
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient release of immunocaptured cells using coiled-coils in a microfluidic device†
Mengen Zhang,
Bin Xu,
Allison Siehr and
Wei Shen*
and
Wei Shen*
Department of Biomedical Engineering, University of Minnesota, Minneapolis, MN 55455, USA. E-mail: shenx104@umn.edu; Tel: +1-612-624-3771
First published on 17th September 2019
Abstract
Label-free and affinity-based cell separation allows highly specific cell capture through simple procedures, but it remains a major challenge to efficiently release the captured cells without changing their structure, phenotype, and function. We report a microfluidic platform for label-free immunocapture of target cells and efficient release of the cells with minimal biochemical and biophysical perturbations. The method capitalizes on self-assembly of a pair of heterodimerizing coiled-coils, A and B. Target cells are captured in microchannels functionalized with an antibody and A and efficiently released by a liquid flow containing B-PEG (a conjugate of B and polyethylene glycol) at a controlled, low shear stress. The released cells have no antibodies attached or endogenous surface molecules cleaved. In a model system, human umbilical vein endothelial cells (HUVECs) were isolated from a mixture of HUVECs and human ovarian carcinoma cells. The capture selectivity, capture capacity, and release efficiency were 96.3% ± 1.8%, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 735 ± 1897 cells per cm2, and 92.5% ± 3.8%, respectively, when the flow was operated at a shear stress of 1 dyn cm−2. The method can be readily adapted for isolation of any cells that are recognizable by a commercially available antibody, and B-PEG is a universal cell-releasing trigger.
735 ± 1897 cells per cm2, and 92.5% ± 3.8%, respectively, when the flow was operated at a shear stress of 1 dyn cm−2. The method can be readily adapted for isolation of any cells that are recognizable by a commercially available antibody, and B-PEG is a universal cell-releasing trigger.
Introduction
Separation of desired cells from heterogeneous cell populations has a wide range of applications in biomedical fields, including disease diagnosis, pathological studies, cell-based therapies, and stem cell biology.1–6 Label-free and affinity-based cell separation, in which target cells expressing a surface biomarker are captured by a matrix material functionalized with a ligand that interacts with the biomarker, allows highly specific cell separation through simple procedures of cell capture, and therefore is attractive.7–13 However, it remains a major challenge to efficiently release the captured cells without changing their structure, phenotype, and function, which is essential for applications in which released cells need to be intact for downstream cultivation and characterization.14,15Various methods have been developed to detach affinity-captured cells, but they suffer from one or more drawbacks, such as having low release efficiency, exposing cells to harsh biochemical or biophysical environments, altering cell surface components, or not easy to be adapted for a wide range of target cells. For example, methods attempting to release cells through breaking ligand–biomarker bonds by using shear force, matrix deformation, or binding competitors for the ligand typically have insufficient cell release efficiencies or involve detrimental conditions to cells,16–20 because the multivalent cell–matrix interactions require a high energy level to be completely disrupted.14,18,21 In addition, the method of using binding competitors is ligand-specific and cell-specific, and cannot be readily adapted for a wide range of target cells. Other cell release strategies include: cleaving the linker between the capture ligand and the matrix surface by chemical, enzymatic, or physical means;13,22–27 degrading the cell surface biomarker enzymatically;28 dissolving the matrix material.29–31 These methods allow efficient cell release, but the released cells have either external molecules attached or endogenous surface molecules cleaved. Such alteration in cell surface components may affect viability, phenotype, and function of sensitive cells. For example, it has been shown that leaving anti-epithelial cell adhesion molecule (EpCAM) antibodies, which are widely used to isolate circulating tumor cells (CTCs), attached on isolated CTCs leads to cytotoxic effects on these cells.32 Many antibodies are known to act as agonists or antagonists, which can change signaling pathways and affect cell viability, phenotype, and function.33–36
We previously reported a molecularly engineered system that could immunocapture target cells with high specificity and efficiently release the captured cells with intact surface components under chemically and physically mild conditions.37 Specifically, this system contains two components: a surface functionalized with a capture antibody and coiled-coil A; a cell-releasing trigger B-PEG, which is a conjugate of coiled-coil B and a polyethylene glycol (PEG) chain. A and B can self-assemble to form heterodimers with high specificity and high affinity.38,39 (Illustration of the sequence and structure of A and B is shown in Fig. S1, ESI.†) However, all the experiments were performed in cell culture wells and the procedures to remove non-captured cells and to release captured cells were operated manually without precise control of the shear force. In addition, under the static condition, mass transfer of B-PEG to reach the bottom of captured cells is a limiting factor. Therefore, the culture wells had to be gently shaken once every 5 min to enhance mass transfer, and 30 min incubation with this frequent shaking was required to sufficiently release cells.
Here we report cell capture in microfluidic channels functionalized with a capture antibody and coiled-coil A and efficient release of the captured cells with B-PEG in a precisely controlled flow condition associated with a small shear stress. A chemical approach suitable for surface modification in microchannels was used to immobilize the capture antibody and A. A model cell mixture containing HUVECs and human ovarian carcinoma cells (OVCAR3 cells) was separated in microchannels functionalized with an anti-CD31 antibody and A. The capture selectivity, capture capacity, and release efficiency of HUVECs were examined under various well-controlled flow conditions. The morphology, viability, and CD31 intactness of released HUVECs were also examined.
Experimental section
Fabrication of microfluidic devices
A master for molding polydimethylsiloxane (PDMS) devices was fabricated on a silicon wafer. Photoresist SU-8 2075 (Microchem) was spin-coated on the wafer at 2820 rpm for 50 seconds to yield a 70 μm-thick layer, followed by a soft bake at 65 °C for 90 min. A transparency printed with the microchannel pattern (50 mm × 2 mm for length × width) at a resolution of 10000 DPI was taped to a piece of glass to serve as a photomask for photolithography. The photomask and the wafer were loaded into a Karl Suss Contact Mask Aligner. The wafer was exposed to UV for 20 s in the aligner, soft baked at 65 °C for 3 min, and hard baked at 95 °C for 7 min. The wafer was developed in propylene glycol monomethyl ether acetate (Sigma-Aldrich) for 7 min, rinsed by isopropyl alcohol for 10 min, and rinsed by water extensively.
To mold a PDMS device against the master, the Sylgard 184 PDMS precursor and the curing agent (Dow Corning) were mixed at a 10:1 weight ratio, cast on the master, vacuumed in a desiccator for 3 h to remove air bubbles, and cured at 37 °C overnight. The PDMS device was peeled off the wafer, and further cured at 80 °C for 3 additional days. The resulting PDMS device had a 50 mm long, 2 mm wide, and 70 μm high microchannel. An inlet and an outlet (1 mm in diameter) were punched near the ends of the microchannel. The PDMS device and a glass slide were sonicated with 70% ethanol and water sequentially, rinsed with water and 100% ethanol, dried with air flow and then in a vacuum desiccator for 4 h, and subjected to plasma treatment for 2 min. The plasma treated PDMS device and glass side were bonded to form a microfluidic device.
Surface modification in microfluidic channels
Immediately after a microfluidic device was assembled, surface modification of the microchannel was performed (Fig. 1). All the reaction steps were conducted at room temperature by circulating the reagents inside the channel using a peristaltic pump. For each reaction or rinsing step, the reagent solution or buffer was loaded in a syringe barrel (serving as a reservoir) and circulated at a flow rate of 440 μL min−1. The surface was first modified with 3 mL of 17% (v/v) 3-glycidoxypropyltrimethoxysilane solution (prepared in 90% ethanol, pH 4.5) for 16 h to introduce epoxy groups. After rinsing with 3 mL of ethanol and 3 mL of PBS sequentially, the microchannel was further modified with 800 μL of 10 μg mL−1 anti-human CD31 antibody solution (eBioScience Inc.) for 4 h to allow immobilization of the antibody through the epoxy–amine reaction. To derivatize the remaining epoxy groups into maleimide groups, the microchannel was modified with 800 μL of 30 mM N-(2-aminoethyl)maleimide trifluoroacetate solution (pH 8.0; Sigma-Aldrich) for 24 h. After rinsing with PBS, the microchannel was further modified with 600 μL of 1 mM cysA solution (pH 7.2, filtered through a 0.2 μm filter) for 24 h to allow immobilization of A through the thiol–maleimide reaction. The cysA solution contained 5 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP, Pierce). The microchannel was rinsed with PBS prior to use.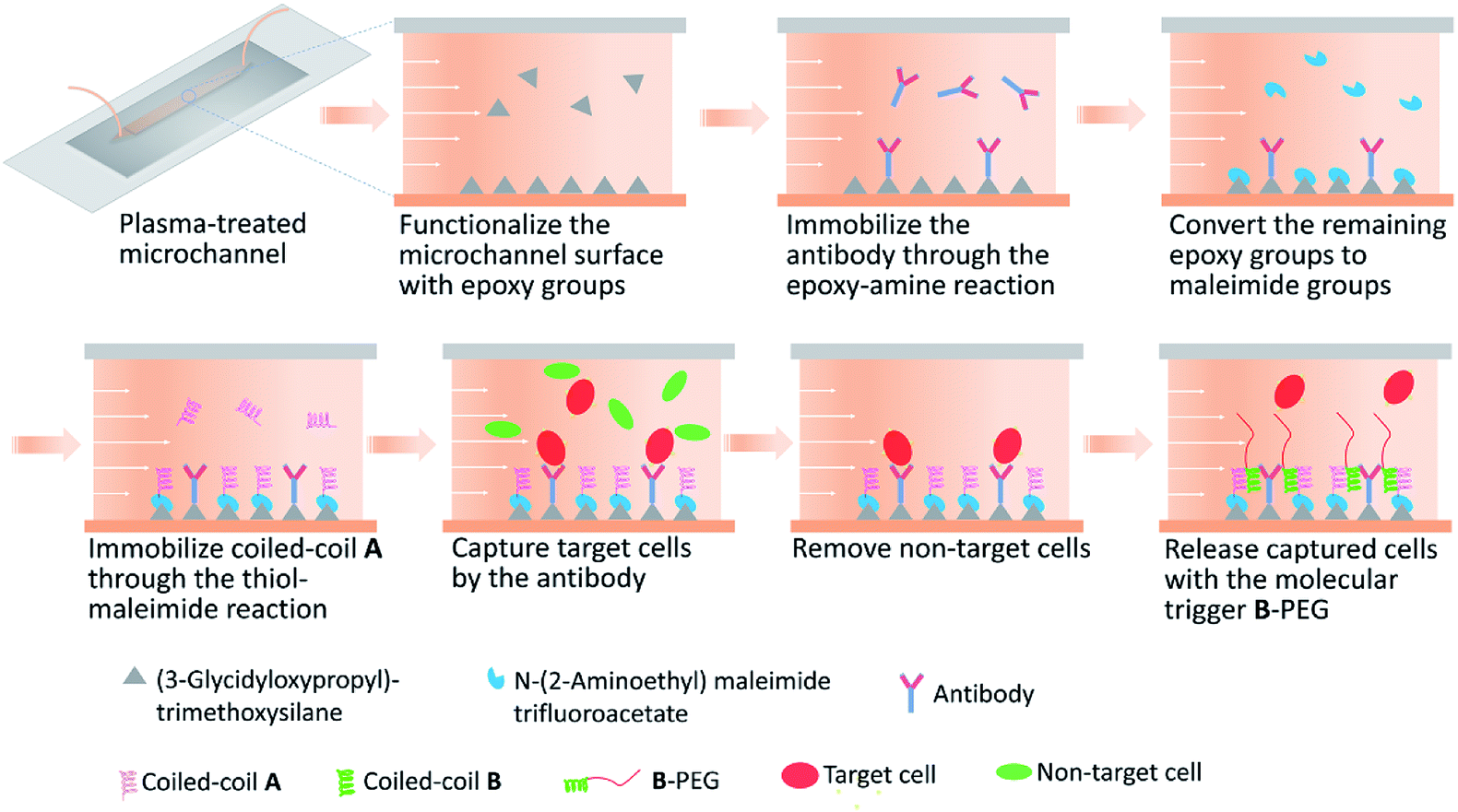 | ||
| Fig. 1 Schematic of surface modification and cell separation in a microfluidic channel. | ||
Preparation of polypeptides and the B-PEG conjugate
Polypeptides (cysA, which contains a cysteine residue at the N-terminus, and Bcys, which contains a cysteine residue at the C-terminus) and the B-PEG conjugate were synthesized and purified as previously reported.37,39 Briefly, the genes encoding cysA and Bcys were constructed in the Qiagen pQE9 expression vector, and cysA and Bcys was each expressed in the E. coli strain SG13009 under control of the bacteriophage T5 promoter. The polypeptides were purified through nickel nitrilotriacetic acid (Ni-NTA) metal-affinity chromatography (Qiagen), and characterized with SDS-polyacrylamide gel electrophoresis and MALDI mass spectrometry. To synthesize B-PEG, a 25 mg mL−1 Bcys solution was prepared in 2 M urea containing 20 mM TCEP (pH 5) and incubated at room temperature for 1 h to reduce disulfide bonds, followed by adjusting the pH value to 7.4. A 150 mg mL−1 PEG-maleimide solution (30 kDa, Laysan Bio Inc.) was mixed with the Bcys solution at a 15:1 molar ratio, and the thiol–maleimide reaction was allowed to proceed under stirring and protection with argon gas for 2 days at room temperature. The reaction mixture was loaded in a column packed with Ni-NTA resin to remove unreacted PEG-maleimide. The B-PEG and unreacted Bcys bound to the Ni-NTA resin were eluted and further separated with thiopropyl-sepharose 6B resin. Purified B-PEG was dialyzed against water at 4 °C for 2 days and dried through lyophilization.
