
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuick construction of a C–N bond from arylsulfonyl hydrazides and Csp2–X compounds promoted by DMAP at room temperature†
Kai Yangab,
Juan-Juan Gaoc,
Shi-He Luo *be,
Han-Qing Wub,
Chu-Ming Pangb,
Bo-Wen Wangb,
Xiao-Yun Chen*d and
Zhao-Yang Wangbe
*be,
Han-Qing Wub,
Chu-Ming Pangb,
Bo-Wen Wangb,
Xiao-Yun Chen*d and
Zhao-Yang Wangbe
aCollege of Pharmacy, Gannan Medical University, Ganzhou, Jiangxi province, P. R. China 341000
bSchool of Chemistry and Environment, South China Normal University, Key Laboratory of Theoretical Chemistry of Environment, Ministry of Education, Guangzhou, Guangdong province, P. R. China 510006. E-mail: pinky_r@163.com
cCollege of Sports and Rehabilitation, Gannan Medical University, Ganzhou, Jiangxi province, P. R. China 34100
dSchool of Environmental and Chemical Engineering, Jiangsu University of Science and Technology, Zhenjiang, Jiangsu province, P. R. China 212003. E-mail: xiaoyun_chen12@163.com
eKey Laboratory of Functional Molecular Engineering of Guangdong Province, School of Chemistry and Chemical Engineering, South China University of Technology, 381 Wushan Road, Guangzhou, Guangdong province, P. R. China 510640
First published on 26th June 2019
Abstract
An efficient approach for C–N bond construction by the coupling reaction of arylsulfonyl hydrazides and Csp2–X compounds is described for the first time with good yields at room temperature. The reaction promoted by the simple base DMAP displays excellent regioselectivity as well as high functional group tolerance with 41 examples. Even for inactive Csp2–Cl compounds, the metal-free transformation also affords a satisfactory yield after prolonging the reaction time, which is comparable to that of the corresponding Csp2–Br compound. The good effect of DMAP and its action mechanism are confirmed by the competitive experiments of reactivity between Cl-substituted and Br-substituted substrates and the single-crystal X-ray analysis of the key intermediate quaternary ammonium salt. Importantly, the application of this method for a gram-scale (even over 10 g) preparation can be accomplished.
Introduction
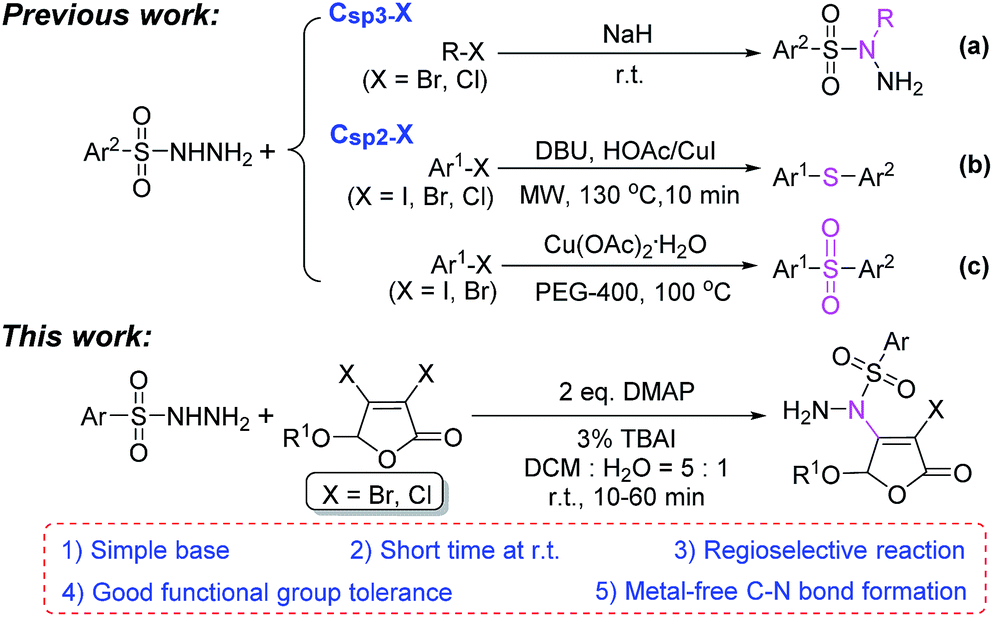
Due to good stability and reactivity, readily available arylsulfonyl hydrazides have attracted considerable attention and been widely used in the synthesis of various natural products and bioactive molecules.1,2 Particularly, arylsulfonyl hydrazides have usually been employed as arylation,3 sulfonylation4 or thioetherification5 agents to construct C–C or C–S bonds for the synthesis of aromatic hydrocarbons, sulfones and thioethers in recent years.For the reaction between arylsulfonyl hydrazides and halides, sulfonyl hydrazides are generally used to react with Csp3–X (I, Br, Cl) compounds in the presence of a strong base such as sodium hydride to form N-substituted sulfonyl hydrazides1,6 (Scheme 1a). However, there are only a few studies on the reaction between sulfonyl hydrazides and less active Csp2–X compounds. The limited number of reports mainly concentrate on the formation of a C–S bond by the copper-catalyzed arylthiolation (Scheme 1b)7 and sulfonylation reaction (Scheme 1c).8
 | ||
| Scheme 1 The reaction of arylsulfonyl hydrazides with halides. | ||
To our knowledge, the reaction of building C–N bond between Csp2–X compounds and sulfonyl hydrazides to form N-substituted sulfonyl hydrazides has not been reported yet in the previous researches. Herein, we reported a metal-free catalytic reaction between sulfonyl hydrazides and non-aromatic Csp2–X (X = Br, Cl) compounds, 3,4-dihalo-2(5H)-furanones, in the presence of readily available organic base DMAP at room temperature (Scheme 1, this work).
