
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: Elevation of USP4 antagonizes oxygen glucose deprivation/reoxygenation-evoked microglia activation and neuroinflammation-mediated neurotoxicity via the TRAF6-NF-κB signaling
Zhaoxia Wang a,
Xinming Lib,
Zhixing Shaoa,
ZhengFanga and
Yueping Zhai*b
a,
Xinming Lib,
Zhixing Shaoa,
ZhengFanga and
Yueping Zhai*b
aNeurology Department, Xi'an City Ninth Hospital, Xi'an 710004, China
bNeurology Department, Xi'an Gaoxin Hospital, No. 16 South Tuanjie Road, Xi'an 710075, China. E-mail: yueping_zhai3@163.com; Tel: +86-029-88333126
First published on 1st August 2019
Abstract
An ischemic stroke is a devastating neurological disease with the typical occurrence of brain ischemia/reperfusion (I/R) injury, and it has high mortality and disability globally. Microglia activation after a stroke results in the release of pro-inflammatory cytokines that can further aggravate brain damage. A recent study confirmed the potential role of ubiquitin-specific peptidase 4 (USP4) in the injury process. Nevertheless, the role and mechanism of USP4 during an ischemic stroke remain elusive. In this research, we simulated an I/R injury by oxygen glucose deprivation/reoxygenation (OGD/R) in vitro and confirmed the obvious down-regulation of USP4 in microglia under OGD/R conditions. Moreover, USP4 elevation antagonized the OGD/R-induced microglia proliferation and activation by suppressing the NO levels and the expression of the microglial marker IBA-1. Additionally, the overexpression of USP4 suppressed the release of microglia activation-induced pro-inflammatory cytokines, including IL-1β, IL-6, and TNF-α. Intriguingly, incubation with the conditioned medium from the microglia under OGD/R conditions induced neurotoxicity by inhibiting cell viability and increasing the LDH release, apoptosis, and caspase-3 activity, which were reversed following USP4 overexpression. Mechanism analysis corroborated that USP4 up-regulation repressed the OGD/R-induced activation of TRAF6-NF-κB signaling. Notably, restoring the TRAF6 signaling ameliorated the suppressive effects of USP4 elevation on microglia activation, inflammation, and the subsequent neuron injury. These findings suggest that USP4 may alleviate ischemic stroke by restraining microglia-mediated neuro-inflammation and neurotoxicity via the TRAF6-NF-κB pathway, due to which it is a promising therapeutic agent against strokes.
Introduction
Stroke is a cerebrovascular disease with one major clinical type being an ischemic stroke.1 Currently, ischemic strokes constitute a worldwide threat to health due to the high mortality and serious disability in adults. Epidemiological data suggest that approximately 16.9 million people suffer from strokes annually, which is predicted to rise to 77 million by 2030.2 Patients who have experienced strokes are usually subjected to a heavy economic burden, and the overall cost is projected to be $2.2 trillion by 2050 in the USA.3 The restoration of the blood supply is a common strategy to reduce more extensive brain injury after an acute stroke. Nevertheless, reperfusion after cerebral ischemia is often accompanied by some disastrous outcomes such as intracranial hemorrhage, edema, and infarction.4 Consequently, a better understanding of the molecular mechanism orchestrating an ischemic stroke is urgent for developing an effective strategy against ischemic strokes.Increasing evidence has recognized the neuroinflammatory response as a predominant contributor for the progression of brain-related diseases, including cerebral ischemia/reperfusion (I/R) injury.5,6 Normally, microglia are the major resident immune cells in the central nervous system and often facilitate host defense. Intriguingly, microglia act as sensors that rapidly activate within a few hours after injury, and they are the major contributors to the inflammatory response during brain I/R injury. Compelling research has confirmed that the excessive activation of microglia leads to deleterious and neurotoxic consequences after an ischemic stroke due to the abundant release of pro-inflammatory cytokines and cytotoxic factors such as interleukin-1β (IL-1β), IL-6, and tumor necrosis factor-α (TNF-α).7–9 Recently, emerging evidence has confirmed that targeting the microglial inflammatory response following I/R can ameliorate brain injury, implying an efficacious and potential therapeutic approach to alleviate strokes.6,10
Microglia activation can induce neuronal inflammation after I/R by activating the inflammation-related pathways, such as nuclear factor-κB (NF-κB) signaling.11 Ubiquitin-specific peptidase 4 (USP4) belongs to the ubiquitin enzyme family that contains a ZF domain and a DUSP domain. Numerous experiments have focused on the effects of USP4 on cancer. Recently, USP4 has become a subject of interest in regulating multiple inflammatory pathways in several diseases. For experiments on nonalcoholic fatty livers, USP4 deletion induces the activation of NF-κB and metabolic dysfunctions, ultimately exacerbating hepatic steatosis and inflammatory responses.12 Furthermore, USP4 may act as a novel therapeutic target to treat Th17-modulated autoimmune diseases.13 Notably, the cessation of USP4 induces microglial activation and subsequent inflammation through NF-κB in secondary spinal cord injury.14 A recent finding has corroborated that the USP4 deficiency exacerbates hepatic I/R injury by enhancing liver inflammation and hepatocyte apoptosis.15 Up to now, the role and mechanism of USP in brain I/R remain poorly elucidated.
In the present study, we sought to elucidate the function of USP4 in simulated cerebral I/R injury in vitro by exposing microglia to oxygen glucose deprivation/reoxygenation (OGD/R). Furthermore, the effects of microglia activation-evoked neurotoxicity were also investigated.
Results
Exposure to OGD/R restrains the expression of USP4 in microglia
To investigate the role of USP4 in cerebral I/R injury, we preliminarily explored the levels of USP4 in OGD/R-simulated I/R in vitro. As presented in Fig. 1A, the prepared cells positively stained with IBA-1 and MAP-2 exhibit microglial and neuronal morphologies, respectively (Fig. 1B). Intriguingly, there was a considerable decrease in the USP4 mRNA levels in microglia under OGD/R conditions compared with that for the control groups (Fig. 1C). Analogously, exposure to OGD/R also induced a concomitant increase in the USP4 protein in microglia (Fig. 1D). Therefore, these results suggest a potential function of USP4 in cerebral I/R damage.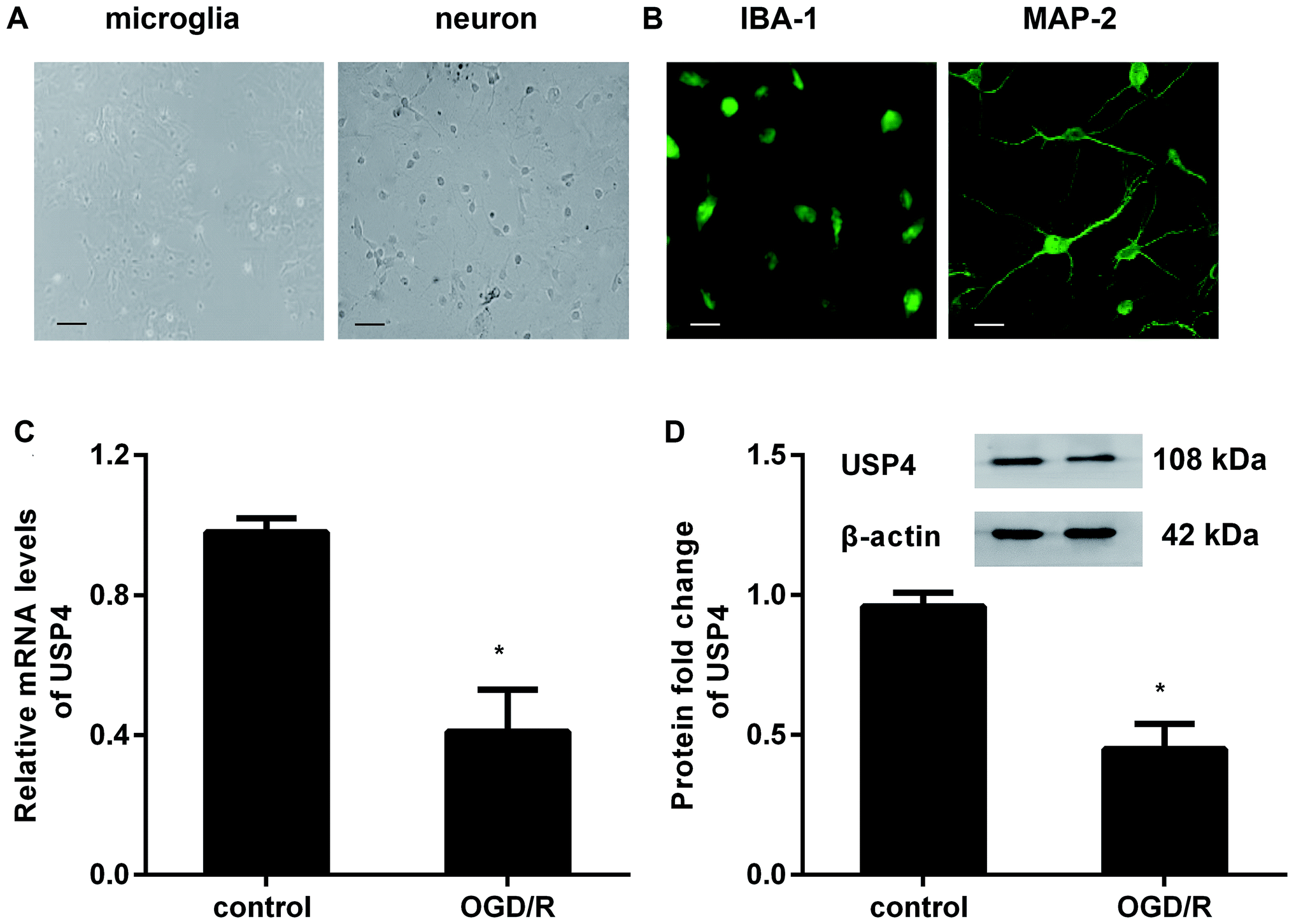 | ||
| Fig. 1 The expression of USP4 decreased in microglia under OGD/R conditions. (A) Microglia and neuronal morphology under light microscopy after 5 days of culture. Scale bars, 40 μM. (B) For identification, cells were separately stained with IBA-1 and MAP-2; then, they were observed by a fluorescence microscope. Scale bars, 10 μM. (C) The isolated microglial cells were incubated with an oxygen glucose deprivation condition for 3 h to simulate an ischemic insult. Then, the cells were exposed to normoxic conditions (95% air, 5% CO2) to mimic reperfusion. The mRNA levels of USP4 were determined by qRT-PCR. (D) The protein expression of USP4 was analyzed by western blotting. *P < 0.05 versus control groups. | ||
Elevation of USP4 inhibits microglia proliferation and activation under OGD/R conditions
To elucidate the involvement of USP4 in brain I/R, the effects of USP4 on microglia proliferation and activation were evaluated. As shown in Fig. 2A, transfection with the recombinant USP4 vector notably increases the transcriptional levels of USP4 in microglia, which is concomitant with similar up-regulation of the USP4 protein expression (Fig. 2B). Intriguingly, exposure to OGD/R increased cell proliferation relative to that of the control groups, which was abrogated following the UPS4 elevation (Fig. 2C). Moreover, enhanced mRNA (Fig. 2D) and protein (Fig. 2E) levels of the microglia marker IBA-1 were determined in the microglia after exposure to OGD/R, which were both suppressed following USP4 overexpression. Additionally, the OGD/R-induced NO production in the microglia was also attenuated after USP4 overexpression (Fig. 2F). These findings indicated the adverse effects of USP4 on microglia proliferation and activation.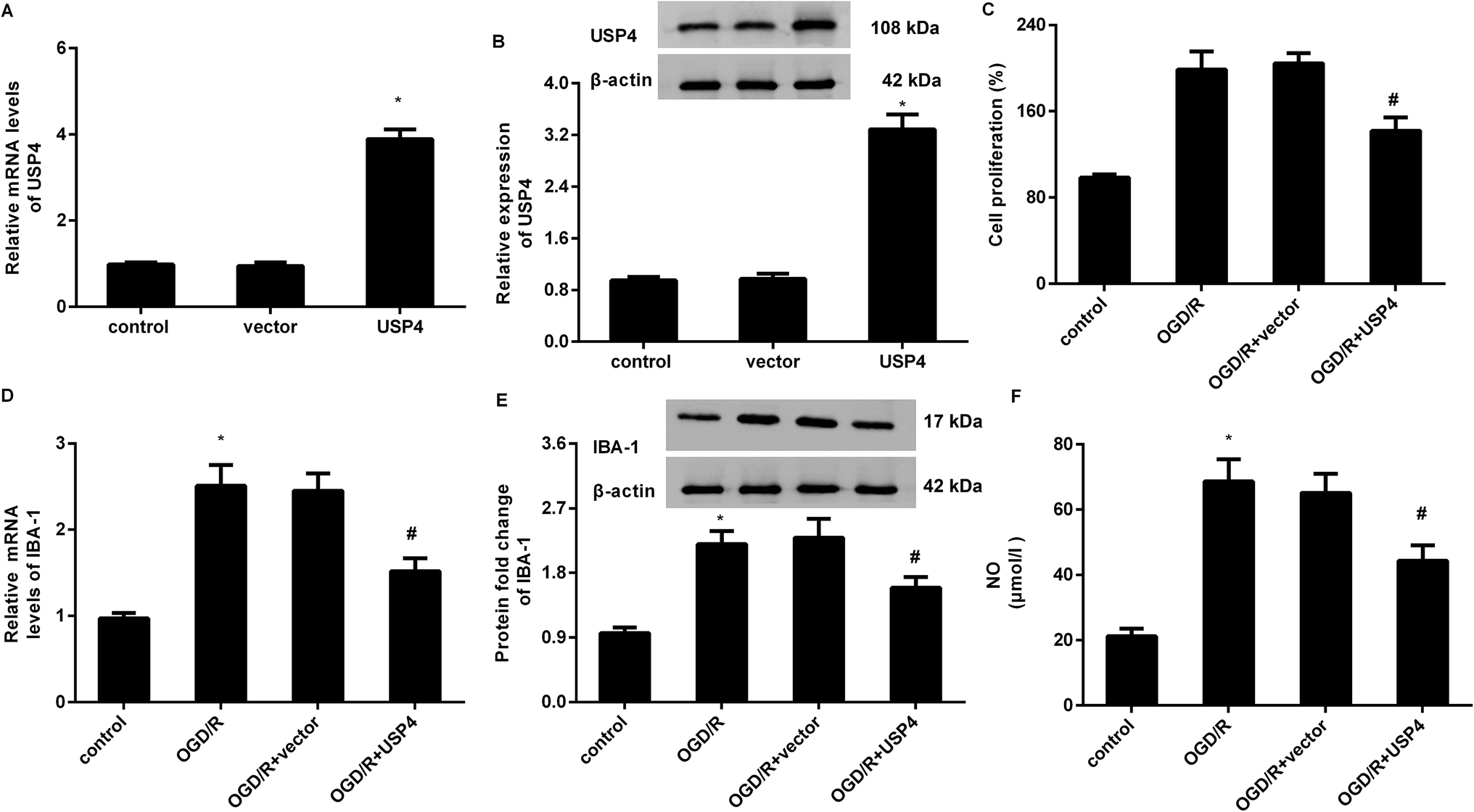 | ||
