
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
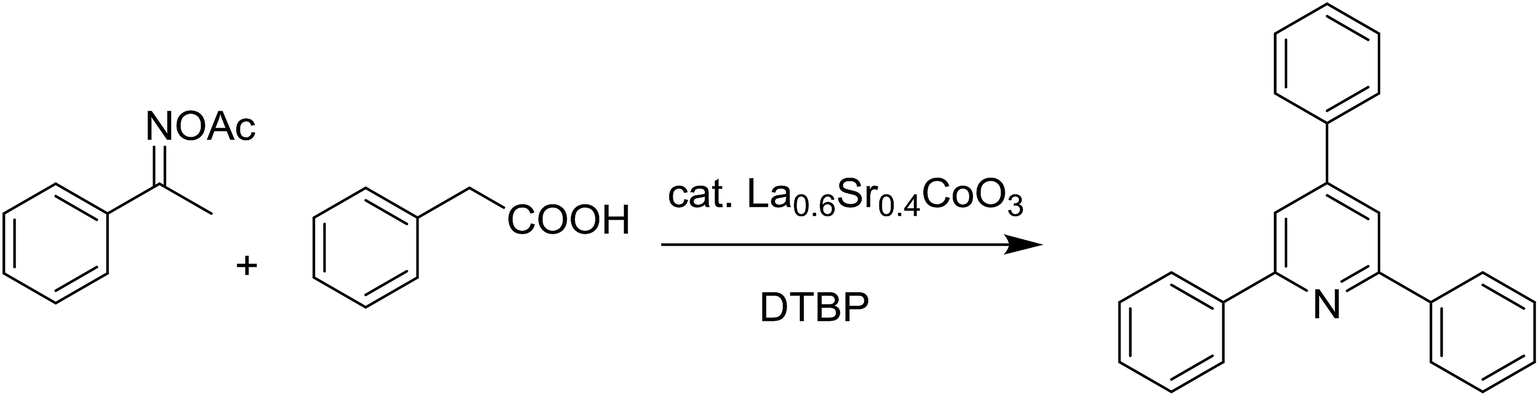
Synthesis of triphenylpyridines via an oxidative cyclization reaction using Sr-doped LaCoO3 perovskite as a recyclable heterogeneous catalyst†
Thu N. M. Le,
Son H. Doan,
Phuc H. Pham ,
Khang H. Trinh,
Tien V. Huynh,
Tien T. T. Tran,
Minh-Vien Le,
Tung T. Nguyen* and
Nam T. S. Phan*
,
Khang H. Trinh,
Tien V. Huynh,
Tien T. T. Tran,
Minh-Vien Le,
Tung T. Nguyen* and
Nam T. S. Phan*
Faculty of Chemical Engineering, HCMC University of Technology, VNU-HCM, 268 Ly Thuong Kiet, District 10, Ho Chi Minh City, Vietnam. E-mail: tungtn@hcmut.edu.vn; ptsnam@hcmut.edu.vn; Fax: +84 8 38637504; Tel: +84 8 38647256 extn 5681
First published on 1st August 2019
Abstract
An La0.6Sr0.4CoO3 strontium-doped lanthanum cobaltite perovskite was prepared via a gelation and calcination approach and used as a heterogeneous catalyst for the synthesis of triphenylpyridines via the cyclization reaction between ketoximes and phenylacetic acids. The transformation proceeded via the oxidative functionalization of the sp3 C–H bond in phenylacetic acid. The La0.6Sr0.4CoO3 catalyst demonstrated a superior performance to that of the pristine LaCoCO3 as well as a series of homogeneous and heterogeneous catalysts. Furthermore, the La0.6Sr0.4CoO3 catalyst was facilely recovered and reused without considerable decline in its catalytic efficiency. To the best of our knowledge, the combination of ketoximes with easily available phenylacetic acids is novel.
1. Introduction
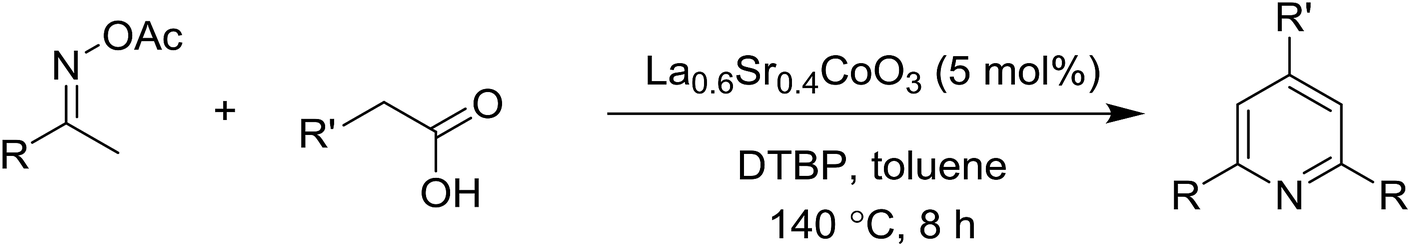
Functionalized pyridines have emerged as a precious class of privileged N-heterocyclic compounds, existing in a wide range of biologically active natural products, medicinal chemicals, agricultural chemicals, and functional organic materials.1–4 Among the numerous pyridines, 2,4,6-triphenylpyridines offer important applications, and the development of effective synthetic approaches for these structures has been considered as a hot topic. The most popular method relies on the three-component cyclocondensation transformation of acetophenones, benzaldehydes, and ammonium acetate under diverse conditions.5 Recently, new synthetic protocols have been developed, in which the utilization of ketoximes as starting materials has been explored for the synthesis of the abovementioned heterocyclic compounds. Ren et al. developed an efficient strategy to achieve 2,4,6-triphenylpyridines via the CuBr-catalyzed coupling of oxime acetates with aldehydes.6 Fu reported the Cu(OTf)2-catalyzed oxidative sp3 C–H coupling of oxime acetates with toluene, benzylamines, and p-toluenesulfonylhydrazones.7 Iron(III) chloride was used for the cyclization of ketoxime carboxylates with N,N-dialkylanilines or aldehyde to afford densely substituted pyridines8,9 (Scheme 1a). The coupling of aryl amines or benzyl halides and aromatic ketones for the synthesis of triarylpyridines is also known.10–12 Certainly, ketoximes have recently gained significant interest as valuable building blocks for numerous important organic structures.13–17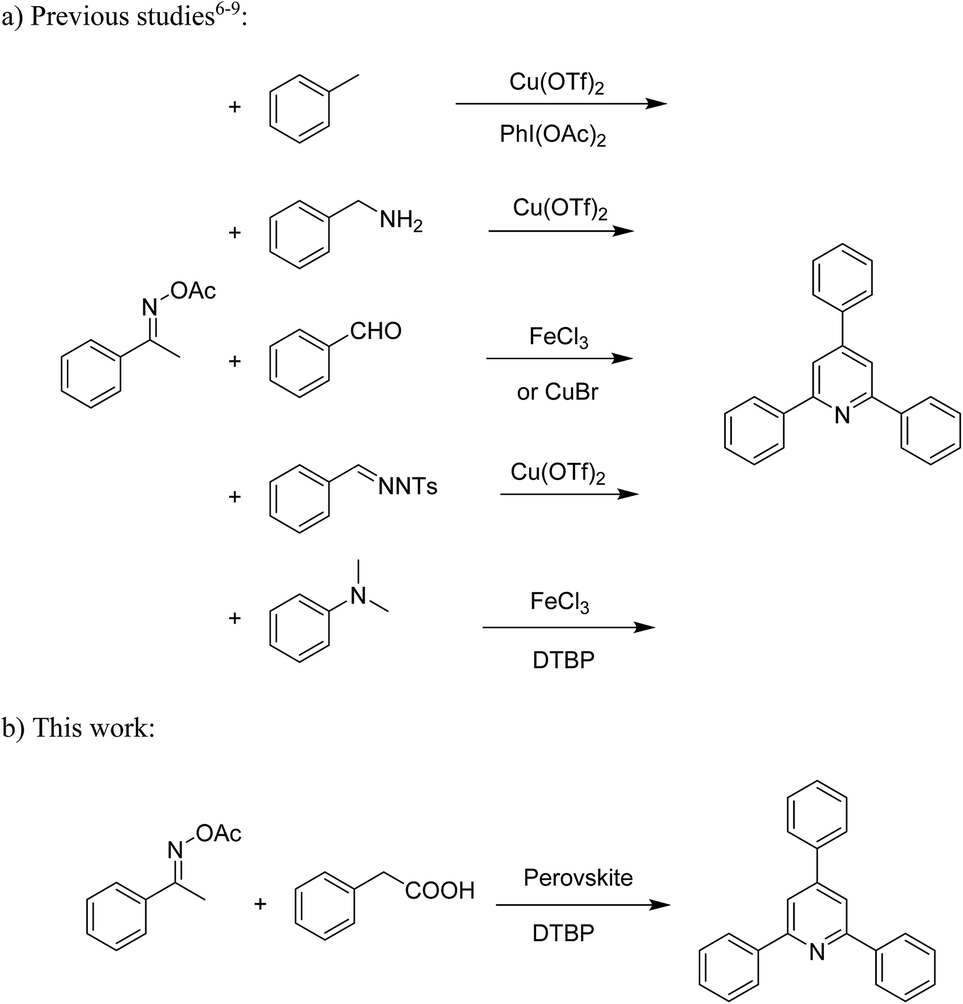 | ||
| Scheme 1 The difference between previous works (a) and this work (b). | ||
Perovskites have attracted extensive interest from researchers worldwide owing to their significant physicochemical properties, such as redox behavior, superconductivity, ferroelectricity, magnetoresistance, and ionic conductivity.18–20 Perovskites are mixed oxides, generally existing in the form of ABO3, where A represents an alkali, alkaline or lanthanide metal, and B stands for a transition metal.21–23 The metal A is responsible for the stability of the perovskite structure, and the substitution of A by other metals affects the valence state of metal B, leading to different catalytic activities.24 Certainly, perovskites have been extensively utilized as heterogeneous catalysts for numerous reduction–oxidation processes.25–31 LaCoO3-based perovskites are one of the most active catalysts for CO oxidation,24,32 NO oxidation,33 syngas conversion,34,35 and hydrogen production.36 Nonetheless, the application of perovskite catalysts in the organic synthesis field is extremely rare in the literature. Additionally, the utilization of easily available phenylacetic acids as green starting materials for organic synthesis via the oxidative functionalization of sp3 C–H bonds has gained considerable attention.37–39 In this work, we report the utilization of a strontium-doped lanthanum cobaltite perovskite as a recyclable heterogeneous catalyst for the synthesis of triphenylpyridines via oxidative cyclization reaction between ketoximes and phenylacetic acids (Scheme 1b). Single oxides or salts of lanthanum, strontium, and cobalt demonstrated a low catalytic performance for the transformation, indicating the importance of the synergistic effect in the perovskite catalyst. To the best of our knowledge, the oxidative cyclization between ketoximes and phenylacetic acids has not been previously mentioned in any literature.
2. Experimental
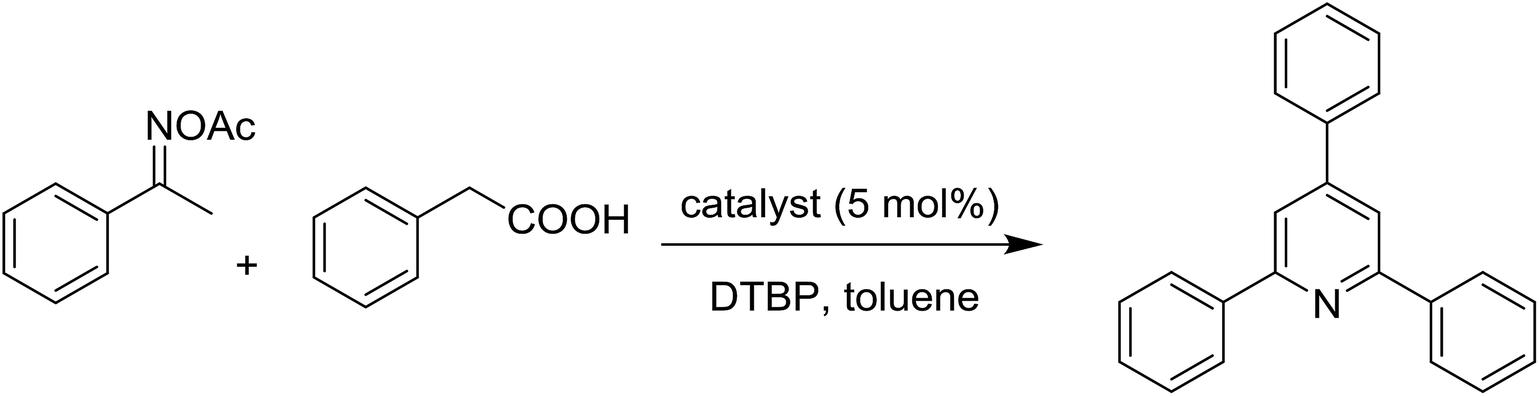
The strontium-doped lanthanum cobaltite perovskite (La0.6Sr0.4CoO3) powder was prepared from La(NO3)3, Sr(NO3)2, and Co(NO3)2 via a gelation and calcination approach, as previously reported in the literature.33 The perovskite was accordingly characterized using several conventional analysis techniques (Fig. S1–S4†). In a representative experiment, a mixture of acetophenone oxime acetate (0.106 g, 0.6 mmol), phenylacetic acid (0.027 g, 0.2 mmol), and diphenyl ether (16 μL, 0.1 mmol) as an internal standard was added to a 12 mL screw-cap pressurized vial containing toluene (2 mL). The reaction mixture was magnetically stirred and heated for 5 min. The La0.6Sr0.4CoO3 perovskite catalyst (2.3 mg, 5 mol%) was then added to the vial. The reaction mixture was magnetically stirred for 2 min to completely disperse the catalyst in the liquid phase, followed by the addition of di-tert-butylperoxide (DTBP, 0.14 mL, 0.6 mmol). The resulting mixture was stirred at 140 °C for 2 h. Subsequently, the reactor tube was back-filled with argon and then stirred at 140 °C for 6 h. The reaction mixture was diluted with ethyl acetate (5 mL) and washed with saturated NaHCO3 solution (5 mL). The organic layer was dried using anhydrous Na2SO4. Reaction yields were recorded from the GC analysis results based on the diphenyl ether internal standard. To isolate the triphenylpyridine product, the ethyl acetate phase was concentrated under reduced pressure, and purified by column chromatography on silica gel with a hexane/ethyl acetate solvent mixture. The triphenylpyridine was consequently confirmed by GC-MS, 1H NMR, and 13C NMR. The strontium-doped lanthanum cobaltite perovskite catalyst was collected from the reaction mixture by centrifugation, washed carefully with ethyl acetate, acetone, and diethyl ether, dried under reduced pressure at room temperature overnight on a Schlenk line, and reutilized in the catalyst recycling studies.3. Results and discussion
The strontium-doped lanthanum cobaltite perovskite was used as a heterogeneous catalyst for the synthesis of 2,4,6-triphenylpyridine via the oxidative cyclization reaction between acetophenone oxime acetate and phenylacetic acid (Scheme 1b). The reaction proceeded in the presence of an oxidant, which is required for the oxidative functionalization of the sp3 C–H bond in phenylacetic acid. Initially, the reaction conditions were optimized to improve the yield of the triphenylpyridine product (Table 1). For the reactions conducted in the liquid phase, the nature of the solvent may be critical, especially when a solid catalyst is employed. Thus, the impact of solvent on the yield of 2,4,6-triphenylpyridine was studied. Running the reaction in chlorobenzene gave 54% yield of the substituted pyridine (entry 1). Heptane was inferior to toluene (entries 2 and 3). It was noted that polar aprotic solvents such as DMF, DMAc, DMSO, and NMP were almost ineffective for the reaction, with less than 5% yield recorded (see the ESI† for details).
