
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransition-metal-free amination phosphoryl azide for the synthesis of phosphoramidates†
Qing Li‡
a,
Xiaohua Sun‡b,
Xiaoqin Yang‡a,
Minghu Wua,
Shaofa Suna and
Xiuling Chen *a
*a
aNon-power Nuclear Technology Collaborative Innovation Center, School of Nuclear Technology and Chemistry & Biology, Hubei University of Science and Technology, Xianning 437100, China. E-mail: cxl828800@163.com; sunshaofa@mail.hbust.com.cn; Fax: +86-715-8338007
bXianning Vocational Technical College, China
First published on 22nd May 2019
Abstract
A facile and efficient approach to phosphoramidates was developed via amination of phosphoryl azides. A variety of phosphoramidates were obtained in one step with good to excellent yields under a mild reaction system. The process uses easily available amines as a N source and offers a new opportunity for P–N bond formation.
Organophosphorus compounds are an important structural feature in pharmaceutical chemistry, biological and materials chemistry.1,2 Among organophosphorus compounds, phosphoramidates are prevalent organic structural motifs widely applied in industry as pharmaceutical prodrugs, medicinal areas that have potent antifungal, antitumor, and anti-HIV activities, and in analytical chemistry to restrain matrix-related ion effects in MALDI-TOF mass spectrometry and improve ionization efficiency. Additionally, phosphoramidates are also important precursors for the synthesis of flame retardants.3–8 Moreover, in recent years, phosphoramidates have become important chiral ligands for several metal-catalyzed reactions.9 For these reasons, the synthesis of phosphoramidates is of great importance. Generally, synthetic procedures for the preparation of phosphoramidates rely on the reaction of phosphorus halides and amines, P(III) as the reagent and corrosive materials but low functional group tolerance and complex operations have limited this application.10 To overcome these problems, alternative methods for phosphoramidates synthesis have been developed.11–16 The Atherton–Todd reaction represents an alternative route for the synthesis of phosphoramidates, but this approach also is far from ideal due to the use of stoichiometric CCl4, which presents a limitation to an environmentally friendly method.11 Recently, a catalyzed oxidative dehydrocoupling of P(O)H bonds with amines reaction has emerged as a green synthetic tool. However, transition metals, halogen or additional additives are required.12 Other methods include a Lewis acid-catalyzed phosphorimidate rearrangement to N,N-disubstituted phosphoramidates, nucleophilic addition of TMSCCl3 to N-phosphinoyl benzaldimines,13 the addition of diarylphosphine oxides to azobenzenes to construct a N–N–P unit,14 and a lanthanum-catalyzed direct double hydrophosphinylation of unactivated nitriles to phosphoramidates.15 Although these protocols have widespread utility, they usually suffer from some intrinsic disadvantages, as they rely on transition metals or are sensitive to water or oxygen.
Recently, transition metal catalyzed C–H phosphoramidation of different substrates with a phosphoryl azide to provide various N-aryl phosphoramidates has been developed.16 However, transition metal catalysts are toxic, especially for use in the drug and pharmaceutical industries. Therefore, a metal-free condition, convenient and green method to synthesize phosphoramidates is still desired. Herein, we report an environmentally friendly approach for the preparation of phosphoramidates via amination of phosphoryl azide (eqn (1)). Our system covers a wide range of amine substrates under mild conditions for preparing highly functionalized phosphoramidates with ease.
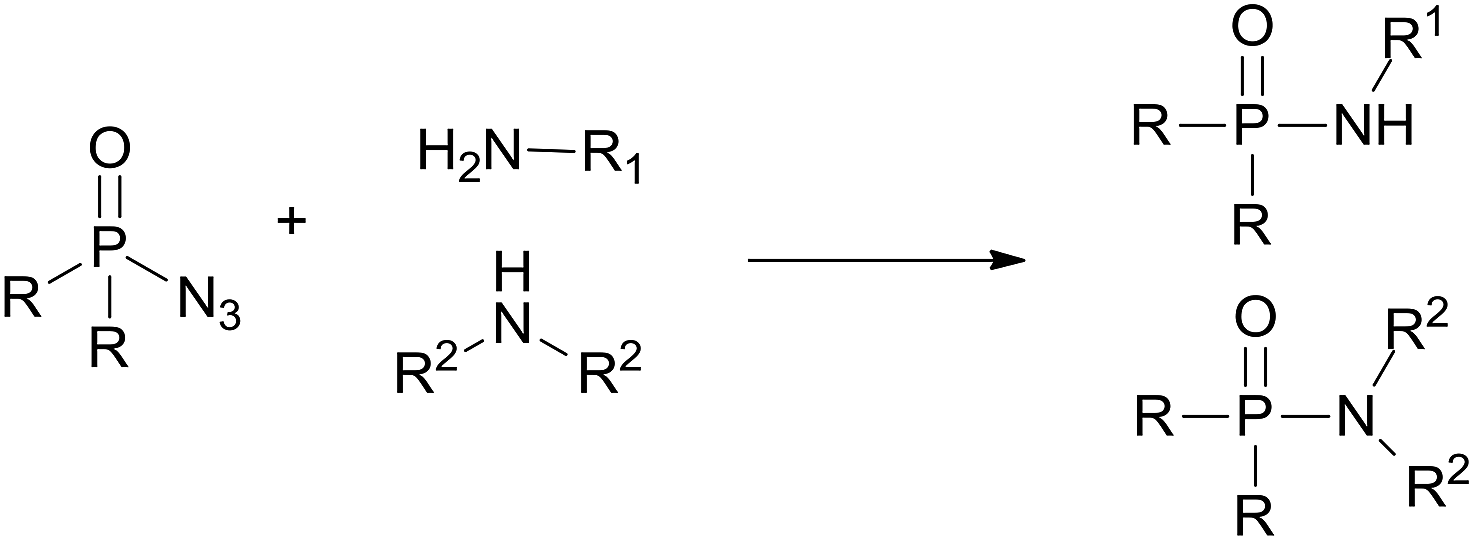 | (1) |
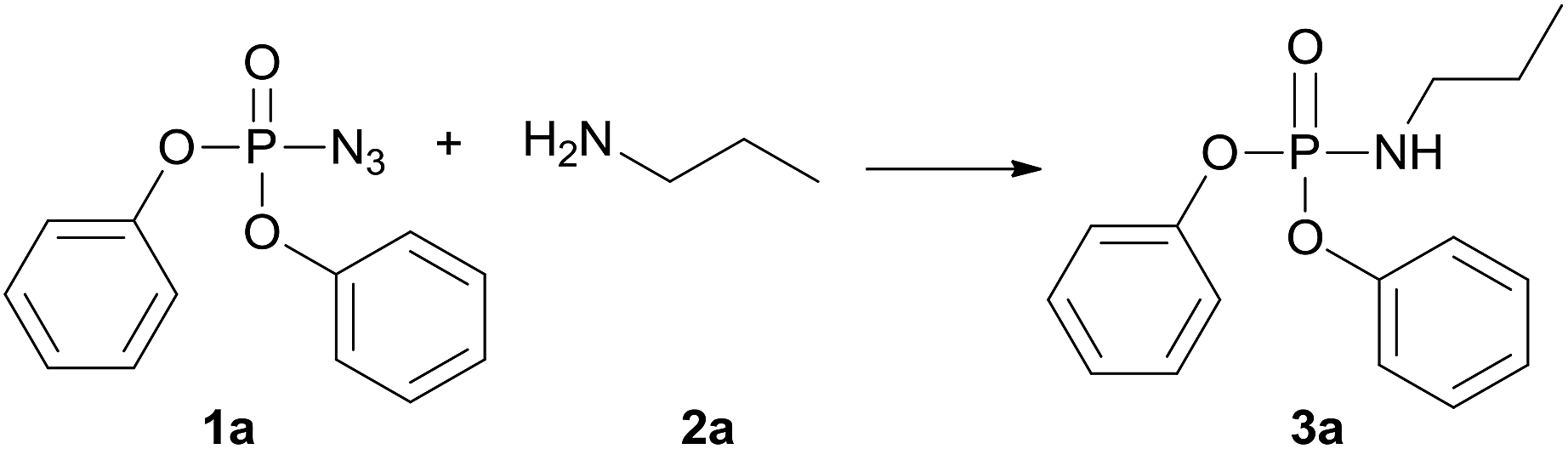
To explore the practicability of this novel amination phosphoryl azide protocol, we choose diphenyl phosphoryl azide (1a) and n-propylamine (2a) as model substrates to optimize the reaction conditions, and the obtained results are compiled in Table 1. First, Cs2CO3 was used as a base to attempt the amination phosphinyl azides reaction when dioxane was used as solvent at 120 °C, and 3a was obtained in 31% yield (Table 1, entry 1). Next, various solvents including THF, toluene, CH3CN, CH3COOC2H5, C2H5OH, DMF, and DMSO were screened (Table 1, entries 2–8), and DMF was found to the best choice to give product 3a in 69% yield (Table 1, entry 8). Then, CuCl or DBU was used instead of Cs2CO3 but lower yields of 3a were isolated (Table 1, entries 9 and 10). The best results were observed when an additional additive such as Cs2CO, CuCl2 or DBU was not added to reaction system, then 3a was obtained in 84% yield (Table 1, entry 11). Thus, it was deduced that external base was not necessary and the starting material amine perhaps acted as an efficient enough base. This amination phosphinyl azide protocol was also dependent on the temperature; for example, when this reaction was performed at 80 °C, 3a was obtained only in 32% yield (Table 1, entry 12) and at 60 °C 3a was not obtained at all (Table 1, entry 13). So, the optimal reaction condition was at 120 °C for 12 h without any additive.
|
|||
|---|---|---|---|
| Entry | Solvent | Additive | Yield of 3ab |
| a Reaction conditions: diphenyl phosphoryl azide 1a (0.2 mmol), n-propylamine 2a (0.4 mmol), base (2.0 equiv.), dioxane (2 mL), in a 25 mL Schlenk tube, 120 °C, 12 h.b Isolated yields.c 80 °C.d 60 °C. | |||
| 1 | Dioxane | Cs2CO3 | 31 |
| 2 | THF | Cs2CO3 | 25 |
| 3 | Toluene | Cs2CO3 | 44 |
| 4 | CH3CN | Cs2CO3 | 51 |
| 5 | CH3COOC2H5 | Cs2CO3 | 36 |
| 6 | C2H5OH | Cs2CO3 | — |
| 7 | DMSO | Cs2CO3 | — |
| 8 | DMF | Cs2CO | 69 |
| 9 | DMF | CuCl2 | 30 |
| 10 | DMF | DBU | 46 |
| 11 | DMF | — | 84 |
| 12c | DMF | — | 32 |
| 13d | DMF | — | — |
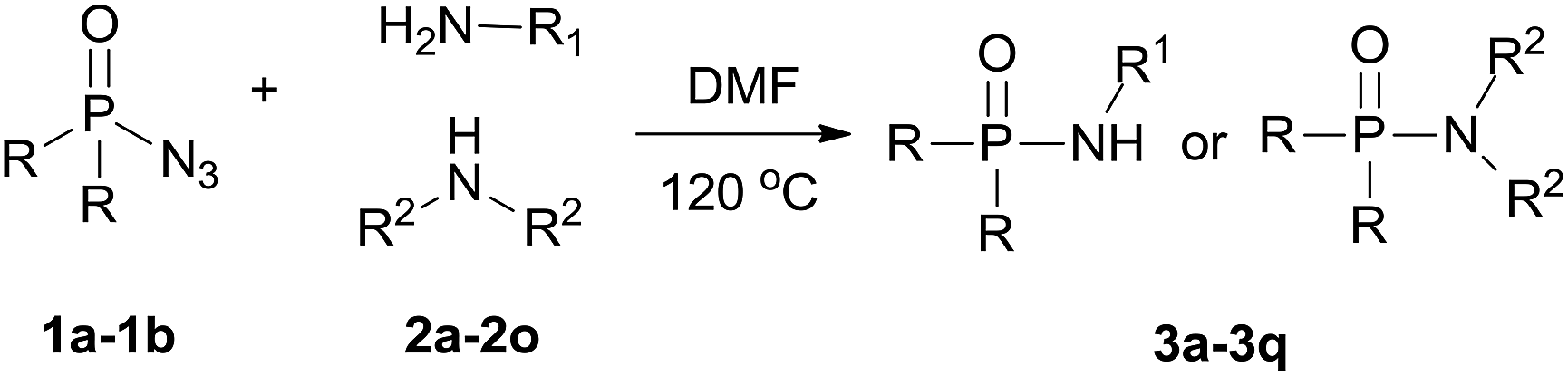
With the optimized reaction conditions in hand, we then evaluated the scope of amination using phosphoryl azides with various amines. As shown in Table 2, substrates with a variety of primary amines and second amines afforded the products in good to excellent yields under the optimum reaction conditions. First, a range of primary amines, such as n-propylamine, n-butylamine, isobutylamine and isopropyl amine, were reacted with diphenyl phosphoryl azide to furnish 3a–3d in excellent yields under the optimized reaction condition. In contrast, tert-butylamine required a longer time of 24 h to afford only a 55% isolated yield of 3e. The reason is that tert-butanol exhibits weak nucleophilicity. Primary amines containing electron-donating substituents, e.g., methoxy (–OCH3) and electron-withdrawing (–CF3), were tolerated and react with diphenyl phosphoryl azide to give good yields of phosphoramidates 3f–3g in 85% and 52%, respectively.
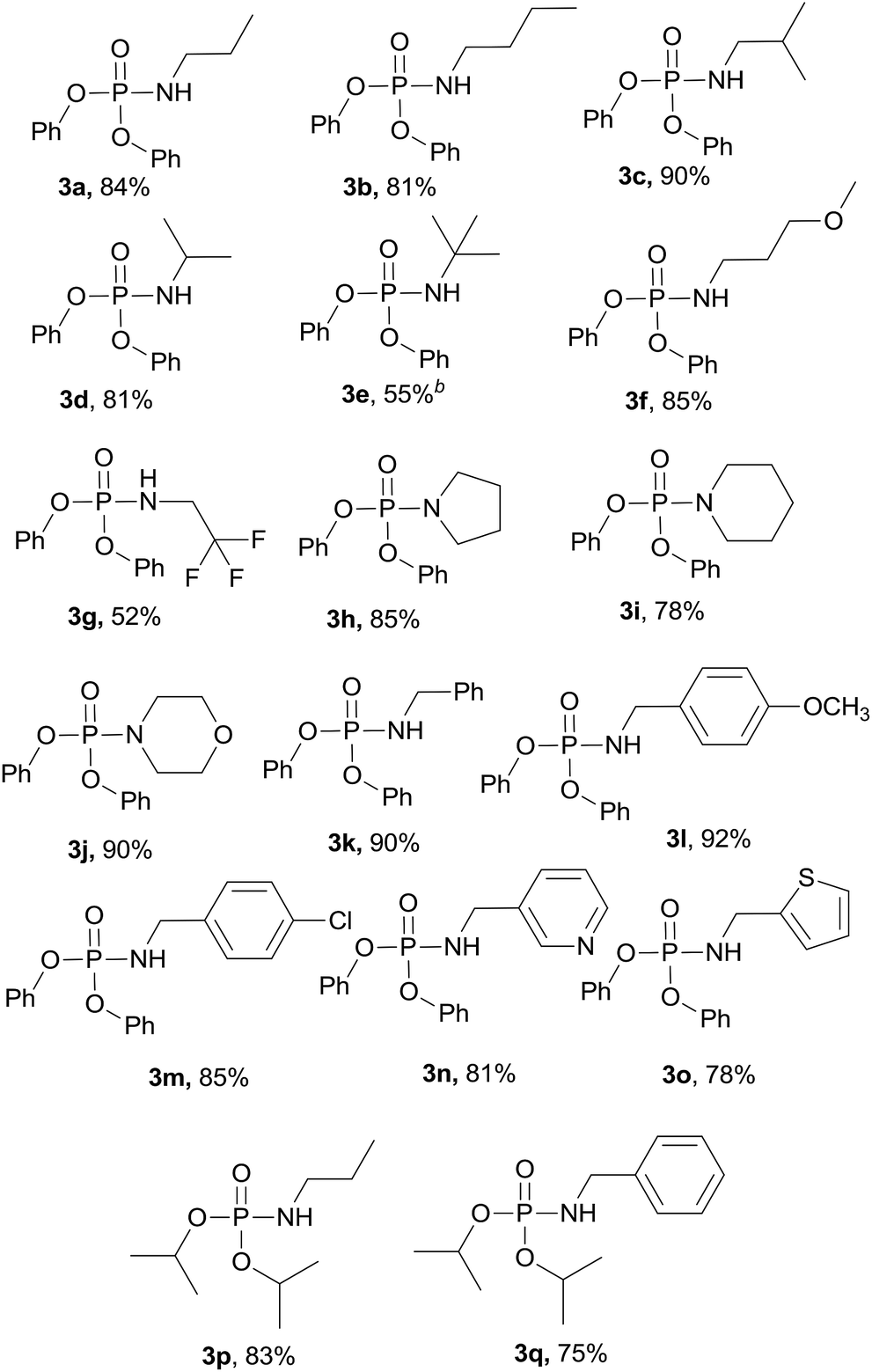
When secondary cyclic amines such as tetrahydropyrrole, piperidine and morpholine were employed, the corresponding phosphoramidates yields turned out to be 85%, 78% and 90%, respectively (3h–3j). A benzylamine bearing an electron-donating substituent (Cl) or electron-withdrawing substituent (OCH3) proceeded smoothly to afford the corresponding products in 92% and 85% isolated yields, respectively, and there was no sharp difference. 3-(Aminomethyl)pyridine and 2-thiophenemethylamine also served as good substrates to produce the corresponding phosphoramidates 3n–3o in excellent yields.
Next, the reaction scope was extended to different phosphoryl azides; for example, diisopropyl phosphoryl azide also reacted with n-propylamine and benzylamine smoothly, affording the desired products 3p and 3q in 83% and 75% yields, respectively.
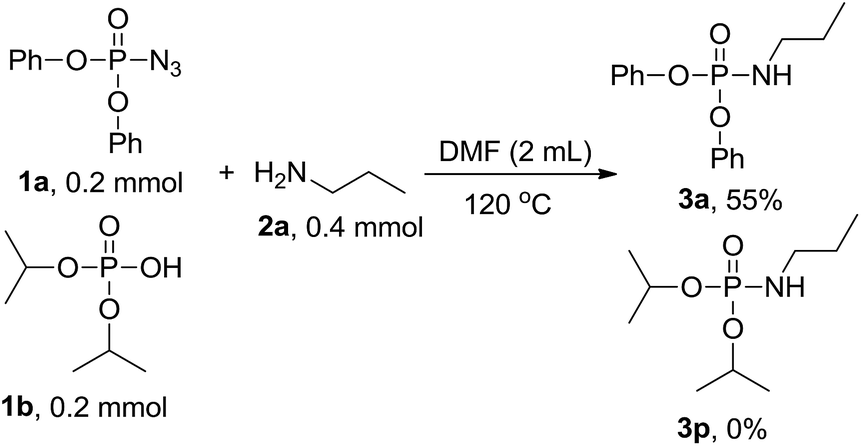
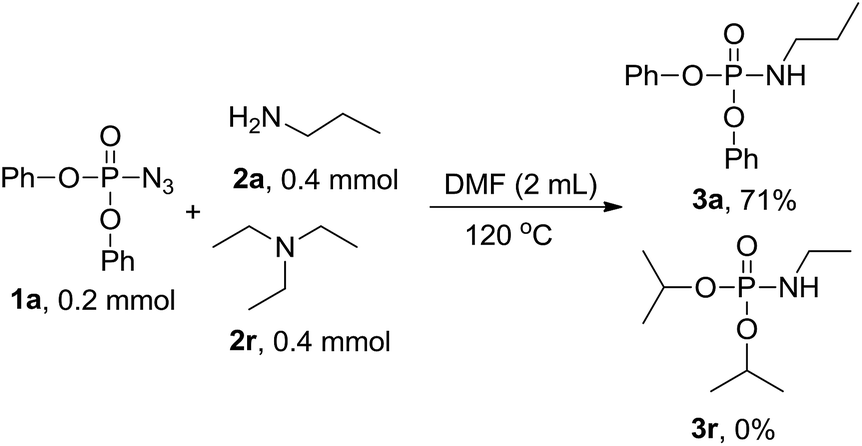
To get more information on the reaction mechanism, the next control experiment was performed with diphenyl phosphoryl azide 1a, diisopropyl hydrogen phosphate 1b and n-propylamine 2a subjected to the reaction under standard conditions. As shown below, 3a was obtained in 55% yield, while 3p was not detected at all (eqn (2)), indicating that phosphoryl azide was not transformed to hydrogen phosphate and then attacked by amine to phosphoramidate, so hydrogen phosphate is not an intermediate. To investigate the sources of nitrogen-atoms and the alkyl group, diphenyl phosphoryl azide 1a, triethylamine 2r and n-propylamine 2a were reacted under the standard conditions. 3a was obtained in 71% yield, whereas 3r was not detected at all, showing that the reaction proceeds via a direct nucleophilic substitution at the P center, with the azide anion serving as the leaving group (eqn (3)).
 | (2) |
 | (3) |
Based on the present results and the reported literature,17 a possible mechanism for the amination of phosphoryl azides in the synthesis of phosphoramidates is suggested in Scheme 1. First, phosphoryl azide 1 is attacked by amine 2 to form intermediate m via a nucleophilic addition, with the azide anion serving as the leaving group by P–N3 bond cleavage affording the desired phosphoramidate 3.
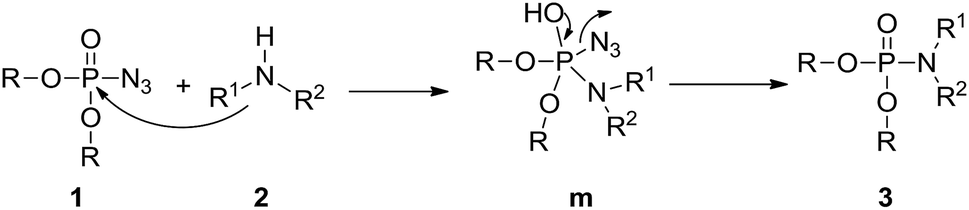 | ||
| Scheme 1 Plausible reaction pathway for the synthesis of phosphoramidate 3. | ||
Conclusions
In summary, we have developed a simple and highly efficient method for the synthesis of phosphoramidates via amination with phosphoryl azides under metal and base free conditions. The presented method exhibits a series of attractive features: good functional-group tolerance, metal and base free conditions and phosphoramidates synthesis via one reaction system. This is a straightforward, efficient and environmentally friendly alternative method for the preparation of industrially useful phosphoramidate derivatives.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by National Natural Science Foundation of China (21603068), and research fund for the doctoral program of Hubei University of Science and Technology (2016-19XB011) are gratefully appreciated.Notes and references
-
(a) J. W. Darrow and D. G. Drueckhammer, J. Org. Chem., 1994, 59, 2976 CrossRef CAS
; (b) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef CAS PubMed
-
(a) C. S. Demmer, N. Krogsgaard-Larsen and L. Bunch, Chem. Rev., 2011, 111, 7981 CrossRef CAS PubMed
-
(a) X. Gao, Z. Tang, M. Lu, H. Liu, Y. Jiang, Y. Zhaoc and Z. Cai, Chem. Commun., 2012, 48, 10198 RSC
-
(a) J. I. Guijarro, J. E. González-Pastor, F. Baleux, J. L. S. Millán, M. A. Castilla, M. Rico, F. Moreno and M. Delepierre, J. Biol. Chem., 1995, 270, 23520 CrossRef CAS PubMed
-
(a) R. A. Dwek, Chem. Rev., 1996, 96, 683 CrossRef CAS PubMed
-
(a) M. J. Berridge and R. F. Irvine, Nature, 1989, 341, 197 CrossRef CAS PubMed
-
(a) K. Wittine, K. Benci, Z. Rajić, B. Zorc, M. Kralj, M. Marjanović, K. Pavelić, E. De Clercq, G. Andrei, R. Snoeck, J. Balzarini and M. Mintas, Eur. J. Med. Chem., 2009, 44, 143 CrossRef CAS PubMed
-
(a) C. Nguyen and A. Kim, Macromol. Res., 2008, 16, 620 CrossRef CAS
-
(a) S. E. Denmark and G. L. Beutner, Angew. Chem., Int. Ed., 2008, 47, 1560 CrossRef CAS PubMed
-
(a) L. D. Quin, A Guide to Organophosphorus Chemistry, Wiley, New York, 2000 Search PubMed
-
(a) F. R. Atherton, H. T. Openshaw and A. R. J. Todd, J. Chem. Soc., 1945, 660 RSC
-
(a) X. Jin, K. Yamaguchi and N. Mizuno, Org. Lett., 2013, 15, 418 CrossRef CAS PubMed
-
(a) I. Wilkening, G. del Signore and C. P. R. Hackenberger, Chem. Commun., 2008, 2932 RSC
- G. Hong, X. Zhu, C. Hu, A. N. Aruma, S. Wu and L. Wang, J. Org. Chem., 2016, 81, 6867 CrossRef CAS
- M. M. I. Basiouny and J. A. R. Schmidt, Organometallics, 2017, 36, 721 CrossRef CAS
-
(a) H. Lu, J. Tao, J. E. Jones, L. Wojtas and X. P. Zhang, Org. Lett., 2010, 12, 1248 CrossRef CAS PubMed
- S. Y. Chow, M. Y. Stevens and L. R. Odell, J. Org. Chem., 2016, 81, 2681 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data for the products. See DOI: 10.1039/c9ra03389k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |