
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent-free synthesis and characterization of allyl chitosan derivatives†
Tatiana A. Akopova *a,
Tatiana S. Deminaab,
Georgii V. Cherkaeva,
Mukhamed A. Khavpacheva,
Kseniya N. Bardakovabc,
Andrey V. Grachevd,
Leonid V. Vladimirovd,
Alexander N. Zelenetskiia and
Petr S. Timashevbcd
*a,
Tatiana S. Deminaab,
Georgii V. Cherkaeva,
Mukhamed A. Khavpacheva,
Kseniya N. Bardakovabc,
Andrey V. Grachevd,
Leonid V. Vladimirovd,
Alexander N. Zelenetskiia and
Petr S. Timashevbcd
aEnikolopov Institute of Synthetic Polymeric Materials, Russian Academy of Sciences, 70 Profsoyuznaya st., Moscow 117393, Russia. E-mail: Akopova@ispm.ru
bInstitute for Regenerative Medicine, Sechenov University, 8-2 Trubetskaya st., Moscow 119991, Russia
cInstitute on Photon Technologies, Federal Scientific Research Centre “Crystallography and Photonics”, Russian Academy of Sciences, 2 Pionerskaya st., Troitsk, Moscow 142190, Russia
dSemenov Institute of Chemical Physics, Russian Academy of Sciences, 4 Kosygina st., Moscow 119991, Russia
First published on 4th July 2019
Abstract
The solvent-free synthesis of allyl-substituted chitosan derivatives through reactive co-extrusion of chitosan powder with allyl bromide at shear deformation was performed. For the structural characterization, FTIR and NMR methods were employed. The results were confirmed by chemical analysis. The total content of allyl substituents from 5 to 50 per 100 chitosan units as a function of the component ratio in the reactive mixtures was revealed. Carrying out the reaction without any additives leads to the selective formation of N-alkylated derivatives, whereas in the presence of alkali the ethers of chitosan were preferentially formed. The results suggest that the proposed approach allows significantly higher yield of products to be obtained at high process speeds and significantly lower reagent consumption as compared with the liquid-phase synthesis in organic medium. The synthesized unsaturated derivatives are promising photosensitive components for use in laser stereolithography for fabrication of three-dimensional biocompatible structures with well-defined architectonics.
Introduction
Polysaccharide chitosan and its derivatives are prospective materials for the creation of biomedical materials in the form of hydrogels, microgranules, nanoparticles, nanofibers, etc.1–4 The presence of primary amine groups in the structure of chitosan determines its solubility in diluted aqueous organic and mineral acids and its unique complex-forming properties. Chitosan exhibits bacteriostatic and antiviral activity, and effectively binds heavy metal ions, proteins and fatty acids.5–7 A number of chitosan derivatives and composites based on it are proposed to produce materials with enhanced solubility, sorption capacity, physical mechanical characteristics and ability to form films, fibres and cross-linked structures.8–11 Recently much attention has been paid to three-dimensional models of different cell cultures formed in the volume of polymer matrices of specified architectonics and offered as biomimetic implants.12–14 Natural polysaccharides, including chitosan and its derivatives, can be successfully used to create similar structures due to their high cellular affinity and ability to control bioresorption.15,16To obtain the materials with a high degree of reproduction of a given morphology with a resolution of up to 200–400 nm the laser stereolithography technology was used.17–19 It is based on the principle of initiating spatial cross-linking of material at two-photon absorption in the highly focused field of femtosecond laser radiation in UV region of spectrum. In order to increase the reactivity of functional groups of polymers in cross-linking reactions when exposed to laser radiation, unsaturated groups were introduced into their structure.20,21
There is a relatively few data in literature on attempts to obtain the unsaturated chitosan derivatives. They are mainly combined with the introduction of vinyl groups into its structure, carried out in an organic solvent medium. For example, Qi et al.22 had obtained the methacrylic derivatives, capable of crosslinking with hydrogels formation under UV radiation. The synthesis of crosslinkable photosensitive product by incorporation of methacrylate groups to prefabricated N-succinyl chitosan was performed by Kufelt et al.23 Shitrit and Bianco-Peled24 had studied the possibility to obtain the acrylated chitosan at its interaction with polyethylene glycol diacrylate (PEGDA) for using as a mucoadhesive drug delivery systems. Photocrosslinkable chitosan carrying vinyl carboxylic acid groups was also synthesized by interaction with maleic anhydride under mild conditions.25 However, the vinyl groups are very active in the processes of chitosan cross-linking via the Michael type reaction, which can lead to decrease in the yield of target products.26 The reactions of chitosan amine groups with anhydrides of unsaturated fatty acids lead mainly to production of insoluble derivatives both in aqueous and common organic solvents, as marked by Zhang and Hirano.27 All the above leads to difficulty of removing the uncrosslinked material from the obtained well-defined 3D structures based on acrylated chitosan derivatives. The groups active in the spatial cross-linking exclusively in sharply focused laser radiation field are represented to us as allyl groups. The side reaction with the free amine groups of the chitosan chain is not typical for their double bond. It is also important that when the amino groups of the polymer are involved in the alkylation reaction, the resulting product is a secondary amine which is capable of protonation in the presence of acids as the primary amino group of chitosan. Therefore, the solubility in aqueous media, which is characteristic for the initial polymer, will not be significantly reduced if the degree of substitution (DS) by hydrophobic moieties is not too high.28
It is known a method for alkylation of chitosan with allyl bromide in PriOH in presence of large excess of alkali,29,30 performed similarly to heterogeneous processes for allyl cellulose derivatives. The authors note the diffusively complicated type of the process due to bad swelling of chitosan both in alkali and PriOH. Homogeneous allylation of cellulose by reacting with allyl bromide was performed in LiCl/DMAc solution containing NaOH powder, and an allyl DS per anhydroglucose unit of 2.80 was found.31 Illy et al.28 carried out the allylation of chitosan in aqueous medium by interaction of water-soluble chitosan oligomers (polymerization degree of 12 units, approx.) with allyl glycidyl ether, and DS was found to be 0.33 in presence of double molar excess of alkylation agent to amine groups. The main disadvantage of homogeneous solution methods is the limited solubility of polymers and, consequently, the low productivity of the processes for obtaining derivatives.
Generally, the production of derivatives of natural polysaccharides, as well as the alkaline deacetylation of chitin with chitosan formation, is always accompanied by preliminary activation of polysaccharides in order to destroy their highly organized supramolecular structure with a complex system of hydrogen bonds preventing their dissolution and melting without decomposition.32 These processes require the use of a large excess of aggressive reagents and solvents followed by their expensive regeneration and often begin as heterogeneous reactions.33–35 Therefore, the development of efficient environmentally friendly methods allowing to increase the availability of polysaccharide functional groups in the processes of their chemical modification is an urgent task.
Mechanochemical synthesis based on joint action of high pressure and shear strains onto solid mixture of reagents, including polymers,36,37 can serve as an alternative method to produce the polysaccharide derivatives. The method allows to avoid any solvents, does not involve the melting of reactive mixtures and, therefore, is a convenient and effective way of target chemical modification of infusible polysaccharides.38–40 Organic solid-state reactions have numerous advantages since they are infinitely high-concentration reactions and proceed much more efficiently and faster than solution reactions in many cases.41 A series of solvent-free mechanochemical organic reactions have been performed by using ball milling process.42 Since organic substances, polymers especially, have low thermal stability and large strain relaxation times, the most appropriate technique for their solid-state modification is twin-screw extrusion, which is capable of fine temperature control at slow continuous deformation of solids.43,44 Shear deformation provides numerous possibilities to circumvent many processing obstacles typical for interaction of hydrophilic polymers and hydrophobic organic reagents.45,46 According to classical theory of mechanochemistry, the shear deformation of solid reagents is accompanied by the formation of active states in the bulk of substance, by disordering of its structure and polarization of molecules.47–50 The consequence is, for example, the intensive polymerization of solid vinyl and cyclic monomers,51 the high mobility of the end functional groups and the ability of heteroceptive links of synthetic polymers and monomers to directional reactions with functional groups of polysaccharides.52,53
Mechanochemical processes, which currently receiving a great deal of interest, are important not only for their high efficiency but also for their simplicity and cleanness. The reactions in these conditions often proceed without the need for any initiators and catalysts, therefore their products are more compatible with human health and the environment. As was shown in our earlier studies, chitosan with high degree of deacetylation,54 its salts and complexes with bioactive organic compounds,55 the acylated and carboxymethylated derivatives56–58 can be produced via the solid-state extrusion. A series of polysaccharide-based new materials have been obtained by application of solid-state co-extrusion to mixtures of polysaccharides like chitosan, chitin or cellulose with synthetic polymers or monomers.59–64
In this study, we developed mechanochemical synthesis of allyl-substituted chitosan derivatives capable of crosslinking with three-dimensional scaffolds formation under UV radiation. For this purpose, regularities of the chitosan reactions in solid state with allyl bromide under shear deformation were revealed. Experiments were performed using solvent-free reactive co-extrusion that may be regarded as more economical and ecologically favorable procedure in chemistry. Techniques such as Nuclear Magnetic Resonance (NMR), Fourier Transform Infrared Spectroscopy (FTIR) and bromometric titration were used for characterization.
Experimental
Materials
Chitosan (molecular weight of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000; degree of acetylation (DA) of 0.15) was prepared from crab chitin (moisture 4.3%, ash content 1.8%, Xiamen Fine Chemical Import & Export Co., LTD, China) through solid-state synthesis in accordance with published procedure.54 Allyl bromide (AB; 99%) of synthetic grade was purchased from Acros Organics (PubChem CID: 7841). Sodium hydroxide of analytical grade (microprills, 99%) was purchased from Merck KGaA (Germany).
000; degree of acetylation (DA) of 0.15) was prepared from crab chitin (moisture 4.3%, ash content 1.8%, Xiamen Fine Chemical Import & Export Co., LTD, China) through solid-state synthesis in accordance with published procedure.54 Allyl bromide (AB; 99%) of synthetic grade was purchased from Acros Organics (PubChem CID: 7841). Sodium hydroxide of analytical grade (microprills, 99%) was purchased from Merck KGaA (Germany).
Preparation of chitosan and its allyl-substituted derivatives
The synthesis was carried out in a pilot twin-screw extruder (Berstorff, Germany) with parallel rotation of screws (d = 40 mm) and controlled heating (4 zones). The extruder is equipped with force elements of screws capable to realize the compression and shear deformation of solid mixtures of reagents. Reagents were fed manually at screw rotation speed of 60 rpm. With a residence time of ca. 2–3 minutes, feed rate of 30 g min−1 was achieved.As a starting component for synthesis of allyl chitosan derivatives the chitin was used. At first stage by means of alkaline deacetylation of chitin at 3-fold molar excess of NaOH and the heating temperature of cylinder body of extruder of 160 °C we obtained chitosan and used it for the synthesis as an intermediate (Sample 1). With this method of carrying out the process it becomes possible to use the chitosan in the most disordered nonequilibrium state, which increases the reactivity of its functional groups under conditions of solid-state synthesis (see Fig. S1†).53 The reactive mixture after chitin deacetylation contained: chitosan 52 ± 2%; sodium hydroxide 25 ± 2%; sodium acetate 23 ± 2%.
The allyl-substituted chitosan derivatives were obtained by repeated treatment of the reactive mixtures after adding of allyl bromide at different ratios during cooling of the extruder working zones to 0–5 °C. The sample of allyl chitosan we had obtained earlier under similar conditions of mechanical action but in the absence of alkali medium65 was used as a reference sample (Sample 2). The molar ratios of allyl bromide (AB) per chitosan unit were 0.5 (for Samples 2 and 3), 1.0 (Sample 4), 1.5 (Sample 5), and 2.0 (Sample 6). Molar content of alkali in reactive mixtures (at preparation of Samples 3–6) was 2 mol of NaOH per chitosan unit.
The products were purified with PriOH followed by dialysis against deionized water (DW) to remove completely the unreacted monomer and alkaline impurities. After 48 h of dialysis the samples were dissolved in 0.1 M HCL and then precipitated with 1 M NaOH. The residues were rinsed with DW up to the absence of a qualitative reaction of flushing waters to chlorine ions and freeze-dried to produce the final product in powder form. To record the NMR spectra, samples were dried in the form of chitosan hydrochloride in vacuum chamber at RT to ensure the dissolution in D2O at measurement.
Characterization of allyl-chitosan samples
1H NMR (300 MHz, D2O) spectra were recorded on a Bruker-Avance II-300 spectrometer at 90 °C. The suppression of the solvent signal was carried out by the standard for spectrometers of this type pulse sequence “zgpr”. To calibrate the scale of chemical shifts the signal of DMSO-d6 (δ = 2.5 ppm) was used. The data of 1H NMR analysis was used to reveal the features of the structure and to calculate the ratio of chitosan and allyl constituents in the synthesized derivatives.FT-IR spectra were recorded on a Bruker Vertex 70 spectrometer (USA). All spectra were initially collected in ATR mode at resolution of 2 or 4 cm−1 by employing an ATR-mono-reflection Gladi ATR (Pike Technologies, USA) accessory equipped with diamond crystal (n = 2.4; angle of incidence 45 deg.). The obtained ATR spectra were converted into IR-absorbance mode. All the spectra presented in this work were recorded and treated using a set of programs: Bruker Opus (version 6.1).
The degree of substitution (DS) per glucosamine unit was determined by titrating the allyl chitosan with bromine in chloroform solution according to standard analysis of the bromine number determination by Knopp method.66
The DS was calculated by the equation:
| Br (%) = 15984x/(167 + 41x), |
984 – molecular weight of Br2 molecule, multiplied by 100; 167 – molecular weight of monomer link of allyl chitosan with DA = 0.15; 41 – molecular weight of allyl group; x – degree of substitution (in parts of unit). When multiplied by 100, the degree of substitution per 100 glucosamine units is obtained.
Results and discussion
NMR analysis
1H NMR spectra of the prepared allyl chitosan samples are presented in Fig. 1, and DS parameters calculated from the spectra are summarized in Table 1. In the figure are marked the spectral ranges used to determine the content of allyl groups in modified chitosan by measuring and comparing the integral intensities of proton signals in structural fragments: H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C
C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) –CH2–O– and H2CC–CH2–NR– (total signal at 5.8 ppm), R = H or All (I); 2CCH–CH2–NR– from 5.3 to 5.6 ppm (II); 2CCH–CH2–O– from 5.0 to 5.2 ppm (III);
–CH2–O– and H2CC–CH2–NR– (total signal at 5.8 ppm), R = H or All (I); 2CCH–CH2–NR– from 5.3 to 5.6 ppm (II); 2CCH–CH2–O– from 5.0 to 5.2 ppm (III); ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) C–NH2 at 3.0–3.1 ppm (IV); 3C–CONH– at 1.9 ppm (V); H-OD – residual signal (when suppressing) the protons of water.
C–NH2 at 3.0–3.1 ppm (IV); 3C–CONH– at 1.9 ppm (V); H-OD – residual signal (when suppressing) the protons of water.
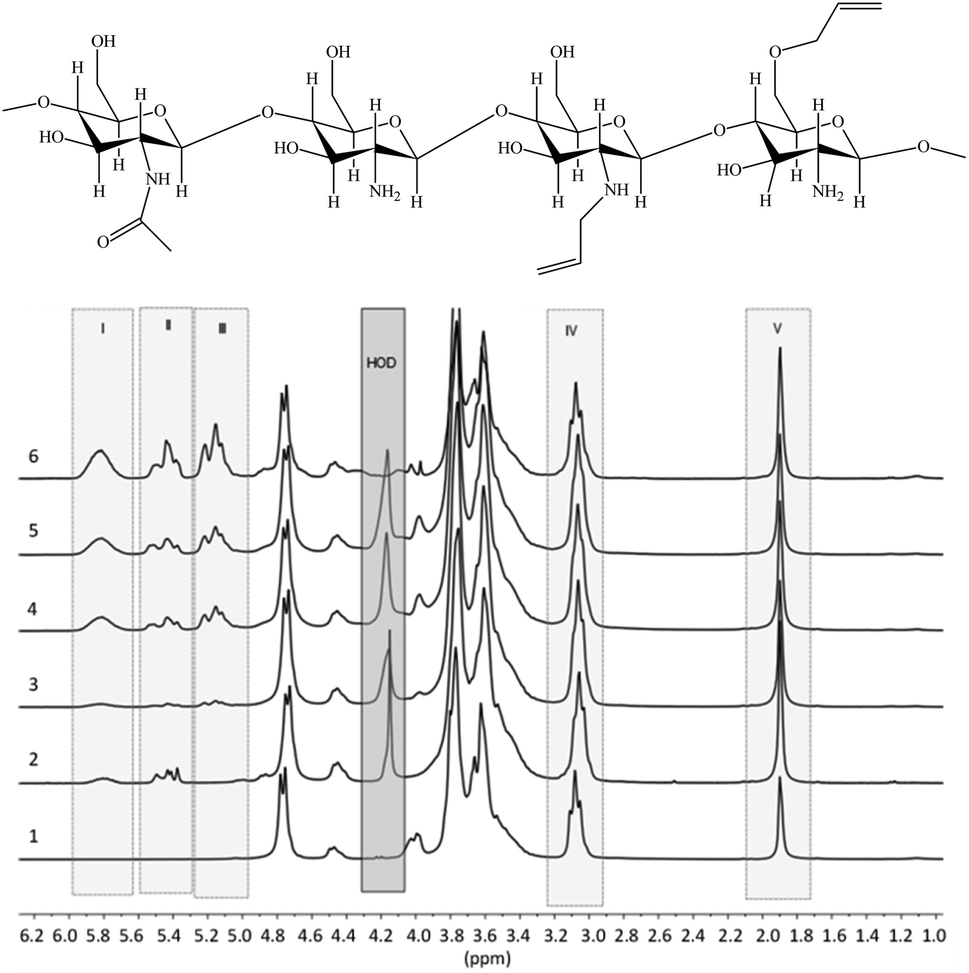 | ||
| Fig. 1 Proposed chemical composition of allyl-modified chitosan chains, and 1H NMR spectra of initial chitosan (1) and a number of allyl chitosan samples (Samples 2–6 in Table 1). | ||
| Samples | Ratio of integral intensity of proton signals in structural fragments I and V | Bromine number (%) | ∑DS | Ratio of N- and O-substituted groups | |
|---|---|---|---|---|---|
| By NMR | By titration | ||||
| a Sample 1 (initial chitosan) according to NMR analysis contains 15% of N-acetylated units (the remains of chitin), the content of which does not change in the chosen conditions of synthesis of derivatives. Ratio of N- and O-substituted groups was calculated by proportion of integral intensity of proton signals in structural fragments II and III. | |||||
| 2 | 0.7 | 7.5 | 0.10 | 0.08 | — |
| 3 | 0.3 | 5.7 | 0.05 | 0.06 | 1:2 |
| 4 | 1.1 | 9.3 | 0.17 | 0.10 | 1:1.8 |
| 5 | 1.4 | 20.8 | 0.21 | 0.23 | 1:1.7 |
| 6 | 3.3 | 41.2 | 0.50 | 0.47 | 1:1.5 |
The results of NMR analysis show that in the absence of NaOH the reaction of allylation proceeds exclusively with amino groups of chitosan (Fig. 2, path 1). Conducting of the process in alkaline medium leads to non-selective formation of O- and N-substituted derivatives of chitosan, meanwhile the content of O-substituted fragments is 1.5–2 times higher. The alkali not only activates the hydroxyl groups of polymer in the nucleophilic substitution reaction, but also binds the halogen ions formed during the reaction (path 2). In the absence of NaOH, the exuded bromine ions can be bound by any of amino groups of chitosan macromolecule, as shown, for example, in Fig. 2, path 1. The total DS of chitosan functional groups is symbatically dependent on the AB content in the reactive mixtures and reaches 0.50 at a double molar excess of alkylation agent with regard to the polymer units.
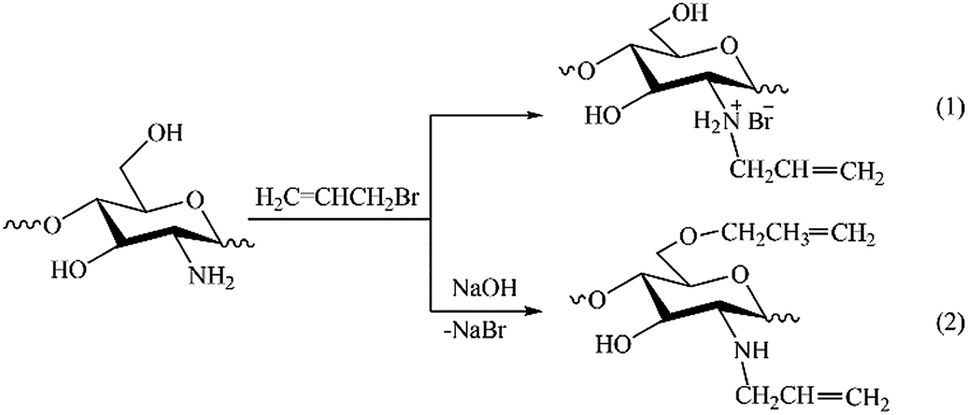 | ||
| Fig. 2 Schemes of reactions occurring at solid-state synthesis in mixtures of chitosan and allyl bromide in the absence of NaOH (1) and in alkaline medium (2). | ||
To confirm the correctness of the signal assignments in 1H NMR spectra of the products, a model mixture consisting of initial chitosan, allyl alcohol and allyl amine with a 5-fold molar excess of chitosan with regard to low-molecular-weight components was prepared. Fig. 3 shows the mixture spectrum, on which the ascriptions of signals of characteristic protons are shown.
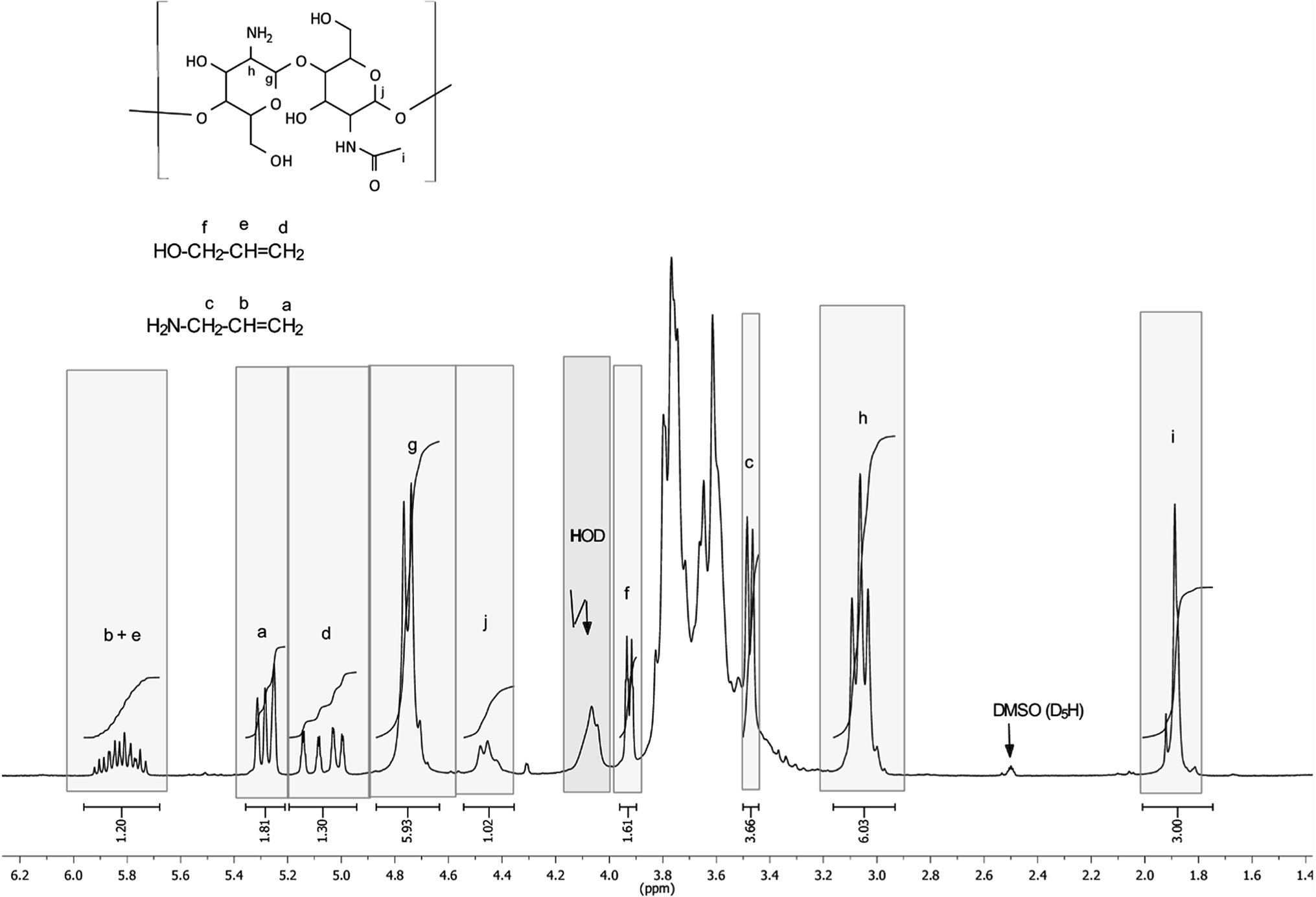 | ||
| Fig. 3 1H NMR spectrum of the model mixture of chitosan with allyl alcohol and allyl amine. The water signal is partially suppressed. | ||
The comparison of 1H NMR spectra of the modified chitosan samples and the model mixture (Fig. 1 and 3) shows that the position of groups of characteristic signals (selected regions) is almost unchanged. The signal groups of terminal vinyl protons in O- and N-allyl fragments do not overlap and have been correctly used for quantitative analysis of ratio of these fragments in the modified chitosan.
The estimation of the number of allyl substituents included in the structure of chitosan, based on determination of the mass of bromine joining to 100 g of polymer during titration, showed a good convergence of results obtained by spectral and chemical methods (Table 1).
FT-IR analysis
Fig. 4 presents the IR-spectra of initial chitosan as well as of the products of its reaction with allyl bromide under conditions of solid state synthesis in presence of NaOH. The high-frequency part (3700–2500 cm−1) of the overview IR-spectrum contains a strong broad band of the stretching vibrations of O–H groups, which overlaps both the doublets of NH2 and CH2 stretching vibrations. As a result, the bands 3356 and 3295 cm−1 of asymmetric and symmetric N–H stretching peaks appear in the spectrum as weak shoulders; whereas the doublet of C–H stretching vibrations can be clearly seen with maximum at 2870 and its high-frequency shoulder at approx. 2997 cm−1. The analysis of the high-frequency region that the relative intensity D[OH]/D[CH] in the spectra of the reaction products consistently decreases with increase of AB evidencing the participation of O–H groups in reaction as well as the appearance of additional alkyl groups (the increase of intensity of the stretching vibrations of C–H bonds). Simultaneously with this effect the intensity of theC–H stretching band of allyl groups (see: peak at 3085 cm−1)67 noticeably increases. At the same time very little (if any) changes could be observed for the stretching vibrations of NH2 groups.
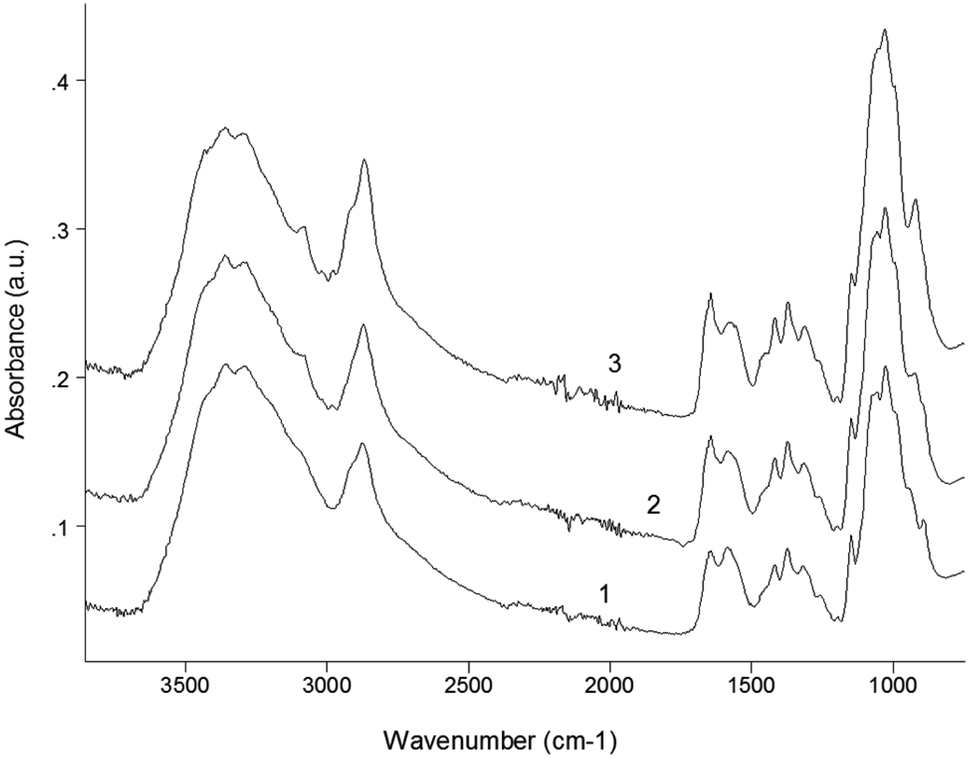 | ||
| Fig. 4 IR-spectra of initial chitosan (1) and allyl chitosan with degree of substitution of 0.2 (2) and 0.5 (3) (Samples 5 and 6 in Table 1, respectively). | ||
To other typical IR-bands of allyl groups are attributed 1645 cm−1 of stretching vibrations of double bonds as well as the 922 cm−1 of C–H deformation.68 Since in the systems under consideration the position of 1645 cm−1 band practically coincides with the Amide-I vibrations of the residual acetamide groups of chitin as well as the intensity of 922 cm−1 band is strong enough and thus is a better match for evaluation of the content of the grafted allyl groups. An analogous band in the spectrum of initial allyl bromide is observed at 927 cm−1.67 The comparison of curves 2 and 3 in this region of spectra shows that the change in molar ratio between AB and chitosan from 1.5 to 2.0 (Samples 5 and 6, respectively) at least doubled the total yield of AB grafting. A very close estimate of the yield of reaction as a function of the components ratio follows from the comparison of the ratio of the relative intensities D[C–O]/D[OH], where D[C–O] is the sum (envelope) of the bands with maximum at 1030 cm−1 of the stretching vibrations of C–O(H) groups as well as other C–O bonds including stretching vibrations of glycosidic bonds in the structure of chitosan macromolecule.
The band of deformation vibrations of NH2 groups in the spectrum of initial chitosan is observed at 1590 cm−1, i.e., in-between the Amide-I and Amide-II bands. The participation of NH2 groups in reaction with AB nonetheless could be seen due to broadening and low-frequency shift towards the Amide-II band (down to 1574 cm−1).
An approximate estimation of the amount of NH2 groups depleted in the reaction with allyl bromide was evaluated by using the ratio D[NH2]/D[1375], where 1375 cm−1 band was employed as the internal standard band according to the literature68 is attributed to overlayed bands of C–N stretching and CH2 deformation vibrations. This well coincides with our conclusion about the character of allylation of chitosan with preferential substitution of hydroxyl groups if the reaction is carried out in the alkaline medium under the above synthesis conditions.
For greater clarity, in the present work we have shown only the IR spectra of samples obtained at high content of allyl bromide in the initial medium (Samples 5 and 6). It should be noted that the observed changes in the IR spectra were characteristic for all the samples studied.
Application aspects
In comparison with the heterogeneous synthesis in organic medium,29 the solid-state synthesis of allyl-substituted derivatives of chitosan is characterized by a significantly lower reagent consumption and a high process speed (few minutes). For introduction in the structure of chitosan of 8–10 unsaturated groups per 100 parts of polymer in conditions of solid-state synthesis it is possible to avoid the use of alkali, while in the liquid-phase process its content should not be less than 0.75 moles per link (PriOH, 70 °C, 4 hours). With the ratio chitosan:NaOH:AB equal to 1:3:2 in the liquid-phase process, the total DS equal to 0.21 against 0.50 in solid-state synthesis is achieved (Table 1, Sample 6). And only the use of reprecipitated chitosan with the maximum disordered structure allowed to increase DS of the product of the liquid-phase synthesis to 0.47. Thus, mechanical activation of the reactive systems under solid-state twin-screw extrusion provide disordering of supramolecular structure of polymer comparable to specially selected conditions of precipitating from a solution to achieve enhanced accessibility of the chitosan functional groups.
It is important for practical use, that all obtained derivatives did not lose the solubility in acidic aqueous media in comparison with the initial polymer. The preliminary studies on usage of obtained unsaturated chitosan derivatives for biomedical application, in particular for fabrication of polymer scaffolds by laser stereolithography, have shown that allyl-substituted derivatives are reactive in the processes of spatial cross-linking under UV radiation (see Fig. S2†).69 At the same time, the introduction of allyl fragments into the structure of chitosan does not change its ability to maintain the adhesion, the spreading and proliferative activity of human mesenchymal stem cells, which was determined by metabolic activity of cells of NCTC L929 line with the use of MTT test.70,71
Conclusions
An original mechanochemical approach has been engaged in this work to synthesize unsaturated derivatives of polysaccharide chitosan which are promising materials for biomedical applications. The most attractive feature of this technique is that the entire modification process proceeds in the solid state via mechanical activation of the reactive systems under shear deformation, and thus does not require any solvents or melting of components. It was revealed that reaction of chitosan allylation at chosen synthesis conditions proceeds in excellent agreement with general regularities of alkylation reactions through bimolecular mechanism of nucleophilic substitution. During the reaction in presence of NaOH the N,O-substituted derivatives with the predominant formation of chitosan ethers are formed nonselectively, whereas amino groups of polymer are reacted exclusively in the absence of alkaline catalysis. FTIR and NMR analyses as well as titration data showed that the total content of allyl substitutes up to 50 per 100 chitosan units can be achieved.The use of allyl-substituted derivatives in laser-induced processes allows to avoid the undesirable polymer cross-linking in non-irradiated areas and to ensure the complete removal of the unnecessary material for formation of the desired structure of polymer scaffold. The obtained materials can be used as the polymer implants with specified internal structure for regenerative medicine purposes, which expands both the field of practical application of chitosan and the range of biocompatible polymers suitable for structuring with the use of laser technologies.
Thus, the proposed solvent-free extrusion process to obtain chitosan derivatives differs from typical liquid-phase methods by high efficiency, simplicity and cleanness. The allyl chitosan derivatives synthesized in this work are effective components for the laser-induced formation of the well-defined polymer scaffolds.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Russian Foundation for Basic Research (grant no. 18-29-17050-mk). NMR spectra registration was performed with the financial support from Ministry of Science and Higher Education of the Russian Federation using the equipment of Collaborative Access Center “Center for Polymer Research” of ISPM RAS.Notes and references
- R. A. A. Muzzarelli, Carbohydr. Polym., 2009, 76, 167–182 CrossRef CAS
.
- M. Prabaharan, J. Biomater. Appl., 2008, 23, 5–36 CrossRef CAS PubMed
- S. K. Shukla, A. K. Mishra, O. A. Arotiba and B. B. Mamba, Int. J. Biol. Macromol., 2013, 59, 46–58 CrossRef CAS PubMed
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef CAS
- R. A. A. Muzzarelli and C. J. Muzzarelli, Adv. Polym. Sci., 2005, 186, 151–202 CrossRef CAS
- A. Verlee, S. Mincke and C. V. Stevens, Carbohydr. Polym., 2017, 164, 268–283 CrossRef CAS PubMed
- M. Rhazi, J. Desbrieres, A. Tolaimate, M. Rinaudo, A. Vottero, P. Alagui and M. El Meray, Eur. Polym. J., 2002, 38, 1523–1530 CrossRef CAS
- F. Suh and H. Matthew, Biomaterials, 2000, 21, 2589–2598 CrossRef PubMed
- I. Younes and M. Rinaudo, Mar. Drugs, 2015, 13, 1133–1174 CrossRef CAS PubMed
- A. Chenite, C. Chaput, D. Wang, C. Combes, M. D. Buschmann, C. D. Hoemann, J. C. Leroux, B. L. Atkinson, F. Binette and A. Selmani, Biomaterials, 2002, 21, 2155–2161 CrossRef
- K. C. Gupta and M. N. V. Ravi Kumar, J. Macromol. Sci., Rev. Macromol. Chem. Phys., 2000, C40, 273–308 CrossRef CAS
- M. Ravi, V. Paramesh, S. R. Kaviya, E. Anuradha and F. D. Solomon, J. Cell. Physiol., 2015, 230, 16–26 CrossRef CAS PubMed
- J. Choi, E. K. Lee, J. Choo, J. Yuh and J. W. Hong, Biotechnol. J., 2015, 10, 1682–1688 CrossRef CAS PubMed
- A. Ovsianikov, V. Mironov, J. Stampfl and R. Liska, Expert Rev. Med. Devices, 2012, 9, 613–633 CrossRef CAS PubMed
- T. S. Demina, D. S. Zaytseva-Zotova, P. S. Timashev, V. N. Bagratashvili, K. N. Bardakova, Ch. Sevrin, E. A. Svidchenko, N. M. Surin, E. A. Markvicheva, C. Grandfils and T. A. Akopova, IOP Conf. Ser.: Mater. Sci. Eng., 2015, 87, 012074 Search PubMed
- F. Croisier and C. Jerome, Eur. Polym. J., 2013, 49, 780–792 CrossRef CAS
- M. Farsari and B. N. Chichkov, Nat. Photonics, 2009, 3, 450–452 CrossRef CAS
- S. Juodkazis, V. Mizeikis and H. Misawa, J. Appl. Phys., 2009, 106, 051101 CrossRef
- D. S. Correa, M. R. Cardoso, V. Tribuzi, L. Misoguti and C. R. Mendonca, IEEE J. Sel. Top. Quantum Electron., 2012, 18, 176–186 CAS
- F. A. M. M. Goncalves, A. C. Fonseca, M. Domingos, A. Gloria, A. C. Serra and J. F. Coelho, Prog. Polym. Sci., 2017, 68, 1 CrossRef CAS
- A. Koroleva, O. Kufelt, S. Schlie-Wolter, U. Hinze and B. Chichkov, Biomed. Eng., 2013, 58, 399–405 Search PubMed
- Z. Qi, J. Xu, Z. Wang, J. Nie and G. Ma, Int. J. Biol. Macromol., 2013, 53, 144–149 CrossRef CAS PubMed
- O. Kufelt, A. El-Tamer, C. Sehring, M. Meißner, S. Schlie-Wolter and B. N. Chichkov, Acta Biomater., 2015, 18, 186–195 CrossRef CAS PubMed
- Y. Shitrit and H. Bianco-Peled, Int. J. Pharm., 2017, 517, 247–255 CrossRef CAS PubMed
- Y. Zhou, C. Zhang, K. Liang, J. Lia, H. Yang, X. Liu, X. Yin, D. Chen and W. Xu, Int. J. Biol. Macromol., 2018, 106, 227 CrossRef CAS PubMed
- H. Sashiwa, N. Yamamori, Y. Ichinose, J. Sunamoto and S. Aiba, Biomacromolecules, 2003, 4, 1250 CrossRef CAS PubMed
- M. Zhang and S. Hirano, Carbohydr. Polym., 1995, 26, 205–209 CrossRef CAS
- N. Illy, M. Robitzer, R. Auvergne, S. Caillol, G. David and B. Boutevin, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 39 CrossRef CAS
- L. A. Nud'ga, V. A. Petrova and M. F. Lebedeva, Russ. J. Appl. Chem., 2003, 76, 1978–1982 CrossRef
- Y. Q. Xia, T. Y. Guo, H. L. Zhao, M. D. Song, B. H. Zhang and B. L. Zhang, J. Sep. Sci., 2007, 30, 1300–1306 CrossRef CAS PubMed
- M. S. Lin and C. S. Huang, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 2303 CrossRef CAS
- S. Salmon and S. M. Hudson, J. Macromol. Sci., Rev. Macromol. Chem. Phys., 1997, C37, 199–276 CAS
- C. K. S. Pillai, W. Paul and C. P. Sharma, Prog. Polym. Sci., 2009, 34, 641–678 CrossRef CAS
- P. R. Rege and L. H. Block, Carbohydr. Res., 1999, 321, 235–245 CrossRef CAS
- T. Heinze, O. A. El Seoud and A. Koschella, in Cellulose Derivatives, Springer Series on Polymer and Composite Materials, Springer, Cham, 2018, vol. 4, pp. 259–292 Search PubMed
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friscic, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steedk and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC
- P. Balaz, M. Achimovicova, M. Balaz, P. Billik, Z. Cherkezova-Zheleva, J. M. Criado, F. Delogu, E. Dutkova, E. Gaffet, F. J. Gotor, R. Kumar, I. Mitov, T. Rojac, M. Senna, A. Streletskii and K. Wieczorek-Ciurowa, Chem. Soc. Rev., 2013, 42, 7571 RSC
- E. V. Prut and A. N. Zelenetskii, Russ. Chem. Rev., 2001, 70, 65–79 CrossRef CAS
- A. M. Dubinskaya, Russ. Chem. Rev., 1999, 68, 637–652 CrossRef CAS
- C. Simionescu and C. V. Oprea, Russ. Chem. Rev., 1988, 57, 283–287 CrossRef
- G. Kaupp, in Organic Solid State Reactions, Top Curr. Chem., ed. F. Toda, Springer, Berlin, Heidelberg, 2005, vol 254, pp. 95–183 Search PubMed
- A. Majee, A. Sarkar, S. Santra, S. K. Kundu, A. Hajra, G. V. Zyryanov, O. N. Chupakhin and V. Charushin, Green Chem., 2016, 18, 4475–4525 RSC
- D. E. Crawford, C. K. G. Miskimmin, A. B. Albadarin, G. Walker and S. L. James, Green Chem., 2017, 6, 1507–1518 RSC
- J. Andersen and J. Mack, Green Chem., 2018, 20, 1435–1443 RSC
- T. Shakhtshneider and V. Boldyrev, in Reactivity of Molecular Solids, ed. E. Boldyreva and V. Boldyrev, John Wiley & Sons, LTD, Chichester, 1999, pp. 271–312 Search PubMed
- W. Xu, M. Wen, J. Yu, Q. Zhang, N. E. Polyakov, A. V. Dushkin and W. Su, Drug Dev. Ind. Pharm., 2018, 44, 1924 CrossRef CAS PubMed
- V. V. Boldyrev and K. Tkáĉova, J. Mater. Synth. Process., 2000, 8, 121–132 CrossRef CAS
- P. Y. Butyagin, Russ. Chem. Rev., 1984, 53, 1025–1038 CrossRef
- M. M. Caruso, D. A. Davis, Q. Shen, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Chem. Rev., 2009, 109, 5755–5798 CrossRef CAS PubMed
- G. W. Wang, Chem. Soc. Rev., 2013, 42, 7668–7700 RSC
- A. A. Zharov, in High-Pressure Chemistry and Physics of Polymers, ed. A. L. Kovarskii, CRC Press, London–Tokyo, 1994, vol. 7, pp. 268–301 Search PubMed
- N. Z. Lyakhov, T. F. Grigorieva, A. P. Barinova and I. A. Vorsina, Russ. Chem. Rev., 2010, 79, 189–203 CrossRef CAS
- T. A. Akopova, A. N. Zelenetskii and A. N. Ozerin, in Focus on Chitosan Research, ed. A. N. Ferguson and A. G. O'Neill, Nova Science Publishers, New York, 2011, vol. 7, pp. 223–254 Search PubMed
- S. Z. Rogovina, T. A. Akopova and G. A. Vikhoreva, J. Appl. Polym. Sci., 1998, 70, 927–933 CrossRef CAS
- A. N. Zelenetskii, T. A. Akopova, N. R. Kildeeva, G. A. Vikhoreva, E. S. Obolonkova and A. A. Zharov, Russ. Chem. Bull., 2003, 52, 2073–2077 CrossRef CAS
- S. Z. Rogovina, G. A. Vikhoreva, T. A. Akopova, I. N. Gorbacheva and S. N. Zelenetskii, Mendeleev Commun., 1998, 6, 107–109 CrossRef
- S. Z. Rogovina, G. A. Vikhoreva and T. A. Akopova, in Chitosan in Pharmacy and Chemistry, ed. R. A. A. Muzzarelli and C. Muzzarelli, ATEC, Italy, 2002, pp. 449–453 Search PubMed
- T. S. Demina, T. A. Akopova, L. V. Vladimirov, A. N. Shchegolichin, A. S. Kechekyan, N. S. Perov, A. O. Chernyshenko and A. N. Zelenetskii, Polym. Sci., Ser. B, 2011, 53, 358–370 CrossRef CAS
- S. Z. Rogovina, T. A. Akopova, G. A. Vikhoreva and I. N. Gorbacheva, Polym. Degrad. Stab., 2001, 73, 557–560 CrossRef CAS
- T. A. Akopova, L. V. Vladimirov, V. A. Zhorin and A. N. Zelenetskii, Polym. Sci., Ser. B, 2009, 51, 124–134 CrossRef
- L. V. Lesnyakova, T. A. Akopova, G. A. Vikhoreva, A. N. Zelenetskii and L. S. Galbraikh, Fibre Chem., 2012, 44, 149–152 CrossRef CAS
- T. A. Akopova, T. S. Demina, A. N. Shchegolikhin, T. S. Kurkin, Ch. Grandfils, N. S. Perov, A. S. Kechekyan and A. N. Zelenetskii, Int. J. Polym. Sci., 2012, 2012, 827967 Search PubMed
- T. S. Demina, T. A. Akopova, L. V. Vladimirov, A. N. Zelenetskii, E. A. Markvicheva and C. Grandfils, Mater. Sci. Eng., C, 2016, 59, 333–338 CrossRef CAS PubMed
- T. S. Demina, K. N. Bardakova, N. V. Minaev, E. A. Svidchenko, A. V. Istomin, G. P. Goncharuk, L. V. Vladimirov, A. V. Grachev, A. N. Zelenetskii and P. S. Timashev, Polymers, 2017, 9, 302 CrossRef PubMed
- T. A. Akopova, P. S. Timashev, T. S. Demina, K. N. Bardakova, N. V. Minaev, V. F. Burdukovskii, G. V. Cherkaev, L. V. Vladimirov, A. V. Istomin, E. A. Svidchenko, N. M. Surin and V. N. Bagratashvili, Mendeleev Commun., 2015, 25, 280–282 CrossRef CAS
- N. D. Cheronis and T. S. Ma, Organic functional group analysis by micro and semimicro methods, Wiley, New York, 1964 Search PubMed
- R. D. McLachlan and R. A. Nyquist, Spectrochim. Acta, Part A, 1988, 24, 103 CrossRef
- L. J. Bellamy, The infra-red spectra of complex molecules, Methuen, London, Wiley, New York, 2nd edn, 1964 Search PubMed
- P. S. Timashev, T. S. Demina, N. V. Minaev, K. N. Bardakova, A. V. Koroleva, O. A. Kufelt, B. N. Chichkov, V. Y. Panchenko, T. A. Akopova and V. N. Bagratashvili, High Energy Chem., 2015, 49, 300–303 CrossRef CAS
- P. S. Timashev, K. N. Bardakova, N. V. Minaev, T. S. Demina, T. A. Mishchenko, E. V. Mitroshina, A. A. Akovantseva, A. V. Koroleva, D. S. Asyutin, L. F. Pimenova, N. A. Konovalov, T. A. Akopova, A. B. Solov'eva, I. V. Mukhina, M. V. Vedunova, B. N. Chichkov and V. N. Bagratashvili, Appl. Biochem. Microbiol., 2016, 52, 508–514 CrossRef CAS
- K. N. Bardakova, T. A. Akopova, A. V. Kurkov, G. P. Goncharuk, D. V. Butnaru, V. F. Burdukovskii, A. A. Antoshin, I. A. Farion, T. M. Zharikova, A. B. Shekhter, V. I. Yusupov, P. S. Timashev and Y. A. Rochev, Mar. Drugs, 2019, 17, 48 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03830b |
| This journal is © The Royal Society of Chemistry 2019 |