|
||||
|---|---|---|---|---|
| Entry | Solvent | Catalyst amount (mol%) | Oxidant | Yieldb (%) |
| a Reaction conditions: acetophenone oxime acetate (0.6 mmol), phenylacetic acid (0.2 mmol), solvent (2 mL), oxidant (0.6 mmol), at 140 °C under air for 2 h and under argon for 6 h.b GC yield.c Reaction at 120 °C.d TBHP in water. DTBP = di-tert-butylperoxide, TBHP = tert-butyl hydroperoxide. | ||||
| 1 | Chlorobenzene | 5 | DTBP | 54 |
| 2 | Heptane | 5 | DTBP | 22 |
| 3 | Toluene | 5 | DTBP | 83 |
| 4c | Toluene | 5 | DTBP | 17 |
| 5 | Toluene | 0 | DTBP | 4 |
| 6 | Toluene | 1 | DTBP | 21 |
| 7 | Toluene | 3 | DTBP | 51 |
| 8 | Toluene | 7 | DTBP | 87 |
| 9 | Toluene | 5 | K2S2O8 | 8 |
| 10 | Toluene | 5 | AgNO3 | 11 |
| 11 | Toluene | 5 | Oxygen | 2 |
| 12d | Toluene | 5 | TBHP | 55 |
With these results, the influence of temperature on the reaction was explored. The reaction was conducted at different temperatures, ranging from room temperature to 140 °C. No product was detected when the temperature was lower than 120 °C, and 17% yield was detected for the reaction performed at 120 °C (entry 4). Additionally, the effect of the catalyst amount on the yield of 2,4,6-triphenylpyridine was investigated. Only 4% yield was detected in the absence of the strontium-doped lanthanum cobaltite perovskite catalyst, indicating that the perovskite was critical for the oxidative cyclization reaction (entry 5). Using 1 mol% catalyst, the reaction afforded 21% yield (entry 6). As expected, increasing the perovskite quantity enhanced the yield of the triphenylpyridine product. Extending the catalyst amount to 3 mol% led to 51% yield, while 87% yield was achieved in the presence of 7 mol% catalyst (entries 7 and 8, respectively).
An oxidant should be present for the oxidative functionalization of the sp3 C–H bond in phenylacetic acid, which facilitates the cyclization.37 Accordingly, the impact of oxidant on the yield of 2,4,6-triphenylpyridine was addressed. The experiment was conducted under standard conditions, using a variety of inorganic and organic oxidants. The reaction using K2S2O8 as the oxidant gave a sluggish crude mixture, with only 8% yield (entry 9). Similarly, AgNO3 and oxygen were not suitable for this reaction (entries 10 and 11, respectively). tert-Butyl hydroperoxide in water was somewhat effective, affording the triphenylpyridine product in 55% yield (entry 12).
Since the synthesis of 2,4,6-triphenylpyridine via oxidative cyclization between acetophenone oxime acetate and phenylacetic acid was conducted in toluene, a critical issue is that the leached species from the strontium-doped lanthanum cobaltite perovskite may be accountable a considerable part of the catalytic efficiency. Thus, to clarify if leaching was serious in this reaction, a control experiment was performed. The reaction was carried out in toluene under argon at 140 °C for 8 h using 3 equivalents of ketoxime and 3 equivalents of DTBP in the presence of 5 mol% catalyst. After the first 160 min reaction time with 47% yield of 2,4,6-triphenylpyridine recorded, the perovskite catalyst was removed by centrifugation. The reaction solution was transferred to a new 12 mL screw-cap pressurized vial. The reaction vial was back-filled with argon and the resulting mixture was subsequently stirred at 140 °C for an additional 320 min. Aliquots were taken at different time intervals, and subsequently analyzed by GC. The GC data provided quantitative information regarding the residual, catalytically active sites in the toluene phase. It was observed that almost no additional 2,4,6-triphenylpyridine was generated in the absence of the strontium-doped lanthanum cobaltite perovskite (Fig. 1). These results verified that the perovskite-catalyzed oxidative cyclization between acetophenone oxime acetate and phenylacetic acid proceeded under truly heterogeneous conditions.
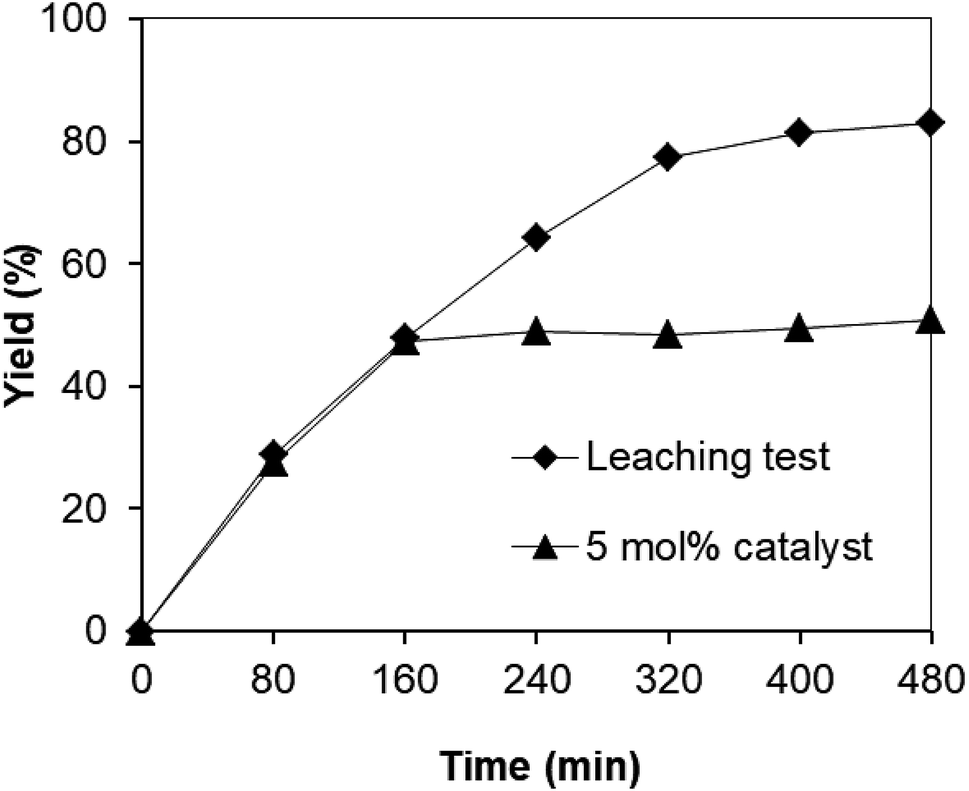 | ||
| Fig. 1 Leaching experiment indicating that no additional 2,4,6-triphenylpyridine was formed after the removal of the catalyst. | ||
To emphasize the advantages of using the strontium-doped lanthanum cobaltite perovskite catalyst, its activity in the oxidative cyclization reaction was compared to that of other catalysts (Table 2). The reaction was conducted in toluene at 140 °C for 8 h, using 3 equivalents of ketoxime and 3 equivalents of DTBP, in the presence of 5 mol% catalyst. First, traditional homogeneous catalysts were utilized for the reaction (entries 1–10). The FeCl3-catalyzed transformation afforded 26% yield, while 6% yield was observed for the reaction using the FeCl2 catalyst. Fe(NO3)3 and FeSO4 displayed a low catalytic performance for the reaction, with 3% and 13% yields being detected, respectively. In the case of copper salts, the use of CuSO4 resulted in 57% yield, while 8% yield was noted for the reaction using a Cu(NO3)2 catalyst. NiSO4 demonstrated a low catalytic performance, although 38% yield was recorded. The three precursors used to synthesize the strontium-doped lanthanum cobaltite perovskite catalyst, including La(NO3)3, Sr(NO3)2, and Co(NO3)2, also exhibited low catalytic activity, forming the expected product in 2%, 39%, and 4% yields, respectively. In a second series of experiments, several heterogeneous catalysts were utilized for the reaction (entries 11–18). Cu(BDC), a copper–organic framework, was active for the reaction, producing the product in 70% yield. The reaction using a ZIF-8A catalyst, a zinc–organic framework, progressed to 60% yield. UiO-66·TFA, a zirconium–organic framework, was less reactive toward the oxidative cyclization reaction, generating the desired product in 40% yield. The single oxides La2O3, CoO, and SrO exhibited low activity, though 38% yield was detected for the case of SrO (entries 14–16). The pristine perovskite, LaCoCo3, was also not very active towards the transformation, although 40% yield was detected (entry 17). Interestingly, using the strontium-doped perovskite La0.6Sr0.4CoO3 as a catalyst for the oxidative cyclization reaction led to 83% yield (entry 18).
|
|||
|---|---|---|---|
| Entry | Homogeneous catalyst | Heterogeneous catalyst | Yieldb (%) |
| a Reaction conditions: acetophenone oxime acetate (0.6 mmol), phenylacetic acid (0.2 mmol), DTBP (0.6 mmol), toluene (2 mL), catalyst (5 mol%), 140 °C, under air for 2 h and under argon for 6 h.b GC yield. | |||
| 1 | FeCl3 | 26 | |
| 2 | FeCl2 | 6 | |
| 3 | Fe(NO3)3 | 3 | |
| 4 | FeSO4 | 13 | |
| 5 | CuSO4 | 57 | |
| 6 | Cu(NO3)2 | 8 | |
| 7 | NiSO4 | 38 | |
| 8 | Co(NO3)2 | 4 | |
| 9 | La(NO3)2 | 2 | |
| 10 | Sr(NO3)2 | 39 | |
| 11 | Cu(BDC) | 70 | |
| 12 | ZIF-8A | 60 | |
| 13 | UiO-66·TFA | 43 | |
| 14 | La2O3 | 19 | |
| 15 | CoO | 11 | |
| 16 | SrO | 38 | |
| 17 | LaCoCo3 | 40 | |
| 18 | La0.6Sr0.4CoO3 | 83 | |
To comprehend the mechanism of the oxidative cyclization reaction, several control experiments were performed, as highlighted in Scheme 2. The presence of (2,2,6,6-tetramethylpiperidin-1-yl)oxy (TEMPO) as a radical scavenger had a significant effect on the formation of benzaldehyde (5) in the first step, which implied the involvement of a radical process in the oxidative decarboxylation of phenylacetic acid (1) (Scheme 2a). In the next two experiments, high conversions to benzaldehyde were achieved, which indicated that 2-hydroxy-2-phenylacetic acid (3) and 2-oxo-2-phenylacetic acid (4) were the likely intermediates of this process (Scheme 2b and c, respectively). When the oxidative cyclization reaction between ketoxime (7) and 2-oxo-2-phenylacetic acid (4) was subsequently carried out under standard conditions, 2,4,6-triphenylpyridine (16) was obtained in 86% yield, suggesting that 2-oxo-2-phenylacetic acid may be an important intermediate (Scheme 2d). The cyclization of the ketoxime (7) and benzaldehyde (5) generated 2,4,6-triphenylpyridine (16) in high yield (92%), which demonstrates that benzaldehyde (5) may be the key intermediate in this reaction (Scheme 2e). The reaction between the ketoxime (7) and benzaldehyde (5) did not proceed in the presence of the antioxidant TEMPO, indicating that a radical process may be involved and that the interaction of TEMPO with the radical species generated in the catalytic cycle may have cease the transformation (Scheme 2f). (Z)-3-Amino-1,3-diphenylprop-2-en-1-one (12) was synthesized and allowed to undergo the cyclization with the ketoxime (7) under two different conditions. The yield of 2,4,6-triphenylpyridine in the cyclization without the La0.6Sr0.4CoO3 catalyst was similar to that of the cyclization using the La0.6Sr0.4CoO3 catalyst (Scheme 2g). The reaction between (1), (7), and (7b) produced 3 products (16, 16b, and 16c) in similar yields under the standard conditions (Scheme 2h). These results suggest that (Z)-3-amino-1,3-diphenylprop-2-en-1-one (12) may be the intermediate in this reaction, and that the transformation of (Z)-3-amino-1,3-diphenylprop-2-en-1-one (12) to 2,4,6-triphenylpyridine (16) could proceed in the absence of the La0.6Sr0.4CoO3 catalyst.
