
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA spherical poly(acrylic acid) brush–enzyme block with high catalytic capacity for signal amplification in digital biological assays†
Yibei Wanga,
Zehang Gaobcd,
Jingwei Yia,
Hongbo Zhoub,
Xiaoxia Fanga,
Hong Xu *a,
Jianlong Zhaob and
Hongchen Gu*a
*a,
Jianlong Zhaob and
Hongchen Gu*a
aSchool of Biomedical Engineering, Med-X Research Institute, Shanghai Jiao Tong University, Shanghai 200030, PR China. E-mail: xuhong@sjtu.edu.cn; hcgu@sjtu.edu.cn
bShanghai Institute of Microsystem and Information Technology, Chinese Academy Science, Shanghai 200030, PR China
cSchool of Information Science and Technology, ShanghaiTech University, Shanghai 201210, China
dUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 30th July 2019
Abstract
Ultrasensitive determination of some ultra-low abundance biological molecules closely related to diseases is currently a wide concern and urgent issue to be addressed. Here, a spherical poly(acrylic acid)–alkaline phosphatase (SP–AKP) signal amplification block using spherical poly(acrylic acid) brush nanoparticles (SP) as the immobilized carriers was designed and synthesized optimally first. The results show that a single SP–AKP with high enzyme binding capacity and high catalytic ability (up to about 4800 effective free AKP per SP–AKP) has much greater fluorescence signal amplification ability than a single free AKP or SiO2–COOH–AKP. Then, a droplet generation microfluidic chip was prepared successfully, and the SP–AKP was loaded and confined in a 14 pL droplet by adjusting its concentration to ensure at most one SP–AKP was encapsulated in each droplet according to Poisson's theory. Finally, the fluorescence signals produced by 4-methylumbelliferyl phosphate (4-MUP) catalyzed via SP–AKP within 6 min were sufficient to be detected by a fluorescence microscope. Thus, the digital signal distribution of “1/0” (signal/background) was obtained, making this SP–AKP signal amplification block a promising enzyme label for potential high sensitivity digital biological detection applications.
Introduction
The enzyme-linked immunosorbent assay (ELISA) is a biological immunoassay technique widely used in clinical analysis and laboratory medicine.1 It utilizes the specific binding of antigen–antibody and the high catalytic ability of enzymes to determine the target concentration rapidly by means of absorbance, fluorescence and chemiluminescence, etc. In recent years, it has been used in the determination of proteins, antigens, antibodies, such as Human Chorionic Gonadotropin (HCG),2 insulin,3 thyrotropin,4 hepatitis B surface antigen (anti-HBV),5 Escherichia coli enterotoxin,6 HIV antibody (anti-HIV),7 etc.However, there are many kinds of biomarkers with extremely low abundance and great potential for clinical applications cannot be detected by current technology. They are like the part of a huge iceberg that sinks under the sea (usually making up more than 80 percent8 of the total volume), so few of them are available for us to know and utilize. For example, some patients with severe neurodegenerative diseases (such as Alzheimer's disease, Parkinson's disease, etc.) have specific “proteins” in their brains (like Aβ1-42, protein tau etc.9–11), due to the Blood–Brain Barrier (BBB), these specific “markers” find it difficult to penetrate the dense BBB into the blood, making their concentrations in the blood as low as femtogram or sub-picograms per milliliter. At present, the most sensitive ELISA detection technology is only at a level of picograms to tens of picograms per milliliter, which makes such “markers” with important clinical diagnostic and prognostic value unable to be applied in clinic.
In addition, digital detection as a single molecule biological detection method with the characteristics of small reaction volume and high throughput is emerging, which uses the microfluidic technology to encapsulate a single immune complex into a large quantity of separate microcavities9,12,13 or microdroplets14–18 for independent signals. Thus, absolute quantitative analysis of the biological samples can be achieved, especially suitable for the detection of ultra-low abundance target biomolecules. Compared with the microarray-based detection, microdroplet-based digital detection is more likely to have low detection limit and wide linear range owing to its infinite number of droplets, becoming a more promising digital detection method. However, the size of the droplet is relatively large (tens of microns) and the fluorescence signal from a single enzyme-catalysed droplet is difficult to detect. Therefore, it has become an urgent issue to improve the loading capacity and total catalytic activity of labelled enzyme in microdroplet immunoassay.
For that matter, efforts have been made by some researchers in adopting various nanoparticle materials as the immobilized carriers of enzymes to enhance the detection sensitivity by amplifying the detection signals with a large quantity of enzymes, such as gold nanoparticles (AuNPs),19 polymer particles (PS microspheres),20 mesoporous silica nanoparticles (MSN),21 magnetic particles.22 However, they have common problems of limited binding capacity and greatly decreased activity of immobilized enzymes. A spherical polyelectrolyte brush is a new kind of material formed when one end of linear polyelectrolyte chains densely attaches to spherical carriers such as nanoparticles.23–26 It has high protein binding capacity, and maintains good biological activity due to the unique three-dimensional space structure formed by soft high-density polyelectrolyte chains grafted on surface.27–29
In this article, we established an effective fluorescence signal amplification enzyme label block based on the spherical poly(acrylic acid) brush (SP) for mircodroplet digital detection. Scheme 1 shows the procedures of the protein immobilization and digital detection. First, alkaline phosphatase (AKP) was covalently immobilized into SP via a “chemical conjugation after electrostatic entrapment” (CCEE) method30 to obtain the signal amplification block SP–AKP, and it reacted with the substrate 4-MUP to produce fluorescein 4-methylumbelliferone (4-MU) which emitted blue fluorescence excited by laser at 360 nm. Second, water-in-oil droplets with a diameter of 30 μm were generated by flow focusing method (two oil phases, one substrate phase and one SP–AKP phase). Moreover, by controlling the concentration of the obtained SP–AKP, each droplet contained at most one SP–AKP particle. After reaction, the digital signals “1” and “0” (“1” for bright droplets that contain the SP–AKP, “0” for dark empty droplets) were observed by a fluorescence microscope and recorded by a CCD camera, and the concentration of the SP–AKP was obtained through quantitative statistics and Poisson distribution formula. In addition, we also made comparative analysis of its catalytic activity to study the signal amplification ability of SP–AKP.
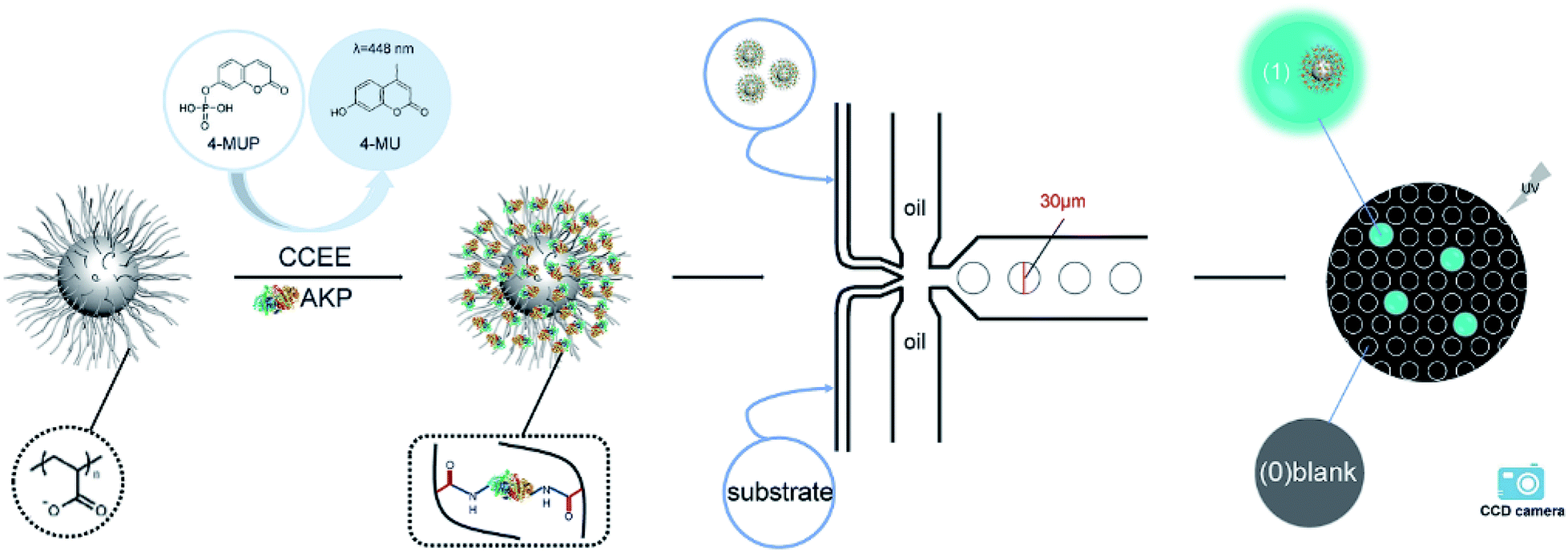 | ||
| Scheme 1 Illustration of the protein immobilization and digital detection. | ||
Experimental section
Materials
Alkaline phosphatase (AKP, from bovine intestinal mucosa) and 4-methylumbelliferyl phosphate (4-MUP) was purchased from Sigma-Aldrich. EDC (N-(3-dimethyl-aminopropyl)-N′-ethylcarbodiimide hydro-chloride) was obtained from Aladdin Reagent (Shanghai) Co., Ltd. NHS (N-hydroxysuccinimide) was obtained from Thermo Scientific. All other reagents used in this work were the products of China National Medicines Group Shanghai Chemical Reagents Company.Tris(hydroxymethyl)aminomethane buffer (Tris–HCl, 0.1 M, pH = 9.0) and 2-(N-morpholino) ethanesulfonic acid buffer (MEST, 10 mM, pH = 5.0, containing 0.05 wt% Tween-20) were used in the protein binding experiments.
SP-300, the SP with high grafting density (σ = 0.42 nm−2) of acrylic acid with a polymerization degree of 300 onto silica nanoparticles with a diameter of 116 nm, and conventional carboxylated SiO2 nanoparticles (SiO2–COOH) with a similar size were both used as immobilized enzyme carriers to evaluate their signal amplification abilities. The synthesis and characterization of SP-300 was done as described recently.31 The preparation of SiO2–COOH was conducted according to the literature.32 Key structural parameters are listed and defined in Table 1 as follows.
| db (nm) | Dc (nm) | Ld (nm) | Ne (μmol g−1) | |
|---|---|---|---|---|
| a SiO2–COOH with the same size as SP-300 was selected as an example.b Diameter of SiO2 core was determined by dynamic light scattering (DLS).c Diameter of SiO2 nanoparticles was determined by dynamic light scattering (DLS).d Lengths of group chains, L = (D − d)/2.e The amount of carboxyl was measured by conductometric titration. | ||||
| SP-300 | 116 | 300 | 92 | 4.425 |
| SiO2–COOH | 121 | 211 | — | 2.513 |
The TEM images of SP-300 and SP–AKP were obtained by a 120 kV transmission electron microscope. The size distributions, PDI and zeta potential of SP-300 and SP–AKP were measured by Zetasizer Nano ZS via dynamic light scattering (DLS) in water.
Protein immobilization
As conventional methods of coupling AKP on SiO2–COOH, 400 μL of 50 mg ml−1 of NHS/EDC solution was added into each reaction system including 0.5 mg SiO2–COOH nanoparticles to activate carboxyl to form NHS ester intermediates. Then 400 μl AKP with different concentration gradients (3, 6, 9, 12, 15 and 18 mg ml−1) were added to the reaction system respectively. The amino groups on the protein AKP attacked NHS intermediates to form amide bonds.CCEE method was adopted to immobilize AKP into the SP, 400 μl AKP with different concentration gradients (3, 6, 9, 12, 15 and 18 mg ml−1) were first entrapped into 0.5 mg SP in each MEST reaction system via electrostatic interaction respectively. Then, the particle–enzyme complexes were washed two times by MEST before the conjugation with 300 μl of 0.5 mM EDC in MEST to react at room temperature for 2 h on a rotary mixer. After conjugation, the particle–enzyme complexes were re-dispersed in 500 μl Tris–HCl.
The protein concentration was quantified by BCA protein quantification method. The protein concentrations in the original protein solution, supernatant, and washing solution in each step were measured to obtain the binding capacity on the particles via subtraction method. The activity of free AKP, SP–AKP and SiO2–COOH–AKP were acquired by microplate reader (SpectraMax_i3). The relative activity of the enzyme was determined by kinetic fluorometry, a fluorescent value was detected every 30 seconds. The relative activity was expressed by the slope of the catalytic curve (RFU s−1).
Droplets generation and signal detection
The droplets generation and detection platform were constructed, and divided into generation device and detection system. The generation device included PDMS chip, injection tubes/needles, vacuum pump and nitrogen pump, and the detection system included fluorescence microscope, mercury lamp, filter, high-speed monochrome CCD camera.The droplets were generated by the flow focusing method (shown in Fig. S1a and b†). AKP/SP–AKP and substrate entered into the two dispersion phases respectively, and the continuous phase (mineral oil and surfactant EM90, Triton X-100) was sheared from both sides to obtain water-in-oil droplets. AKP/SP–AKP reacted with the substrate in droplets at room temperature.
The detection of droplets was mainly achieved by optical photography. Blue fluorescence signal of 448 nm was generated under near-ultraviolet light of a 130 W short-arc mercury lamp with 25% light power and a 380–400 nm cut-off filter. Fluorescence images were captured by the high-speed monochrome CCD camera under the fluorescence microscope in a very short time (exposure time of 100 ms and bandpass filter of 424–469 nm).
Results and discussion
Covalent protein immobilization on SP
Due to the large numbers of carboxyl functional groups on poly(acrylic acid) (PAA) chains, traditional NHS/EDC progress33 seems to be a good choice for protein immobilizations in SP. But previous experiments have shown several drawbacks of the NHS/EDC process used in 3D materials like SP listed as follows: (I) the inner sites of the SP could not be fully utilized due to the steric hindrance. Therefore, the protein binding capacity in covalent immobilization is only less than simple electrostatic adsorption. (II) it is possible that EDC/NHS activation reaction lead to the collapse of polymer chains from stretching state, which is because that the negative charge of carboxyl ionization on PAA chains would turn into a neutral NHS state, losing the advantage of three-dimensional structure of the SP. (III) NHS ester hydrolysis might bring some side reactions, resulting in a complex and uncontrollable coupling process.30To solve above problems, a tailored–designed bioconjugation method entitled “chemical conjugation after electrostatic entrapment” (CCEE) method was developed previously by our group30 and was used in this study. First, the SP was synthesized by RAFT polymerization with a 116 nm SiO2 core and the chain length of the PAA was 92 nm. As a comparison, conventional carboxylated SiO2 nanoparticles (SiO2–COOH) with a similar core size were synthesized via modified Stöber method32 and the surface carboxyl groups were obtained by grafting PAA polymers with average molecular weight of 5000 onto the surface of SiO2 nanoparticles. Key structural parameters of SP and SiO2–COOH are listed in Table 1. As is reported, electrostatic adsorption is the main factor affecting the binding capacity during the immobilization process,30 and AKP could be entrapped into the 3D structure of SP in MEST buffer of pH 5.0 via electrostatic interaction (the isoelectric point of SP and AKP are 4.0 and 5.7 respectively). Then, the obtained particle–protein intermediate complexes were separated by centrifugal and dispersed in MEST by ultrasonic to remove the excess unadsorbed proteins. Next, EDC was added to form amide bonds by covalently conjugating carboxyl groups on SP with residual amino groups of AKP enzyme. Finally, the SP–enzyme complexes (SP–AKP) were washed and dispersed in Tris–HCl buffer for further application.
Fig. 1a and b shows the TEM images of SP and the SP–AKP after covalent immobilization of AKP by CCEE method. The SiO2 core presented dark black can be observed clearly around with light grey PAA chains shell. When undergoing TEM characterization, the SP-300/SP–AKP particles were on copper grid in a dry state, and PAA chains grafted onto the silica nanoparticles collapsed into polymer shell, forming a certain gap between the SP particles; after coupling with AKP, the thickness of polymer shell around the SiO2 core increased distinctly, and the distance between the SP particles widened (shown in Fig. 1b and inset images). This is due to a large number of enzyme molecules acting as cross-linking agents combined onto the PAA chains, preventing the chains from collapsing and exhibiting an increase in thickness in the dry state. It proves that the enzyme molecules were successfully immobilized into the inner parts of the SP, making full use of the sites on the PAA chains.
 | ||
| Fig. 1 TEM images of SP (a) before and (b) after covalent immobilization with AKP by CCEE (with a close-up of one particle at top left). (c) The corresponding DLS size distributions of particles SP-300 and SP–AKP. (d) Photos of the SP–AKP in aqueous suspension (left), after centrifugation and the fluorescent products (right). | ||
Fig. 1c shows the size distributions of SP-300 and SP–AKP particles by dynamic light scattering (DLS). They are approximately 300 nm in size with very low polydispersity index (PDI) in pure water (PDI < 0.08 and PDI < 0.02 respectively), indicating that the as-synthesized SP-300 and SP–AKP nanoparticles have monodisperse size with less aggregations. In the process of CCEE method, electrostatic adsorption is the dominant reaction,30 the flexible chain segments can constantly adjust the binding positions of enzyme molecules to make full use of the sites and achieve efficient binding. In addition, Fig. 1d shows the state of SP–AKP in Tris–HCl buffer (pH 9.0) with excellent dispersion stability, which could be easily separated from the buffer after centrifugation for 6 min.
Catalytic effect of SP–AKP
After adding substrate 4-MUP into SP–AKP (1 mg ml−1), ultra-bright blue fluorescence could be observed by the naked when excited under 360 nm (shown in Fig. 1d), demonstrating that the AKP molecules on SP maintain high catalytic capacity. According to the previous researches, the enzymes immobilized on the carriers would be partially inactivated due to deformation. In order to study the catalytic effect, we measured the catalytic activity of AKP and SP–AKP at different 4-MUP concentrations and studied the variation of catalytic rate with varying substrate concentration, which allowed us to determine the kinetic parameters for respective enzymes.As shown in Fig. 2a and b, the catalytic rates of free AKP and SP–AKP first both increase linearly with the increasing substrate concentration, then tend to balance and reach a relatively stable rate. By fitting the data points with the Michaelis–Menten equation, the Michaelis–Menten constant (Km) of free AKP and SP–AKP are determined to be 10.96 μM and 16.84 μM respectively. Since their Km values are relatively close, the substrate affinity of SP–AKP does not decline too much compared with free AKP. In addition, the turnover number (Kcat = Vmax/[E], [E] is the concentration of AKP or SP–AKP) is 6.91 s−1 for AKP and 2.11 s−1 for SP–AKP respectively, thus, we can draw a conclusion that the relative activity of SP–AKP decrease to about 30.6% of free AKP.
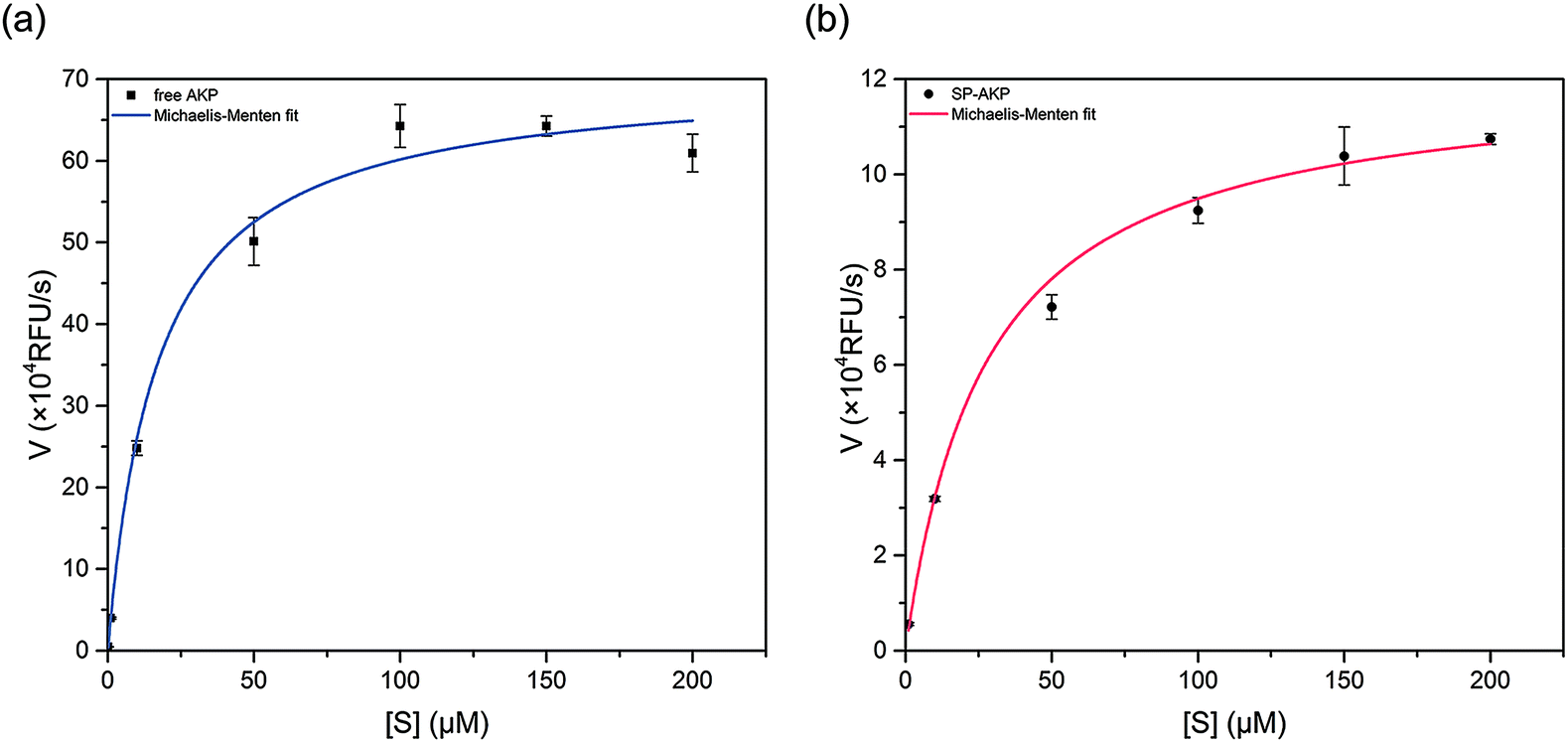 | ||
| Fig. 2 Dependence of the catalytic rate of (a) free AKP and (b) SP–AKP on 4-MUP concentration. The activity was measured in the presence of 0.1 M Tris–HCl and 0.5 mM MgCl2 at pH 9.0. Data points were fitted to the Michaelis–Menten equation to give a Km of free AKP and the SP–AKP of 10.96 μM and 16.84 μM respectively. | ||
Establishment of signal amplification block based on SP–AKP
Since AKP on SP maintains relatively high catalytic activity and substrate affinity, the catalytic capacity of SP–AKP was studied in depth to prove the feasibility of the established fluorescent signal amplification enzyme label block. In the meantime, to further verify the signal amplification effect, we compared the SP–AKP with conventional SiO2–COOH–AKP in a similar size in terms of protein binding capacity and relative activity. As shown in Fig. 3, the binding capacity of SP–AKP increases with the increasing added amount of AKP, and it can be well controlled. Besides, the relative total activity of SP–AKP and SiO2–COOH–AKP both increases with the increasing binding capacity, and the maximum relative total activity are 4.90 × 105 RFU s−1, 4.57 × 103 RFU s−1, respectively. Fig. 4b shows SP and the SiO2–COOH can be combined with up to about 1800 μg AKP per mg SP (∼16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 AKP per SP) and 800 μg AKP per mg SiO2–COOH (∼6000 AKP per nanoparticle) respectively, on these conditions, the relative total activity of SiO2–COOH–AKP is only 1% of SP–AKP. Furthermore, at the similar binding amount (794.24 μg mg−1 SP and 779.57 μg mg−1 SiO2–COOH), the relative total activity of SiO2–COOH–AKP is about 50 times lower than SP–AKP. In conclusion, one single SP can combine a large number of AKP (up to about 4800 effective free AKP) and makes the total activity of a single SP–AKP much higher than that of a single AKP, which means this SP–AKP has great potential to be a signal amplification enzyme label block.
000 AKP per SP) and 800 μg AKP per mg SiO2–COOH (∼6000 AKP per nanoparticle) respectively, on these conditions, the relative total activity of SiO2–COOH–AKP is only 1% of SP–AKP. Furthermore, at the similar binding amount (794.24 μg mg−1 SP and 779.57 μg mg−1 SiO2–COOH), the relative total activity of SiO2–COOH–AKP is about 50 times lower than SP–AKP. In conclusion, one single SP can combine a large number of AKP (up to about 4800 effective free AKP) and makes the total activity of a single SP–AKP much higher than that of a single AKP, which means this SP–AKP has great potential to be a signal amplification enzyme label block.
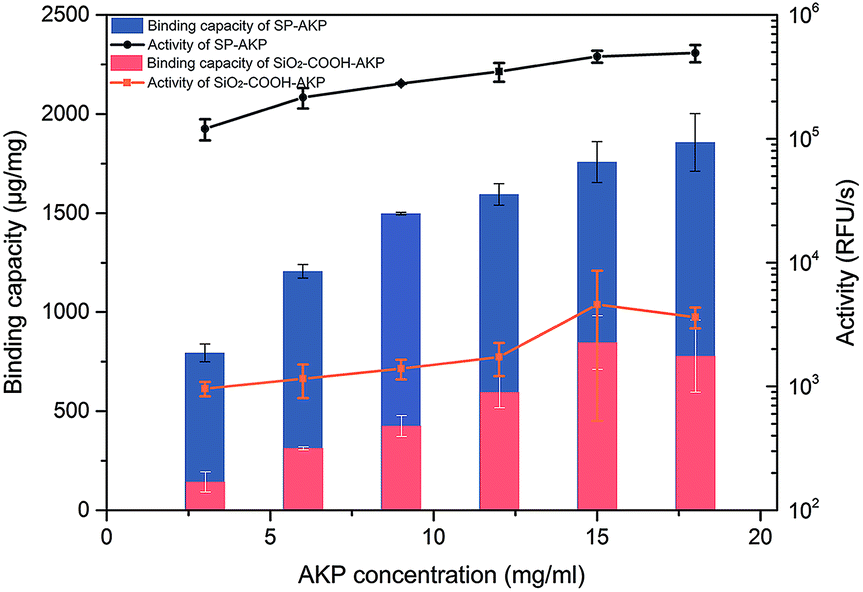 | ||
| Fig. 3 Binding capacity and activity of SP–AKP and SiO2–COOH–AKP and in dependence of concentration of AKP. Histogram shows binding capacity and line chart shows activity. | ||
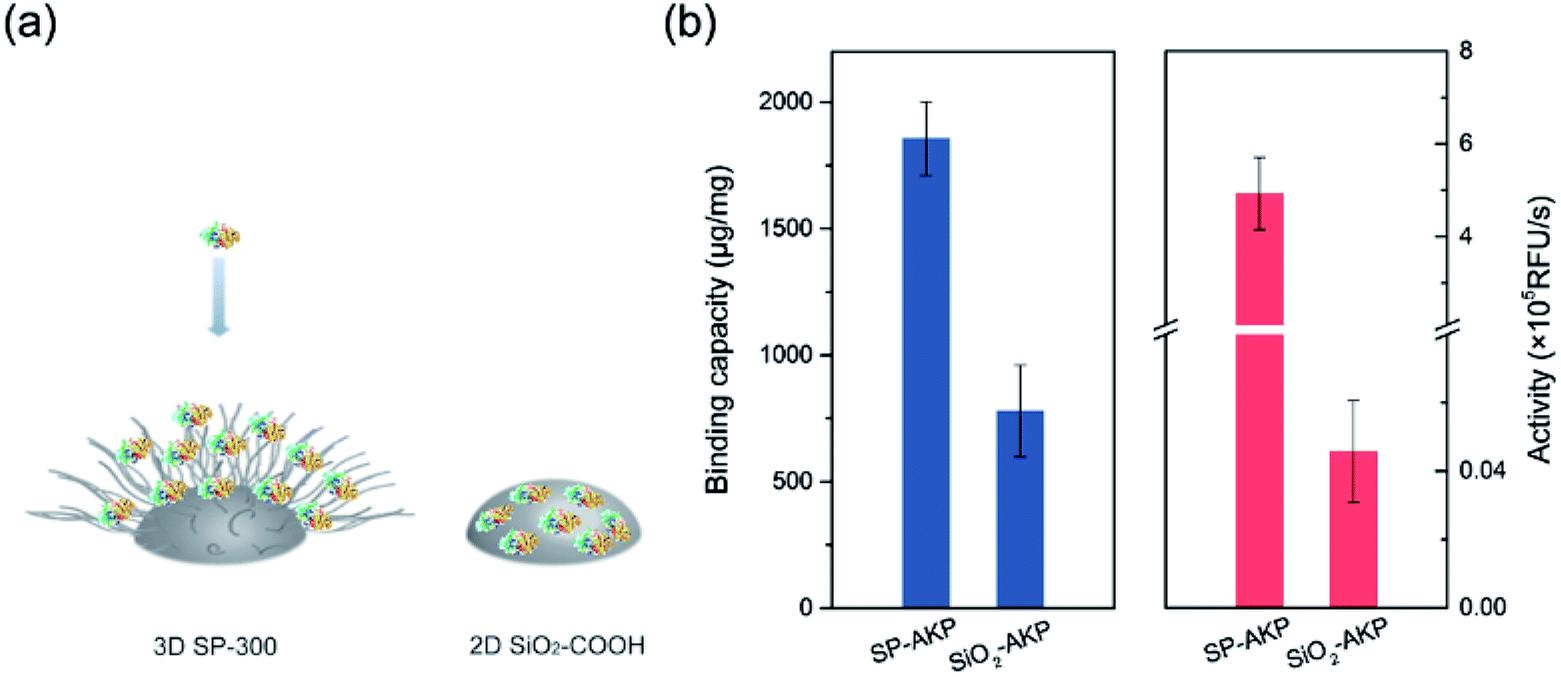 | ||
| Fig. 4 (a) Comparison on protein immobilization mechanisms between 3D SP-300 and 2D SiO2–COOH. (b) Comparison of SP–AKP and SiO2–COOH–AKP in terms of protein binding capacity and activity. | ||
The above results prove that the SP can immobilize a large number of enzymes and maintain higher enzyme activity than the traditional silica nanoparticles with hard surface, which endows the SP–AKP with a strong fluorescence signal amplification ability. This is because the hard surface structure of the conventional carboxylated SiO2 particles (SiO2–COOH) limits the binding capacity of the enzyme to only one layer of enzymes on its surface, and the enzymes are prone to deformation. On the contrary, SP has a special 3D spatial structure, as well as soft long PAA chains with abundant carboxyl groups (shown in Fig. 4a), and thus the enzymes can maintain certain natural structures and catalytic activity. Circular dichroism (CD) of free AKP and SP–AKP further indicates the immobilized enzymes in SP maintains good protein structures (shown in Fig. S2†).
In general, fluorescence products catalysed by SP–AKP are usually dispersed in the solution. However, conventional bioassays that uses enzyme labels, such as ELISA, are typically reacted in large volume wells (i.e., ∼100 μL), which means millions of enzymes at femto molar concentration can be detected through existing detection techniques. Therefore, when the abundance of biomolecules in the reaction system is extremely low, it is difficult to be detected in final system. In recent years, microdroplets produced by microfluidic chip technology characterized by small volume, fast production speed, non-diffusion of samples, cross-contamination avoiding between samples, stable reaction conditions and rapid mixing have been widely used in the biomedical fields such as DNA, protein, enzyme detection and drug delivery. Thanks to microfluidic technology, the reaction is confined to picoliter-volume droplets, and the fluorescent signals produced by a single target in a short time are possible to be detected by a fluorescence microscope as the concentration of fluorescent molecules can be much higher.34
Therefore, we constructed a microdroplet generation and detection platform suitable for the SP–AKP signal amplification block, which produced a large quantity of uniform and stable water-in-oil microdroplets. Single signal amplification block based on single SP–AKP nanoparticle could be separated from each other to form an independent microreactor to realize digital fluorescent signal expression. Thus, microdroplets were produced by flow focusing method. The substrate and enzyme entered from two different water dispersed phase channels at the same time, while the continuous oil phase (mineral oil and surfactant) turned around the cross channel, and squeezed two water dispersed phase to form droplets with the diameter of 30 μm. By keeping the concentration of substrate equivalent, the average number of SP–AKP in a single droplet can be changed by adjusting the concentration of SP–AKP. According to the Poisson distribution9,34 (eqn (1), P(X): the probability that there are exactly X SP–AKP nanoparticles per droplet, λ: the average number of SP–AKP nanoparticles per droplet), at very low concentration, most droplets have no SP–AKP, a few droplets contain one SP–AKP, and very few ones contain more than one SP–AKP.
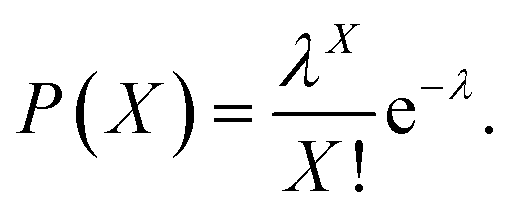 | (1) |
Fig. 5 depicts the fluorescence images of free AKP (λ = 0.1, the average number of free AKP molecules per droplet) and SP–AKP (λ = 0.1 in theory, the average number of SP–AKP particles per droplet). There are obvious and stable digital signal “1/0” in SP–AKP system, while no such signals in free AKP system even if the catalytic time was extended to 1 h (the grey value of all droplets in Fig. 5a are close to the background), and the signal-to-background ratio of a single SP–AKP is much higher than a single enzyme. It means that the fluorescence signals generated by a single enzyme could not be detected, while the SP–AKP, similarly as a single labelled building block, can produce sufficient fluorescence molecules in a 14 pL droplet to over the detection limit of CCD we used here in a very short time due to there are up to about 16000 AKP molecules inside one SP–AKP (corresponding to 4800 effective free AKP), demonstrating the necessity of signal amplification in droplet-based digital bioassay.
 | ||
| Fig. 5 The fluorescence images of (a) free AKP (λ = 0.1) and (b) the SP–AKP (λ = 0.1) in 14 pL droplets after a 6 min incubation with 0.25 mM 4-MUP. The exposure time was 100 ms. The brightness and contrast of two images were automatically adjusted by ImageJ software to clearly distinguish all droplets. | ||
Moreover, we analysed the fluorescence intensity of signal “1/0” droplets, and statistics are shown in Fig. S3 and Table S1.† It is verified to satisfy the Poisson distribution. The total number of droplets is 6096, P(X = 0) = 91.04%, and the actual λ is 0.0939 calculated by Poisson distribution formula which is almost consistent with theoretical feeding. Thus, according to this discrete probability distribution, we can obtain λ through P(X = 0), and then the concentration of SP–AKP can be obtained according to the volume of droplet.
Conclusions
In summary, a sensitive immunoassay signal amplification system (SP–AKP) based on the spherical poly(acrylic acid) brush (SP) has been developed by a CCEE method, with the characteristics of uniform, stable, good dispersion and easy separation. The results show that SP have higher enzyme binding capacity than traditional nanoparticles with hard surface, such as SiO2–COOH, and the relative total activity of SP–AKP is 100 times higher than SiO2–COOH–AKP. And then we built a microdroplet platform to separate the particle–enzyme block from each other, and obtained a considerable number of uniform droplets. The fluorescent signal distribution of “1/0” expressed in droplets further verifies the single SP–AKP with high catalytic capacity and remarkable fluorescence signal amplification ability in comparison to the single free AKP and SiO2–COOH–AKP. These results indicate that the established SP–AKP signal amplification block is a promising label material for ultra-sensitive digital biological detection.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is supported by the National Natural Science Foundation of China (Grant No. 21874091), and SJTU funding (Grant No. 17X10090003).Notes and references
- R. M. Lequin, Clin. Chem., 2005, 51, 2415–2418 CrossRef CAS PubMed.
- H. T. Kuo, J. Z. Yeh, C. M. Jiang and M. C. Wu, J. Immunol. Methods, 2012, 381, 32–40 CrossRef CAS PubMed.
- M. J. Macdonald and J. P. Gapinski, Metab., Clin. Exp., 1989, 38, 450–452 CrossRef CAS PubMed.
- A. E. Ohwovoriole and T. J. Wilkin, J. Immunol. Methods, 1988, 115, 99–104 CrossRef CAS PubMed.
- O. E. Tsitsilonis, A. Thrasyvoulides, A. Balafas, J. F. Voutsas, M. Papamichail and P. Lymberi, J. Pharm. Biomed. Anal., 2004, 34, 811–822 CrossRef CAS PubMed.
- S. Shan, D. Liu, Q. Guo, S. Wu, R. Chen, K. Luo, L. Hu, Y. Xiong and W. Lai, J. Dairy Sci., 2016, 99, 7025–7032 CrossRef CAS.
- G. Filice, L. Soldini, P. Orsolini, E. Razzini, R. Gulminetti, D. Campisi, L. Chiapparoli, E. Cattaneo and G. Achilli, Microbiologica, 1991, 14, 185–194 CrossRef CAS.
- Erenna SMC, https://www.instrument.com.cn/netshow/SH101341/down_826643.htm, 2017.
- D. M. Rissin, D. H. Wilson and D. C. Duffy, The Immunoassay Handbook, 4th edn, 2013, vol. 1, ch. 4, pp. 223–242 Search PubMed.
- M. J. Savage, K. Juliya, W. Abigail, T. Katherine, K. Rachel, C. M. Tanesha, J. W. Maxwell, N. G. Hatcher, S. J. Haugabook and W. Guoxin, J. Neurosci., 2014, 34, 2884–2897 CrossRef CAS.
- R. Tarawneh, D. Head, S. Allison, V. Buckles, A. M. Fagan, J. H. Ladenson, J. C. Morris and D. M. Holtzman, JAMA Neurol., 2015, 72, 656–665 CrossRef PubMed.
- K. Leirs, P. Tewari Kumar, D. Decrop, E. Perez-Ruiz, P. Leblebici, B. Van Kelst, G. Compernolle, H. Meeuws, L. Van Wesenbeeck, O. Lagatie, L. Stuyver, A. Gils, J. Lammertyn and D. Spasic, Anal. Chem., 2016, 88, 8450–8458 CrossRef CAS PubMed.
- L. Chang, D. M. Rissin, D. R. Fournier, T. Piech, P. P. Patel, D. H. Wilson and D. C. Duffy, J. Immunol. Methods, 2012, 378, 102–115 CrossRef CAS PubMed.
- M. L. S. Haakan and N. Joensson, Angew. Chem., 2009, 121, 2556–2559 CrossRef.
- L. Mazutis, J. Gilbert, W. L. Ung, D. A. Weitz, A. D. Griffiths and J. A. Heyman, Nat. Protoc., 2013, 8, 870–891 CrossRef CAS PubMed.
- Z. Guan, Y. Zou, M. Zhang, J. Lv, H. Shen, P. Yang, H. Zhang, Z. Zhu and C. James Yang, Biomicrofluidics, 2014, 8, 014110 CrossRef PubMed.
- A. Rakszewska, J. Tel, V. Chokkalingam and W. T. S. Huck, NPG Asia Mater., 2014, 6, e133 CrossRef CAS.
- S. Tian, Z. Zhang, J. Chen, M. Du, Z. Li, H. Yang, X. Ji and Z. He, Talanta, 2018, 186, 24–28 CrossRef CAS PubMed.
- Y. Zhou, X. L. Tian, Y. S. Li, F. G. Pan, Y. Y. Zhang, J. H. Zhang, L. Yang, X. R. Wang, H. L. Ren and S. Y. Lu, Biosens. Bioelectron., 2011, 26, 3700–3704 CrossRef CAS PubMed.
- Y. Li, F. Gao, W. Wei, J. B. Qu, G. H. Ma and W. Q. Zhou, J. Mol. Catal. B: Enzym., 2010, 66, 182–189 CrossRef CAS.
- C. Lei, C. Xu, A. Nouwens and C. Yu, J. Mater. Chem. B, 2016, 4, 4975–4979 RSC.
- M. Tudorache and C. Bala, Sensors, 2008, 8, 7571–7580 CrossRef CAS PubMed.
- M. Ballauff, Prog. Polym. Sci., 2007, 32, 1135–1151 CrossRef CAS.
- A. Weiss, X. Guo and M. Ballauff, Macromolecules, 1999, 6043–6046 Search PubMed.
- Q. de Robillard, X. Guo, M. Ballauff and T. Narayanan, Macromolecules, 2000, 33, 9109–9114 CrossRef CAS.
- X. Guo and M. Ballauff, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2001, 64, 051406 CrossRef CAS.
- A. Wittemann, B. Haupt and M. Ballauff, Phys. Chem. Chem. Phys., 2003, 6, 1671–1677 RSC.
- S. Rosenfeldt, A. Wittemann, M. Ballauff, E. Breininger, J. Bolze and N. Dingenouts, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2004, 70, 061403 CrossRef CAS PubMed.
- G. Jackler, A. Wittemann, M. Ballauff and C. Czeslik, Spectroscopy, 2004, 18, 289–299 CrossRef CAS.
- Z. Qu, K. Chen, H. Gu and H. Xu, Bioconjugate Chem., 2014, 25, 370–378 CrossRef CAS PubMed.
- Z. Qu, F. Hu, K. Chen, Z. Duan, H. Gu and H. Xu, J. Colloid Interface Sci., 2013, 398, 82–87 CrossRef CAS PubMed.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- S. Sam, L. Touahir, A. J. Salvador, P. Allongue, J. N. Chazalviel, A. C. Gougetlaemmel, d. V. C. Henry, A. Moraillon, F. Ozanam and N. Gabouze, Langmuir, 2010, 26, 809–814 CrossRef CAS.
- D. M. Rissin and D. R. Walt, Nano Lett., 2006, 6, 520 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03404h |
| This journal is © The Royal Society of Chemistry 2019 |