Cell maintenance
HUVECs were maintained in EGM™-2 BulletKit™ (Lonza). The GFP+ OVCAR3 were maintained in RMPI-1640 medium (Life Technologies) supplemented with 10% fetal bovine serum (FBS, Atlanta Biologicals), 1% penicillin–streptomycin (Life Technologies), and 2 mM Glutamax (Life Technologies). Both media were changed every 2 days.Cell capture and release
HUVECs were stained with CellTracker™ Orange CMTMR dye (ThermoFisher Scientific) by incubating with culture medium containing 2 μM dye for 30 min at 37 °C. The suspensions of the stained HUVECs and the GFP+ OVCAR3 cells were prepared in serum-free EGM medium and mixed at a 1:1 ratio. The cell density in the mixture was 2 million per mL for each cell type. The cell mixture was loaded into each microchannel and incubated at 37 °C for 30 min to allow cell capture (the PDMS side of the channel was at the bottom, so cells were captured on the PDMS surface), followed by removal of non-captured cells with a flow of serum-free medium. Flow rates were controlled at 11, 21, and 42 μL min−1 to generate shear stresses of 0.5, 1, and 2 dyn cm−2, respectively;23 the durations were 4, 2, and 1 min, respectively, to keep the volume of the liquid used to remove non-captured cells the same under different shear stresses. The captured cells were imaged with a 5× objective on a Zeiss Observer Z1 inverted fluorescence microscope for quantification of capture selectivity and capture capacity.
Release of captured cells was performed with a flow of serum-free medium containing 200 μM or 400 μM B-PEG at a flow rate corresponding to a shear stress of 1 dyn cm−2. The durations for the release experiments were 10 min for 400 μM B-PEG and 20 min for 200 μM B-PEG, so that the same amount of B-PEG was used. Each microchannel was imaged with a 5× objective before and after the release experiment to determine the release efficiency. A control in which a 400 μM PEG solution (30 kDa) was used to release captured cells at 1 dyn cm−2 for 10 min was conducted. In addition, release of captured HUVECs with a flow of serum-free medium not containing B-PEG or PEG at various shear stresses in the range of 2–10 dyn cm−2 was also examined (the duration was 1 min). All the experiments and controls were repeated at least 3 times using independently fabricated microchannel devices.
Assessment of morphology and viability of released cells
The suspension of the cells released from one microchannel was mixed with fresh EGM medium at a 1:1 volume ratio and replated in an FBS-coated well in a 96-well plate (the well was coated by incubating with 200 μL of FBS at 37 °C for 1 h). Half hour after replating, half of the medium in the well was replaced with fresh EGM medium. One hour after replating, all the medium in the well was replaced with fresh EGM medium. The morphology of the cells 1 h, 3 h, and overnight after replating was examined through bright-field microscopy. After overnight culture, the cells were stained with a live cell tracker CellTracker™ Orange CMTMR dye and imaged on a Zeiss Observer Z1 inverted fluorescence microscope to evaluate cell viability.
Assessment of intactness of CD31 on released cells by flow cytometry
To examine whether released cells had intact CD31, captured and released HUVECs were fixed in 1% paraformaldehyde (Sigma-Aldrich) for 5 min, blocked in 1% bovine serum albumin (Fisher) for 30 min, and stained with the anti-CD31 primary antibody (eBioScience Inc., 1:300 dilution) for 1 h and a goat anti-mouse AlexaFluor488 secondary antibody (Invitrogen, 1:500 dilution) for 45 min sequentially or stained with only the secondary antibody (no primary antibody staining). HUVECs not subject to the capture and release processes were stained in the same manner as a control. All steps were conducted at room temperature. Stained cells were analyzed by a BD LSR II flow cytometer (BD Biosciences, San Jose, CA). Cells were first gated on the basis of forward scatter (FSC) and side scatter (SSC). The fluorescence intensity of gated cells was then analyzed using a GFP channel (excitation with a 488 nm laser; emission detected using 505LP and 525/50 nm filters). Flow cytometry data were analyzed with FlowJo (software version 10, Ashland, OR).
Statistical analysis
Data are reported as mean ± standard deviation (SD) of the mean. Unpaired t-test was used for statistical analysis. A p value of <0.05 is considered to be statistically significant.Results and discussion
Fabrication of microfluidic devices and surface modification of the microchannels
Microfluidic devices having a 50 mm long, 2 mm wide, and 70 μm high microchannel were fabricated by bonding a molded PDMS replica to a glass slide. Both the PDMS and the glass slide need to be plasma treated for efficient bonding, and therefore all surface modification steps were conducted in the microchannel after a device was assembled to avoid damaging functional chemical groups. The chemical approach illustrated in Fig. 1 was used to immobilize a capture antibody and coiled-coil A in a microchannel. The microchannel surface was first derivatized with a silane coupling agent 3-glycidoxypropyltrimethoxysilane to introduce amine-reactive epoxy groups, which allowed immobilization of the antibody.40 After the remaining epoxy groups were derivatized to maleimide groups with N-(2-aminoethyl)maleimide trifluoroacetate, cysA (containing a cysteine residue) was immobilized on the surface through the thiol–maleimide reaction. This approach to modifying the microchannel surface was robust, allowing highly reproducible experimental results.We previously reported cell capture and release in culture wells functionalized with a capture antibody and A.37 In that study, the culture well surface was derivatized with sulfosuccinimidyl 6-(4′-azido-2′-nitrophenylamino)hexanoate (Sulfo-SANPAH) to introduce an amine-reactive N-hydroxysuccinimide (NHS) ester group, which allowed antibody immobilization and other downstream surface modifications. We observed large variations when using commercial Sulfo-SANPAH from different lots or from different manufacturers, probably due to the poor stability of the NHS ester group. In addition, surface derivatization with Sulfo-SANPAH, which contains an NHS ester group and a photoactivatable nitrophenyl azide, requires exposure to UV light. Therefore, surface derivatization with Sulfo-SANPAH in a microchannel requires sufficient transmittance of UV light through the materials used to build the device. This may restrict materials selection when the technology is adapted for various applications in the future. In contrast, the chemical approach reported in the present study is robust and applicable in devices made of materials with all kinds of optical properties.