| Fig. 2 Overexpression of USP4 suppressed microglia proliferation and activation under OGD/R conditions. (A) Microglia were transfected with the recombinant USP4 vector or empty vector and the transcriptional levels of USP4 mRNA were analyzed. (B) Western blotting was performed to determine the corresponding protein levels of USP4. (C) Cells transfected with USP4 plasmids were exposed to OGD/R and the cell proliferation was then evaluated using an MTT assay. (D and E) The effects on microglia marker IBA mRNA (D) and protein expression (E) were detected. (F) The contents of NO were measured using commercial kits. *P < 0.05 vs. control groups, #P < 0.05 vs. OGD/R groups. | ||
USP4 overexpression antagonized microglia activation-evoked inflammatory response
The microglial activation-evoked inflammatory response has extraordinary roles in the development of the brain I/R injury.9,10 We next elucidated the function of USP4 in the microglia activation-induced inflammation. As shown in Fig. 3A, exposure to OGD/R increases the transcriptional levels of pro-inflammatory cytokines IL-1β (Fig. 3A), which is concomitant with the elevation of IL-6 (Fig. 3B) and TNF-α (Fig. 3C) mRNA levels. Notably, USP4 overexpression antagonized OGD/R-induced IL-1β, IL-6, and TNF-α transcriptional levels (Fig. 3A–C). Intriguingly, the OGD/R treatment promoted the release of IL-1β (Fig. 3D), IL-6 (Fig. 3E), and TNF-α (Fig. 3F) from microglia, which were offset after USP4 overexpression.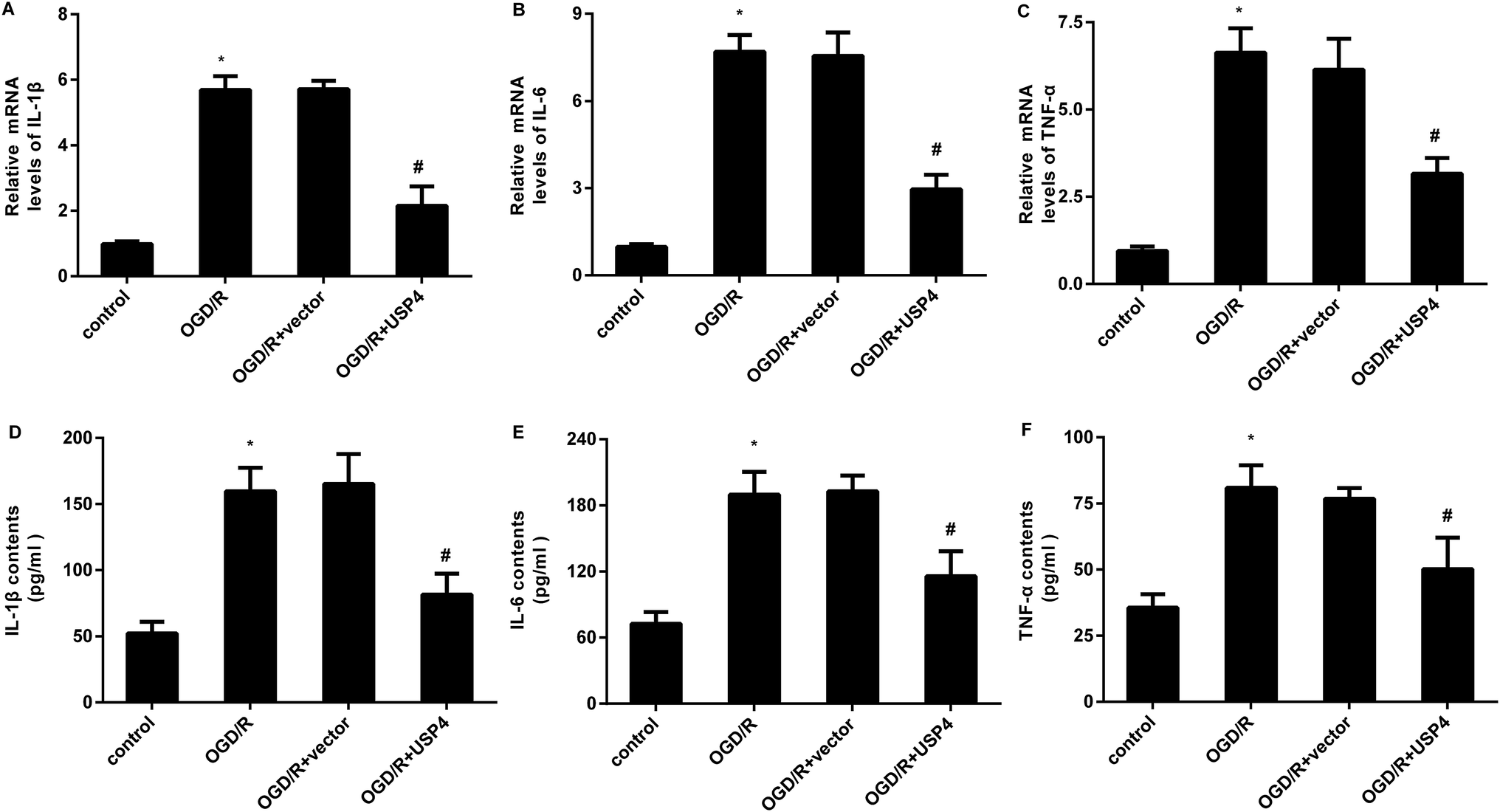 | ||
| Fig. 3 USP4 elevation inhibited the OGD/R-induced inflammatory response in microglia. Cells were transfected with pcDNA-USP4 plasmids prior to OGD/R exposure. Then, the transcriptional levels of IL-1β (A), IL-6 (B) and TNF-α (C) were determined by qRT-PCR. (D–F) An ELISA assay was conducted to measure the concentrations of IL-1β (A), IL-6 (B) and TNF-α (C) in USP4-overexpressed microglia under OGD/R conditions. *P < 0.05, #P < 0.05. | ||
Up-regulation of USP4 ameliorates neurotoxicity induced by conditioned medium (CM) from OGD/R-activated microglia
The activation of microglia aggravates neuronal injury during cerebral I/R by releasing cytotoxic factors such as IL-1β and NO.11,16 Therefore, we further analyzed the function of USP4 in microglia-triggered neurotoxicity. Toxic CM from the OGD/R-treated microglia obviously restrained neuronal viability (Fig. 4A), which was negated when the neurons were incubated with CM from the USP4-overexpressed microglia under OGD/R conditions. Furthermore, exposure to CM from the OGD/R groups facilitated LDH release from the neurons (Fig. 4B), cell apoptosis (Fig. 4C) and caspase-3 activity (Fig. 4D), indicating neurotoxic injury under microglia activation. Importantly, the elevation of USP4 notably ameliorated the adverse effects of CM from the OGD/R-stimulated microglia on LDH release, cell apoptosis, and caspase-3 activity (Fig. 4B–D).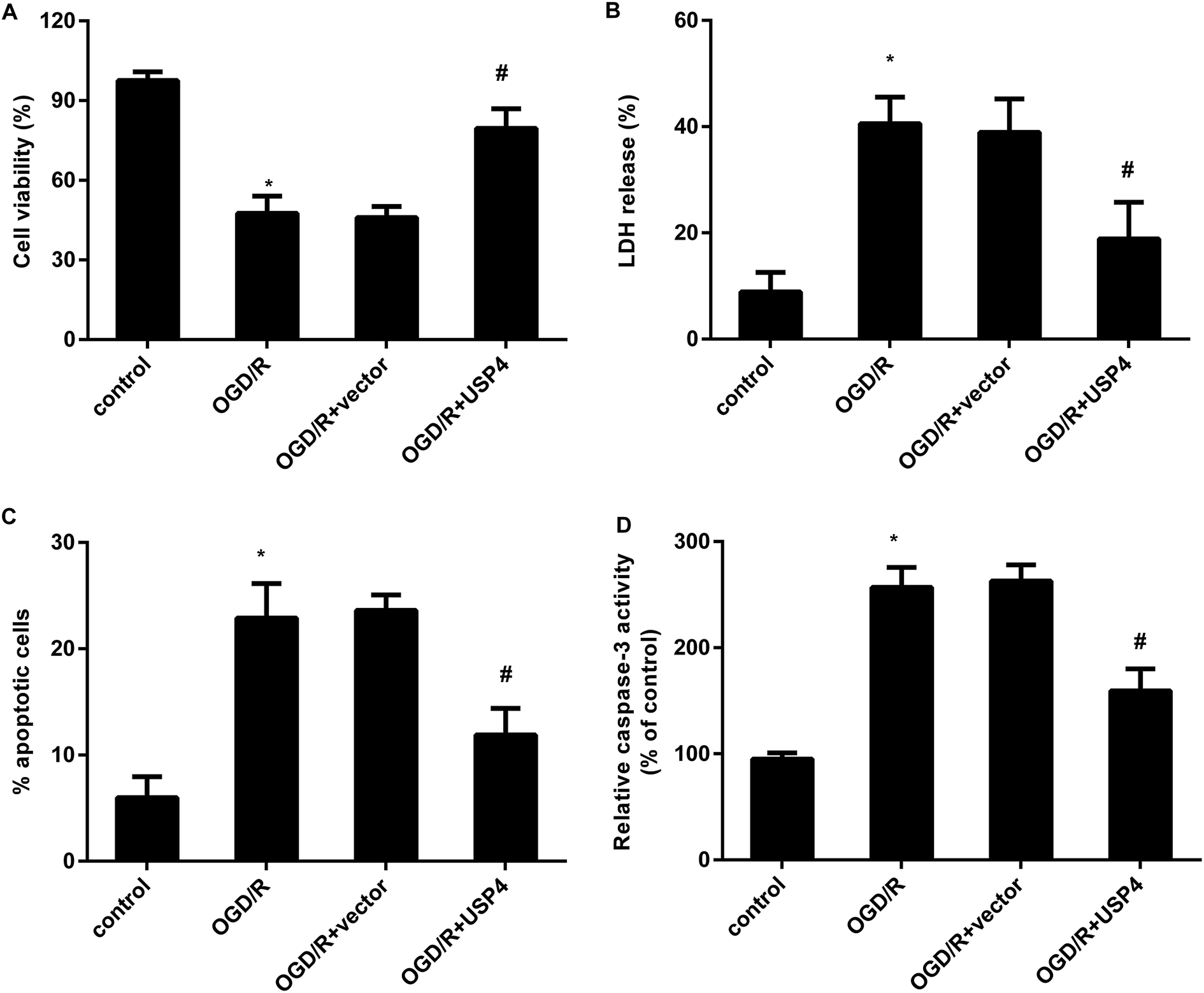 | ||
| Fig. 4 Enhancement of USP4 alleviated neurotoxicity induced by incubation with a conditioned medium (CM) from OGD/R-activated microglia. (A) The conditioned medium (CM) was collected from OGD/R-activated microglia that were pre-transfected with pcDNA-USP4 vectors. Then, the primary neurons were incubated with CM from microglia. Cell viability was evaluated using an MTT assay. (B) LDH release was then tested using a colorimetric assay kit. (C) A flow cytometer was applied to analyze cell apoptosis. (D) The effects on caspase-3 activity were also assessed. *P < 0.05, #P < 0.05. | ||
Overexpression of USP4 suppresses the OGD/R-evoked activation of the TRAF6-NF-κB signaling in microglia
Increasing evidence has confirmed that the activation of TRAF6-NF-κB signaling is involved in inflammatory responses and ischemia/reperfusion injuries.17,18 To elaborate the mechanism underlying the USP4-mediated function in OGD/R-treated microglia, we detected the activation of the TRAF6-NF-κB signaling. As shown in Fig. 5A–C, OGD/R administration increases the expressions of TRAF6 (Fig. 5A and B) and p-p65 NF-κB (Fig. 5A and C) levels, and these increases were offset following the USP4 elevation, indicating the inhibitory effects of USP4 on the OGD/R-activated TRAF6-NF-κB pathway.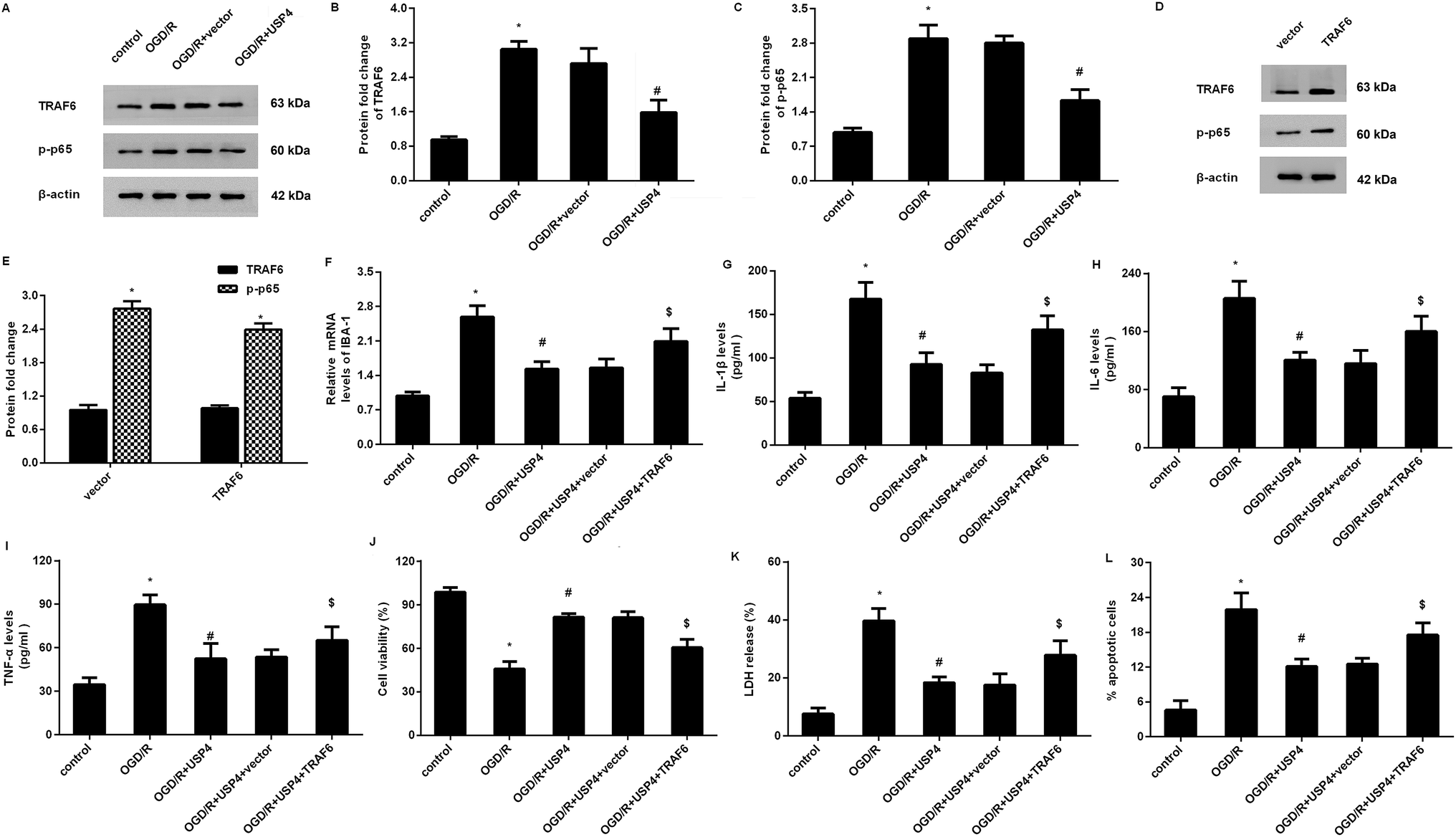 | ||
| Fig. 5 USP4 regulated microglia activation-induced inflammation and neuron injury by blocking the TRAF6-NF-κB signaling. (A) After transfection with the recombinant USP4 vectors, microglia were exposed to OGD/R. Then, the expression of TRAF6, p-p65 NF-κB was detected by western blotting. (B and C) The corresponding quantified analysis of TRAF6, p-p65 NF-κB was evaluated by the Image J software. (D and E) Effects of pcDNA-TRAF6 transfection on the protein levels of TRAF6 and p-p65NF-κB. (F) The mRNA levels of IBA-1 were measured by qRT-PCR. (G–I) The concentrations of IL-1β (G), IL-6 (H) and TNF-α (I) were detected by an ELISA assay. (J–L) The subsequent effects on cell viability (J), LDH release (K) and apoptosis (L) were also evaluated. *P < 0.05 vs. control groups, #P < 0.05 vs. OGD/R groups, and $P < 0.05 vs. OGD/R + USP4 groups. | ||
Restoring the TRAF6 signaling overturns the effects of USP4 on microglia activation and neurotoxicity under OGD/R conditions
To further elucidate whether the TRAF6 signaling was responsible for the USP4-mediated function in OGD/R-treated microglia, we re-activated this signaling by transfection with recombinant pcDNA-TRAF6 plasmids as evidence that the pcDNA-TRAF6 transfection enhanced the expressions of TRAF6 and p-p65 NF-κB (Fig. 5D and E). Intriguingly, the reactivation of the TRAF6 pathway reversed the expression of IBA-1 mRNA in USP-treated cells under OGD/R conditions (Fig. 5F). Moreover, the suppressive effects of the USP4 elevation on the OGD/R-induced production of IL-1β (Fig. 5G), IL-6 (Fig. 5H), and TNF-α (Fig. 5I) were negated after reactivating the TRAF6 signaling. Additionally, incubation with CM from USP4-overexpressed microglia under OGD/R conditions elevated neuronal viability (Fig. 5J) as well as suppressed the LDH release (Fig. 5K) and apoptosis (Fig. 5L). In striking contrast, these changes were reversed after reactivating the TRAF6 signaling.Discussion
Cerebral I/R is a common clinical phenotype in the pathological process of ischemic stroke and it is the second leading cause of death worldwide.19 Intriguingly, targeting microglial inflammation has become a subject of interest in ischemic stroke therapy.5,20,21 In the current research, we utilized OGD/R to mimic I/R in vivo and confirmed the down-regulation of USP4 in OGD/R-exposed microglia. Importantly, the up-regulation of USP4 suppressed microglia proliferation and activation under OGD/R conditions. Moreover, USP4 elevation ameliorated microglia activation-evoked pro-inflammatory cytokine (IL-1β, IL-6, and TNF-α) production and neuron injury by inhibiting the TRAF6-NF-κB signaling. Cumulatively, the results of the current study indicate that USP4 may suppress microglia activation-induced inflammation and neurotoxicity under simulated I/R conditions, due to which it is a promising therapeutic target for ischemic stroke.Neuroinflammation is a critical contributor to the progression of a stroke.22,23 During this process, the rapid activation and proliferation of microglia are the characterizations of inflammatory responses in brain ischemic stroke.16,24 Intriguingly, recent research has revealed the involvement of USP4 in inflammatory progression. For instance, USP4 has been validated to be a novel therapeutic target for Th17-modulated autoimmune diseases.13 To elucidate the function of USP4 in cerebral I/R, we detected the expression levels of USP4 in microglia upon OGD/R. Notably, the OGD/R exposure induced down-regulation of USP4 in microglia. Furthermore, the treatment with OGD/R also increased microglia proliferation and activation. In vivo, increasing evidence has substantiated the activation and proliferation of microglia in infarct cortical regions after an ischemic stroke.16 Of note, the overexpression of USP4 suppressed microglia proliferation, NO production, and the expression of the microglial marker IBA-1, indicating the adverse effects of USP4 on microglia activation. More importantly, the USP4 elevation restrained the release of microglia activation-evoked pro-inflammatory cytokines, including IL-1β, IL-6, and TNF-α. Therefore, these findings imply that USP4 up-regulation may restrain microglia-induced neuroinflammation under simulated ischemic stroke conditions.
