
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The CuFe2O4@SiO2@ZrO2/SO42−/Cu nanoparticles: an efficient magnetically recyclable multifunctional Lewis/Brønsted acid nanocatalyst for the ligand- and Pd-free Sonogashira cross-coupling reaction in water†
Mohammad Ali Nasseri *,
Seyyedeh Ameneh Alavi,
Milad Kazemnejadi and
Ali Allahresani
*,
Seyyedeh Ameneh Alavi,
Milad Kazemnejadi and
Ali Allahresani
Department of Chemistry, Faculty of Science, University of Birjand, P. O. Box 97175-615, Birjand, Iran. E-mail: manaseri@birjand.ac.ir
First published on 3rd July 2019
Abstract
Herein, the synthesis and application of copper-incorporated sulfated zirconium oxide supported on CuFe2O4 NPs (CuFe2O4@SiO2@ZrO2/SO42−/Cu NPs) as a novel Lewis/Brønsted acid nanocatalyst were studied for the Sonogashira C–C cross-coupling reaction. The fabricated CuFe2O4@SiO2@ZrO2/SO42−/Cu catalyst exhibited efficient activity for a large variety of aryl iodides/bromides and, most importantly, aryl chlorides in water and in the presence of NaOH as a base in short reaction times. The catalyst was fully characterized by FTIR, TG-DTG, VSM, XRD, EDX, FE-SEM and TEM analyses. A synergetic effect could be considered to have arisen from the various Lewis acid and Brønsted acid sites present in the catalyst. The efficient incorporation of copper into zirconia provided a robust highly stable hybrid, which prevented any metal leaching, whether from the magnetite moiety and/or Cu sites in the reaction mixture. Moreover, the catalyst was successfully recovered from the mixture by a simple external magnet and reused for at least 9 consecutive runs. Zero metal leaching, stability, consistency with a variety of substrates, fast performance, cost-effectiveness, environmental friendliness, and preparation with accessible and cheap materials are some of the advantages and highlights of the current protocol.
1. Introduction
The Sonogashira C–C cross-coupling reaction is one of the most applicable types of C–C cross-couplings, which involves the coupling of vinyl or aryl halides or triflates with terminal alkynes (Csp2–Csp);1,2 since its vital application for the construction of complex biological and pharmaceutical molecules from simple precursors, the Sonogashira reaction has had significant importance in the field of synthetic organic chemistry.3 The reaction was first developed using Pd and Cu as a catalyst and a co-catalyst, respectively (Scheme 1). Since the discovery of this reaction, various methodologies have been developed to resolve its impediments. Palladium is a toxic, rare and expensive transition metal that is mainly used along with toxic and air-sensitive phosphine ligands.4 More importantly, the presence of copper as a co-catalyst promotes the Glaser-type homo-coupling of terminal acetylenes to generate a by-product (Scheme 1); thus, various attempts have been developed to perform the reaction under copper-free and Pd-free conditions in a mild, safe, ecofriendly, and cost-effective manner; in this regard, one strategy is the use of cheaper and safer alternative transition metals including Ni,5 Cu,6 Fe,7 and Co.8 Among these, the potential of Cu for application in the C–N as well as C–C cross coupling reactions is well-known;9 moreover, several achievements have been reported for the Cu-catalyzed Sonogashira reaction; the recent examples include the use of Cu2O/RGO,10 Cu/Mn bimetallic,9 CuI/PPh3/K2CO3,11 CuI/K3PO4/1,4-dioxane,12 and Au·CuFe2O4@silica as catalysts for this reaction.2 Recently, Sun and coworkers13 have reported the application of a Cu-MOF derived from two-phase Cu/Cu2O-rGO as an efficient catalyst for the Sonogashira reaction.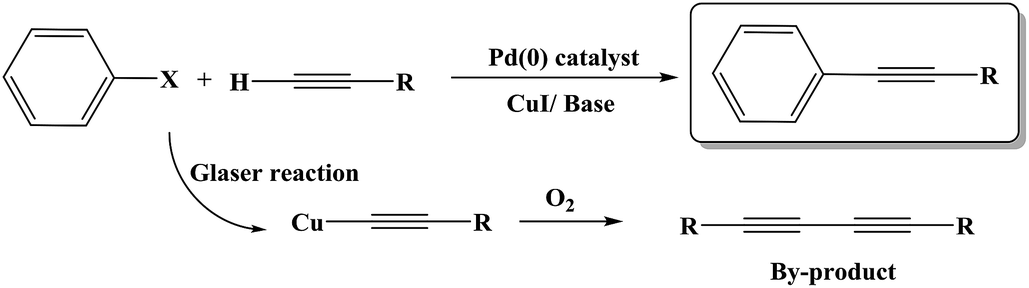 | ||
| Scheme 1 Traditional Sonogashira C–C cross coupling reaction and its possible diyne by-product formation. | ||
However, various drawbacks, including harsh reaction conditions, long reaction times, use of expensive and toxic materials, lack of selectivity, lack of environmental sustainability, and low reaction yields, especially for aryl chlorides, are still present in most of the reported protocols; therefore, the development of a promising alternative method is required.
Zirconia is one of the most well-known promising solid acids with significant catalytic activity. It is widely used as an efficient acid catalyst in oil refineries and petrochemical industries for processes such as hydrocarbon conversion, alkylation, cracking, Friedel–Crafts acylation, esterification and isomerization;14,15 moreover, the activity of zirconia can be largely promoted by its treatment with sulfate groups, and as a result, sulfated zirconium oxide (ZrO2/SO42−) is obtained. The high thermal stability, outstanding catalytic activity, high acidity, stability in various organic solvents, and durability under harsh reaction conditions are some of the notable and applicable properties of sulfated zirconia that make it a suitable support for more modifications (an objective of this study) and/or catalytic aspects.16 Various catalytic activities, such as towards benzylation,16 multicomponent reactions,17 and synthesis of dioxane,18 of ZrO2/SO42− have been reported in the literature; in addition, heterogeneous solid supports can be magnetized by magnetic nanoparticles (MNPs) to make these supports magnetically recoverable;19 moreover, due to their high aspect ratio, MNPs can strongly improve the catalytic activity of a catalyst.3
In this study, we introduced copper-incorporated sulfated zirconium oxide supported on CuFe2O4 nanoparticles as an efficient, recyclable and durable magnetic nanocatalyst for the first time for the C–C cross coupling reaction of phenylacetylene with aryl iodides, aryl bromides and aryl chlorides under mild reaction conditions. The present system not only benefits from the durable ZrO2/SO42− solid support, but also the magnetic CuFe2O4 magnetic core in the catalyst provides suitable recyclability to the catalyst via an external magnet.
2. Experimental
2.1. Instrumentation and materials
All chemicals were freshly purchased from Sigma and Merck or Fluka Chemical Companies with no further purification. All solvents were distilled under a N2 atmosphere and dried before use. The reaction progress was monitored by thin layer chromatography (TLC). The FTIR spectra were obtained via the JASCO FT/IR 4600 spectrophotometer using KBr pellets. The 1H NMR (250 MHz) and 13C NMR (62.9 MHz) spectroscopies were performed by the Bruker Avance DPX-250 spectrometer in CDCl3 and DMSO-d6 as solvents, respectively. TMS was used as an internal standard. Mass spectrometry was performed using the Thermolyne 79300 model tube furnace equipped with the MKS gas analyzer coupled to a quadrupole mass selective detector. The scanning electron microscopy images (FE-SEM) were obtained by the TESCAN MIRA3 apparatus. Transmission electron microscopy (TEM) was conducted using the Philips EM208 microscope at 100 kV. The magnetic behavior of the samples was investigated using the Lake Shore Cryotronics 7407 vibrating sample magnetometer (VSM) at room temperature. EDX spectroscopy was performed using a field-emission scanning electron microscope (FESEM, JEOL 7600F), equipped with an X-ray energy dispersive spectrometer obtained from Oxford instruments. The TGA of the samples was performed using NETZSCH STA 409 PC/PG under a N2 atmosphere at the heating rate of 10 °C min−1 in the temperature range of 25–850 °C. Metal leaching studies were performed using the VARIAN VISTA-PRO CCD simultaneous ICP-OES instrument as well as the inductively coupled plasma mass spectrometer (ICP-MS) Thermo Elemental VG PQ ExCell.2.2. Preparation of the CuFe2O4@SiO2 MNPs
The CuFe2O4 NPs were prepared according to a previously reported procedure.20 Cu(NO3)2 (10 mmol, 1.9 g) and Fe(NO3)3 (20 mmol, 4.8 g) were dissolved in water (75 mL) and ultrasonicated for 30 min. Then, NaOH (25 mL, 1 N) was added dropwise to the abovementioned solution until a reddish-black sediment was formed. The reaction mixture was stirred at 90 °C for 2 h. The reddish-black precipitate was washed with water (2 × 25 mL) and EtOH (2 × 25 mL) until the pH of the solution was adjusted to 7.0. The sediment was separated, dried in a vacuum oven for 12 h, and then calcined in a furnace at 700 °C for 5 h at the heating rate of 20 °C min−1. The CuFe2O4@SiO2 MNPs were synthesized using the sol–gel method. CuFe2O4 (2.0 g, 8.5 mmol) was ultrasonically dispersed in ethanol (25 mL) for 2 h at 60 °C, and then, aqueous NaOH (10% w/w, 10 mL) was added to the mixture followed by stirring at room temperature for 30 min. Then, tetraethoxyorthosilicate (TEOS, 1.0 mL) was added to the mixture, and stirring was continued for further 24 h. The CuFe2O4@SiO2 MNPs were separated from the solution by an external magnetic field, washed with water (3 × 5 mL) and EtOH (2 × 5 mL), and then dried under vacuum for 48 h. The resultant CuFe2O4@SiO2 MNPs were calcined at 800 °C for 4 h at the heating rate of 20 °C min−1.2.3. Preparation of the CuFe2O4@ZrO2/SO42−/Cu NPs
Sulfated zirconium oxide was prepared according to a previously reported protocol with slight modification.21,22 At first, ZrCl4 (2.3 g, 10 mmol) as a precursor was dissolved in 10 mL deionized water. The ammonia solution (10 mL, 1 N) was added dropwise for 30 min until the pH was adjusted to 11. The resulting suspension was aged for 24 h at room temperature. Then, the obtained white sediment was washed with deionized water using centrifugation (5 × 10 mL) until the solution was neutralized (pH = 7). The resultant Zr(OH)4 was dried at 100 °C for 24 h. The isolated yield for Zr(OH)4 was found to be 96% (1.5 g). Subsequently, Zr(OH)4 (1.0 g) was immersed in an (NH4)2SO4 solution (2.5 g in 50 mL distilled water) with the mixing ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, Zr:S. The solution was dried at 100 °C for 24 h followed by calcination at 650 °C for 3 h in a furnace at the heating rate of 10 °C min−1. The resultant ZrO2/SO42− (1.0 g) was impregnated in the Cu(OAc)2 solution (0.1 g in 50 mL distilled water) to obtain the Cu content of 10 wt% (theoretically). The mixture was dried at 100 °C for 12 h and subsequently calcined at 400 °C for 3 h under an air atmosphere at the heating rate of 10 °C min−1. The obtained ZrO2/SO42−/Cu was washed with deionized water (3 × 10 mL) and then dried in an oven (50 °C) for 8 h. The prepared ZrO2/SO42−/Cu NPs were supported on CuFe2O4@SiO2 magnetic nanoparticles. A mixture of CuFe2O4@SiO2 NPs (0.2 g) in H2O:EtOH (15 mL, 1:3 v/v) was sonicated for 10 min at room temperature. A pre-dispersed ethanolic solution of ZrO2/SO42− (0.4 g) was added to the abovementioned mixture followed by sonication for additional 10 min at room temperature. NaOH 10% w/w (15 mL) was added dropwise to the solution for 30 min under sonication. The solution was stirred for 24 h. The resultant CuFe2O4@ZrO2/SO42−/Cu NPs were separated by an external magnetic, washed with deionized water and dried in an oven for 12 h. The complete route for the preparation of the catalyst is shown in Scheme 2.
:3, Zr:S. The solution was dried at 100 °C for 24 h followed by calcination at 650 °C for 3 h in a furnace at the heating rate of 10 °C min−1. The resultant ZrO2/SO42− (1.0 g) was impregnated in the Cu(OAc)2 solution (0.1 g in 50 mL distilled water) to obtain the Cu content of 10 wt% (theoretically). The mixture was dried at 100 °C for 12 h and subsequently calcined at 400 °C for 3 h under an air atmosphere at the heating rate of 10 °C min−1. The obtained ZrO2/SO42−/Cu was washed with deionized water (3 × 10 mL) and then dried in an oven (50 °C) for 8 h. The prepared ZrO2/SO42−/Cu NPs were supported on CuFe2O4@SiO2 magnetic nanoparticles. A mixture of CuFe2O4@SiO2 NPs (0.2 g) in H2O:EtOH (15 mL, 1:3 v/v) was sonicated for 10 min at room temperature. A pre-dispersed ethanolic solution of ZrO2/SO42− (0.4 g) was added to the abovementioned mixture followed by sonication for additional 10 min at room temperature. NaOH 10% w/w (15 mL) was added dropwise to the solution for 30 min under sonication. The solution was stirred for 24 h. The resultant CuFe2O4@ZrO2/SO42−/Cu NPs were separated by an external magnetic, washed with deionized water and dried in an oven for 12 h. The complete route for the preparation of the catalyst is shown in Scheme 2.
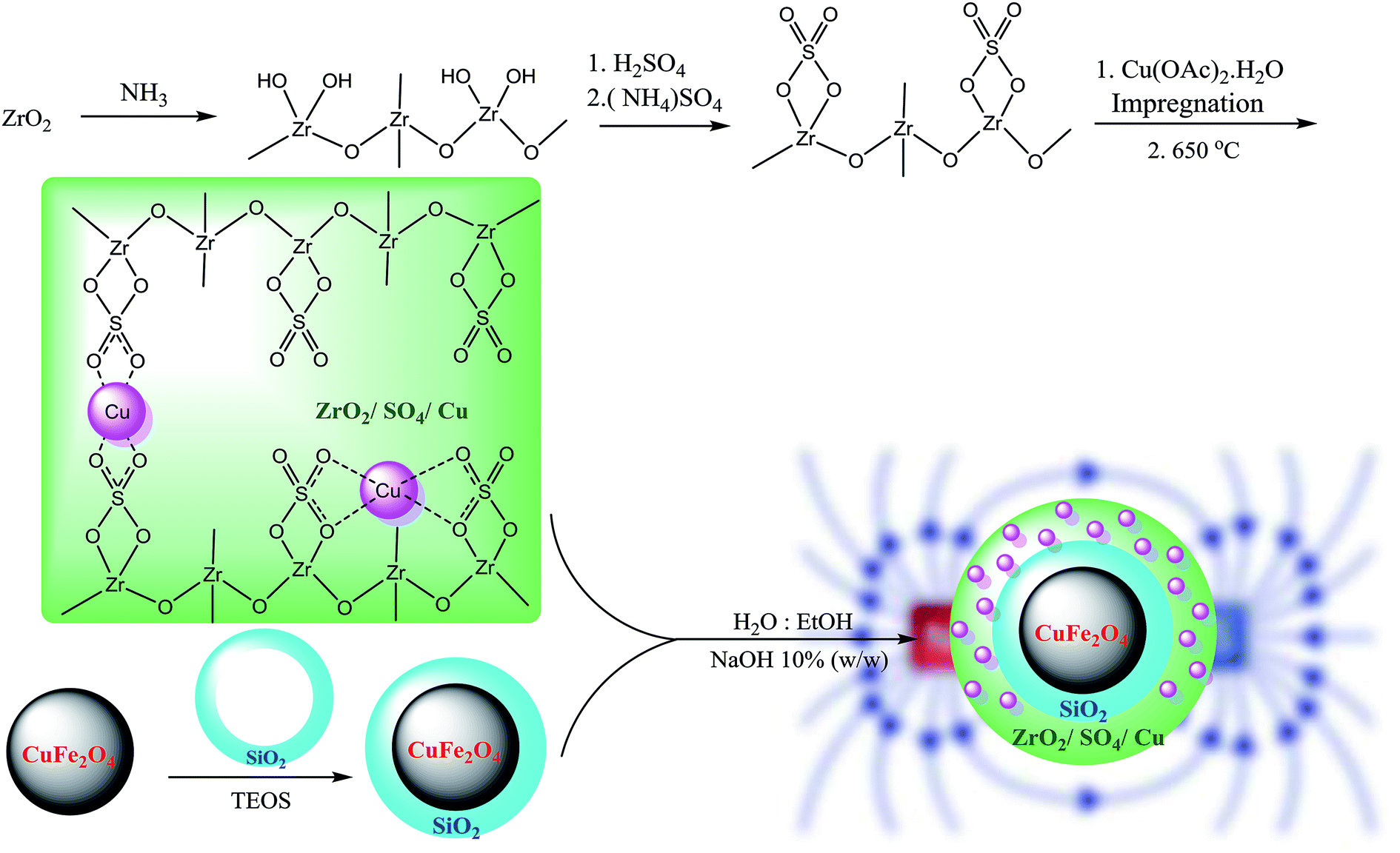 | ||
| Scheme 2 Synthesis of CuFe2O4@SiO2@ZrO2/SO42−/Cu nanoparticles. | ||
2.4. General procedure for the Sonogashira coupling reaction using the CuFe2O4@SiO2@ZrO2/SO42−/Cu nanoparticles
A mixture of phenylacetylene (1.5 mmol), aryl halide (1.0 mmol), the CuFe2O4@SiO2@ZrO2/SO42−/Cu magnetic nanocatalyst (0.005 g, 0.3 mol% Cu), NaOH (1.0 mmol) and water (1.0 mL) was stirred at 60 °C in an oil bath. The reaction progress was monitored by TLC. Upon completion of the reaction, the catalyst was magnetically removed, and the mixture was extracted with 10 mL of Et2O. The combined organic layers were dried over MgSO4, and then, the solvent was removed under reduced pressure. The desired pure coupling product was obtained by flash chromatography of the crude product.3. Results and discussion
3.1. Catalyst characterization
The FTIR spectra of Zr(OH)4, ZrO2/SO42−, ZrO2/SO42−/Cu, and CuFe2O4@SiO2@ZrO2/SO42−/Cu are shown in Fig. 1A(a). The broad and resolved peak at 3403 cm−1 related to the O–H stretching vibration confirmed the hydration of zirconium chloride using ammonia. Moreover, the absorption peaks related to Zr–O–Zr could be seen at 640–750 cm−1 (Fig. 1A(a)). The FTIR spectrum of ZrO2/SO42− demonstrated characteristic peaks at 1143, 1044, and 994 cm−1 (as a shoulder), which corresponded to the asymmetric or symmetric stretching vibrations of the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O or S–O bonds (Fig. 1A(b)).23 These vibrations were characteristic for the bidentate sulfate ions coordinated to a metal cation. The series of peaks at 467–747 cm−1 were assigned to the Zr–O–Zr asymmetric stretching vibrations.24 Moreover, the broad peak at 3421 cm−1 and the medium peak at 1636 cm−1 were assigned to the O–H stretching and bending vibrations of the adsorbed and/or coordinated water by the sulfate groups, respectively.25
O or S–O bonds (Fig. 1A(b)).23 These vibrations were characteristic for the bidentate sulfate ions coordinated to a metal cation. The series of peaks at 467–747 cm−1 were assigned to the Zr–O–Zr asymmetric stretching vibrations.24 Moreover, the broad peak at 3421 cm−1 and the medium peak at 1636 cm−1 were assigned to the O–H stretching and bending vibrations of the adsorbed and/or coordinated water by the sulfate groups, respectively.25
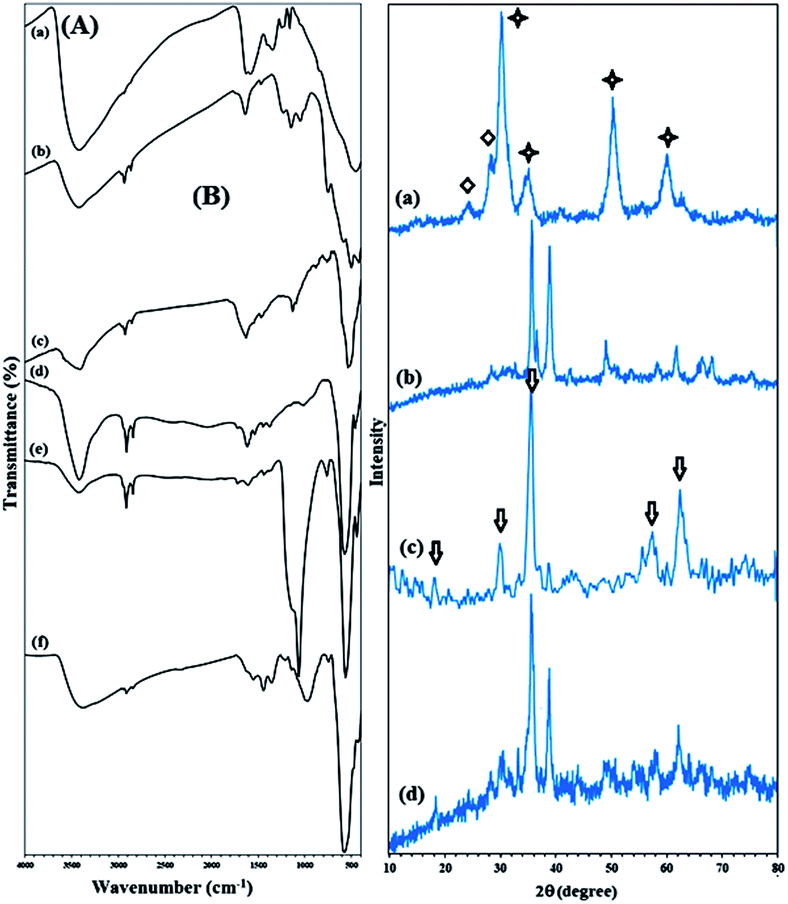 | ||
| Fig. 1 (A) FTIR spectra of (a) Zr(OH)4, (b) ZrO2/SO42−, (c) ZrO2/SO42−/Cu, (d) CuFe2O4, (e) CuFe2O4@SiO2, and (f) CuFe2O4@SiO2@ZrO2/SO42−/Cu. (B) XRD pattern of (a) ZrO2/SO42−, (b) ZrO2/SO42−/Cu, (c) CuFe2O4, and (d) CuFe2O4@SiO2@ZrO2/SO42−/Cu. The star indicates a tetragonal structure, and the diamond represents a monoclinic structure. | ||
A sharp peak near 500 cm−1 was attributed to the Cu–O stretching vibration, demonstrating that the incorporation of the Cu ions took place through the oxygen atoms of the sulfated ions in ZrO2/SO42−/Cu; moreover, this subsequently confirmed the successful coordination of the Cu cations to the catalyst framework (Fig. 1A(c)).26 The stretching vibrations related to Zr–O–Zr were covered due to this strong absorption.
In the CuFe2O4 FTIR spectrum, two absorptions at 1629 and 3435 cm−1 represented the H–O–H bending and free O–H stretching vibrations, respectively, due to the water molecules adsorbed on the surface of the CuFe2O4 NPs with high aspect ratio.27 The two absorption bands at 476 and 590 cm−1 were assigned to the Cu–O and Fe–O stretching vibrations, respectively (Fig. 1A(d)).27,28 The strong absorption at 1093 cm−1 (Si–O vibrations) confirmed the successful coating of the CuFe2O4 NPs with a silica shell (Fig. 1A(e)).
The presence of vibration bands at 421, 575, and 870–1148 cm−1, which were due to Fe–O, Cu–O, and Si–O–Si, respectively, demonstrated that ZrO2/SO42−/Cu was successfully supported on CuFe2O4@SiO2 (Fig. 1A(f)). In addition, the presence of several bands with medium intensity in the 1361–1641 cm−1 region was allocated to the ZrO2/SO42− stretching vibrations (Fig. 1A(f)).
The XRD patterns of ZrO2/SO42−, ZrO2/SO42−/Cu, and CuFe2O4@SiO2@ZrO2/SO42−/Cu are shown in Fig. 1B. ZrO2/SO42− demonstrated three characteristic peaks with strong intensities at 2θ = 30.4°, 50.3° and 60.2°, which represented the tetragonal structure of ZrO2/SO42− with high crystallinity (JCPDS 17-0923) (Fig. 1B(a)).21,29 More precisely, a mixture of the monoclinic and tetragonal phases was observed in the spectrum (Fig. 1B(a), stars and diamonds represent a tetragonal and monoclinic structure, respectively) that was in agreement with the reported ZrO2/SO42 crystal structure.21,29 The peaks with lower intensities at 2θ = 24.1° and 28.3° were assigned to the monoclinic structure of zirconia.
Note that the presence of sulfated groups does not lead to a phase change of zirconia; this is may be due to strong interactions between zirconia and the sulfate ions.29 On the other hand, based on the presence of prominent peaks related to the trigonal structure of ZrO2/SO42−, it could be concluded that the impregnation of the sulfate ions showed a strong effect on the phase modification of zirconia from a thermodynamically more stable monoclinic phase to a metastable tetragonal phase. The incorporation of Cu into the structure of ZrO2/SO42− caused a small shift of the peaks related to the sulfated zirconia crystal structure at 2θ = 31.7°, 35.6°, 38.8°, 48.4°, 58.2°, 61.8°, 66.0°, and 68.08°. However, the peaks at 2θ = 35.6°, 38.8°, 48.4°, 66.0°, and 68.08° matched well with the indices (002), (111), (202), (311), and (113), respectively, which corresponded to the thermally prepared CuO powder structure (Fig. 1B(b)).30,31 Furthermore, the peaks with much lower intensities near to baseline indicating the crystal structure of the sulfated zirconium oxide (Fig. 1B(b)). The results suggested that the Cu–O bond between the sulfate groups in ZrO2/SO42−/Cu and the Cu ions was completely in agreement with its corresponding FTIR spectrum. The XRD pattern of the CuFe2O4 NPs represented Bragg's reflections at 2θ = 18.6°, 30.2°, 35.5°, 57.8°, and 62.80° (arrows in Fig. 1B(c)) corresponding to their indices (101), (200), (211), (321) and (400). These reflections were consistent with the tetragonal crystal structure of CuFe2O4 (JCPDS card no. 34-0425), in agreement with those reported in the literature (Fig. 1B(c)).2,27,28 Fig. 1B(d) shows the crystal structure of the catalyst CuFe2O4@SiO2@ZrO2/SO42−/Cu. The peak intensities for the CuFe2O4 crystal structure were reduced when ZrO2/SO42− was supported on the CuFe2O4@SiO2 surface. The results confirmed that the functionalization of CuFe2O4 did not lead to a phase change of ZrO2/SO42. Moreover, the presence of a sharp peak at 2θ = 38.2° demonstrated the incorporation of Cu into the catalyst. The peaks at 2θ = 60.0°, 50.3°, 30.2°, and 50.7° may be assigned to the ZrO2/SO42− crystal structure.
The preparation of ZrO2/SO42− was confirmed by the presence of the Cu, Zr, O, and S elements, which were detected by EDX analysis (Fig. 2a). Moreover, the presence of these elements in the catalyst was investigated and confirmed by EDX analysis. As shown in Fig. 2b, the elements Zr, Cu, O, S, Fe, and Si were detected in the catalyst. The thermal behavior of ZrO2/SO42− and the catalyst is shown in Fig. 3a. ZrO2/SO42− showed a significant thermal stability, and only a 7.5% weight loss was observed in the temperature range of 25–1000 °C (Fig. 3a). This degradation occurred in four steps, where the first and second steps were assigned to the loss of the physically adsorbed water from the catalyst surface (0.26% weight loss at 210 °C), and the escape of the trapped water in the catalyst network by sulfate groups (1.19% weight loss at 350 °C), respectively. The third weight loss in the temperature range of 530–665 °C was related to the oxidation of copper and the formation of CuO.32 The weight loss that appeared in the temperature range of 680–860 °C was due to the decomposition of sulfate as well as structural OH− groups.33–35 The decoration of ZrO2/SO42−/Cu on CuFe2O4@SiO2 improved the thermal stability of the catalyst, and only 6.5% weight loss was observed until 1000 °C. The first weight loss with a mild slope, which lasted till 780 °C, was due to the loss of the adsorbed water in the crystal structure of the catalyst. The subsequent weight loss was attributed to the decomposition of incorporated Cu and sulfate groups with a 4.0% weight loss (Fig. 3b).
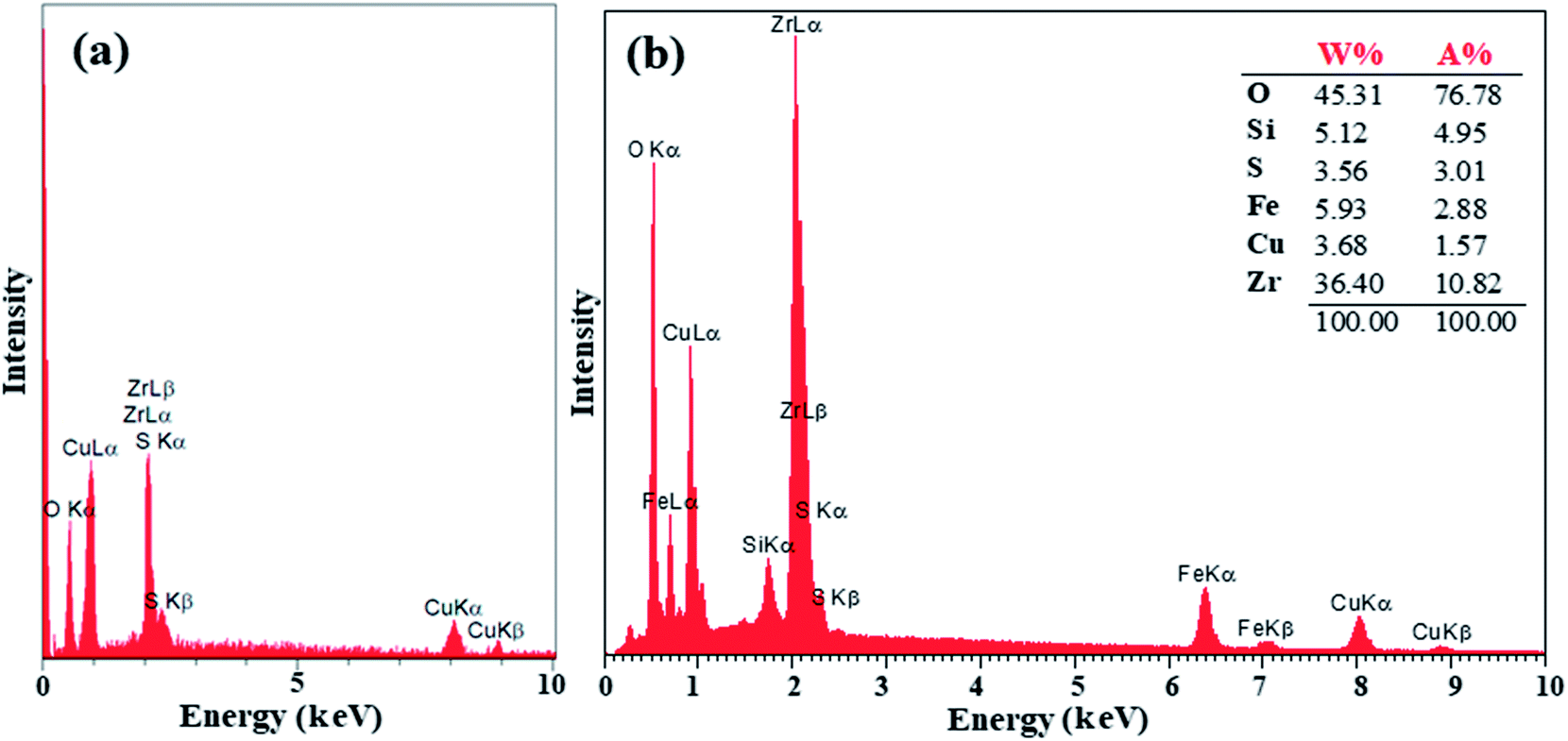 | ||
| Fig. 2 EDX spectra of (a) ZrO2/SO42/Cu and (b) CuFe2O4@SiO2@ZrO2/SO42−/Cu. | ||
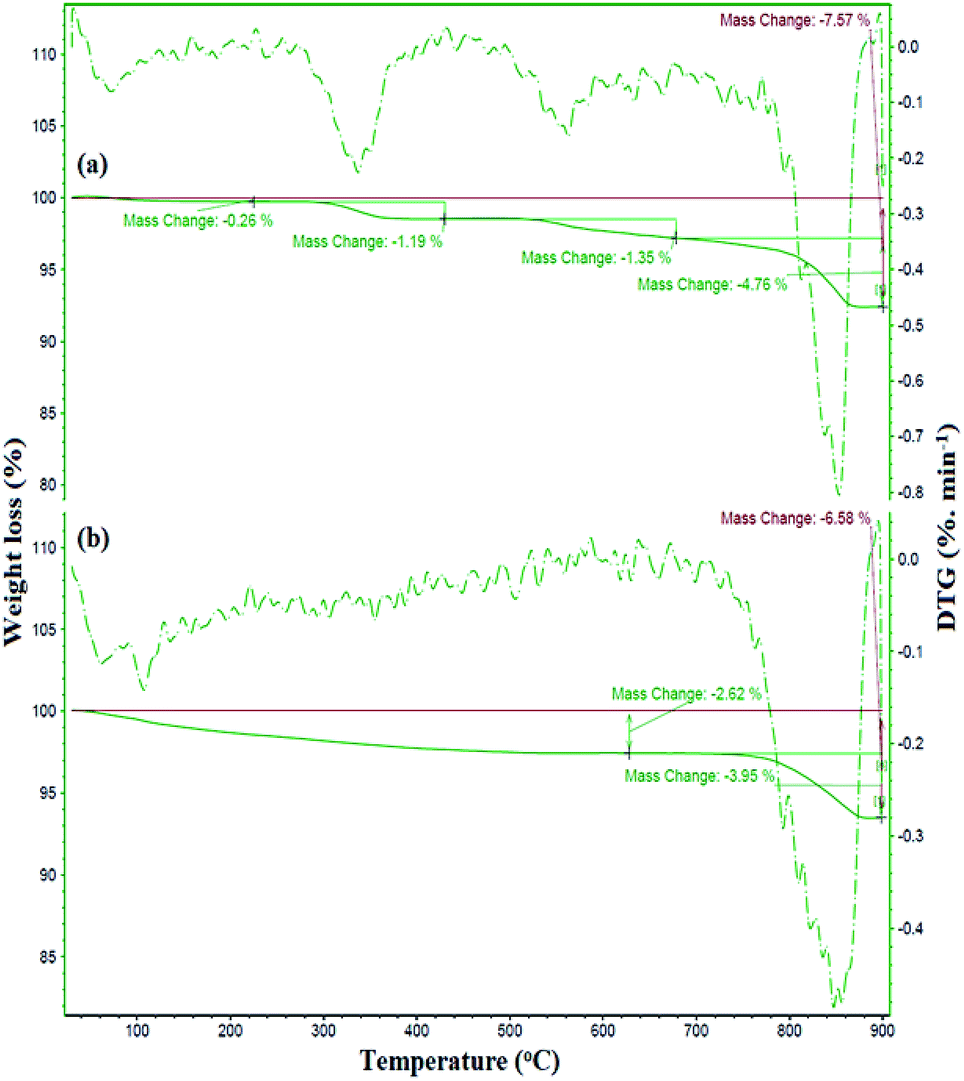 | ||
| Fig. 3 TGA-DTG curves of (a) ZrO2/SO42−/Cu and (b) CuFe2O4@SiO2@ZrO2/SO42−/Cu. | ||
The magnetic properties of CuFe2O4 and CuFe2O4@SiO2@ZrO2/SO42−/Cu were studied by VSM analysis (Fig. 4). As shown in Fig. 4, the samples represented a superparamagnetic behavior with no hysteresis loops in their spectra. The saturation magnetization for CuFe2O4 was found to be 24.6 emu g−1 (Fig. 4a). This amount was largely reduced to 10.1 emu g−1 for CuFe2O4@SiO2@ZrO2/SO42−/Cu; this strongly confirmed its surface functionalization (Fig. 4b). However, there was sufficient magnetic response for the complete separation of nanoparticles from the mixture. The inset figures show the immediate separation of CuFe2O4@SiO2@ZrO2/SO42−/Cu from the mixture in 120 seconds under an applied external magnetic field after the dispersion of CuFe2O4@SiO2@ZrO2/SO42−/Cu.
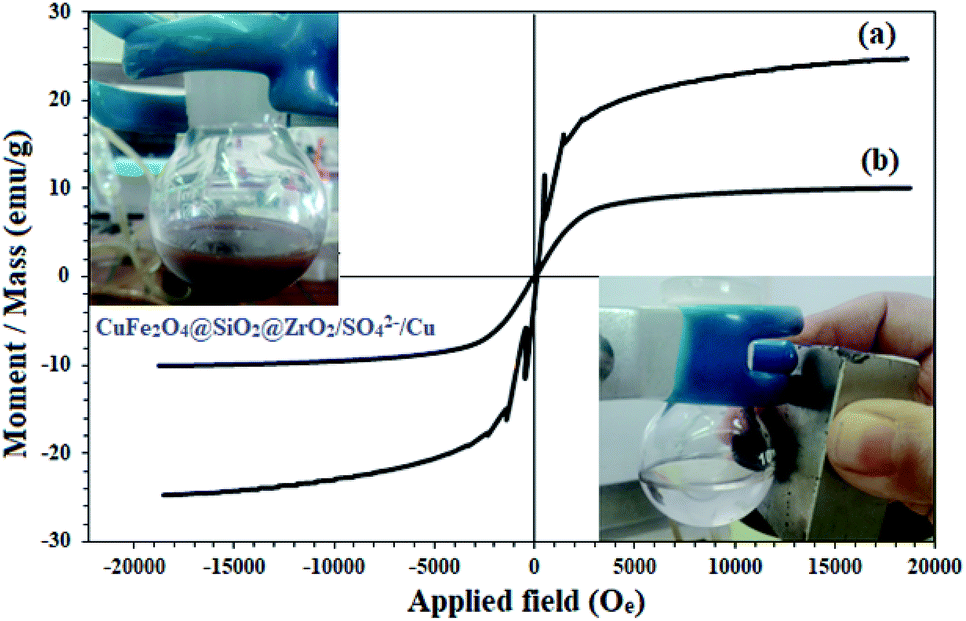 | ||
| Fig. 4 Magnetic behavior of (a) CuFe2O4 and (b) CuFe2O4@SiO2@ZrO2/SO42−/Cu. | ||
The morphology and shape of ZrO2/SO42−/Cu and CuFe2O4@SiO2@ZrO2/SO42−/Cu were studied by the SEM and TEM techniques. The SEM images of ZrO2/SO42−/Cu and CuFe2O4@SiO2@ZrO2/SO42−/Cu represented an irregularly shaped amorphous morphology that could be due to the expected agglomeration of activated ZrO2/SO42− by the inclusion of transition metals (Fig. 5a and b).36 By comparing their TEM images (Fig. 5c and d), this agglomeration was more clearly observed. According to Fig. 5c and d, the particles had the average size of 15 nm and 40 nm for ZrO2/SO42−/Cu and CuFe2O4@SiO2@ZrO2/SO42−/Cu, respectively.
 | ||
| Fig. 5 (a) FE-SEM and (b) TEM images of ZrO2/SO42−/Cu. (c) FE-SEM and (d) TEM images of CuFe2O4@SiO2@ZrO2/SO42−/Cu. | ||
3.2. Optimization of reaction parameters
To find premium reaction conditions for the Sonogashira C–C coupling reaction, the reaction of iodobenzene with phenylacetylene was chosen as the model reaction. The effects of the reaction parameters, such as the type of base, reaction temperature, solvent, and catalyst amount, were studied. The results are presented in Table 1. The reaction obviously proceeded in polar-protic solvents such as EtOH, MeOH and water (Table 1, entries 1, 8, and 12). These results were in agreement with the structure of the catalyst containing hydrophilic groups as well as the mechanism proposed in the next section. Other solvents provided low to moderate yields. The highest efficiency was obtained in water after reaction for 30 min in the presence of 0.005 g of catalyst (entry 12, 92%). There was no satisfactory conversion under solvent-free conditions (Table 1, entry 11). NaOH and KOH were found to be efficient bases for this transformation (Table 1, entries 12 and 13). Moreover, 60 °C and 0.005 g of the catalyst were the premium temperature and catalyst amount for the model reaction, respectively (Table 1, entries 12, 21–26).| Entry | Solvent | Base | Catalyst (g) | T (°C) | t (min) | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | EtOH | NaOH | 0.005 | 60 | 30 | 80 |
| 2 | CH3CN | NaOH | 0.005 | 60 | 30 | 69 |
| 3 | THF | NaOH | 0.005 | 60 | 30 | 40 |
| 4 | CH2Cl2 | NaOH | 0.005 | 60 | 30 | 50 |
| 5 | Toluene | NaOH | 0.005 | 60 | 30 | 75 |
| 6 | CHCl3 | NaOH | 0.005 | 60 | 30 | Trace |
| 7 | DMSO | NaOH | 0.005 | 60 | 30 | 89 |
| 8 | MeOH | NaOH | 0.005 | 60 | 30 | 88 |
| 9 | Dioxane | NaOH | 0.005 | 60 | 30 | 45 |
| 10 | DMF | NaOH | 0.005 | 60 | 30 | 75 |
| 11 | Solvent-free | NaOH | 0.005 | 60 | 30 | Trace |
| 12 | H2O | NaOH | 0.005 | 60 | 30 | 92 |
| 13 | H2O | KOH | 0.005 | 60 | 30 | 92 |
| 14 | H2O | K2CO3 | 0.005 | 60 | 30 | 66 |
| 15 | H2O | K3PO4 | 0.005 | 60 | 30 | 90 |
| 16 | H2O | NaOAC | 0.005 | 60 | 30 | 90 |
| 17 | H2O | LiHMDS | 0.005 | 60 | 30 | 55 |
| 18 | H2O | Et3N | 0.005 | 60 | 30 | 76 |
| 19 | H2O | HMTA | 0.005 | 60 | 30 | 54 |
| 20 | H2O | t-BuOK | 0.005 | 60 | 30 | 88 |
| 21 | H2O | NaOH | 0.001 | 60 | 30 | 77 |
| 22 | H2O | NaOH | 0.003 | 60 | 30 | 82 |
| 23 | H2O | NaOH | 0.01 | 60 | 30 | 90 |
| 24 | H2O | NaOH | 0.005 | R.T. | 30 | 88 |
| 25 | H2O | NaOH | 0.005 | 80 | 30 | 92 |
| 26 | H2O | NaOH | 0.005 | Ref. | 30 | 93 |
The scope of the reaction was investigated and extended with a variety of aryl halides and phenylacetylene in the presence of CuFe2O4@SiO2@ZrO2/SO42−/Cu NPs as the catalyst under the previously obtained optimum conditions. As shown in Table 2, the method tolerated various substrates bearing either electron-donating and/or electron-withdrawing substituents, and high-to-excellent yields were obtained for all substrates (Table 2). Generally, the substrates with electron-withdrawing substituents provide higher efficiencies than others in terms of time and yield (Table 2, for example entries 5, 7, and 13). Moreover, iodide as a leaving group accelerated the reaction than Br or Cl. The results are in agreement with an oxidative addition/reductive elimination mechanism, which has been discussed hereinafter.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R | X | 9 | Time (min) | Yielda (%) | TON | TOF (h−1) |
| a Reaction conditions: aryl halide (1.0 mmol), phenylacetylene (1.5 mmol), NaOH (1.0 mmol), H2O (2 mL), catalyst (0.3 mol% Cu), and 60 °C. | |||||||
| 1 | H | I | 9a | 30 | 92 | 326 | 562 |
| 2 | H | Cl | 9b | 100 | 88 | 293 | 177 |
| 3 | H | Br | 9c | 45 | 92 | 307 | 409 |
| 4 | 4-Me | I | 9d | 30 | 94 | 313 | 626 |
| 5 | 4-CO2H | I | 9e | 20 | 89 | 297 | 873 |
| 6 | 4-NH2 | Br | 9f | 55 | 76 | 253 | 275 |
| 7 | 2-NO2 | I | 9g | 20 | 90 | 300 | 909 |
| 8 | 3-Me | I | 9h | 60 | 82 | 273 | 273 |
| 9 | 2-NH2 | Cl | 9i | 150 | 69 | 230 | 92 |
| 10 | 4-CO2H | Br | 9j | 70 | 78 | 260 | 224 |
| 11 | 4-SMe | Br | 9k | 90 | 58 | 193 | 128 |
| 12 | 3-NH2 | Cl | 9l | 180 | 70 | 233 | 77 |
| 13 | 4-CN | Br | 9m | 25 | 92 | 307 | 730 |
| 14 | 4-COH | I | 9n | 65 | 88 | 293 | 271 |
| 15 | 4-OMe | I | 9o | 55 | 88 | 293 | 318 |
| 16 | 3-SMe | Cl | 9p | 110 | 68 | 227 | 124 |
| 17 | 4-NO2 | I | 9q | 30 | 98 | 326 | 196 |
| 18 | 2-SMe | Cl | 9r | 140 | 72 | 240 | 103 |
| 19 | 4-NO2 | Br | 9s | 45 | 97 | 323 | 430 |
3.3. Mechanistic study
At first, we investigated the model reactions in the presence of CuFe2O4, CuFe2O4@SiO2, ZrCl4, ZrO2/SO42−, and ZrO2/SO42−/Cu as control experiments. The corresponding results are summarized in Table 3. As shown in the Table, ZrCl4, (NH4)2SO4, ZrO2/SO42−, and Cu(OAc)2 did not afford any coupling products under the reaction conditions. Interestingly, the CuFe2O4 NPs demonstrated catalytic activity for the reaction (Table 3, entry 6). This activity was reduced after coating of these NPs with a silica shell; this was further evidence for the catalytic activity of CuFe2O4 NPs (Table 3, entries 6 and 7). Previously, Gholinejad and coworkers2 have reported a possible interference of CuFe2O4@silica as a catalyst towards the Sonogashira coupling reaction; this is in agreement with the results obtained from the control experiments. ZrO2/SO42−/Cu provided a 76% isolated yield (Table 3, entry 8). The results suggested that (i) the catalytic activity of CuFe2O4@SiO2/ZrO2/SO42−/Cu originated due to the incorporation of copper into the catalyst, and (ii) a synergetic effect could be considered to have originated from the various functionalities of the catalyst that promoted the coupling reaction; accordingly, the yield obtained using CuFe2O4@SiO2/ZrO2/SO42−/Cu is roughly the sum of the yields obtained using CuFe2O4@SiO2 and ZrO2/SO42−/Cu separately (Table 3, entries 7 and 8); this finding suggests that both parts of the final catalyst contribute to the catalytic activity.
|
||
|---|---|---|
| Entry | Catalyst | Yield (%) |
| 1 | No catalyst | No reaction |
| 2 | ZrCl4 | No reaction |
| 3 | (NH4)2SO4 | No reaction |
| 4 | ZrO2/SO42- | No reaction |
| 5 | Cu(OAC)2 | No reaction |
| 6 | CuFe2O4 | 26% |
| 7 | CuFe2O4@SiO2 | 15% |
| 8 | ZrO2/SO42−/Cu | 76% |
| 9 | CuFe2O4@SiO2/ZrO2/SO42−/Cu | 92% |
Based on the abovementioned observations, we suggested the most possible mechanistic pathway for this method. According to the results obtained from the control experiments as well as the literature,10–12,37 there were several catalytic active sites on the surface of CuFe2O4@SiO2, ZrCl4, and ZrO2/SO42−. Scheme 3 shows a plausible structure for CuFe2O4@SiO2, ZrCl4, and ZrO2/SO42−. The coordinated Cu and zirconium were efficient active Lewis sites. Moreover, water was coordinated through an interconversion reaction between free sulfate groups on the catalyst, and this provided active Brønsted acid sites. The presence of water as a solvent promoted the active Brønsted acid sites (Scheme 3); this explained the high catalytic activity of the catalyst with water as a solvent. Due to the presence of these catalytic active sites in CuFe2O4@SiO2, ZrCl4, and ZrO2/SO42−, a synergetic effect could be speculated for this catalyst, arising from the Cu sites, Zr sites, sulfate groups,38 coordinated water,21 and CuFe2O4.2 A plausible structure for the catalyst is shown in Scheme 4, which is in agreement with the characterization data as well as the structure proposed in literature.2,21,38 In the first step of the proposed mechanism, Cu-acetylide (Scheme 4, intermediate I) was formed via oxidative addition with the participation of a base. This addition could be mediated by electron transfer from zirconium to copper (from Cu I to Cu II for example, see Scheme 3). To prove this claim, the Sonogashira reaction was performed in the presence of CuSO4 under the same reaction conditions. No coupling products were found in the mixture. However, it could be concluded that the presence of zirconium in the catalyst was mandatory for electron transfer. A water molecule was formed during this transformation. The hydrophilic nature of the catalyst surface arising from the sulfate groups increased the solubility of the base. Due to interconversion between sulfate groups (Schemes 3 and 4), the cation of the base (Na+ in this case) could be coordinated to Zr, and the reaction was accelerated. This step was supported by the Cu-catalyzed Sonogashira reactions9,11,12,37 as well as our observations in the control experiments. In the next step, another catalyst molecule formed the π-complex intermediate II, which generated a positive charge in acetylene and thus favored a nucleophilic attack on the electron-rich aryl halide. Due to the presence of the coordinated cation on the catalyst surface (Na+ in this case), a four-membered intermediate II, as shown in Scheme 4, was generated. The intermediate II underwent reductive elimination and led to the formation of a C–C coupling product as well as a NaX salt. The catalyst was regenerated for the next cycle (Scheme 4).
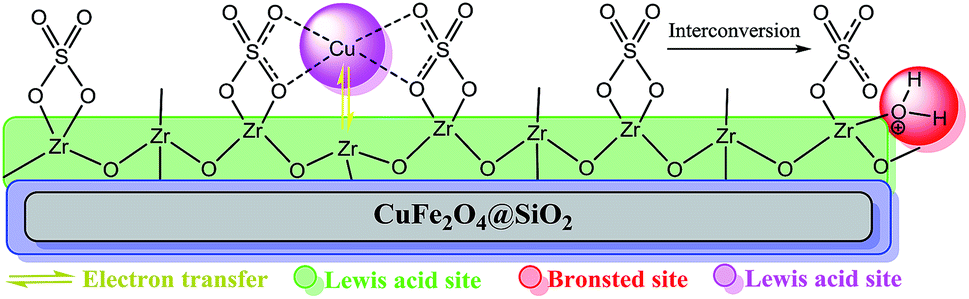 | ||
| Scheme 3 The most possible structure and various acid functionalities of CuFe2O4@SiO2@ZrO2/SO42−/Cu. | ||
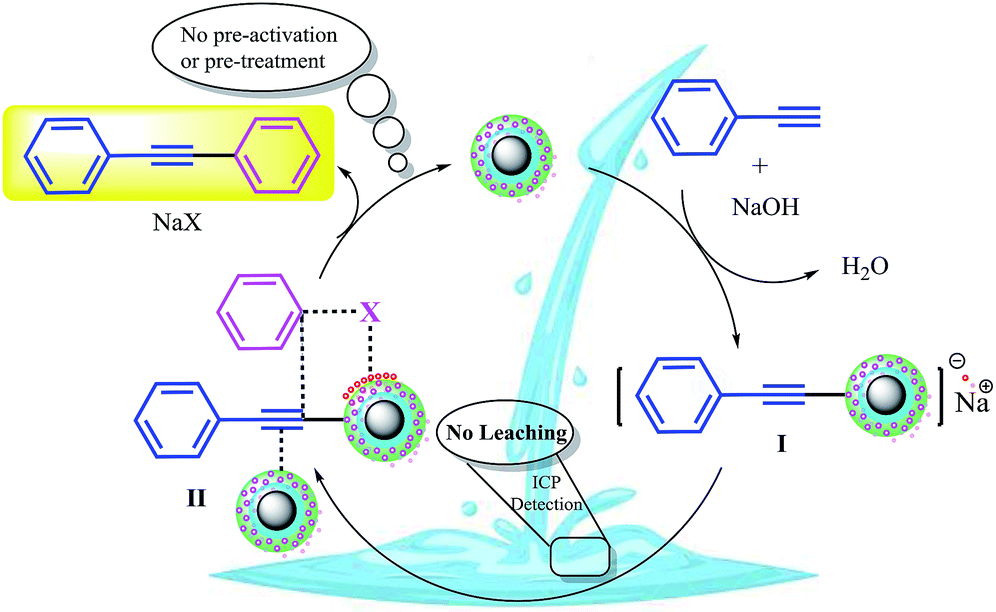 | ||
| Scheme 4 A tentative mechanism for the Sonogashira cross-coupling reactions catalyzed by CuFe2O4@SiO2@ZrO2/SO42−/Cu. | ||
According to the proposed catalyst structure shown in Scheme 4, the catalyst can provide a suitable medium for conducting the reaction in water. As shown in Scheme 5, with respect to the nanocomposite structure of the catalysts, organic compounds were introduced into the catalyst by removing water, which contained catalytic active sites including copper and zirconium; as the concentration and number of effective collisions increased, the reaction proceeded with high efficiency. This structure not only addressed the concerns about the transfer of mass in an aqueous medium, but was also consistent with the high efficiencies achieved for the Sonogashira compounds in this study. In the end, the desired product was removed from the medium. This rigid intermediate also prevented the formation of diyne by-products, which were produced by the coupling of two equivalents of Cu-acetylide in the presence of molecular oxygen (Scheme 1, Glaser type reaction).6
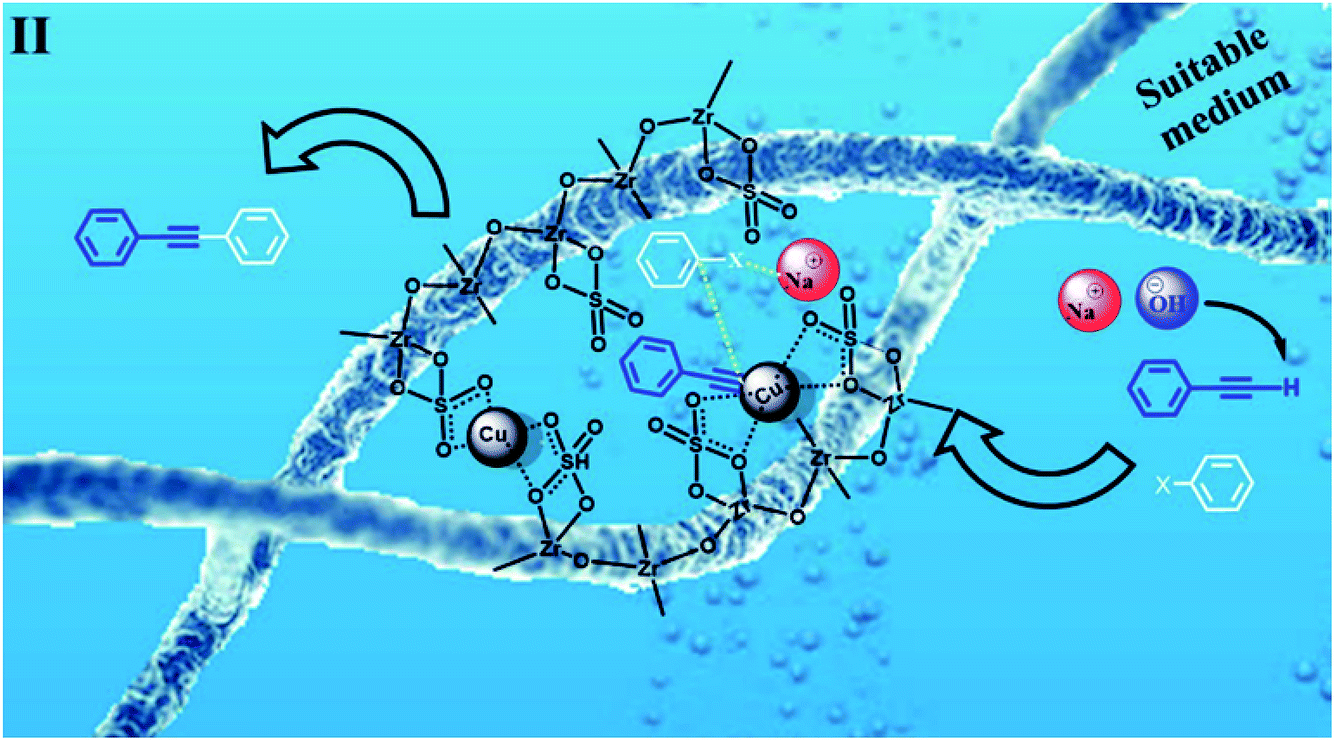 | ||
| Scheme 5 Intermediate II in Scheme 3. A proposed scheme for the possible interaction of the materials in the C–C Sonogashira coupling in water in the presence of CuFe2O4@SiO2@ZrO2/SO42−/Cu. | ||
3.4. Recoverability studies
Stability, durability and, consequently, recyclability of a heterogeneous catalyst are prominent and important factors from economical, energy saving, and environmental points of view;39–41 the rigid inorganic structure of the sulfated zirconium oxide solid support along with the magnetic properties of the CuFe2O4 moieties made the catalyst recoverable and reusable and minimized any metal leaching. The recyclability of the catalyst was investigated in the Sonogashira cross-coupling reaction of phenylacetylene and iodobenzene in the presence of NaOH at 60 °C. The catalyst was recovered in each cycle, washed with EtOH (2 × 5 mL) and reused in the next run without any purification or pre-activation. Fig. 6a shows the corresponding results for nine consecutive runs, and an insignificant loss in efficiency (catalyst yield and reaction yield) was observed.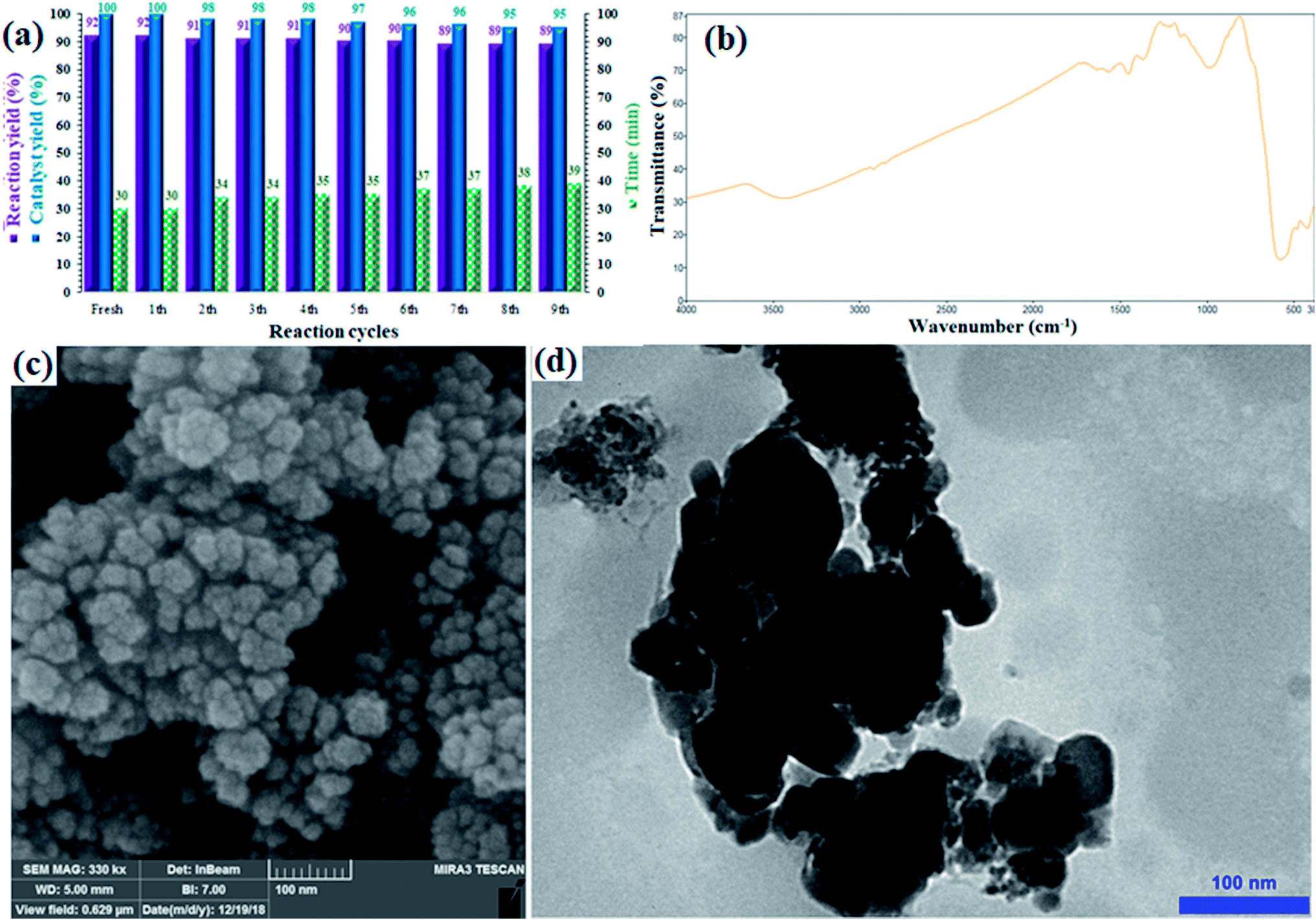 | ||
| Fig. 6 (a) Recovery and reusability of CuFe2O4@SiO2@ZrO2/SO42−/Cu in the Sonogashira reaction of iodobenzene with phenylacetylene under optimized reaction conditions. (b) FTIR spectrum, (c) FE-SEM, and (d) TEM images of the recovered catalyst after the 9th run. | ||
The yield of the coupling product reached 89% (3% loss) after the 9th run. There was also a very intransigent increase in the reaction time until the 9th cycle. The results suggested a rigid and durable structure for CuFe2O4@SiO2@ZrO2/SO42−/Cu as a heterogeneous nano-catalyst. Furthermore, to elucidate the chemical structure as well as stability of the catalyst, the catalyst recovered after the 9th run was studied by FTIR, FE-SEM and TEM analyses (Fig. 6b–d). After comparing the FTIR spectrum of the recovered catalyst with the corresponding FTIR spectrum of the fresh catalyst, it was determined that the structure of the catalyst remained intact during the recycles (Fig. 6b). Moreover, the FE-SEM and TEM images of the recovered catalyst revealed that the morphology of the nanoparticles was the same as revealed in the corresponding images of the fresh catalyst (Fig. 6c and d), respectively. No agglomeration or increase in the particle size was observed even after nine consecutive recycles. Note that the catalyst did not show any detectable metal leaching even after the 9th run. ICP analysis of the residue obtained from the mixture after the 9th run was performed to separately investigate the presence of Fe, Cu, and Zr; for each experiment, a negligible amount of these elements was detected, which confirmed the heterogeneous nature as well as durability of the catalyst during the reactions (Table S1†).
The heterogeneous nature of the catalyst was studied by a hot filtration test.42 The aforementioned model reaction was applied for this test. The catalyst was magnetically removed after 10 min of the reaction (30% yield, GC analysis). The reaction was allowed to proceed, and the conversion was investigated after 2 h by GC. The reaction conversion reached 33%, which confirmed that CuFe2O4@SiO2@ZrO2/SO42−/Cu operated heterogeneously in the mixture, and no metal leaching took place during the reaction.
We compared the catalytic activity of CuFe2O4@SiO2@ZrO2/SO42−/Cu with those reported for the Sonogashira coupling reaction of phenyl acetylene with 4-Me-iodobenzene, 4-NO2-bromobenzene, and 4-MeO-iodobenzene. As shown in Table 4, the present methodology was superior to all the reported catalytic systems in terms of time, catalyst amount and yield of the reaction. Evidently, the reaction conditions were very mild, and the heterogeneous catalyst compromised some advantages such as easy preparation and recycling, minimum metal contamination and economic friendliness.
|
|||||||
|---|---|---|---|---|---|---|---|
| Run | X | R | Catalyst | Condition | Time (h) | Yield (%) | Ref. |
| a GQD = graphene quantum dots.b CS = chitosan.c AO = amidoxime. | |||||||
| 1 | I | 4-Me | PdCu@GQD@Fe3O4 (Pd 0.3 mol%, Cu 0.35 mol%)a | Toluene or DMA/DABCO/50 °C | 24 | 91 | 1 |
| 2 | Au·CuFe2O4@silica | DMA/t-BuOK/115 °C | 48 | 96 | 2 | ||
| 3 | MgO@PdCu (Pd 0.05 mol%, Cu 0.01 mol%) | DMF/DABCO/60 °C | 24 | 97 | 43 | ||
| 4 | CuI (0.2 mol%) PPh3 (4 mol%) | H2O/K2CO3/Ar/140 °C | 24 | 93 | 11 | ||
| 5 | Pd/Fe3O4 NPs (0.2 mol%) | DMF/piperidine/110 °C | 24 | 83 | 44 | ||
| 6 | CuFe2O4@SiO2@ZrO2/SO42−/Cu (0.3 mol% Cu) | H2O/NaOH/60 °C | 30 min | 94 | This work | ||
| 7 | Br | 4-NO2 | Pd-CS (0.1% mol)b | EtOH/H2O/K2CO3/65 °C | 8 | 100 | 45 |
| 8 | Pd/Fe3O4 NPs (0.2 mol%) | DMF/piperidine/110 °C | 24 | 73 | 44 | ||
| 9 | CuFe2O4@SiO2@ZrO2/SO42−/Cu (0.3 mol% Cu) | H2O/NaOH/60 °C | 45 min | 97 | This work | ||
| 10 | I | 4-MeO | Fe3O4/AO/Pd (0.1 mol%)c | DMF/Et3N/80 °C | 0.5 | 98 | 46 |
| 11 | Pd/Fe3O4 NPs(0.2 mol%) | DMF/piperidine/110 °C | 24 | 90 | 44 | ||
| 12 | CuFe2O4@SiO2@ZrO2/SO42−/Cu (0.3 mol% Cu) | H2O/NaOH/60 °C | 55 | 88 | This work | ||
4. Conclusion
Herein, copper was incorporated into sulfated zirconium oxide (ZrO2/SO42−/Cu) supported on copper ferrite nanoparticles (CuFe2O4 NPs); the resultant compound was found to be an efficient magnetically durable catalyst for the Sonogashira reaction in water. The catalyst demonstrated high efficiency not only for aryl iodides but also for aryl bromides and aryl chlorides. Note that the catalytic activity of the modified sulfated zirconium oxide in the organic synthesis has been rarely studied. The catalyst has a monoclinic-tetragonal mixed crystal structure, high thermal stability until 1000 °C, and a 10 emu g−1 saturation magnetization with a 40 nm average size and an irregular shape. The catalyst was further characterized by the EDX and FTIR analyses. This magnetic nanocatalyst could be recycled for at least 9 consecutive runs without any notable loss in activity. The study on the recovered catalyst revealed the high stability and durability of the proposed catalyst. The control experiments completely rule out the synergetic effect of ZrO2/SO42−/Cu and CuFe2O4@SiO2, which leads to the incredible catalytic activity of the proposed catalyst; in literature, the interference of CuFe2O4@SiO2 in the Sonogashira reaction has also been demonstrated. An electron-transfer between Cu and Zr metal sites could be responsible for the proposed oxidative addition and reductive elimination mechanism, in agreement with literature. Furthermore, an interconversion between the sulfate ions on the catalyst surface mediated/facilitated the function of the base in water via adsorption of the cation. The current methodology can indeed replace the expensive Pd-based catalytic systems with highly toxic and expensive phosphine ligands to catalyze the Sonogashira cross-coupling reactions. The use of water as a solvent, short reaction time, high efficiency and absence of by-products are other advantages of the abovementioned catalyst.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the University of Birjand for providing financial support.References
- M. Gholinejad, J. Ahmadi, C. Nájera, M. Seyedhamzeh, F. Zareh and M. Kompany-Zareh, ChemCatChem, 2017, 9, 1442–1449 CrossRef CAS.
- M. Gholinejad, J. Ahmadi and C. Nájera, ChemistrySelect, 2016, 1, 384–390 CrossRef CAS.
- A. Sardarian, M. Kazemnejadi and M. Esmaeilpour, Dalton Trans., 2019, 48, 3132–3145 RSC.
- T. Lauterbach, M. Livendahl, A. Rosellón, P. Espinet and A. M. Echavarren, Org. Lett., 2010, 12, 3006–3009 CrossRef CAS PubMed.
- K. E. Poremba, N. T. Kadunce, N. Suzuki, A. H. Cherney and S. E. Reisman, J. Am. Chem. Soc., 2017, 139, 5684–5687 CrossRef CAS PubMed.
- S. Yugandar, S. Konda and H. Ila, Org. Lett., 2017, 19, 1512–1515 CrossRef CAS PubMed.
- A. K. Sharma, W. C. Sameera, M. Jin, L. Adak, C. Okuzono, T. Iwamoto, M. Kato, M. Nakamura and K. Morokuma, J. Am. Chem. Soc., 2017, 139, 16117–16125 CrossRef CAS PubMed.
- M. Corpet and C. Gosmini, Chem. Commun., 2012, 48, 11561–11563 RSC.
- D. R. Pye, L. J. Cheng and N. P. Mankad, Chem. Sci., 2017, 8, 4750–4755 RSC.
- B. Wang, Y. Wang, X. Guo, Z. Jiao, G. Jin and X. Guo, Catal. Commun., 2017, 101, 36–39 CrossRef CAS.
- Y. Liu, V. Blanchard, G. Danoun, Z. Zhang, A. Tlili, W. Zhang and M. Taillefer, ChemistrySelect, 2017, 2, 11599–11602 CrossRef CAS.
- S. Asha, A. M. Thomas, S. M. Ujwaldev and G. Anilkumar, ChemistrySelect, 2016, 1, 3938–3941 CrossRef CAS.
- W. Sun, L. Gao, X. Sun and G. Zheng, Dalton Trans., 2018, 47, 5538–5541 RSC.
- X. Li, K. Nagaoka, L. J. Simon, R. Olindo, J. A. Lercher, A. Hofmann and J. Sauer, J. Am. Chem. Soc., 2005, 127, 16159–16166 CrossRef CAS PubMed.
- A. Galadima, R. P. K. Wells and J. A. Anderson, Appl. Petrochem. Res., 2012, 1, 35–43 CrossRef CAS.
- C. Khatri, M. K. Mishra and A. Rani, Fuel Process. Technol., 2010, 91, 1288–1295 CrossRef CAS.
- B. M. Reddy, P. M. Sreekanth and P. Lakshmanan, J. Mol. Catal. A: Chem., 2005, 237, 93–100 CrossRef CAS.
- V. S. Marakatti, G. V. Shanbhag and A. B. Halgeri, Appl. Catal., A, 2013, 451, 71–78 CrossRef CAS.
- S. Ghiami, M. A. Nasseri, A. Allahresani and M. Kazemnejadi, React. Kinet., Mech. Catal., 2019, 126, 383–398 CrossRef CAS.
- S. Swami, A. Agarwala and R. Shrivastava, New J. Chem., 2016, 40(11), 9788–9794 RSC.
- R. F. Perez, S. J. Canhaci, L. E. Borges and M. A. Fraga, Catal. Today, 2017, 289, 273–279 CrossRef CAS.
- K. Yang, H. Li, S. Zhao, S. Lai, W. Lai, Y. Lian and W. Fang, Ind. Eng. Chem. Res., 2018, 57, 3855–3865 CrossRef CAS.
- M. K. Mishra, B. Tyagi and R. V. Jasra, J. Mol. Catal. A: Chem., 2004, 223, 61–65 CrossRef CAS.
- F. Heshmatpour and R. B. Aghakhanpour, Adv. Powder Technol., 2012, 23, 80–87 CrossRef CAS.
- D. Sarkar, D. Mohapatra, S. Ray, S. Bhattacharyya, S. Adak and N. Mitra, Ceram. Int., 2007, 33, 1275–1282 CrossRef CAS.
- A. S. Ethiraj and D. J. Kang, Nanoscale Res. Lett., 2012, 7, 70 CrossRef PubMed.
- J. Kurian and M. J. Mathew, J. Magn. Magn. Mater., 2018, 451, 121–130 CrossRef CAS.
- W. Ponhan and S. Maensiri, Solid State Sci., 2009, 11, 479–484 CrossRef CAS.
- G. Shi, F. Yu, Y. Wang, D. Pan, H. Wang and R. Li, Renewable Energy, 2016, 92, 22–29 CrossRef CAS.
- D. P. Volanti, D. Keyson, L. S. Cavalcante, A. Z. Simões, M. R. Joya, E. Longo, J. A. Varela, P. S. Pizani and A. G. Souza, J. Alloys Compd., 2008, 459, 537–542 CAS.
- A. Othonos and M. Zervos, Nanoscale Res. Lett., 2011, 6, 622 CrossRef.
- H. Sharghi, S. Ebrahimpourmoghaddam and M. M. Doroodmand, J. Organomet. Chem., 2013, 738, 41–48 CrossRef CAS.
- N. Liu, X. Guo, A. Navrotsky, L. Shi and D. Wu, J. Catal., 2016, 342, 158–163 CrossRef CAS.
- F. Lonyi and J. Valyon, J. Therm. Anal. Calorim., 1996, 46, 211–218 CrossRef CAS.
- K. Parida, V. Quaschning, E. Lieske and E. Kemnitz, J. Mater. Chem., 2001, 11, 1903–1911 RSC.
- A. K. Aboul-Gheit, F. K. Gad, G. M. Abdel-Aleem, D. S. El-Desouki, S. M. Abdel-Hamid, S. A. Ghoneim and A. H. Ibrahim, Egypt. J. Pet., 2014, 23, 303–314 CrossRef.
- A. Sagadevan and K. C. Hwang, Adv. Synth. Catal., 2012, 354, 3421–3427 CrossRef CAS.
- S. V. Jadhav, K. M. Jinka and H. C. Bajaj, Appl. Catal., A, 2010, 390, 158–165 CrossRef CAS.
- M. Kazemnejadi and A. R. Sardarian, RSC Adv., 2016, 6, 91999–92006 RSC.
- M. Kazemnejadi, A. Shakeri, M. Nikookar, R. Shademani and M. Mohammadi, R. Soc. Open Sci., 2018, 5, 171541 CrossRef CAS PubMed.
- M. Kazemnejadi, A. Shakeri, M. Mohammadi and M. Tabefam, J. Iran. Chem. Soc., 2017, 14, 1917–1933 CrossRef CAS.
- M. Kazemnejadi, M. Nikookar, M. Mohammadi, A. Shakeri and M. Esmaeilpour, J. Colloid Interface Sci., 2018, 527, 298–314 CrossRef CAS PubMed.
- M. Gholinejad, M. Bahrami, C. Nájera and B. Pullithadathil, J. Catal., 2018, 363, 81–91 CrossRef CAS.
- M. Nasrollahzadeh, S. M. Sajadi, A. Rostami-Vartooni and M. Khalaj, J. Mol. Catal. A: Chem., 2015, 396, 31–39 CrossRef CAS.
- S. Frindy, A. Primo, M. Lahcini, M. Bousmina, H. Garcia and A. El Kadib, Green Chem., 2015, 17, 1893–1898 RSC.
- H. Veisi, A. Sedrpoushan, B. Maleki, M. Hekmati, M. Heidari and S. Hemmati, Appl. Organomet. Chem., 2015, 29, 834–839 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR, 13C-NMR and mass spectra of coupling products. See DOI: 10.1039/c9ra03406d |
| This journal is © The Royal Society of Chemistry 2019 |