Results and discussion
Optimization of reaction conditions
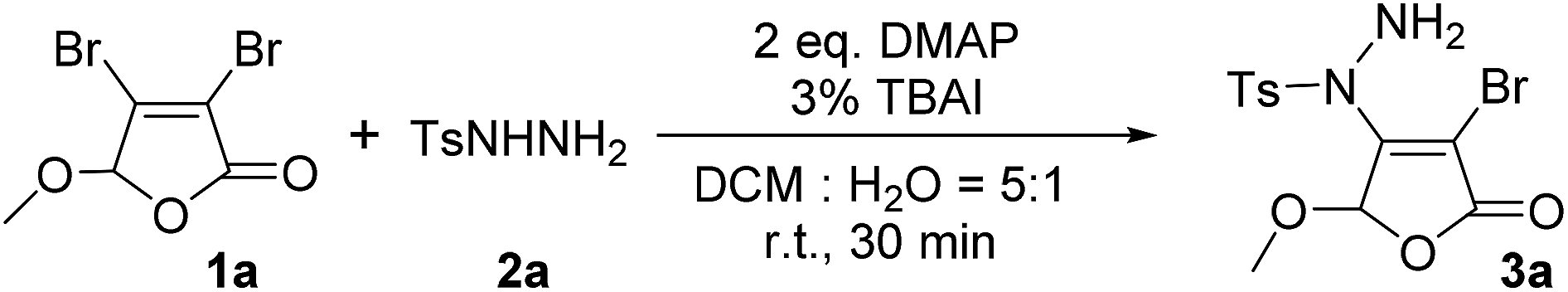
For the first time, we provide a new approach for C–N bond construction by the coupling reaction of Csp2–X compounds and sulfonyl hydrazides. More importantly, this approach is also adapted to the less active Csp2–Cl compounds, which can afford comparable yield to Csp2–Br compounds. Moreover, the 2(5H)-furanone moiety is frequently found in natural products9 and drug molecules,10,11 which makes the synthesis of 2(5H)-furanone compounds with polyfunctional groups pharmaceutically valuable. Thus, we choose 3,4-dihalo-2(5H)-furanones as non-aromatic Csp2–X (X = Br, Cl) reactants in this investigation.Initially, the condition parameters were optimized by selecting the reaction of 3,4-dibromo-5-methoxy-2(5H)-furanone 1a and p-toluene sulfohydrazide 2a as the model reaction. As shown in Table 1, the reaction does not occur in the absence of catalyst (Entry 1). While using copper salt (CuI), 1,10-phenanthroline (Phen) and sodium carbonate (Na2CO3) as the catalytic system (Entry 2), the reaction still does not occur. Further changing the base to potassium fluoride (KF), the target product 3a can be obtained with 30% yield (Entry 3). Even without the addition of CuI and Phen, 3a can also be obtained with 32% yield (Entry 4). Thus, we did not use CuI and Phen any more in the following screening. Controlling the other conditions, different organic bases were also investigated (Entries 4–7). Obviously, using 4-dimethylaminopyridine (DMAP) gives the best yield of 3a, which is up to 53% (Entry 5).
|
||||
|---|---|---|---|---|
| Entry | Cat. (or additive) (eq.) | 1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2a :2a |
Solvent | Yieldb (%) |
| a Reaction conditions: all reactions were performed with 1a (0.5 mmol), 2a (0.6 mmol) and solvent (3 mL), at room temperature (r.t.), 30 min.b Isolated yield. | ||||
| 1 | — | 1:1.2 |
DMF | Trace |
| 2 | CuI (0.1) Phen (0.1) Na2CO3 (2.0) | 1:1.2 |
DMF | Trace |
| 3 | CuI (0.1) Phen (0.1) KF (2.0) | 1:1.2 |
DMF | 30 |
| 4 | KF (2.0) | 1:1.2 |
DMF | 32 |
| 5 | DMAP (2.0) | 1:1.2 |
DMF | 53 |
| 6 | DABCO (2.0) | 1:1.2 |
DMF | 49 |
| 7 | DBU (2.0) | 1:1.2 |
DMF | 38 |
| 8 | DMAP (2.0) | 1:1.2 |
EtOH | 45 |
| 9 | DMAP (2.0) | 1:1.2 |
MeCN | 65 |
| 10 | DMAP (2.0) | 1:1.2 |
THF | 52 |
| 11 | DMAP (2.0) | 1:1.2 |
DCM | 78 |
| 12 | DMAP (2.0) | 1:1.2 |
Toluene | 37 |
| 13 | DMAP (2.0) | 1:1.2 |
DCM:H2O = 5:1 |
62 |
| 14 | DMAP (2.0), TBAI (0.03) | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1.2 :1.2 |
DCM:H2O = 5:1 |
82 |
| 15 | DMAP (2.0), TBAI (0.03) | 1:1.2 |
DCE:H2O = 5:1 |
76 |
| 16 | DMAP (2.0), TBAI (0.03) | 1:1.2 |
Toluene:H2O = 5:1 |
66 |
| 17 | DMAP (2.0), TBAI (0.03) | 1:1.2 |
DCM:H2O = 1:1 |
70 |
| 18 | DMAP (1.0), TBAI (0.03) | 1:1.2 |
DCM:H2O = 5:1 |
58 |
| 19 | DMAP (1.5), TBAI (0.03) | 1:1.2 |
DCM:H2O = 5:1 |
69 |
| 20 | DMAP (2.5), TBAI (0.03) | 1:1.2 |
DCM:H2O = 5:1 |
78 |
| 21 | DMAP (2.0), TBAI (0.03) | 1:1 |
DCM:H2O = 5:1 |
75 |
| 22 | DMAP (2.0), TBAI (0.03) | 1.2:1 |
DCM:H2O = 5:1 |
79 |
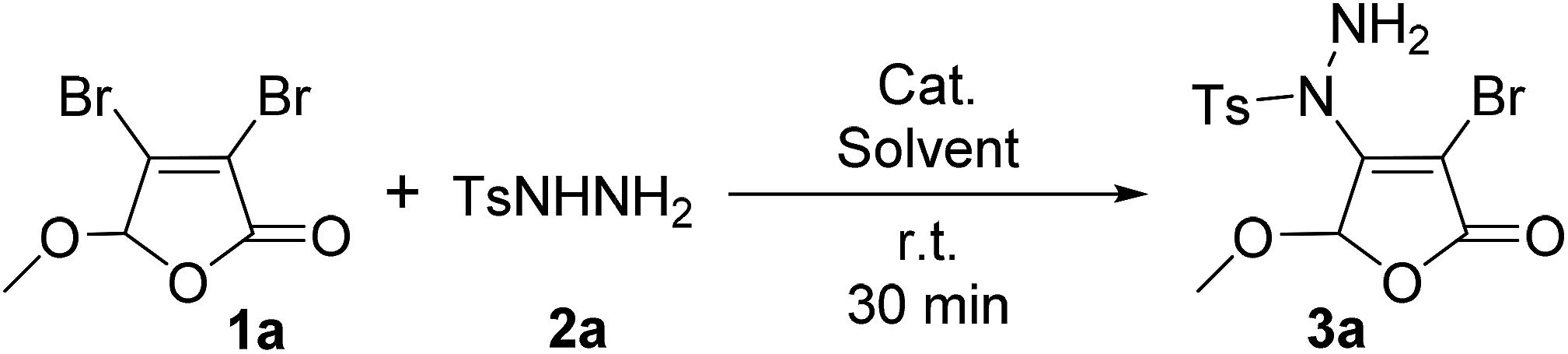
The optimization of solvents was further carried out (Table 1, Entries 8–12). The results indicate that dichloromethane (DCM) is found to be the best in single solvent system and the yield of 3a can be 78% (Entry 11). We also discussed the effect of mixed solvent system. In DCM–H2O with a volume ratio of 5:1, the yield of 3a is decreased to 62% (Entry 13). However, after the addition of 3% tetrabutylammonium iodide (TBAI) as a phase transfer catalyst, the yield of 3a is markedly increased to 82% (Entry 14). Compared with other organic solvent and water with a volume ratio of 5:1 as the mixed solvent (Entries 14–16), the yield of 3a in DCM–H2O is the best (Entry 14). However, with the increase of water fraction in this mixed solvent, the yield of 3a is decreased to 70% (Entry 17).
It can be learnt from Entries 14 and 18–20 in Table 1 that the amount of base also plays an important role in this reaction system. The optimized dosage of DMAP is 2.0 eq. (Entry 14). In addition, the feed ratio of reactants was discussed and the results were collected in Entries 14 and 21–22. It can be found that when the feed ratio of 1a and 2a is 1:1.2, the yield of 3a is the highest value of 82% (Entry 14). Altering the reaction temperature and time, no better changes can be achieved as monitored by TLC. Thus, the optimized reaction conditions were identified as 2.0 eq. DMAP, 3 mol% of TBAI, 1a:2a = 1:1.2, a mixture of DCM and H2O (v:v = 5:1) as the solvent at room temperature for 30 min.
Scope of reaction substrates
Under the optimized reaction conditions, we further studied the scope of the reaction substrate with respect to 5-substituted 3,4-dibromo-2(5H)-furanones and sulfonyl hydrazides, respectively. The results are summarized in Table 2. Interestingly, all reactions of 5-substituted 3,4-dibromo-2(5H)-furanones 1 and p-toluene sulfohydrazide (2a) occur in high yields (70–86%, 3a–3j). Especially, the substituted 2(5H)-furanones with 5-alkoxy, such as methoxy, ethoxy, n-butoxy, n-hexyloxy (HexO), isopropoxy, cyclohexyloxy (CyO), can give the products in good yields (78–86%, 3a–3g). For example, 82% of 3a (methoxy) was isolated, while 3f (isopropoxy) is obtained in 81% yield. Even for the larger CyO, the product 3g was also successfully obtained and the yield is as high as 78%. Thus, the steric hindrance effect of 5-substituted group on the yield is not particularly obvious.
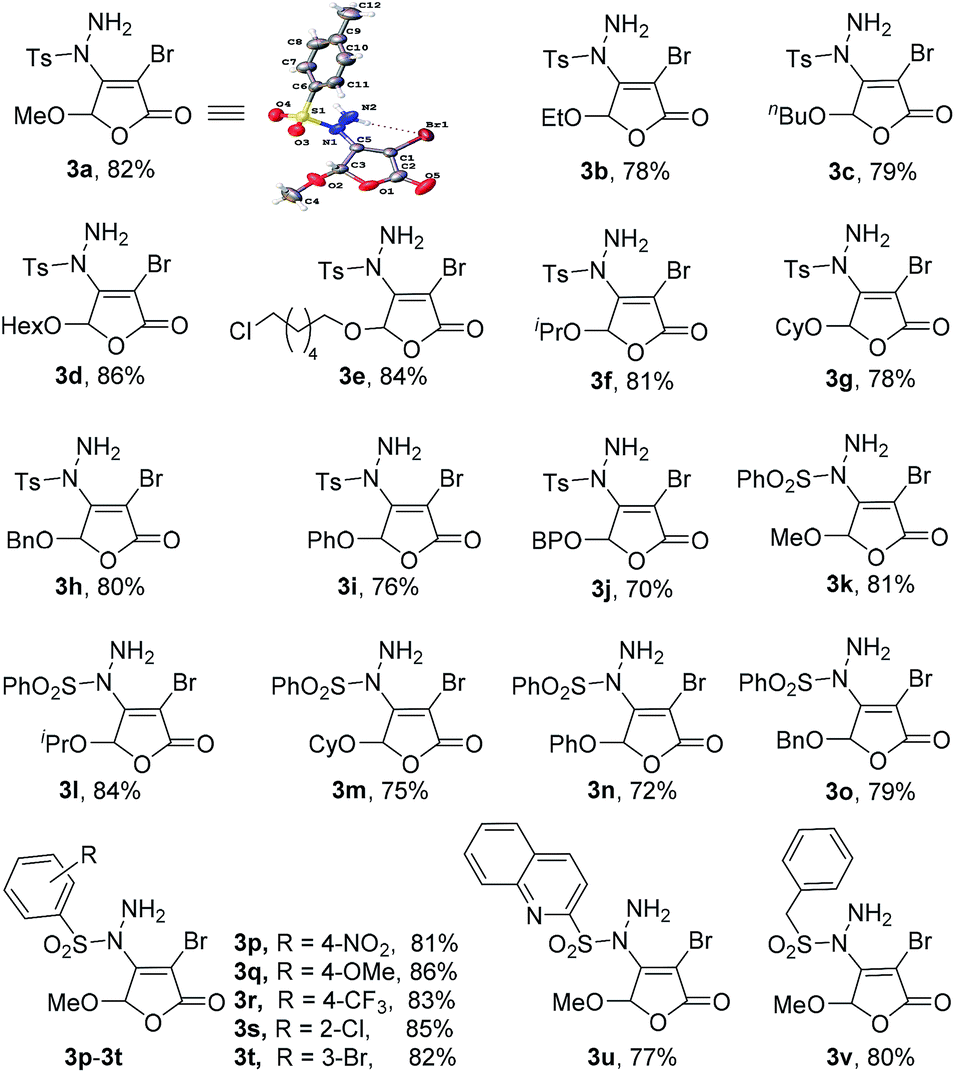
Noteworthily, the catalytic process is highly chemoselective in a case where 2(5H)-furanone contains more than one potentially reactive group. For example, for the substrate of 6-chlorohexyloxy, the sulfonyl hydrazide is highly selective to react with Csp2–Br at the C-4 of 2(5H)-furanone to form 3e, instead of reacting with Csp3–Cl at the 5-substituted group. Moreover, when 5-substituted group is benzyloxy (BnO), the yield of 3h can also reach to 80% as expected. For the substituted 2(5H)-furanones with aryloxy, e.g., phenoxy, biphenyloxy (BPO), the relatively lower yields are obtained (70–76%, 3i–3j). Further, a series of 5-substituted 3,4-dibromo-2(5H)-furanones as substrates 1 were explored in the reaction with phenylsulfonyl hydrazide (2b). The reaction also has good tolerance on different functional groups (e.g., alkoxy, aryloxy) in good yields (72–84%, 3l–3o).
In addition, many arylsulfonyl hydrazides 2 with electron-donating (e.g., methoxy) or electron-withdrawing (e.g., halogen, nitro, trifluoromethyl) groups can react with 3,4-dibromo-5-methoxy-2(5H)-furanone 1a to produce the corresponding products 3p–3t in good yields (81–86%). As anticipated, heteroaryl sulfonyl hydrazide is also suitable in the protocol with satisfactory result (3u). Importantly, benzyl sulfonyl hydrazide, as a kind of alkyl-based sulfonyl hydrazides can be successfully applied (3v). All the obtained products 3a–3v have been characterized by 1H and 13C NMR spectroscopic measurements including the single crystal X-ray structure of 3a,12 which can be seen in ESI† for details.
Reactivity of 3,4-dichloro-2(5H)-furanones
On the basis of above investigation on 3,4-dibromo-2(5H)-furanones, we further studied the reactivity of the lower active 3,4-dichloro-2(5H)-furanones with arylsulfonyl hydrazides. To our delight, good productivity for these 3,4-dichloro-2(5H)-furanones can also be gained by prolonging reaction time to 30–60 minutes, as shown in Table 3.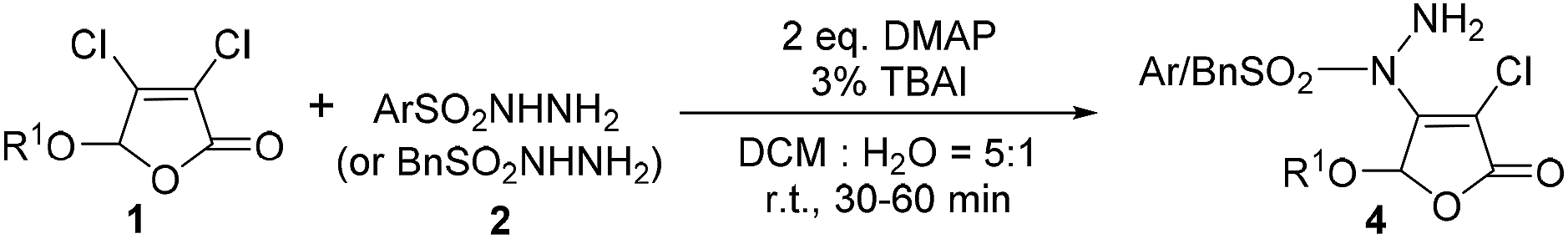
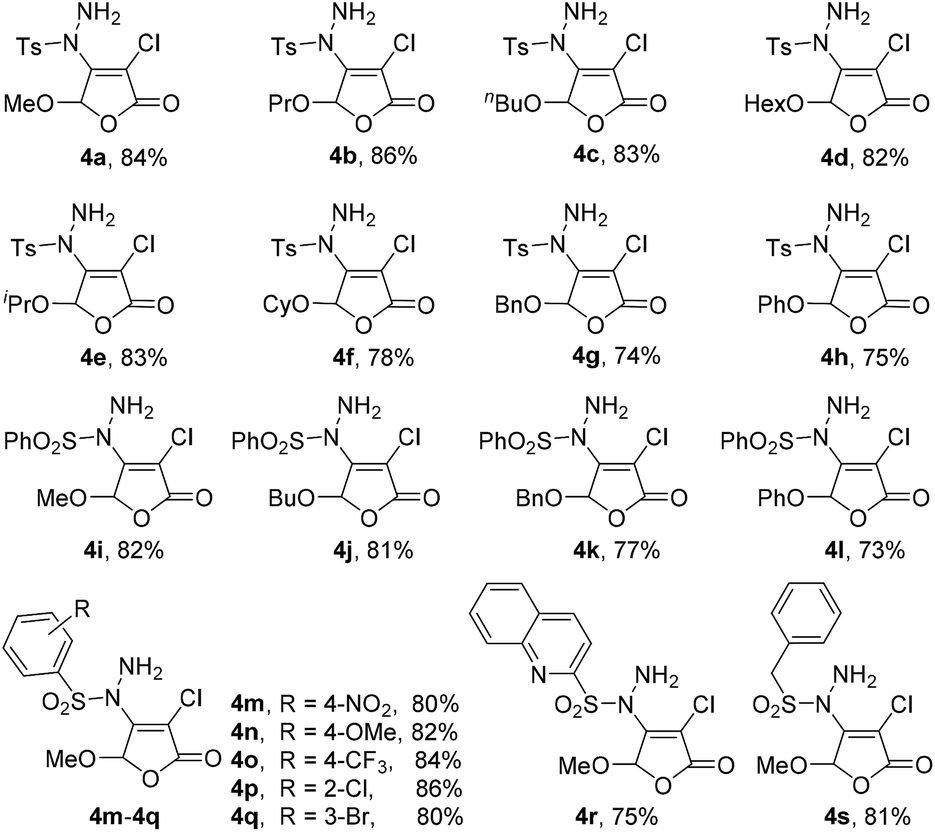
The experimental results reveal that the gratifying yields can be obtained from both substrates bearing alkoxy and aryloxy moieties as 5-substituted group. For the methoxy, propoxy, n-butoxy, HexO, isopropoxy, CyO, the yield is from 78% to 86% (4a–4f); for BnO (4g) and phenoxy (4h), the yields are 74% and 75%, respectively. Similarly, 5-substituted 3,4-dichloro-2(5H)-furanones can also be reacted with phenylsulfonyl hydrazide (2b) in yield of 73% to 82% (4i–4l). Especially, when arylsulfonyl hydrazides 2 containing different functional groups, such as alkoxy, halogen, nitro and CF3, are utilized to react with 3,4-dichloro-5-methoxy-2(5H)-furanone 1, the corresponding products can be acquired with satisfactory yields (4m–4q). It is worth noting that quinoline-2-sulfonohydrazide and benzyl sulfonyl hydrazide are also suitable reaction partners for this novel transformation (4r–4s).
Generally, the activity of the chlorinated substrates is lower than that of the brominated substrates.11a,11c However, the above results indicate that, comparing with 3,4-dibromo-2(5H)-furanones, the yields given by 3,4-dichloro-2(5H)-furanones are not reduced. The serial competitive experiments show that though 3,4-dibromo-2(5H)-furanones have higher activity and faster reaction rate to complete the reaction in a shorter time, the comparable yields can be obtained from the 3,4-dichloro-2(5H)-furanones after prolonging reaction time once the amount of sulfonyl hydrazide is enough also (see ESI† for details). These indicate that the activity of DMAP is so high that it is very easy for DMAP to form intermediate with 3,4-dihalo-2(5H)-furanones (which is involved in the reaction mechanism, and will be discussed in the following). Hence, the reactivity of both 3,4-dibromo-2(5H)-furanones and the 3,4-dichloro-2(5H)-furanones can give close reaction yields.
Regioselectivity of reaction
The protocol shows excellent regioselectivity in the reaction. As shown in Scheme 2, we did not find the primary nitrogen substituted product 3-I. Even increased the ratio of 1a:2a to 4:1, neither 3-I nor N,N′-disubstituted product 3-II was detected. This means the primary nitrogen in arylsulfonyl hydrazide does not participate in the reaction under our conditions. At the same time, the product 3-III cannot be observed under the standard conditions (even 1a:2a = 1:4). It indicates that the bromine at the C-3 position of 3,4-dibromo-2(5H)-furanone 1a is less active and does not participate in the reaction yet. Therefore, for both kinds of reactants, 3,4-dihalo-2(5H)-furanones and arylsulfonyl hydrazides, this transformation is highly regioselective.
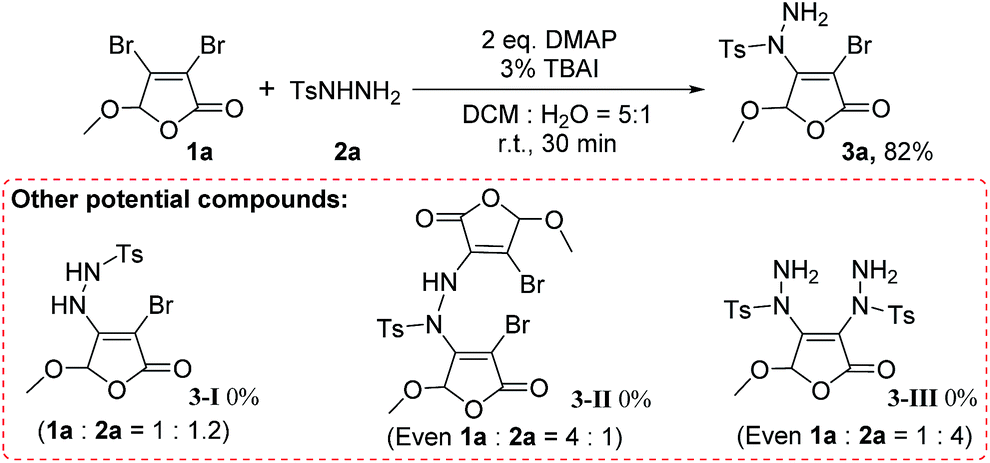 | ||
| Scheme 2 Regioselectivity of reaction. | ||
Plausible reaction mechanism
In order to gain more insight into our reaction mechanism, two control experiments were performed using 2.0 eq. radical inhibitor 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) or 2,6-di-tert-butyl-4-methylphenol (BHT) under the standard conditions (Scheme 3, eqn (1)). It can be found that the desired product 3a is still obtained in 78% and 82% yields, respectively. These observations demonstrate that a radical process should be excluded in this transformation.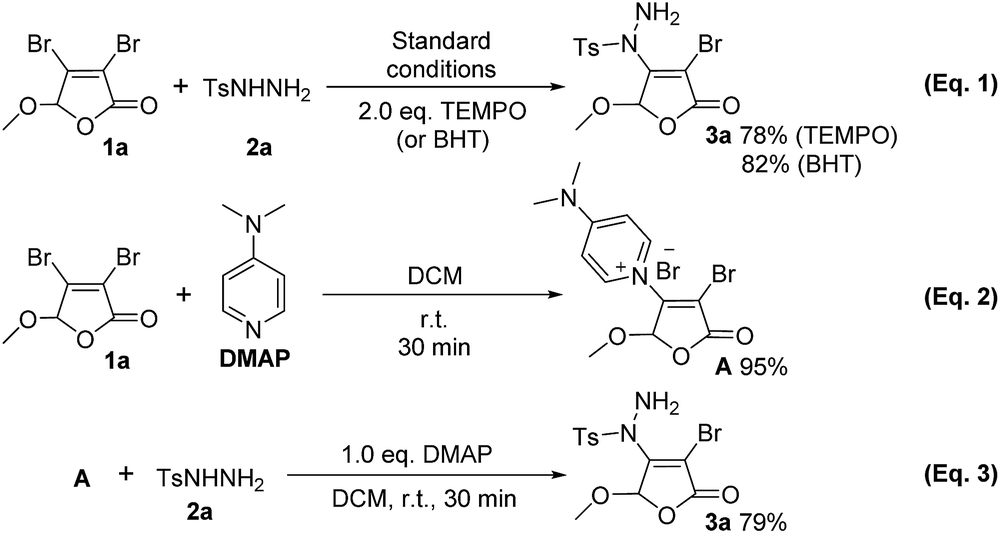 | ||
| Scheme 3 Control experiments. | ||
Organocatalysis plays an increasingly significant role in organic synthesis recently.13 DMAP has been widely used in organic reaction as catalyst14,15 or base.16 During our experiments, quaternary ammonium salt A was surprisingly found (its chemical structure was confirmed by X-ray,12 see ESI† for details). Furthermore, stirring 3,4-dibromo-5-methoxy-2(5H)-furanone 1a and DMAP at room temperature for 30 min, the isolated yield of compound A is 95% (Scheme 3, eqn (2)). Compound A can further react with p-toluene sulfohydrazide 2a to give the final product 3a in isolated yield of 79% (eqn (3)). These imply that compound A should be a key intermediate during this process.
On the basis of the above experimental results and previous reports,17 the secondary nitrogen of sulfonyl hydrazides is regarded as a nonbasic amine maintaining its nucleophilicity. Thus, a possible nucleophilic substitution process based on the promotion mechanism of DMAP15,18 is proposed as illustrated in Scheme 4.
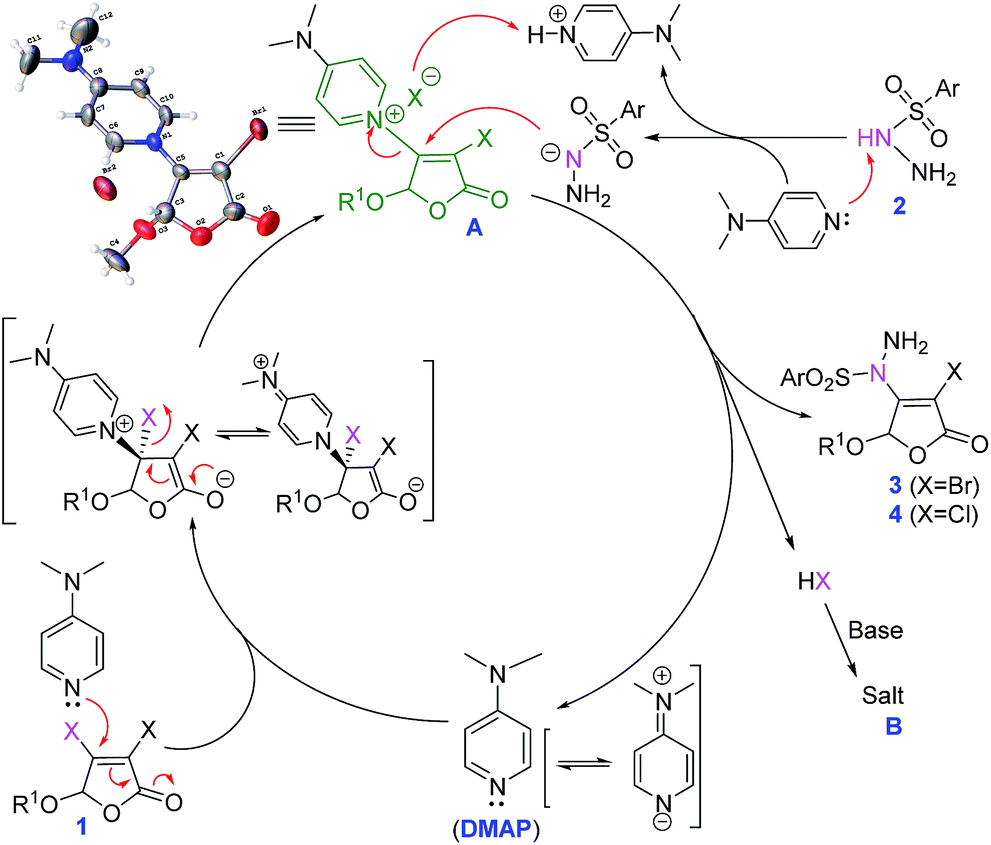 | ||
| Scheme 4 Plausible reaction mechanism. | ||
Firstly, 3,4-dihalo-2(5H)-furanone 1 reacts with DMAP to form the intermediate A by Michael addition–elimination. Then, A is attacked by sulfonyl hydrazides 2, followed by the regeneration of DMAP and the formation of product 3 (or 4). At the same time, HX as leaving compound is absorbed by DMAP to form salt B, which is transferred to the aqueous phase by TBAI as phase transfer catalyst, further promoting the conversion. Therefore, DMAP plays dual roles as suitable organic base and key catalyst.
Scale-up application
It is significant to apply such a metal-free catalytic, effective, and easy-to-operate method into practical use, such as large-scale or industrial preparation. Using 3,4-dibromo-5-methoxy-2(5H)-furanone 1a and p-toluene sulfohydrazide 2a as model substrates for large-scale reaction under the standard conditions, the results are shown in Table 4.
|
||||
|---|---|---|---|---|
| Entry | Amount of 1a | Amount of 2a | Product 3a (g) | Yielda (%) |
| a Isolated yield.b 1 h.c 2 h.d Amount of 1 (5.0 mmol) and 2 (6.0 mmol). | ||||
| 1 | 0.1349 g (0.5 mmol) | 0.1116 g (0.6 mmol) | 0.1544 | 82 |
| 2 | 1.3493 g (5 mmol) | 1.1163 g (6 mmol) | 1.5042 | 80 |
| 3 | 4.0783 g (15 mmol) | 3.3489 g (18 mmol) | 4.3995 | 78 |
| 4b | 8.1567 g (30 mmol) | 6.6978 g (36 mmol) | 8.4613 | 75 |
| 5c | 13.5945 g (50 mmol) | 11.1629 g (60 mmol) | 13.9123 | 74 |
 |
||||
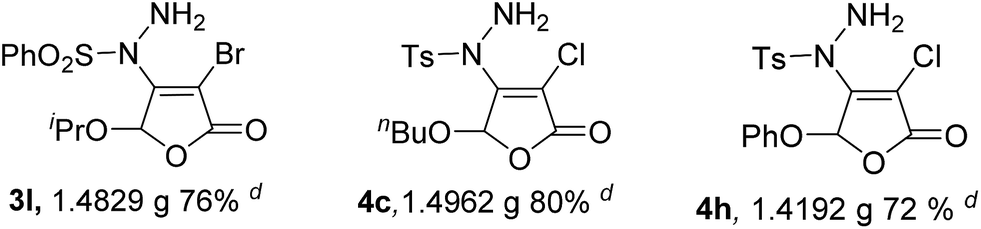
It can be found that with the increase of the amount of reactant 1a from 0.5 to 50 mmol stepwise, the yield of product 3a is gradually decreased. However, the yield of product 3a can still reach to 74% (13.9123 g, Entry 5) even when the amount of 1a is expanded by 100 times. Similarly, the other products, such as 3l, 4c and 4h, can also be obtained by large scale preparation with good yields.
The above results not only may provide a new approach for the development of 2(5H)-furanone derivatives with potentially effective bioactivities. In addition, due to the existence of many reactive functional groups in these sulfonyl hydrazide products, the concise synthesis can be further extended in the future. For example, the free amino group on the sulfonyl hydrazide moiety may react with aldehydes and acyl halides to construct new carbon–nitrogen double bond or amide bond, which will promote the accessibility to the polyfunctionalized drug molecules.
Conclusions
In summary, the C–N coupling reaction of Csp2–X compounds with arylsulfonyl hydrazides was achieved for the first time with good yields. The protocol shows excellent reaction site selectivity, which can realize coupling reaction of arylsulfonyl hydrazides at the secondary nitrogen atom, as well as good functional group tolerance for 2(5H)-furanone substrates, involving in a variety of complex structures, such as ester, ether, acetal units, lactone, aryl rings and C–X, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds. Importantly, the readily available Csp2–X compounds, including low-reactive Csp2–Cl compounds, are also suitable for this DMAP promoted method. Furthermore, the low cost of the reagents and wide range of substrates make this novel discovery as a powerful route for the synthesis of 2(5H)-furanone derivatives. Owing to the biological activity of sulfonyl hydrazides,19 the more derivatization synthesis and further biological activity assay are still in progress.
C bonds. Importantly, the readily available Csp2–X compounds, including low-reactive Csp2–Cl compounds, are also suitable for this DMAP promoted method. Furthermore, the low cost of the reagents and wide range of substrates make this novel discovery as a powerful route for the synthesis of 2(5H)-furanone derivatives. Owing to the biological activity of sulfonyl hydrazides,19 the more derivatization synthesis and further biological activity assay are still in progress.
Experimental
General information
Melting point was performed on an X-5 digital melting point apparatus without correcting. 1H, 13C and 19F NMR spectra were collected on a Varian AS600 spectrometer in CDCl3 or D2O using tetramethylsilane (TMS) as an internal standard. High-resolution mass spectra (HRMS) were obtained with a LCMS-IT-TOF mass spectrometer. Single-crystal X-ray analysis was obtained using Bruker APEX2 Smart CCD. TLC was performed by using commercially prepared 100–400 mesh silica gel plates (GF254) and visualization was effected at 254 nm. All reagents and solvents were purchased from commercial sources and used without further purification. Different 5-alkoxy (aryloxy) 3,4-dihalo-2(5H)-furanone intermediates were synthesized according to the literature procedure.11Experimental procedure for compounds 3a–3v
The mixture of 3,4-dibromo-2(5H)-furanone 1 (0.50 mmol), DMAP (2.0 eq.), TBAI (3 mol%) and sulfonyl hydrazide 2 (0.60 mmol) in DCM:H2O (3 mL, v:v = 5:1) was stirred at room temperature for 10–30 min. After the completion of the reaction, the reaction mixture was quenched with H2O (15 mL) and extracted with DCM (3 × 15 mL). Then, the organic layer was dried over anhydrous Na2SO4. After filtration and evaporation of the solvents under reduced pressure, the crude product was purified by column chromatography on silica gel to afford desired product.
Experimental procedure for compounds 4a–4s
The mixture of 3,4-dichloro-2(5H)-furanone 1 (0.50 mmol), DMAP (2.0 eq.), TBAI (3 mol%) and sulfonyl hydrazide 2 (0.60 mmol) in DCM:H2O (3 mL, v:v = 5:1) was stirred at room temperature for 30–60 min. After the completion of the reaction, the reaction mixture was quenched with H2O (15 mL) and extracted with DCM (3 × 15 mL). Then, the organic layer was dried over anhydrous Na2SO4. After filtration and evaporation of the solvents under reduced pressure, the crude product was purified by column chromatography on silica gel to afford desired product.
Experimental procedure for intermediate A
The mixture of 3,4-dibromo-5-methoxy-2(5H)-furanone 1 (0.50 mmol), and DMAP (2.0 eq.), in DCM (3 mL) was stirred at room temperature for 30 min. Once the reaction was complete, the evaporation of the solvents under reduced pressure gave a white solid. Then, the solid was recrystallized with DCM, and the pure intermediate A was obtained.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Guangdong Natural Science Foundation (No. 2014A030313429), National Natural Science Foundation of China (No. 21602085), Natural Science Foundation of Jiangsu Province (No. BK20160551), Guangzhou Science and Technology Project Scientific Special (General Items, No. 201607010251), Open Fund of the Key Laboratory of Functional Molecular Engineering of Guangdong Province in SCUT (2017kf01) and Guangdong Provincial Science and Technology Project (No. 2017A010103016) is greatly appreciated.References
- N. E. Campbell and G. M. Sammis, Angew. Chem., Int. Ed., 2014, 53, 6228 CrossRef CAS PubMed.
- (a) K. Namba, K. Takeuchi, Y. Kaihara, M. Oda, A. Nakayama, A. Nakayama, M. Yoshida and K. Tanino, Nat. Commun., 2015, 6, 8731 CrossRef PubMed; (b) Z.-X. Huang, C.-P. Wang and G.-B. Dong, Angew. Chem., Int. Ed., 2016, 55, 5299 CrossRef CAS PubMed; (c) Q. Sun, L.-G. Li, L.-Y. Liu, Q.-Q. Guan, Y. Yang, Z.-G. Zha and Z.-Y. Wang, Org. Lett., 2018, 20, 5592 CrossRef CAS PubMed.
- (a) F. L. Yang, X. T. Ma and S. K. Tian, Chem.–Eur. J., 2012, 18, 1582 CrossRef CAS PubMed; (b) O. Y. Yuen, C. M. So and F. Y. Kwong, RSC Adv., 2016, 6, 27584 RSC; (c) Y. H. Shang, Appl. Organomet. Chem., 2018, 32, e4484 CrossRef.
- (a) S.-Y. Li, X. Li, F. Yang and Y.-J. Wu, Org. Chem. Front., 2015, 2, 1076 RSC; (b) Y. Zhao, Y.-L. Lai, K.-S. Du, D.-Z. Lin and J.-M. Huang, J. Org. Chem., 2017, 82, 9655 CrossRef CAS PubMed; (c) Z.-H. Peng, X. Zheng, Y.-J. Zhang, D.-L. An and W.-R. Dong, Green Chem., 2018, 20, 1760 RSC; (d) X.-T. Zhu, Q.-L. Lu, X. Wang, T.-S. Zhang, W.-J. Hao, S.-J. Tu and B. Jiang, J. Org. Chem., 2018, 83, 9890 CrossRef CAS PubMed; (e) L.-X. Liu, K. Sun, L.-B. Su, J.-Y. Dong, L. Cheng, X. D. Zhu, C. T. Au, Y. B. Zhou and S. F. Yin, Org. Lett., 2018, 20, 4023 CrossRef CAS PubMed; (f) J.-B. Ni, Y. Xue, L.-P. Sun and A. Zhang, J. Org. Chem., 2018, 83, 4598 CrossRef CAS PubMed; (g) B. Yao, T. Miao, P. Li and L. Wang, Org. Lett., 2019, 21, 124 CrossRef CAS PubMed.
- (a) G. Y. Zhang, S. S. Lv, A. Shoberu and J. P. Zou, J. Org. Chem., 2017, 82, 9801 CrossRef CAS PubMed; (b) Q. Yu, Y. Yang, J.-P. Wan and Y. Liu, J. Org. Chem., 2018, 83, 11385 CrossRef CAS PubMed; (c) Y.-S. Bao, X.-Q. Yang, Q.-F. Zhou and F. Yang, Org. Lett., 2018, 20, 1966 CrossRef CAS PubMed; (d) Z. Yang, Y.-Q. Yan, A. Li, J.-S. Liao, L. Zhang, T. Yang and C.-S. Zhou, New J. Chem., 2018, 42, 14738 RSC; (e) Y.-L. Hou, L.-Y. Zhu, H. Hu, S.-W. Chen, Z.-F. Li, Y. J. Liu and P. Gong, New J. Chem., 2018, 42, 8752 RSC.
- (a) T. J. Donohoe, L. P. Fishlock, J. A. Basutto, J. F. Bower, P. A. Procopiou and A. L. Thompson, Chem. Commun., 2009, 3008 RSC; (b) T. J. Donohoe, J. F. Bower, J. A. Basutto, L. P. Fishlock, P. A. Procopiou and C. K. A. Callens, Tetrahedron, 2009, 65, 8969 CrossRef CAS.
- N. Singh, R. Singh, D. S. Raghuvanshi and K. N. Singh, Org. Lett., 2013, 15, 5874 CrossRef CAS PubMed.
- (a) X.-M. Wu and Y. Wang, New J. Chem., 2018, 42, 10953 RSC; (b) I. V. Ukrainets, A. A. Tkach, V. V. Kravtsova and S. V. Shishkina, Chem. Heterocycl. Compd., 2008, 44, 677 CrossRef CAS.
- (a) B. Mao, M. Fananas-Mastral and B. L. Feringa, Chem. Rev., 2017, 117, 10502 CrossRef CAS PubMed; (b) M. Yoritate, Y. Takahashi, H. Tajima, C. Ogihara, T. Yokoyama, Y. Soda, T. Oishi, T. Sato and N. Chida, J. Am. Chem. Soc., 2017, 139, 18386 CrossRef CAS PubMed; (c) W.-Y. Qi, J.-X. Zhao, W.-J. Wei, K. Gao and J.-M. Yue, J. Org. Chem., 2018, 83, 1041 CrossRef CAS PubMed; (d) S. Fujita, K. Nishikawa, T. Iwata, T. Tomiyama, H. Ikenaga, K. Matsumoto and M. Shindo, Chem.–Eur. J., 2018, 24, 1539 CrossRef CAS PubMed.
- (a) N. Grimblat, T. S. Kaufman and A. M. Sarotti, Org. Lett., 2016, 18, 6420 CrossRef CAS PubMed; (b) P. Yang, M. Yao, J. Li, Y. Li and A. Li, Angew. Chem., Int. Ed., 2016, 55, 6964 CrossRef CAS PubMed; (c) Q. A. Castillo, J. Triana, J. L. Eiroa, L. Calcul, E. Rivera, L. Wojtas, J. M. Padron, L. Boberieth, M. Keramane, E. Abel-Santos, L. A. Baez and E. A. Germosen, J. Nat. Prod., 2016, 79, 907 CrossRef CAS PubMed; (d) X. Li, P. H.-Y. Cheong and R. G. Carter, Angew. Chem., Int. Ed., 2017, 56, 1704 CrossRef CAS PubMed; (e) B. Srinivas, D. S. Reddy, N. A. Mallampudi and D. K. Mohapatra, Org. Lett., 2018, 20, 6910 CrossRef CAS PubMed; (f) M.-X. Wei, J. Zhang, F.-L. Ma, J.-Y. Yu, W. Luo and X.-Q. Li, Eur. J. Med. Chem., 2018, 155, 165 CrossRef CAS PubMed.
- (a) Y.-C. Wu, S.-H. Luo, W.-J. Mei, L. Cao, H.-Q. Wu and Z.-Y. Wang, Eur. J. Med. Chem., 2017, 139, 84 CrossRef CAS PubMed; (b) L. Cao, S.-H. Luo, H.-Q. Wu, L.-Q. Chen, K. Jiang, Z.-F. Hao and Z.-Y. Wang, Adv. Synth. Catal., 2017, 359, 2961 CrossRef CAS; (c) Y.-C. Wu, L. Cao, W.-J. Mei, H.-Q. Wu, S.-H. Luo, H.-Y. Zhan and Z.-Y. Wang, Chem. Biol. Drug Des., 2018, 92, 1232 CrossRef CAS PubMed; (d) L. Cao, J.-X. Li, H.-Q. Wu, K. Jiang, Z.-F. Hao, S.-H. Luo and Z.-Y. Wang, ACS Sustainable Chem. Eng., 2018, 6, 4147 CrossRef CAS; (e) S.-H. Luo, K. Yang, J.-Y. Lin, J.-J. Gao, X.-Y. Wu and Z.-Y. Wang, Org. Biomol. Chem., 2019, 17, 5138 RSC; (f) H.-Q. Wu, K. Yang, S.-H. Luo, X.-Y. Wu, N. Wang, S.-H. Chen and Z.-Y. Wang, Eur. J. Org. Chem., 2019 DOI:10.1002/ejoc.201900749.
- CCDC-1885760 (for 3a) and 1885761 (for A), contain the supplementary crystallographic data for this paper..
- (a) Y. Qin, L. H. Zhu and S. Z. Luo, Chem. Rev., 2017, 117, 9433 CrossRef CAS PubMed; (b) Y.-B. Wang and B. Tan, Acc. Chem. Res., 2018, 51, 534 CrossRef CAS PubMed; (c) T. Chanda and J. C.-G. Zhao, Adv. Synth. Catal., 2018, 360, 2 CrossRef CAS; (d) W.-M. Mao, W. Shi, J. Li, D.-Y. Su, X.-M. Wang, L. Zhang, L. Pan, X. Wu and H.-X. Wu, Angew. Chem., Int. Ed., 2019, 58, 1106 CrossRef CAS PubMed; (e) Q.-Q. Yang, X. Yin, X.-L. He, W. Du and Y.-C. Chen, ACS Catal., 2019, 9, 1258 CrossRef CAS.
- (a) A. Sakakura, K. Kawajiri, T. Ohkubo, Y. Kosugi and K. Ishihara, J. Am. Chem. Soc., 2007, 129, 14775 CrossRef CAS PubMed; (b) J. S. Quesnel, A. Fabrikant and B. A. Arndtsen, Chem. Sci., 2016, 7, 295 RSC; (c) Y. Ren, L.-G. Meng, T. Peng, L.-J. Zhu and L. Wang, Adv. Synth. Catal., 2018, 360, 3176 CrossRef CAS; (d) B. Wang, Y. Wei, Z. J. Li, L. Pan and Y. S. Li, ChemCatChem, 2018, 10, 5287 CrossRef CAS.
- L. W. Zeng, Z. C. Lai and S. L. Cui, J. Org. Chem., 2018, 83, 14834 CrossRef CAS PubMed.
- (a) L. Y. Zhu, P. P. Yan, L. Y. Zhang, Z. T. Chen, X. F. Zeng and G. F. Zhong, RSC Adv., 2017, 7, 51352 RSC; (b) J. Peng, J.-J. Xiang, H.-J. Wang, F.-B. Li, Y.-S. Huang, L. Liu, C. Y. Liu, A. M. Asiri and K. A. Alamry, J. Org. Chem., 2017, 82, 9751 CrossRef CAS PubMed; (c) X. J. Zhang, A. Y. Xia, H. Y. Chen and Y. H. Liu, Org. Lett., 2017, 19, 2118 CrossRef CAS PubMed; (d) H.-H. Kong, H.-L. Pan and M.-W. Ding, J. Org. Chem., 2018, 83, 12921 CrossRef CAS PubMed.
- (a) K. Namba, I. Shoji, M. Nishizawa and K. Tanino, Org. Lett., 2009, 11, 4970 CrossRef CAS PubMed; (b) T. J. Donohoe, L. P. Fishlock, J. A. Basutto, J. F. Bower, P. A. Procopiou and A. L. Thompson, Chem. Commun., 2009, 3008 RSC; (c) K. Namba, Y. Kaihara, H. Yamamoto, H. Imagawa, K. Tanino, R. M. Williams and M. Nishizawa, Chem.–Eur. J., 2009, 15, 6560 CrossRef CAS PubMed; (d) T. J. Martin, V. G. Vakhshori, Y. S. Tran and O. Kwon, Org. Lett., 2011, 13, 2586 CrossRef CAS PubMed.
- (a) P. L. Lagueux-Tremblay, A. Fabrikant and B. A. Arndtsen, ACS Catal., 2018, 8, 5350 CrossRef CAS; (b) A. Verma, S. Jana, C. D. Prasad, A. Yadav and S. Kumar, Chem. Commun., 2016, 52, 4179 RSC; (c) A. Polley, G. Bairy, P. Das and R. Jana, Adv. Synth. Catal., 2018, 360, 4161 CrossRef CAS.
- (a) S. Riaz, I. U. Khan, M. Bajda, M. Ashraf, Qurat-ul-Ain, A. Shaukat, T. U. Rehman, S. Mutahir, S. Hussain, G. Mustafa and M. Yar, Bioorg. Chem., 2015, 63, 64 CrossRef CAS PubMed; (b) A. Kamal, M. N. A. Khan, K. S. Reddy and K. Rohini, Bioorg. Med. Chem., 2007, 15, 1004 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data and 1H, 13C spectra for all compounds and data of single-crystal X-ray analysis. CCDC 1885760 and 1885761. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra03403j |
| This journal is © The Royal Society of Chemistry 2019 |