Selective cell capture
A cell mixture containing HUVECs (stained with CellTracker™ Orange CMTMR dye) and GFP+ OVCAR3 cells at a 1:1 ratio was introduced in a microchannel functionalized with an anti-CD31 antibody and coiled-coil A and allowed for incubation for 30 min, followed by introduction of a continuous flow of serum-free medium at a flow rate of 11 μL min−1 to remove non-captured cells. The shear stress imposed on the cells at the bottom surface was 0.5 dyn cm−2 according to τ = 6μQ/(WH2), where τ is the shear stress, μ is the dynamic viscosity of the fluid, Q is the flow rate, and W and H are the width and height of the microchannel.23 The images acquired before and after introduction of the flow revealed that HUVECs, which expressed CD31 on the surface, were selectively captured, and a slow flow associated with a shear stress of 0.5 dyn cm−2 was sufficient to remove the majority of OVCAR3 cells, which did not express CD31 (Fig. 2a and b). A control experiment in which the cell mixture was introduced in a microchannel functionalized with A alone showed that few cells of either type adhered (Fig. 2c), suggesting that selective capture of HUVECs shown in Fig. 2b was specifically mediated by the immobilized antibody. In another control experiment in which the cell mixture was introduced in a microchannel with an unmodified surface, considerable number of cells of both types remained adherent after the flow of serum-free medium was introduced (Fig. 2d), suggesting that the unmodified microchannel surface interacted with both cell types non-specifically and a shear stress of 0.5 dyn cm−2 was not sufficient to break these non-specific interactions. These experiments and controls suggested that the A co-immobilized on the surface prevented non-specific cell–surface interactions and that the microchannel surface used in Fig. 2a and b was thoroughly modified with the capture antibody and A, as patches of the unmodified surface would allow OVCAR3 cells to be captured through non-specific interactions.
 | ||
| Fig. 2 Capture of HUVECs (red) from a mixture of HUVECs and OVCAR3 cells (green). (a) The mixture was injected into a microchannel functionalized with an anti-CD31 antibody and coiled-coil A. (b) Non-captured cells in (a) was rinsed off with a slow flow of medium imposing a shear stress of 0.5 dyn cm−2 for 4 min, revealing selective capture of HUVECs. (c) In a control microchannel functionalized with A alone, few cells of either type adhered after rinsing. (d) In a control microchannel with an unmodified surface, both cell types adhered after rinsing. The scale bars are 200 μm. | ||
Capture selectivity, which is defined as the number of captured HUVECs divided by the total number of cells remaining in a channel after a flow was introduced to remove non-captured cells, was examined. The images acquired before and after removal of non-captured cells revealed that the capture selectivity was 93.9% ± 2.1%, 96.3% ± 1.8%, and 95.9% ± 1.9% when non-captured cells were removed at 0.5, 1, and 2 dyn cm−2, respectively, in microchannels functionalized with the anti-CD31 antibody and coiled-coil A (Fig. 3a). The high capture selectivity suggests that the molecular design and the chemical approach used in the study result in a microfluidic system that allows highly selective immunocapture of target cells. The values of capture selectivity at the various shear stresses are not statistically different, suggesting that a shear stress of 0.5 dyn cm−2 was sufficient to remove the cells not captured by the antibody and the cells captured by the antibody would not detach at 2 dyn cm−2.
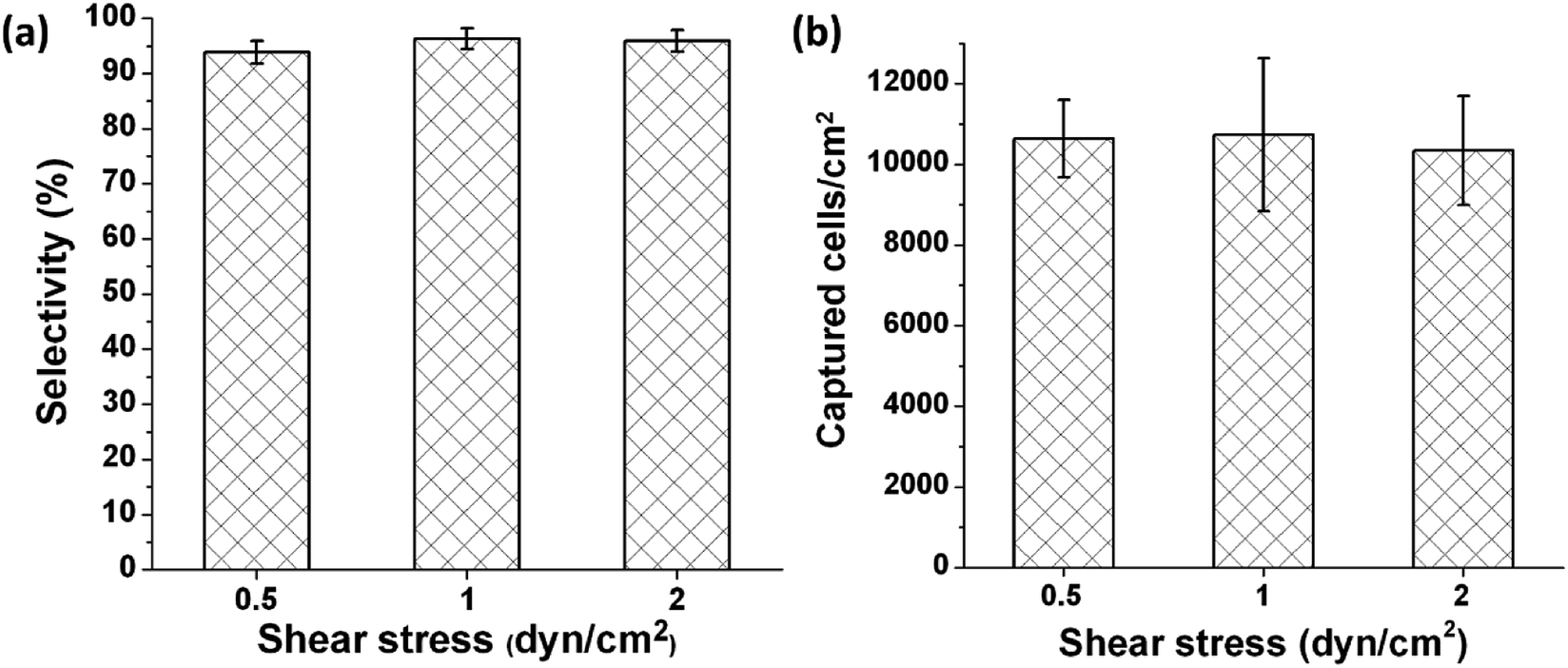 | ||
| Fig. 3 Capture selectivity (a) and capture capacity (b) of HUVECs in microchannels functionalized with an anti-CD31 antibody and coiled-coil A when non-captured cells were removed with a flow of medium imposing various shear stresses. The durations of the flow were 4, 2, and 1 min for 0.5, 1, and 2 dyn cm−2, respectively, to keep the volume of the liquid used to remove non-captured cells the same in different conditions. Error bars represent standard deviations, n = 4. | ||
Capture capacity, which is defined as the number of captured HUVECs in unit area, was quantified on the basis of the images acquired after non-captured cells were removed. The capture capacity was 10637 ± 961 cells per cm2, 10735 ± 1897 cells per cm2, and 10345 ± 1353 cells per cm2 when non-captured cells were removed at 0.5, 1, and 2 dyn cm−2, respectively (Fig. 3b). These values of capture capacity are not statistically different, further confirming that the cells captured by the antibody would not detach at 2 dyn cm−2.
Cell release
To release the HUVECs immunocaptured in a microchannel, a serum-free EGM medium containing 400 μM B-PEG was allowed to flow through the channel at a shear stress of 1 dyn cm−2 for 10 min. Most of the captured cells were released (Fig. 4a and b). In a control experiment in which a medium containing 400 μM PEG (30 kDa) was allowed to flow through the channel, only a few cells were released (Fig. 4c and d). The PEG size in the control was the same as that in B-PEG (30 kDa). These results suggest that the coiled-coil B played an essential role in efficiently releasing the immunocaptured HUVECs at a low shear stress. Heterodimerization between the A immobilized on the surface and the B in B-PEG brought B-PEG to the surface, and the PEG chains adopted extended conformations to repel and release the cells. In contrast, there is no driving force for unmodified PEG molecules to adhere to the surface, and therefore PEG molecules could not release the captured cells efficiently. It should also be noted that releasing cells with a flow of B-PEG solution in microchannels accelerated cell release as compared with that under a static condition. In our previous study in which cells were captured in culture wells, the samples had to be incubated with B-PEG for at least 30 min and gently shaken once every 5 min to sufficiently release captured cells.37 In contrast, captured cells were sufficiently released within 10 min when the B-PEG solution flowed through microchannels at a shear stress of 1 dyn cm−2, most likely because the convective flow enhanced mass transfer.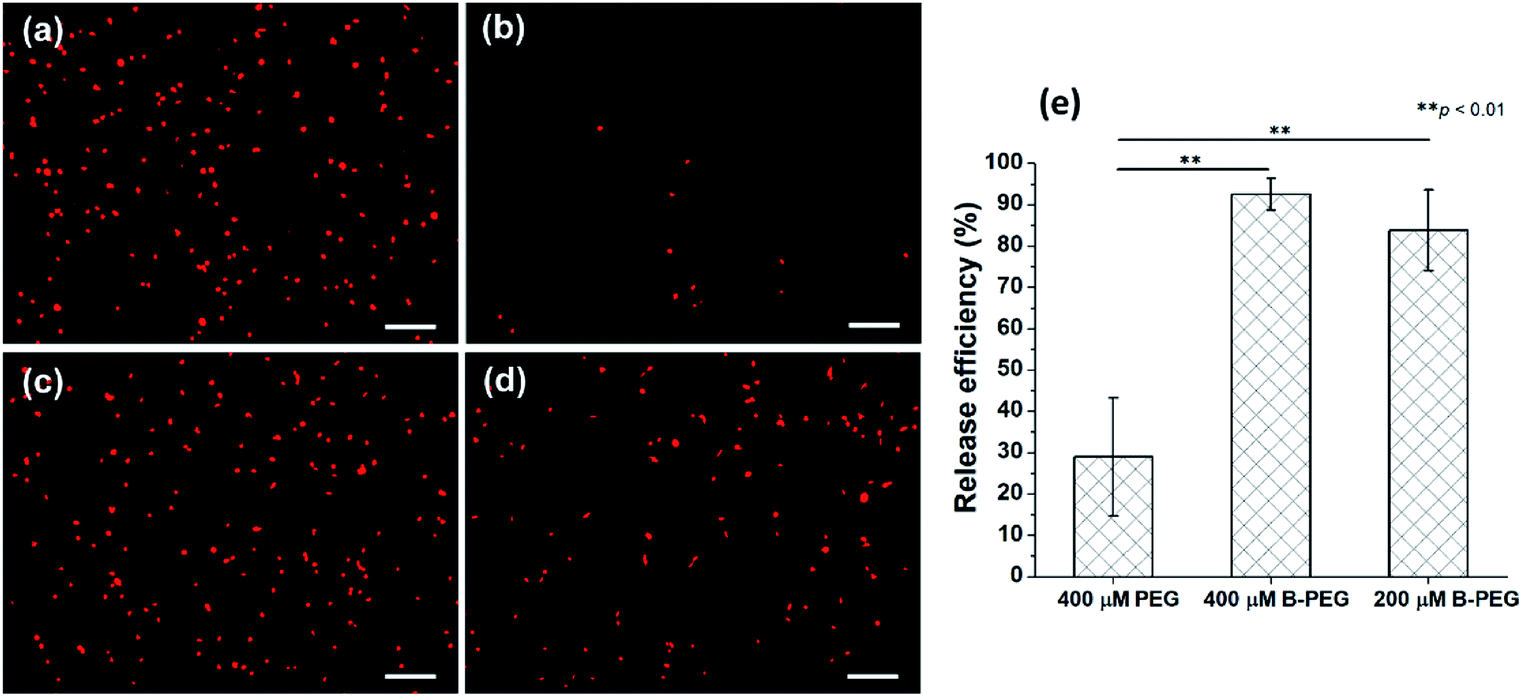 | ||
| Fig. 4 Release of HUVECs captured in microchannels functionalized with an anti-CD31 antibody and coiled-coil A. The release experiments were conducted at a flow rate imposing a shear stress of 1 dyn cm−2. The durations for the release experiments were 10 min for 400 μM B-PEG or PEG and 20 min for 200 μM B-PEG. (a) and (c) HUVECs captured in microchannels. (b) The captured HUVECs in (a) were efficiently released with a flow of medium containing 400 μM B-PEG. (d) The captured HUVECs in (c) were not efficiently released with a flow of medium containing 400 μM PEG. (e) Quantitative analysis of the release efficiency of captured HUVECs. Error bars represent standard deviations, n = 3. **p < 0.01, Student's t-test. The scale bars are 200 μm. | ||
The release efficiency, which is defined as (the number of captured HUVECs − the number of HUVECs remaining in the channel after cell release)/the number of captured HUVECs, was quantified on the basis of the images acquired before and after the release experiment. The release efficiency was 92.5% ± 3.8% when the cells were released with a medium containing 400 μM B-PEG at 1 dyn cm−2 (Fig. 4e). In contrast, only 28.9% ± 14.3% of captured HUVECs were released when a medium containing 400 μM PEG flowed through the channel in the control experiment. Cell release with a medium containing 200 μM B-PEG at a shear stress of 1 dyn cm−2 for 20 min was also performed (a 20 min duration was used to keep the same total amount of B-PEG), and the release efficiency was 83.8% ± 9.8%, slightly lower than that with a releasing medium containing 400 μM B-PEG.
To further confirm that B-PEG played an essential role in efficiently releasing the immunocaptured HUVECs at a low shear stress, control experiments were conducted in which a serum-free medium not containing B-PEG flowed through the channel to release captured HUVECs at various shear stresses. The release efficiencies were 7.7% ± 6.7%, 30.5% ± 4.8%, 62.4% ± 16.8%, and 64% ± 17.7% at shear stresses of 2, 5, 7, and 10 dyn cm−2, respectively (Fig. 5). Without B-PEG, the release efficiency at 5 dyn cm−2 was approximately one third of that with a releasing medium containing B-PEG at 1 dyn cm2; even at 10 dyn cm−2, the release efficiency was only approximately two thirds of that with a releasing medium containing B-PEG at 1 dyn cm−2.
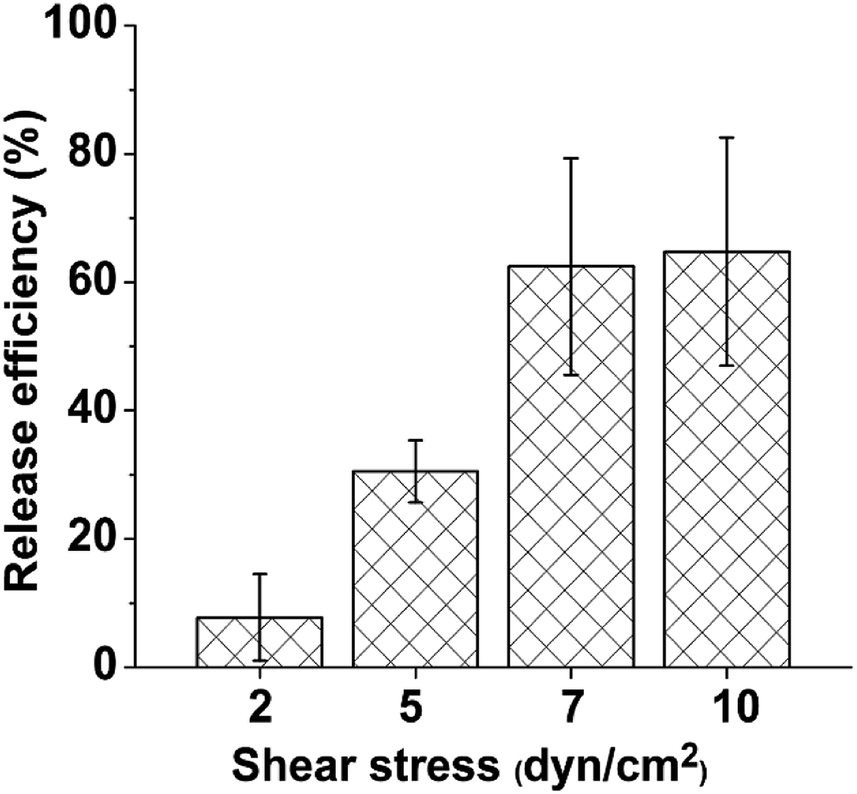 | ||
| Fig. 5 Release efficiency of captured HUVECs with a flow of medium not containing B-PEG at various shear stresses. The duration of the release experiments was 1 min. Error bars represent standard deviations, n = 3. | ||
It is expected that the densities of immobilized antibody and A and the ratio between them are important for the success of both cell capture and cell release. The procedures reported in the experimental section to modify the surface of microchannels were determined after we tested a variety of parameters. In particular, the antibody concentration and the reaction time for the step of antibody immobilization, which are expected to determine the ratio between immobilized antibody and A, are essential. We observed that lower antibody concentration and shorter reaction time at this step impaired cell capture, and higher antibody concentration and longer reaction time hurt cell release. Future studies will include systematic tuning of the densities of immobilized antibody and A and the ratio between them, characterization of these material properties, and examination of how these properties affect cell capture and release.
Morphology and viability of released cells
To examine the morphology and viability of released HUVECs, non-stained HUVECs were mixed with GFP+ OVCAR3 cells and allowed to be captured in a microchannel functionalized with the anti-CD31 antibody and A. After the non-captured cells were removed with a flow of medium at 0.5 dyn cm−2, the captured HUVECs were released with a flow of medium containing 400 μM B-PEG at a shear stress of 1 dyn cm−2 for 10 min. The cell recovery rate quantified by dividing the number of collected cells by the expected number of released cells (using a release efficiency of 93%) was 71%. The suspension of the released cells was mixed with fresh EGM medium at a 1:1 volume ratio and replated in an FBS-coated well in a 96-well plate. To remove B-PEG from the medium, half medium was replaced with fresh EGM medium at half hour after replating, and all the medium was replaced with fresh EGM medium at 1 h after replating, when cell attachment was obvious (Fig. 6a). At 3 h after replating, the cells exhibited cobblestone morphology (Fig. 6b), typical for statically cultured HUVECs. After overnight culture, all the cells were alive as revealed from staining with the CellTracker™ Orange CMTMR dye (Fig. 6c and d). Few green fluorescent OVCAR3 cells were observed in Fig. 6d, which is a merged fluorescence image of red and green channels.
 | ||
| Fig. 6 Culture of released HUVECs. (a–c) The released cells 1 h (a), 3 h (b), and overnight (c) after replating. (d) The fluorescence image of the released cells cultured overnight and stained with a live cell tracker CellTracker™ Orange CMTMR dye. The scale bars are 200 μm. | ||
Intactness of CD31 on released cells
Flow cytometry was used to evaluate the intactness of CD31 after the separation process. HUVECs subjected to the capture and release processes were stained with anti-CD31 primary antibody and a fluorescently labelled secondary antibody sequentially, or stained with only the secondary antibody. HUVECs not subjected to the separation process were stained in the same manner as a control. Gating on the basis of FSC and SSC showed 97.4% of the events were HUVECs for the control and 95% of the events were HUVECs for the samples subjected to the separation process, suggesting that the separation process minimally affected the cell integrity (Fig. S2a and b, ESI†). Gated HUVECs were then analyzed for their fluorescence distribution. Separated HUVECs stained with both primary and secondary antibodies showed a similar fluorescence distribution to control cells (Fig. S2c, ESI†), suggesting that CD31 receptors were not lost during cell separation, as the fluorescence distribution peak would be located to the left of the control peak if CD31 receptors were lost. Separated HUVECs stained with only the secondary antibody exhibited a negligible fluorescent signal similar to that of control HUVECs (Fig. S2d, ESI†), suggesting that the anti-CD31 antibody used for cell capture did not attach on the cell surface during cell release, as a strong fluorescence signal would be observed if they did. These results confirmed the intactness of CD 31on released cells.The method reported here allows label-free cell separation through highly specific immunocapture, and it can be readily adapted for a wide range of target cells that are recognizable by a commercially available antibody. The captured cells can be released efficiently at a well-controlled, low shear stress in the microfluidic device without being exposed to any harsh conditions. The released cells have intact surface components, with neither external molecules attached nor endogenous surface molecules cleaved. This is in contrast to previously reported cell release methods after immunocapture. In most of these methods, cells are released with the capture antibody attached.22–26,30,31 In some studies, non-specific proteases, such as trypsin, were used to degrade the cell surface biomarker to release cells.28 It has been shown that both types of alteration in the cell surface components (having antibodies attached or having endogenous cell surface molecules cleaved) may change viability, phenotype, and function of sensitive cells.32–36,41
Conclusions
We report a microfluidic platform capable of isolating target cells from heterogeneous cell populations through highly specific immunocapture and efficiently releasing the captured cells at a controlled flow rate associated with a small shear stress. The method is label-free and performed under mild conditions, and the released cells have intact biochemical components at the surface, with no external molecules attached nor endogenous molecules cleaved. The collective merits of this cell separation method are enabled by a molecular engineering strategy harnessing a pair of heterodimerizing coiled-coils A and B, with A co-immobilized on the surface with the capture antibody and B conjugated with PEG to form a molecular trigger B-PEG for cell release. The method can be adapted for isolation of any cells recognizable by a commercially available antibody, and B-PEG is a universal cell-releasing trigger.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Science Foundation (CBET-1134148) for financial support of this work and thank Professor Sundaram Ramakrishnan in the Department of Pharmacology at the University of Minnesota for providing GFP+ OVCAR3 cells. We also thank the IMV core facility and Professor Louis Mansky at the University of Minnesota for allowing use of the BD LSRII Flow Cytometer.References
- J. W. Chan, D. K. Lieu, T. Huser and R. A. Li, Anal. Chem., 2009, 81, 1324–1331 CrossRef CAS PubMed.
- X. Cheng, D. Irimia, M. Dixon, K. Sekine, U. Demirci, L. Zamir, R. G. Tompkins, W. Rodriguez and M. Toner, Lab Chip, 2007, 7, 170–178 RSC.
- A. Gros, M. R. Parkhurst, E. Tran, A. Pasetto, P. F. Robbins, S. Ilyas, T. D. Prickett, J. J. Gartner, J. S. Crystal, I. M. Roberts, K. Trebska-McGowan, J. R. Wunderlich, J. C. Yang and S. A. Rosenberg, Nat. Med., 2016, 22, 433–438 CrossRef CAS PubMed.
- S. Nagrath, L. V. Sequist, S. Maheswaran, D. W. Bell, D. Irimia, L. Ulkus, M. R. Smith, E. L. Kwak, S. Digumarthy, A. Muzikansky, P. Ryan, U. J. Balis, R. G. Tompkins, D. A. Haber and M. Toner, Nature, 2007, 450, 1235–1239 CrossRef CAS PubMed.
- E. Heitzer, M. Auer, C. Gasch, M. Pichler, P. Ulz, E. M. Hoffmann, S. Lax, J. Waldispuehl-Geigl, O. Mauermann, C. Lackner, G. Hofler, F. Eisner, H. Sill, H. Samonigg, K. Pantel, S. Riethdorf, T. Bauernhofer, J. B. Geigl and M. R. Speicher, Cancer Res., 2013, 73, 2965–2975 CrossRef CAS PubMed.
- K. Schriebl, S. Lim, A. Choo, A. Tscheliessnig and A. Jungbauer, Biotechnol. J., 2010, 5, 50–61 CrossRef CAS PubMed.
- A. Kumar, F. M. Plieva, I. Y. Galaev and B. Mattiasson, J. Immunol. Methods, 2003, 283, 185–194 CrossRef CAS PubMed.
- E. Mandrusov, A. Houng, E. Klein and E. F. Leonard, Biotechnol. Prog., 1995, 11, 208–213 CrossRef CAS PubMed.
- J. Miwa, Y. Suzuki and N. Kasagi, J. Microelectromech. Syst., 2008, 17, 611–622 CAS.
- A. Sin, S. K. Murthy, A. Revzin, R. G. Tompkins and M. Toner, Biotechnol. Bioeng., 2005, 91, 816–826 CrossRef CAS PubMed.
- L. B. Ujam, R. H. Clemmitt, S. A. Clarke, R. A. Brooks, N. Rushton and H. A. Chase, Biotechnol. Bioeng., 2003, 83, 554–566 CrossRef CAS PubMed.
- K. Wang, M. K. Marshall, G. Garza and D. Pappas, Anal. Chem., 2008, 80, 2118–2124 CrossRef CAS PubMed.
- Z. Zhang, N. Chen, S. Li, M. R. Battig and Y. Wang, J. Am. Chem. Soc., 2012, 134, 15716–15719 CrossRef CAS PubMed.
- J. Hubble, Trends Biotechnol., 1997, 15, 249–255 CrossRef CAS.
- Q. Zheng, S. M. Iqbal and Y. Wan, Biotechnol. Adv., 2013, 31, 1664–1675 CrossRef PubMed.
- R. E. Nordon and K. Schindhelm, Artif. Organs, 1997, 21, 107–115 CrossRef CAS PubMed.
- L. S. Cheung, X. Zheng, A. Stopa, J. C. Baygents, R. Guzman, J. A. Schroeder, R. L. Heimark and Y. Zohar, Lab Chip, 2009, 9, 1721–1731 RSC.
- M. B. Dainiak, A. Kumar, I. Y. Galaev and B. Mattiasson, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 849–854 CrossRef CAS PubMed.
- Y. Wan, Y. Liu, P. B. Allen, W. Asghar, M. A. Mahmood, J. Tan, H. Duhon, Y. T. Kim, A. D. Ellington and S. M. Iqbal, Lab Chip, 2012, 12, 4693–4701 RSC.
- D. D. Putnam, V. Namasivayam and M. A. Burns, Biotechnol. Bioeng., 2003, 81, 650–665 CrossRef CAS PubMed.
- J. Hubble, Immunol. Today, 1997, 18, 305–306 CrossRef CAS PubMed.
- H. Liu, X. Liu, J. Meng, P. Zhang, G. Yang, B. Su, K. Sun, L. Chen, D. Han, S. Wang and L. Jiang, Adv. Mater., 2013, 25, 922–927 CrossRef CAS PubMed.
- U. A. Gurkan, T. Anand, H. Tas, D. Elkan, A. Akay, H. O. Keles and U. Demirci, Lab Chip, 2011, 11, 3979–3989 RSC.
- S. Hou, H. Zhao, L. Zhao, Q. Shen, K. S. Wei, D. Y. Suh, A. Nakao, M. A. Garcia, M. Song, T. Lee, B. Xiong, S. C. Luo, H. R. Tseng and H. H. Yu, Adv. Mater., 2013, 25, 1547–1551 CrossRef CAS PubMed.
- H. Zhu, J. Yan and A. Revzin, Colloids Surf., B, 2008, 64, 260–268 CrossRef CAS PubMed.
- S. Jeon, W. Hong, E. S. Lee and Y. Cho, Theranostics, 2014, 4, 1123–1132 CrossRef CAS PubMed.
- B. Wildt, D. Wirtz and P. C. Searson, Nat. Protoc., 2010, 5, 1273–1280 CrossRef CAS PubMed.
- U. Dharmasiri, S. Balamurugan, A. A. Adams, P. I. Okagbare, A. Obubuafo and S. A. Soper, Electrophoresis, 2009, 30, 3289–3300 CrossRef CAS PubMed.
- B. D. Plouffe, M. A. Brown, R. K. Iyer, M. Radisic and S. K. Murthy, Lab Chip, 2009, 9, 1507–1510 RSC.
- A. Hatch, G. Hansmann and S. K. Murthy, Langmuir, 2011, 27, 4257–4264 CrossRef CAS PubMed.
- A. M. Shah, M. Yu, Z. Nakamura, J. Ciciliano, M. Ulman, K. Kotz, S. L. Stott, S. Maheswaran, D. A. Haber and M. Toner, Anal. Chem., 2012, 84, 3682–3688 CrossRef CAS PubMed.
- M. Münz, A. Murr, M. Kvesic, D. Rau, S. Mangold, S. Pflanz, J. Lumsden, J. Volkland, J. Fagerberg, G. Riethmüller, D. Rüttinger, P. Kufer, P. A. Baeuerle and T. Raum, Cancer Cell Int., 2010, 10, 44 CrossRef PubMed.
- A. Mijares, D. Lebesgue, G. Wallukat and J. Hoebeke, Mol. Pharmacol., 2000, 58, 373–379 CrossRef CAS PubMed.
- E. S. Vitetta and J. W. Uhr, Cancer Res., 1994, 54, 5301–5309 CAS.
- A. I. Machuca-Parra, A. A. Bigger-Allen, A. V. Sanchez, A. Boutabla, J. Cardona-Velez, D. Amarnani, M. Saint-Geniez, C. W. Siebel, L. A. Kim, P. A. D'Amore and J. F. Arboleda-Velasquez, J. Exp. Med., 2017, 214, 2271–2282 CrossRef CAS PubMed.
- U. B. Hagemann, L. Gunnarsson, S. Geraudie, U. Scheffler, R. A. Griep, H. Reiersen, A. R. Duncan and S. M. Kiprijanov, PLoS One, 2014, 9, e103776 CrossRef PubMed.
- M. Zhang and W. Shen, Macromol. Biosci., 2017, 17, 1600330 CrossRef PubMed.
- W. Shen, K. Zhang, J. A. Kornfield and D. A. Tirrell, Nat. Mater., 2006, 5, 153–158 CrossRef CAS PubMed.
- B. Liu, Y. Liu, J. J. Riesberg and W. Shen, J. Am. Chem. Soc., 2010, 132, 13630–13632 CrossRef CAS PubMed.
- B. Xu, A. Magli, Y. Anugrah, S. J. Koester, R. C. R. Perlingeiro and W. Shen, Biomaterials, 2018, 183, 54–66 CrossRef CAS PubMed.
- K. Tsuji, M. Ojima, K. Otabe, M. Horie, H. Koga, I. Sekiya and T. Muneta, Cell Transplant., 2017, 26, 1089–1102 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03871j |
| This journal is © The Royal Society of Chemistry 2019 |