It is a fact that excessive microglia activation will induce the release of abundant pro-inflammatory cytokines (e.g., IL-1β) and cytotoxic factors (e.g., NO), which may contribute to neuron injuries and expand brain injuries after an ischemic stroke.9,22,24 In this study, our findings confirmed that the conditioned medium from activated microglia upon OGD/R resulted in neurotoxicity by suppressing neuronal proliferation and increasing the LDH release, cell apoptosis, and caspase-3 activity. Analogous to these results,7,8 previously reported results have also demonstrated microglia activation-induced ischemia-like neuron damage. Of interest, the up-regulation of USP4 dramatically reversed the suppressive effects of the conditioned medium from OGD/R-treated microglia on neuron injury. These data suggest that USP4 may ameliorate brain I/R injury by attenuating the microglia activation-evoked inflammatory response and subsequent neurotoxicity. An emerging study has corroborated that the deficiency of USP4 exacerbates hepatic I/R injury by promoting a hepatic inflammatory response.15
We next elaborated the mechanism underlying the function of USP4 in microglia-mediated neuron injury and substantiated that USP4 inhibited the activation of the TRAF6-NF-κB signaling in OGD/R-treated microglia. The nuclear factor-κB (NF-κB) pathway has critical roles in inflammation and immune response and can be activated during the process of microglia activation-evoked neuronal inflammation after brain I/R.10,11 Many studies have confirmed the aberrant activation of the TRAF6-NF-κB pathway during the progression of I/R.17,25 For instance, the up-regulation of TRAF6 increases neuron death through neuroinflammation and neuro-oxidative signals and ultimately exacerbates the ischemic stroke.26 The blockage of the TRAF6 and NF-κB pathways by miR-125 antagonizes inflammation and protects the liver from I/R injury.17 In rats, the inhibition of the TRAF/NF-κB pathway after middle cerebral artery occlusion (MCAO) can notably attenuate neuroinflammation and brain infarction.18 Notably, this study found that reactivating the TRAF6 pathway negated the effects of USP4 on microglial inflammation and subsequent neuron injuries.
Collectively, this study highlighted that the USP4 elevation suppressed microglia activation-evoked inflammation and neuron injury by inhibiting the TRAF6-NF-κB pathway. Therefore, these findings indicate that the USP4 administration may ameliorate an ischemic stroke by abrogating the microglia-induced inflammatory response and neuron damage, affording a promising strategy to fight strokes. Nevertheless, the current research only elucidated the roles of USP4 in vitro. Does USP4 exert the ideal function in strokes in vivo? Is the TRAF6-NF-κB pathway involved in USP4-mediated efficacy in strokes? Are other signaling pathways involved in this process? All these problems will be investigated in our future work.
Materials and methods
Isolation and culture of microglia and neurons
Primary microglia and neurons were prepared from the cerebral cortices of postnatal day-1 Sprague–Dawley rats (the Center of Laboratory Animals, the Fourth Military Medical University) as previously described.9 All experimental protocols were conducted according to the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. Briefly, cortices were chopped and digested with 0.25% trypsin after mechanical disruption. For microglia isolation, the dissociated cells were seeded in flasks pre-coated with poly-L-lysine (Sigma-Aldrich, St. Louis, MO, USA) and cultured in a medium that was replaced every 3 days with antibiotic-free minimum Eagle's medium (Gibco, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS). Approximately 10 days later, microglial cells were harvested by shaking the flasks in a table concentrator at 250 rpm for 0.5 h at 37 °C. The purities of the isolated microglia exceeded 95%, as demonstrated by detecting the microliga-specific marker IBA-1.For neuron isolation, the dissociated cells were plated into plates that were pretreated with poly-L-lysine. Then, the cells were cultured in high glucose DMEM medium supplemented with 10% FBS, 10% horse serum, 2 mM glutamine, and antibiotics (100 U mL−1 penicillin and 100 mg mL−1 streptomycin) (Sigma). After incubation for 24 h, the culture medium was changed to DMEM medium (feeding medium) containing 5% horse serum, 100 U mL−1 penicillin, 100 mg mL−1 streptomycin, 2 mM glutamine, 0.01% N2, and 0.04% B27 (Gibco). Three days later, 10 μM cytosine arabinoside (Sigma) was supplemented into the medium for 24 h to prevent the growth of non-neuronal cells. The culture medium was replaced with the feeding medium every 3 days. Approximately 10 days later, over 95% cells were identified as neurons by determining the neuron-specific marker MAP-2. All cells were maintained in a humidified incubator with 5% CO2 at 37 °C.
Construction of the recombinant plasmids
Total RNA from microglia was prepared using an RNeasy Mini kit (Qiagen, Valencia, CA, USA). The cDNA was then synthesized based on the extracted RNA using the SuperScript II First Strand Synthesis System (Invitrogen, Carlsbad, CA, USA). Afterwards, rat full-length USP4 and TRAF6 cDNA were amplified by PCR, followed by digestion with restriction enzymes. Then, the prepared cDNA was inserted into the pcDNA3.1(+) vectors (Invitrogen) to construct the recombinant pcDNA-USP4, pcDNA-TRAF6 plasmids. Microglial cells were then seeded into 6-well plates and grown to 70–80% confluence. Cells were subsequently transfected with the recombinant plasmids or empty vector using Lipofectamine 2000 reagents (Invitrogen). The efficacy was evaluated by western blotting.Microglia exposure to oxygen glucose deprivation/reoxygenation (OGD/R) model
To simulate the ischemia/reperfusion injury in vivo, cells were exposed to OGD/R in vitro. In brief, cells were cultured in glucose-free DMEM medium in a chamber containing 1% O2, 95% N2, and 5% CO2 for 3 h to mimic ischemic insult. Afterwards, the cells were returned to the normoxic condition (95% air, 5% CO2) for 12 h in a regular medium to mimic reperfusion. All cells were incubated at 37 °C.Quantitative real-time RT-PCR (qRT-PCR)
The transcriptional levels of USP4, IBA-1, IL-1β, IL-6, and TNF-α were detected by the qRT-PCR assay. All reactions were conducted according to the manufacturer's instructions provided by the SYBR Premix Ex TaqTM II Kit (TaKaRa, Dalian, China). qRT-PCR was performed on an ABI Prism7500 sequence detection system (Applied Biosystems) at 95 °C for 10 min; then, we conducted 40 cycles at 95 °C for 10 s, 58.5 °C for 30 s, and 72 °C for 30 s. The specific primers were synthesized by Shanghai Sangon Co., Ltd (Shanghai, China) as follows: USP4 (sense, 5′-CCAGCCTCAGAAGAAGAAGAAG-3′; anti-sense, 5′-CAAGGGTCTCCATGGTAGTAAAG-3′) and IBA-1 (sense, 5′- TCCGAGGAGACGTTCAGTTA-3′; anti-sense, 5′-GTTGGCTTCTGGTGTTCTTTG-3′). The primers for IL-1β, IL-6, and TNF-α were synthesized as previously reported.16 To normalize the expression of these genes, β-actin was applied as an internal standard. The relative expression of the target genes was calculated using 2−ΔΔCt.Western blotting assay
Microglial cells were collected and homogenized in RIPA lysis buffer with protease inhibitor, and the total protein concentration was analyzed by the BCA protein assay kit (Beyotime, Shanghai, China). Equal amounts of protein samples were loaded and separated by 12% SDS–PAGE. Then, the target protein was transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA, USA). Following the incubation with 5% non-fat milk to interdict the non-specific binding, the membranes were incubated with primary antibodies against rat USP4 (Santa Cruz), IBA-1, TRAF6, and p-p65 NF-κB (obtained from Abcam, Cambridge, UK, USA) at 4 °C overnight. After rinsing with TBST, the HRP-conjugated secondary antibodies were supplemented into membranes for further incubation for 2 h. The densities of the binding bands were then visualized using the ECL reagent (Beyotime) and quantified with the Gel Doc™ XR imaging system (Bio-Rad Laboratories, Hercules, CA, USA) and Image J software (National Institutes of Health, Bethesda, MD, USA).Determination of nitric oxide (NO) levels
Before exposure to the OGD/R conditions, microglia were pretreated with pcDNA-USP4 or pcDNA-TRAF6. Then, the contents of NO were measured using a commercial NO assay kit (Invitrogen). Briefly, cells were incubated with the Griess reagent for 15 min at room temperature. The concentration of NO produced in the microglia was then assessed by detecting the absorbance at 540 nm and referencing to a freshly prepared standard curve of nitrite.ELISA assay for inflammatory cytokine measurement
The cells transfected with pcDNA-USP4 plasmids and pcDNA-TRAF6 were exposed to OGD/R. Then, the cells were collected and centrifuged. The concentrations of IL-1β, IL-6, and TNF-α in supernatants were measured by the commercial ELISA kits (Boster Co., Ltd., Wuhan, China). All protocols were carried out according to the manufacturers' instructions.Conditioned media (CM) from microglia and neuron treatment
The microglia were pretreated with pcDNA-USP4 vectors or pcDNA-TRAF6 prior to OGD/R exposure for 12 h. Then, the cell supernatants were collected after centrifugation. The primary neurons were incubated with the prepared conditioned medium (CM) from the microglia for 24 h.MTT assay
To detect microglial cell proliferation and neuron viability, cells under various conditions were gathered. Afterwards, 100 μL of MTT (Beyotime) was supplemented into each well for further incubation for 4 h. Then, 100 μL of a formazan solution was added to completely solubilize the formazan precipitate at 37 °C for 4 h. The absorbance at 570 nm was then detected to evaluate cell proliferation and viability via a spectrophotometer.Detection of lactate dehydrogenase (LDH) release
Neurons were incubated with the conditioned medium from various microglia. Then, the cells were mixed with 50 μL of the LDH release reagent for 2 h. The cells were subsequently centrifuged for 5 min, and the LDH release was then assessed using an LDH detection kit (Beyotime). The optical density at 490 nM was captured using a microplate reader. All procedures were conducted according to the manufacturer's instructions.Cell apoptosis evaluation by a flow cytometer
Cell apoptosis was analyzed by double Annexin V/PI staining. Briefly, after culture in CM from the microglia, the neurons were washed with PBS. Subsequently, the cells were centrifuged and re-suspended in 195 μL binding buffer. Then, the cells were incubated with Annexin V-FITC (5 μL) and 10 μL of PI (Beyotime) for 20 min. All samples were transferred to flow-cytometry tubes and analyzed by a flow cytometer (BD Biosciences, USA) to detect cell apoptosis.Measurement of caspase-3 activity
After treatment with the indicated conditions, the cells were collected and lysed with lysis buffer under a cold condition for 15 min. The lysates were then centrifuged at 4 °C. Approximately 10 min later, all specimens were incubated with 10 μL of specific caspase-3 substrate Ac-DEVD-pNA (2 nM) for 2 h. All protocols were performed according to the recommendations of a commercial Caspase-3 Activity Assay Kit (Beyotime). Optical densities at 405 nm were measured to analyze the activity of caspase-3.Statistical analysis
All data were obtained from at least three independent experiments and expressed as the mean ± SD. SPSS 19.0 (SPSS Inc.) was used to analyze all data. Statistical analyses were evaluated by a Student t-test for two groups and one-way analysis of variance (ANOVA) followed by post hoc Student–Newman–Keuls test for the differences of three or more groups. P < 0.05 was considered significant.Funding
This research did not receive any specific grants from funding agencies in the public, commercial, or not-for-profit sectors.Conflicts of interest
There are no conflicts of interest concerning this article.Acknowledgements
I'd like to express my sincere thanks to all those who have given me a hand in writing this manuscript.References
- P. D. Schellinger, M. Kaste and W. Hacke, An update on thrombolytic therapy for acute stroke, Curr. Opin. Neurol., 2004, 17, 69–77 CrossRef CAS PubMed.
- Y. Bejot, B. Daubail and M. Giroud, Epidemiology of stroke and transient ischemic attacks: Current knowledge and perspectives, Rev. Neurol., 2016, 172, 59–68 CrossRef CAS PubMed.
- B. M. Demaerschalk, H. M. Hwang and G. Leung, US cost burden of ischemic stroke: a systematic literature review, Am. J. Manag. Care, 2010, 16, 525–533 Search PubMed.
- Z. Nagy and S. Nardai, Cerebral ischemia/reperfusion injury: From bench space to bedside, Brain Res. Bull., 2017, 134, 30–37 CrossRef CAS PubMed.
- J. Godinho, A. B. de Sa-Nakanishi, L. S. Moreira, R. M. W. de Oliveira, C. H. Huzita, J. C. P. Mello, A. O. F. da Silva, C. V. Nakamura, I. S. Previdelli, M. Ribeiro and H. Milani, Ethyl-acetate fraction of Trichilia catigua protects against oxidative stress and neuroinflammation after cerebral ischemia/reperfusion, J. Ethnopharmacol., 2018, 221, 109–118 CrossRef CAS PubMed.
- A. Partoazar, S. Nasoohi, S. M. Rezayat, K. Gilani, S. E. Mehr, A. Amani, N. Rahimi and A. R. Dehpour, Nanoliposome containing cyclosporine A reduced neuroinflammation responses and improved neurological activities in cerebral ischemia/reperfusion in rat, Fundam. Clin. Pharmacol., 2017, 31, 185–193 CrossRef CAS PubMed.
- B. Zhao, H. Wang, C. X. Li, S. W. Song, S. H. Fang, E. Q. Wei and Q. J. Shi, GPR17 mediates ischemia-like neuronal injury via microglial activation, Int. J. Mol. Med., 2018, 42, 2750–2762 CAS.
- X. Zeng, H. Ren, Y. Zhu, R. Zhang, X. Xue, T. Tao and H. Xi, Gp91phox (NOX2) in Activated Microglia Exacerbates Neuronal Damage Induced by Oxygen Glucose Deprivation and Hyperglycemia in an In Vitro Model, Cell. Physiol. Biochem., 2018, 50, 783–797 CrossRef CAS PubMed.
- X. Y. Zhang, X. R. Wang, D. M. Xu, S. Y. Yu, Q. J. Shi, L. H. Zhang, L. Chen, S. H. Fang, Y. B. Lu, W. P. Zhang and E. Q. Wei, HAMI 3379, a CysLT2 receptor antagonist, attenuates ischemia-like neuronal injury by inhibiting microglial activation, J. Pharmacol. Exp. Ther., 2013, 346, 328–341 CrossRef CAS PubMed.
- H. Xu, W. Qin, X. Hu, S. Mu, J. Zhu, W. Lu and Y. Luo, Lentivirus-mediated overexpression of OTULIN ameliorates microglia activation and neuroinflammation by depressing the activation of the NF-kappaB signaling pathway in cerebral ischemia/reperfusion rats, J. Neuroinflammation, 2018, 15, 83–100 CrossRef PubMed.
- D. Su, Y. Cheng, S. Li, D. Dai, W. Zhang and M. Lv, Sphk1 mediates neuroinflammation and neuronal injury via TRAF2/NF-kappaB pathways in activated microglia in cerebral ischemia reperfusion, J. Neuroimmunol., 2017, 305, 35–41 CrossRef CAS PubMed.
- Y. Zhao, L. Gao, L. Xu, R. Tong, N. Lin, Y. Su, Y. Yan, Y. Gao, J. He, L. Kong, A. Yuan, Y. Zhuge, F. Wang and J. Pu, Ubiquitin-specific protease 4 is an endogenous negative regulator of metabolic dysfunctions in nonalcoholic fatty liver disease, Hepatology, 2018, 68, 897–917 CrossRef CAS PubMed.
- J. Yang, P. Xu, L. Han, Z. Guo, X. Wang, Z. Chen, J. Nie, S. Yin, M. Piccioni, A. Tsun, L. Lv, S. Ge and B. Li, Cutting edge: Ubiquitin-specific protease 4 promotes Th17 cell function under inflammation by deubiquitinating and stabilizing RORgammat, J. Immunol., 2015, 194, 4094–4097 CrossRef CAS PubMed.
- X. Jiang, M. Yu, Y. Ou, Y. Cao, Y. Yao, P. Cai and F. Zhang, Downregulation of USP4 Promotes Activation of Microglia and Subsequent Neuronal Inflammation in Rat Spinal Cord After Injury, Neurochem. Res., 2017, 42, 3245–3253 CrossRef CAS PubMed.
- J. Zhou, T. Qiu, T. Wang, Z. Chen, X. Ma, L. Zhang and J. Zou, USP4 deficiency exacerbates hepatic ischaemia/reperfusion injury via TAK1 signalling, Clin. Sci., 2019, 133, 335–349 CrossRef CAS PubMed.
- R. Zhou, Z. Yang, X. Tang, Y. Tan, X. Wu and F. Liu, Propofol protects against focal cerebral ischemia via inhibition of microglia-mediated proinflammatory cytokines in a rat model of experimental stroke, PLoS One, 2013, 8, e82729 CrossRef PubMed.
- Z. Huang, D. Zheng, J. Pu, J. Dai, Y. Zhang, W. Zhang and Z. Wu, MicroRNA-125b protects liver from ischemia/reperfusion injury via inhibiting TRAF6 and NF-kappaB pathway, Biosci., Biotechnol., Biochem., 2019, 83, 829–835 CrossRef CAS PubMed.
- G. Wu, D. W. McBride and J. H. Zhang, Axl activation attenuates neuroinflammation by inhibiting the TLR/TRAF/NF-kappaB pathway after MCAO in rats, Neurobiol. Dis., 2018, 110, 59–67 CrossRef CAS PubMed.
- V. L. Feigin, M. H. Forouzanfar, R. Krishnamurthi, G. A. Mensah, M. Connor, D. A. Bennett, A. E. Moran, R. L. Sacco, L. Anderson, T. Truelsen, M. O'Donnell, N. Venketasubramanian, S. Barker-Collo, C. M. Lawes, W. Wang, Y. Shinohara, E. Witt, M. Ezzati, M. Naghavi and C. Murray, Global and regional burden of stroke during 1990-2010: findings from the Global Burden of Disease Study 2010, Lancet, 2014, 383, 245–254 CrossRef.
- R. Guruswamy and A. ElAli, Complex Roles of Microglial Cells in Ischemic Stroke Pathobiology: New Insights and Future Directions, Int. J. Mol. Sci., 2017, 18(pii), E496 CrossRef PubMed.
- Y. Chen, S. J. Won, Y. Xu and R. A. Swanson, Targeting microglial activation in stroke therapy: pharmacological tools and gender effects, Curr. Med. Chem., 2014, 21, 2146–2155 CrossRef CAS PubMed.
- X. Yang, T. Asakawa, S. Han, L. Liu, W. Li, W. Wu, Y. Luo, W. Cao, X. Cheng, B. Xiao, H. Namba, C. Lu, Q. Dong and L. Wang, Neuroserpin Protects Rat Neurons and Microglia-Mediated Inflammatory Response Against Oxygen-Glucose Deprivation- and Reoxygenation Treatments in an In Vitro Study, Cell. Physiol. Biochem., 2016, 38, 1472–1482 CrossRef CAS PubMed.
- M. Steinlin, Neuroinflammation in Ischemic Pediatric Stroke, Semin. Pediatr. Neurol., 2017, 24, 201–206 CrossRef PubMed.
- A. Sapkota, B. P. Gaire, K. S. Cho, S. J. Jeon, O. W. Kwon, D. S. Jang, S. Y. Kim, J. H. Ryu and J. W. Choi, Eupatilin exerts neuroprotective effects in mice with transient focal cerebral ischemia by reducing microglial activation, PLoS One, 2017, 12, e0171479 CrossRef PubMed.
- X. He, Y. Zheng, S. Liu, S. Shi, Y. Liu, Y. He, C. Zhang and X. Zhou, MiR-146a protects small intestine against ischemia/reperfusion injury by down-regulating TLR4/TRAF6/NF-kappaB pathway, J. Cell. Physiol., 2018, 233, 2476–2488 CrossRef CAS PubMed.
- T. Li, J. J. Qin, X. Yang, Y. X. Ji, F. Guo, W. L. Cheng, X. Wu, F. H. Gong, Y. Hong, X. Y. Zhu, J. Gong, Z. Wang, Z. Huang, Z. G. She and H. Li, The Ubiquitin E3 Ligase TRAF6 Exacerbates Ischemic Stroke by Ubiquitinating and Activating Rac1, J. Neurosci., 2017, 37, 12123–12140 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |