metallic Ag and Mn3O4 as well as some CO2. The enigmatic CO2 is an intercalated gas in the manganese oxide matrix instead of the expected chemisorbed carbonate form. Compound 1 is a mild and efficient oxidant toward benzylic alcohols (unsubstituted, 2-MeO, 2-NO2 and 4-NO2). A solvent- and temperature-dependent oxidation takes place in all reactions with an almost quantitative yield of benzaldehydes. Chloroform is found to be the best solvent. The reaction of benzyl alcohol in CCl4 and benzene results in benzaldehyde and benzoic acid, whereas in benzene diphenyl formation occurs due to the oxidative coupling of benzoic acid and benzene in the presence of metallic silver catalyst.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research within project No. VEKOP-2.3.2-16-2017-00013 and GINOP-2.2.1-15-2017-00084 was supported by the European Union and the State of Hungary, co-financed by the European Regional Development Fund. N. V. M. and P. B. are grateful for the Hungarian Scientific Research Found (K-124544 and K-115762). I. M. Szilágyi thanks for a János Bolyai Research Fellowship of the Hungarian Academy of Sciences and a ÚNKP-18-4-BME-238 grant supported by the New National Excellence Program of the Ministry of Human Capacities, Hungary. An NRDI K 124212 and an NRDI TNN_16 123631 grants are acknowledged. The research reported in this paper was supported by the Higher Education Excellence Program of the Ministry of Human Capacities in the frame of Nanotechnology and Materials Science Research Area of Budapest University of Technology (BME FIKP-NAT). József Magyari gratefully acknowledges the Hungarian Academy of Sciences (MTA) Domus Hungarica Grant for the research support.Notes and references
- (a) H. Firouzabadi, B. Vessal and M. Naderi, Tetrahedron Lett., 1982, 23(17), 1847–1850 CrossRef CAS; (b) A. J. Bischoff, B. M. Nelson, Z. L. Niemeyer, M. S. Sigman and M. Movassaghi, J. Am. Chem. Soc., 2017, 139(43), 15539–15547 CrossRef CAS PubMed; (c) B. E. Haines, B. M. Nelson, J. M. Grandner, J. Kim, K. N. Houk, M. Movassaghi and D. G. Musaev, J. Am. Chem. Soc., 2018, 140(41), 13375–13386 CrossRef CAS PubMed; (d) A. K. Jain, Meena and D. Kumar, International Journal of Emerging Research in Management and Technology, 2014, 3(3), 17–22 Search PubMed; (e) J. Banerji, K. K. Banerji, L. Kótai, D. Sharma and P. K. Sharma, J. Indian Chem. Soc., 2011, 88(12), 1879–1886 CAS.
- (a) A. S. R. Anjaneyulu, P. Umasundari and C. V. M. Sastry, Indian J. Chem., 1986, 25B(9), 955–956 CAS; (b) H. Firouzabadi and A. Sardarian, Synth. Commun., 1983, 13(10), 863–866 CrossRef CAS; (c) J. Banerji, L. Kotai and K. K. Banerji, Indian J. Chem., 2009, 48A(6), 797–800 CAS; (d) D. G. Lee, e-EROS Encyclopedia of Reagents for Organic Synthesis, 2001, pp. 1–2 Search PubMed; (e) A. V. Gulevskaya, B. U. W. Maes, C. Meyers, W. A. Herrebout and B. J. van der Veken, Eur. J. Chem., 2006, 5305–5314 CAS; (f) R. Chandra, A. Sarkar and N. Biswas, Proc. Indian Natl. Sci. Acad., Part A, 1994, 60(2), 465–470 CAS; (g) H. Firouzabadi and A. R. Sardarian, Synthesis, 1986, 946–948 CrossRef CAS.
- (a) O. V. Serduke, A. V. Gulevskaya, A. F. Pozharskii, Z. A. Starikova and I. A. Profatilova, Tetrahedron, 2006, 62(4), 652–661 CrossRef CAS; (b) A. L. Gulevskaya, A. F. Pozharskii and L. V. Lomachenkova, Khim. Geterotsikl. Soedin., 1990, 1575 CAS; (c) A. V. Gulevskaya, B. U. W. Maes and C. Meyers, Synlett, 2007, 71–74 CAS; (d) D. V. Besedin, A. V. Gulevskaya and A. F. Pozharskii, Mendeleev Commun., 2000, 150 CrossRef CAS.
- (a) J. Banerji, L. Kótai, P. K. Sharma and K. K. Banerji, Eur. Chem. Bull., 2012, 1(5), 135–140 CAS; (b) T. Purohit, J. Banerji, L. Kótai, I. Sajo, K. K. Banerji and P. K. Sharma, J. Indian Chem. Soc., 2012, 89(8), 1045–1052 CAS; (c) A. Agrawal, R. Sailani, P. Sharma, C. L. Khandelwal and P. D. Sharma, Oxid. Commun., 2016, 39(2), 1273–1281 CAS.
- Lv. Ying, C. Yang and Z. Wang, Process for preparation of quinazoline derivatives and intermediates thereof, PCT Int. Appl., 2013, WO 2013060023A1.
- S. Ghosh, S. S. Acharyya, S. K. Sharma and R. Bal, Catal. Sci. Technol., 2016, 6(12), 4644–4654 RSC.
- (a) L. Kotai, I. Gács, I. E. Sajó, P. K. Sharma and K. K. Banerji, Trends Inorg. Chem., 2009, 11, 25–104 CAS; (b) L. Kotai, J. Fodor, E. Jakab, I. Sajo, P. Szabo, F. Lonyi, J. Valyon, I. Gacs, Gy. Argay and K. K. Banerji, Transition Met. Chem., 2006, 31(1), 30–34 CrossRef CAS; (c) L. Kotai, I. Sajo, J. Fodor, P. Szabo, E. Jakab, Gy. Argay, S. Holly, I. Gacs and K. K. Banerji, Transition Met. Chem., 2005, 30(8), 939–943 CrossRef CAS.
- I. E. Sajó, G. B. Kovács, T. Pasinszki, P. A. Bombicz, Z. May, I. M. Szilágyi, A. Jánosity, K. K. Banerji, R. Kant and L. Kótai, J. Coord. Chem., 2018, 71(16–18), 2884–2904, DOI:10.1080/00958972.2018.1493464.
- (a) T. Klobb, C. R. Hebd. Seances Acad. Sci., 1894, 118, 1271–1273 Search PubMed; (b) T. Klobb, Bull. Soc. Chim., 1894, 11, 604–609 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- A. Kálmán, L. Párkányi and Gy. Argay, Acta Crystallogr., Sect. B: Struct. Sci., 1993, 49, 1039–1049 CrossRef.
- J. C. Dyason, P. C. Healy, L. M. Engelhardt and A. H. White, Aust. J. Chem., 1985, 38(9), 1325–1328 CrossRef CAS.
- G. A. Bowmaker, Effendy, K. C. Lim, B. W. Skelton, D. Sukarianingsih and A. H. White, Inorg. Chim. Acta, 2005, 358, 4342–4350 CrossRef CAS.
- R. Macy, J. Am. Chem. Soc., 1925, 47, 1031–1036 CrossRef CAS.
- K. Nilsson and A. Oskarsson, Acta Chem. Scand., Ser. A, 1982, 36(7), 605–610 CrossRef.
- T. Kocsis, J. Magyari, I. E. Sajó, T. Pasinszki, Z. Homonnay, I. M. Szilágyi, A. Farkas, Z. May, H. Effenberger, S. Szakáll, R. P. Pawar and L. Kótai, J. Therm. Anal. Calorim., 2018, 132, 493–502 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- (a) V. Petrusevski and K. Trencevski, Croat. Chem. Acta, 1986, 59(4), 867–881 CAS; (b) V. Petrusevski and B. Soptrayanov, J. Mol. Struct., 1988, 175, 349–354 CrossRef CAS; (c) D. Y. Wu, B. Ren, Y. X. Jiang, X. Xu and Z. Q. Tian, J. Phys. Chem. A, 2002, 106, 9042 CrossRef CAS.
- (a) Y. Bando and S. Tagakura, Theor. Chim. Acta, 1968, 9, 210–221 CrossRef CAS; (b) J. Neugebauer and E. J. Baerends, J. Phys. Chem., 2005, 109, 1168–1179 CrossRef CAS PubMed; (c) S. L. Holt and C. J. Ballhausen, Theor. Chim. Acta, 1967, 7, 313–320 CrossRef CAS; (d) P. Boopalachandran and J. Laane, Chem. Phys. Lett., 2008, 462, 178–182 CrossRef CAS.
- J. Koerbl, Mikrochim. Acta, 1956, 44(11), 1705–1721 CrossRef.
- K. P. Sinha and A. P. B. Sinha, J. Chem. Phys., 1957, 61, 758–761 CrossRef CAS.
- (a) F. Buciuman, F. Patcas, R. Craciun and D. R. T. Zahn, Phys. Chem. Chem. Phys., 1999, 1, 185–190 RSC; (b) F. A. Al Sagheer, M. A. Hasan, L. Pasupulety and M. I. Zaki, J. Mater. Sci. Lett., 1999, 18, 209–211 CrossRef CAS; (c) S. Music, M. Ristic and S. Popovic, J. Mol. Struct., 2009, 924–926, 243–247 CrossRef CAS; (d) C. M. Julien, M. Massot and C. Poinsignon, Spectrochim. Acta, Part A, 2004, 60, 689–700 CrossRef CAS; (e) W. B. White and V. G. Keramidas, Spectrochim. Acta, Part A, 1972, 28, 501–509 CrossRef CAS; (f) F. Davar, F. Mohandes and M. Salavati-Niasari, Inorg. Chim. Acta, 2009, 362, 3663–3668 CrossRef CAS; (g) A. S. Povarennikh, Konst. Svoistva Miner., 1979, 13, 53–78 Search PubMed.
- Z. W. Chen, J. K. L. Lai and C. H. Shek, J. Non-Cryst. Solids, 2006, 352, 3285–3289 CrossRef CAS.
- (a) T. Larbi, K. Doll and T. Manoubi, J. Alloys Compd., 2016, 688, 692–698 CrossRef CAS; (b) M. Ishii, M. Nakahira and T. Yamanaka, Solid State Commun., 1972, 11, 209–212 CrossRef CAS.
- L. Biernacki and S. Pokrzywnicki, J. Therm. Anal. Calorim., 1999, 55, 227–232 CrossRef CAS.
- C. Y. Chen, J. Y. Zeng and H. M. Lee, Inorg. Chim. Acta, 2007, 360(1), 21–30 CrossRef CAS.
- A. Gorgeu, C. R. Hebd. Seances Acad. Sci., 1892, 114, 912–915 Search PubMed.
- (a) R. P. Westerdahl and P. J. Leader, Inorg. Nucl. Chem. Lett., 1969, 5, 199–201 CrossRef CAS; (b) A. J. Hegedus and K. Martin, Microchim. Acta, 1966, 833–852 CrossRef CAS; (c) A. J. Hegedus and A. B. Kiss, Acta Chim. Acad. Sci. Hung., 1967, 51, 251–254 CAS.
- Y. Sawada and K. Manabe, J. Therm. Anal., 1991, 37, 1657–1663 CrossRef CAS.
- T. Brusentsova, R. E. Peale, D. Maukonen, G. E. Harlow, J. S. Boesenberg and D. Ebel, Am. Mineral., 2010, 95, 1515–1522 CrossRef CAS.
- K. Nakamoto, Infrared and Raman spectra of inorganic and coordination compounds, Part A and B, Wiley Intersci Publ, New York, 5th edn, 1997 Search PubMed.
- (a) S. L. Brock, N.-G. Duan, Z. R. Tian, O. Giraldo, H. Zhou and S. L. Suib, Chem. Mater., 1998, 10, 2619–2628 CrossRef CAS; (b) I. Knopf, T. Ono, M. Temprado, D. Tofana and C. C. Cummins, Chem. Sci., 2014, 5, 1772–1776 RSC.
- J. Liebig, Ann. Chem. Pharm., 1856, 8(1), 132–139 CrossRef.
- J. Kan, S. Huang, J. Lin, M. Zhang and W. Su, Angew. Chem., Int. Ed., 2015, 54, 2199–2203 CrossRef CAS PubMed.
- S. M. Jörgenson, J. Prakt. Chem., 1886, 33, 489 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1879263. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra03230d |
| This journal is © The Royal Society of Chemistry 2019 |