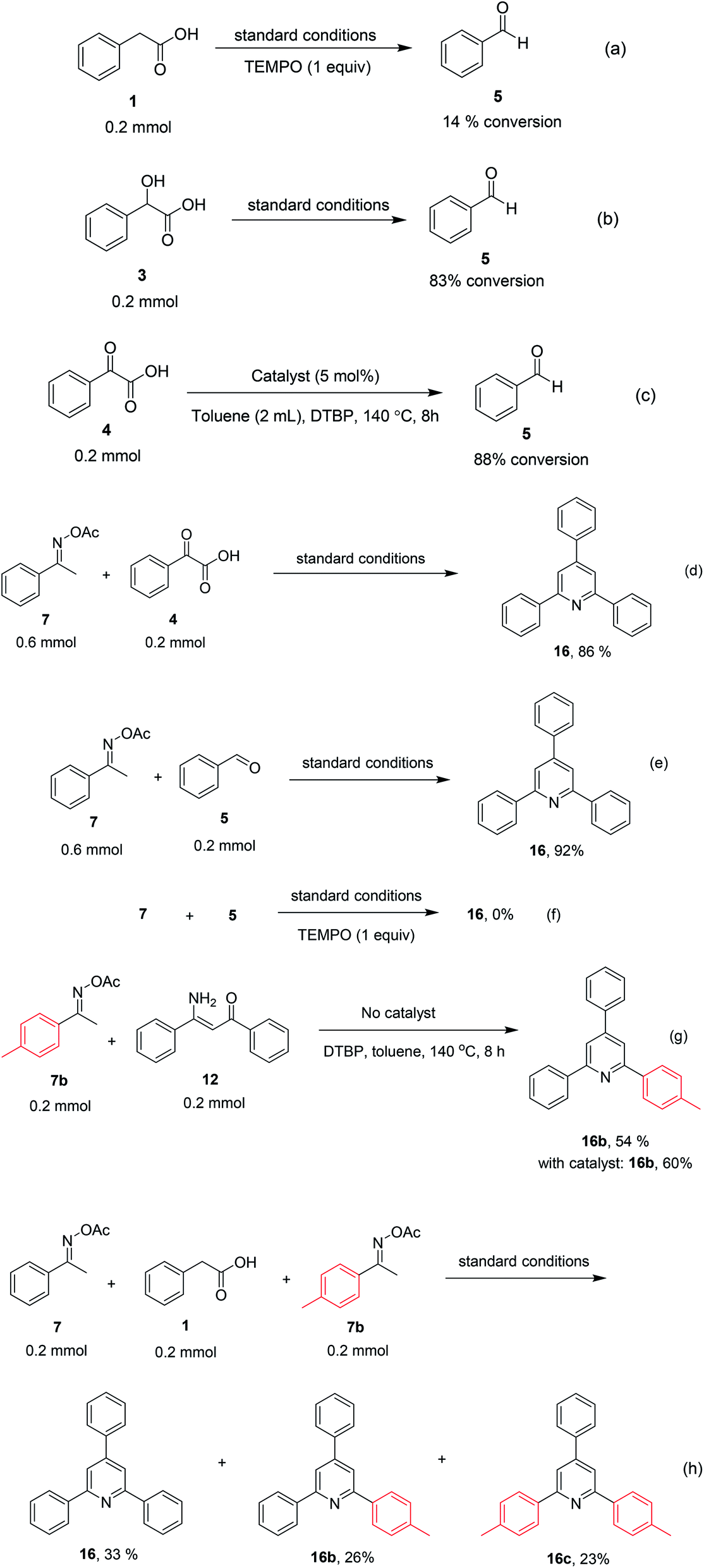 | ||
| Scheme 2 Control experiments. | ||
Based on the aforementioned results and previous reports,8,37,40–43 a plausible mechanism is proposed in Scheme 3. The first step is the aerobic oxidation of phenylacetic acid (1) to benzaldehyde (5) via the formation of 2-hydroxy-2- phenylacetic acid (3) and 2-oxo-2-phenylacetic acid (4), followed by the single electron transfer (SET) process between benzaldehyde and Mn+1 to generate benzoyl radical (6). It should be noted that a benzylic radical (2) is formed in this process. The next step is the reductive cleavage of the N–O bond of the ketoxime (7) by Mn via a single electron transfer (SET) to achieve the Mn+1 species and imine radical (8), which undergo rapid coordination to Mn to give the imino-Mn+1 intermediate (9). Tautomerization of the imino-Mn+1 intermediate (9) forms the enamino-Mn+1 intermediate (10), which reacts with the benzoyl radical (6) to generate a new radical (11). This radical undergoes a single electron transfer (SET) process, resulting in the formation of intermediate (12) upon regeneration of the Mn species. Subsequently, the condensation of the intermediate (12) with a second molecule of the ketoxime (7) produces the intermediate (13) and releases NH2OAc. Tautomerization of intermediate (13) affords intermediate (14), followed by the intramolecular cyclization of intermediate (14), generating intermediate (15). Finally, the dehydration of intermediate (15) produces 2,4,6-triphenylpyridine (16). In this catalytic cycle, M is the cobalt species in the La0.6Sr0.4CoO3 catalyst. It should be noted that using CoO as a catalyst for the oxidative cyclization reaction resulted in a very low yield (entry 15, Table 2). The pristine perovskite, LaCoCo3, was also not very active towards the transformation (entry 17, Table 2), while utilizing the strontium-doped perovskite, La0.6Sr0.4CoO3 as the catalyst afforded 80% yield (entry 18, Table 2). It was previously reported that substituting the La(III) site by Sr(II) site in the perovskite will improve the mobility of lattice oxygen, thus significantly enhancing the redox activity of the catalyst.24,44 Certainly, the synergistic effect in the La0.6Sr0.4CoO3 catalyst is critical for the oxidative cyclization reaction.
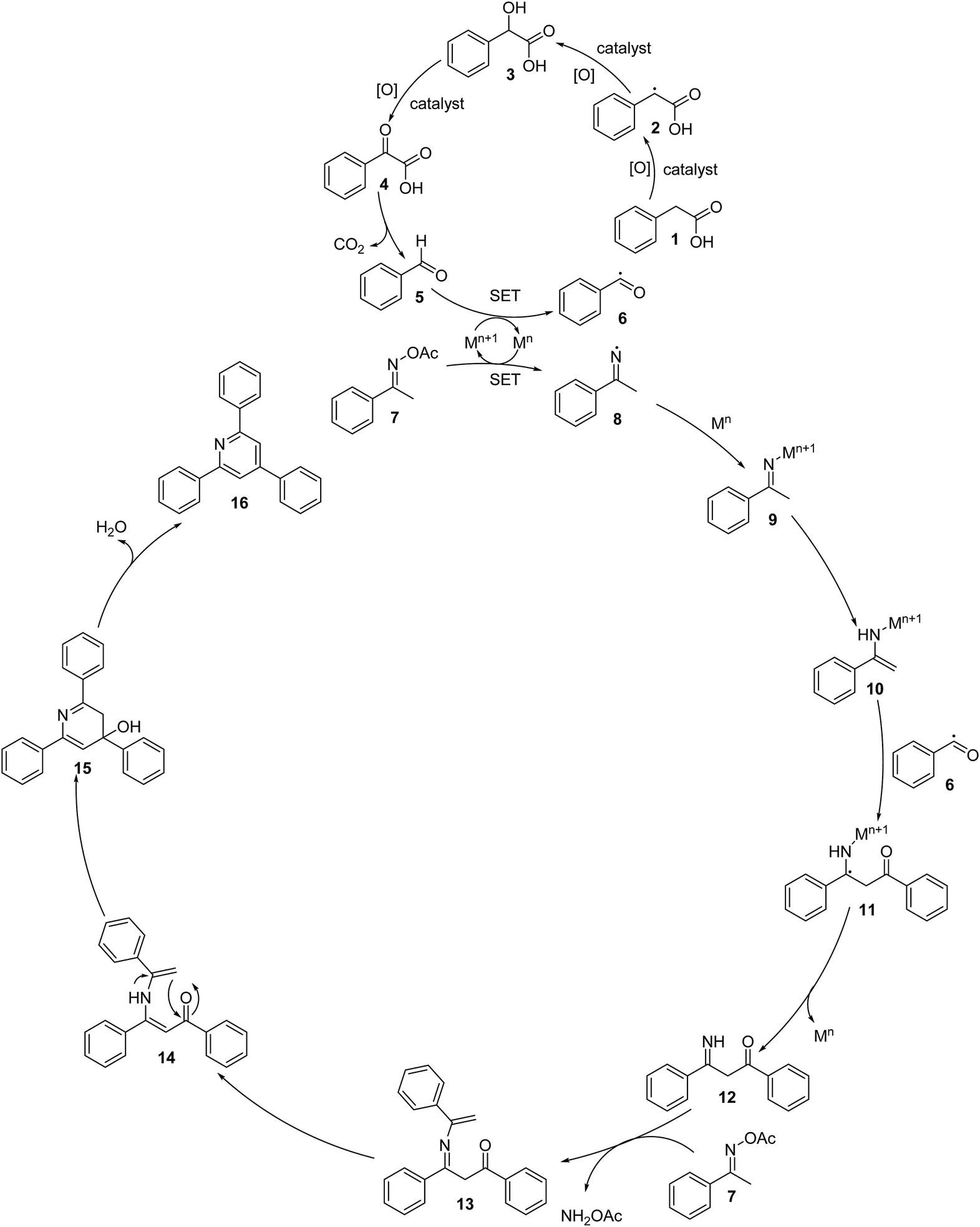 | ||
| Scheme 3 Proposed reaction mechanism. | ||
One critical aspect that must be addressed for the oxidative cyclization reaction using the strontium-doped lanthanum cobaltite perovskite catalyst is the possibility of catalyst reutilization. In the best case, it should be possible to recover and reutilize the solid catalyst many times before it ultimately becomes totally deactivated. Accordingly, the reutilization of the La0.6Sr0.4CoO3 catalyst was investigated for the synthesis of 2,4,6-triphenylpyridine via the oxidative cyclization reaction between acetophenone oxime acetate and phenylacetic acid under the standard conditions. The reaction was conducted in toluene at 140 °C for 8 h using 3 equivalents of ketoxime and 3 equivalents of DTBP, in the presence of 5 mol% catalyst. At the end of each catalytic run, the La0.6Sr0.4CoO3 catalyst was collected from the reaction mixture by centrifugation, washed carefully with ethyl acetate, acetone, and diethyl ether. The recovered catalyst was dried under reduced pressure at room temperature overnight on a Schlenk line, and reutilized for the next experiment. The experimental results indicated that the La0.6Sr0.4CoO3 catalyst could be reutilized several times in the oxidative cyclization reaction. At the fifth catalytic run, 82% yield of 2,4,6-triphenylpyridine was recorded (Fig. 2). Moreover, the recovered La0.6Sr0.4CoO3 catalyst was characterized via XRD and FT-IR analyses, and the results were compared with that of the new strontium-doped perovskite. The XRD (Fig. 3) and FT-IR (Fig. 4) observations revealed that the La0.6Sr0.4CoO3 catalyst was stable in the synthesis of 2,4,6-triphenylpyridine via the oxidative cyclization reaction under the standard conditions.
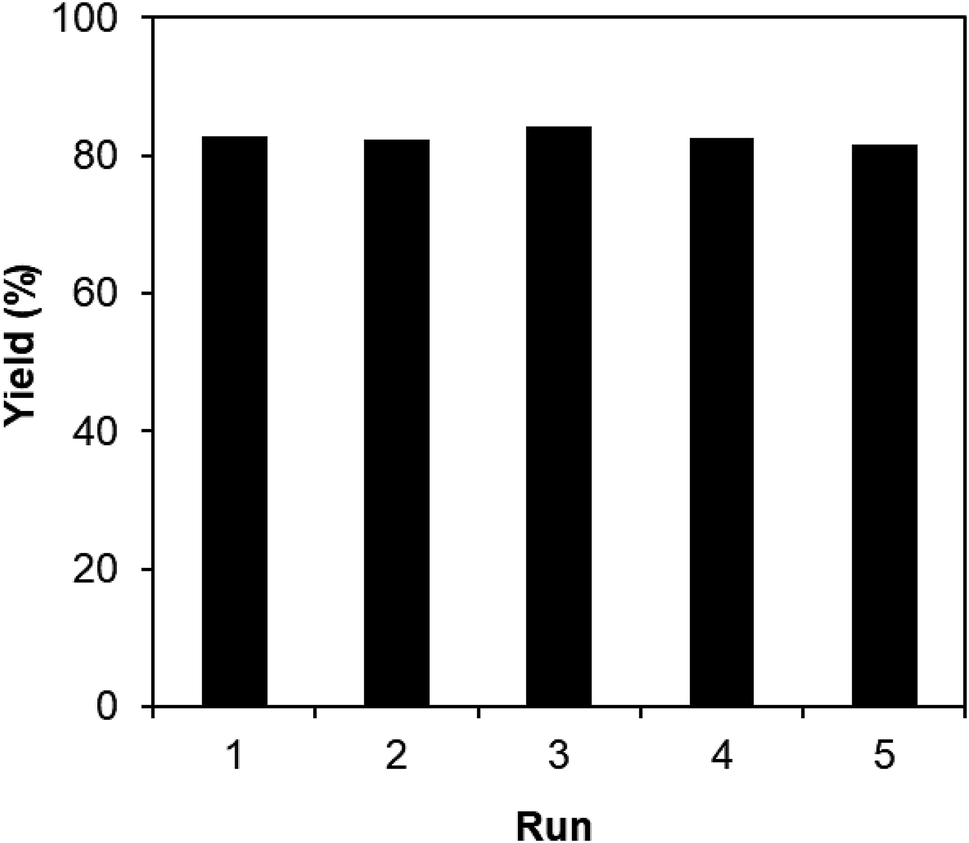 | ||
| Fig. 2 Reutilization of the La0.6Sr0.4CoO3 catalyst. | ||
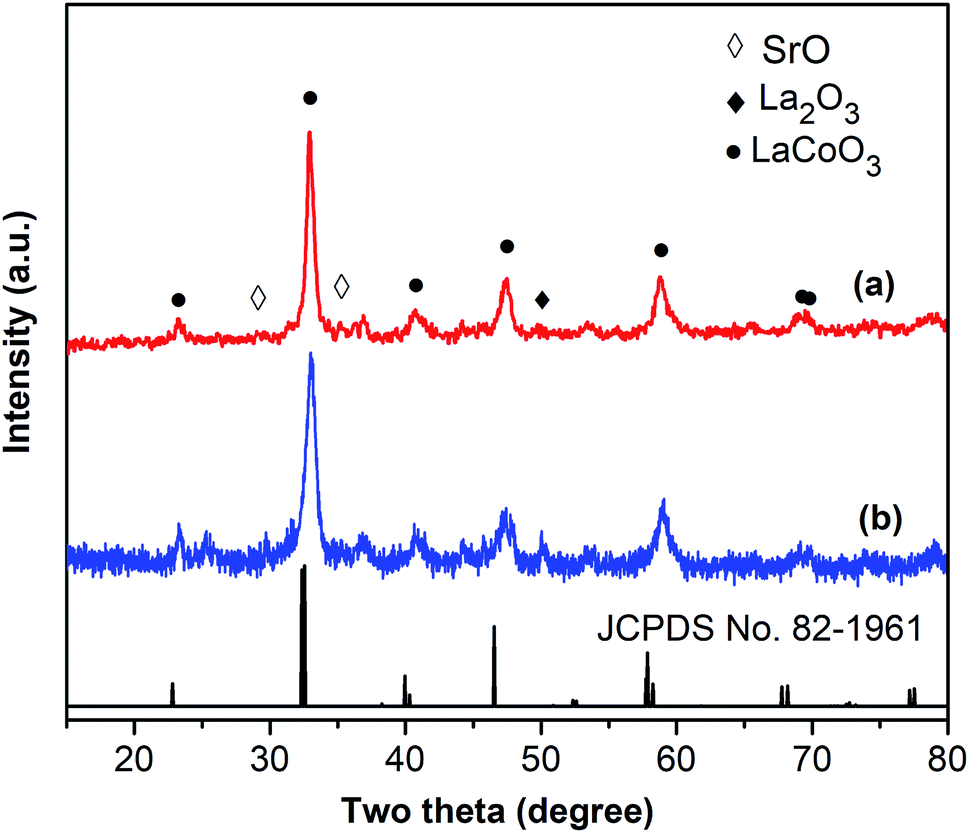 | ||
| Fig. 3 XRD results for the fresh (a) and reutilized (b) La0.6Sr0.4CoO3 perovskite catalysts. | ||
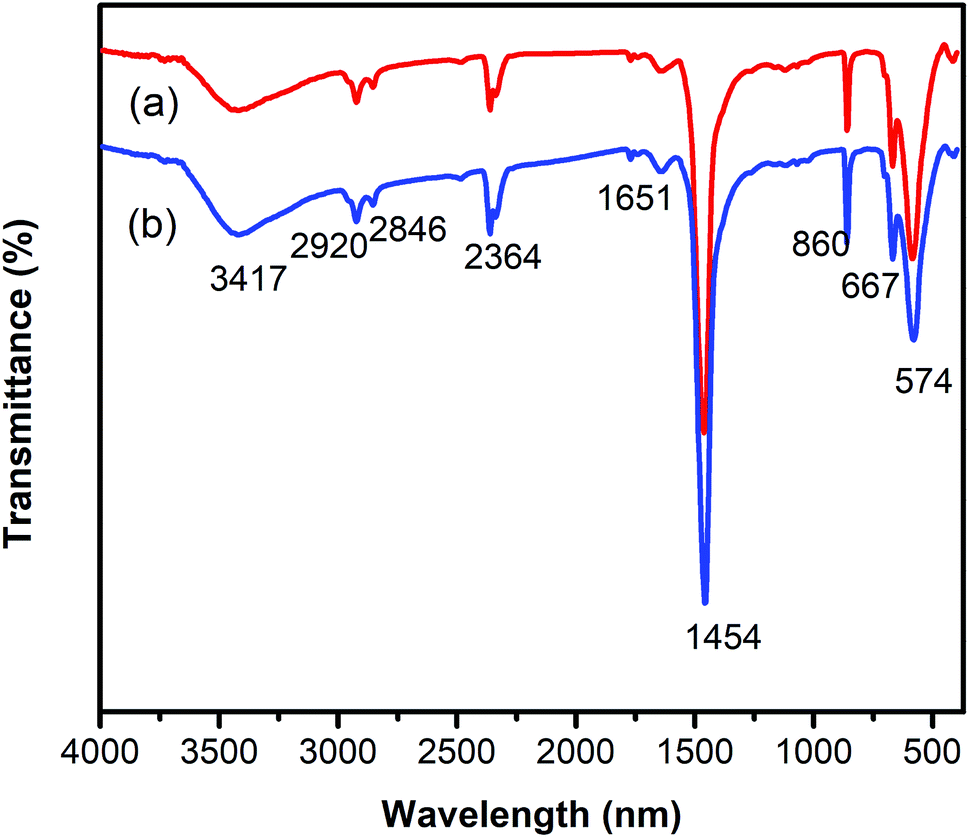 | ||
| Fig. 4 FT-IR observations for the fresh (a) and reutilized (b) La0.6Sr0.4CoO3 perovskite catalysts. | ||
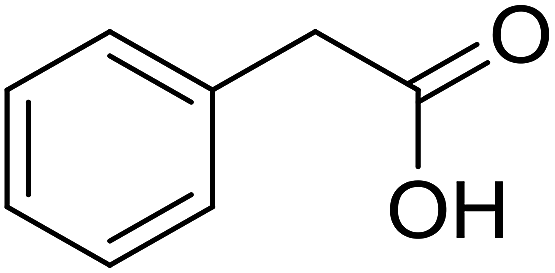
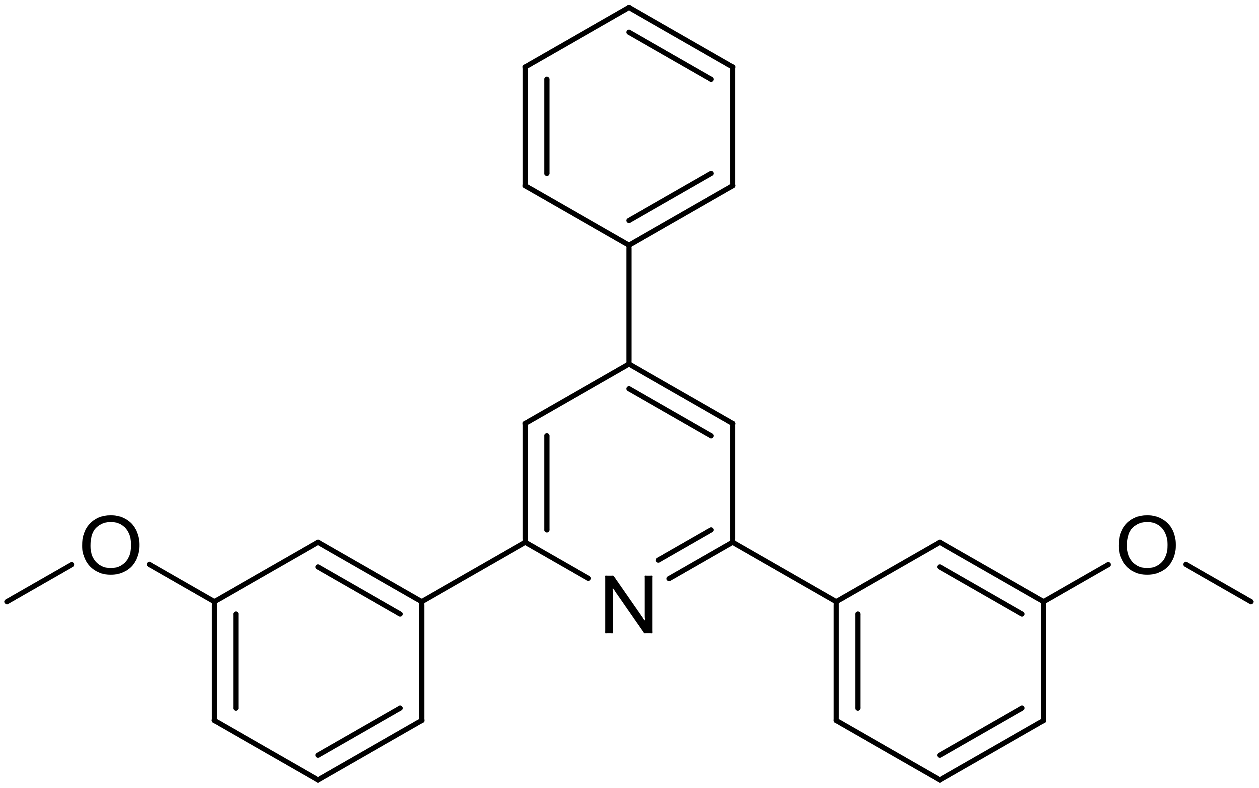
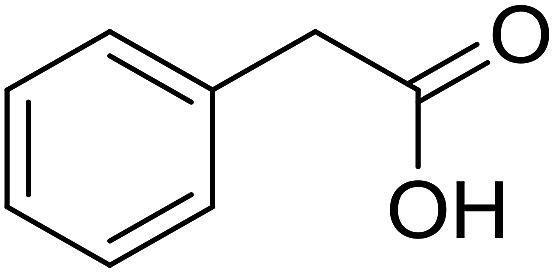
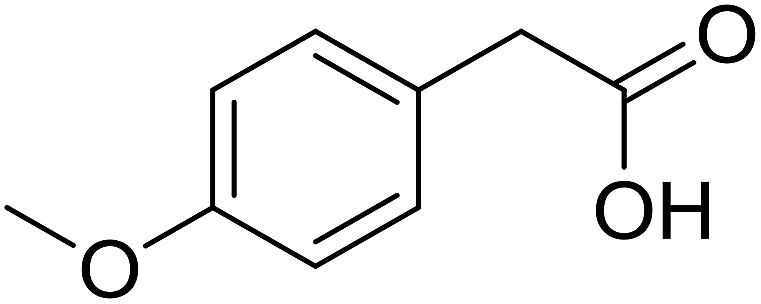
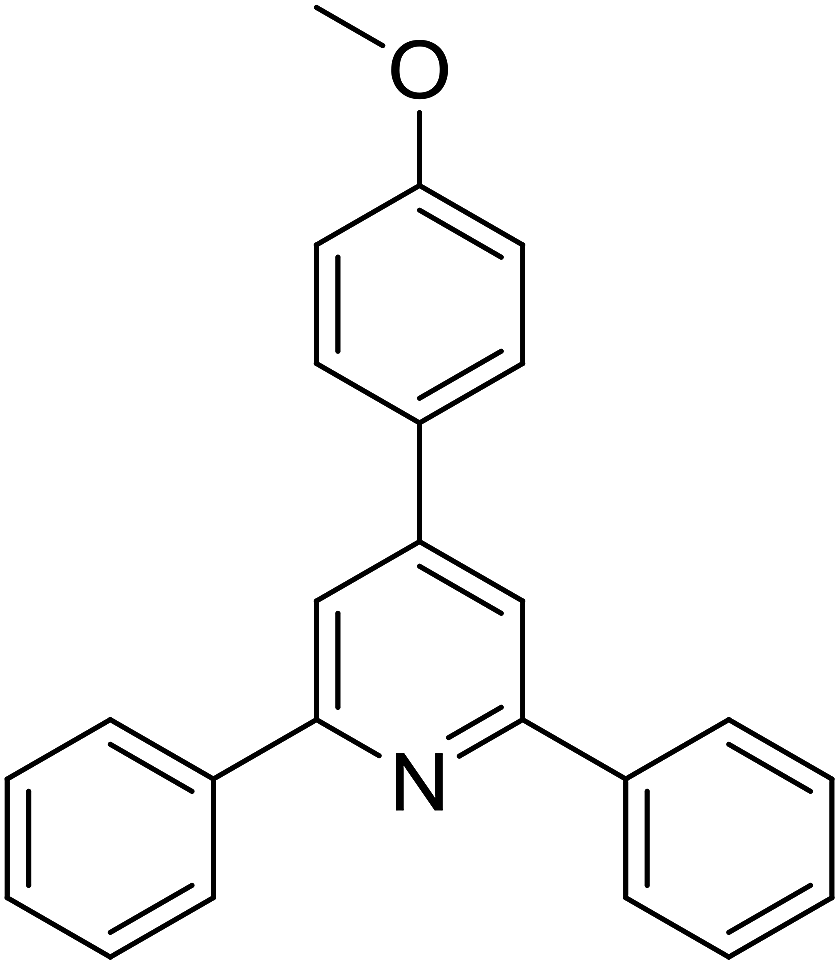
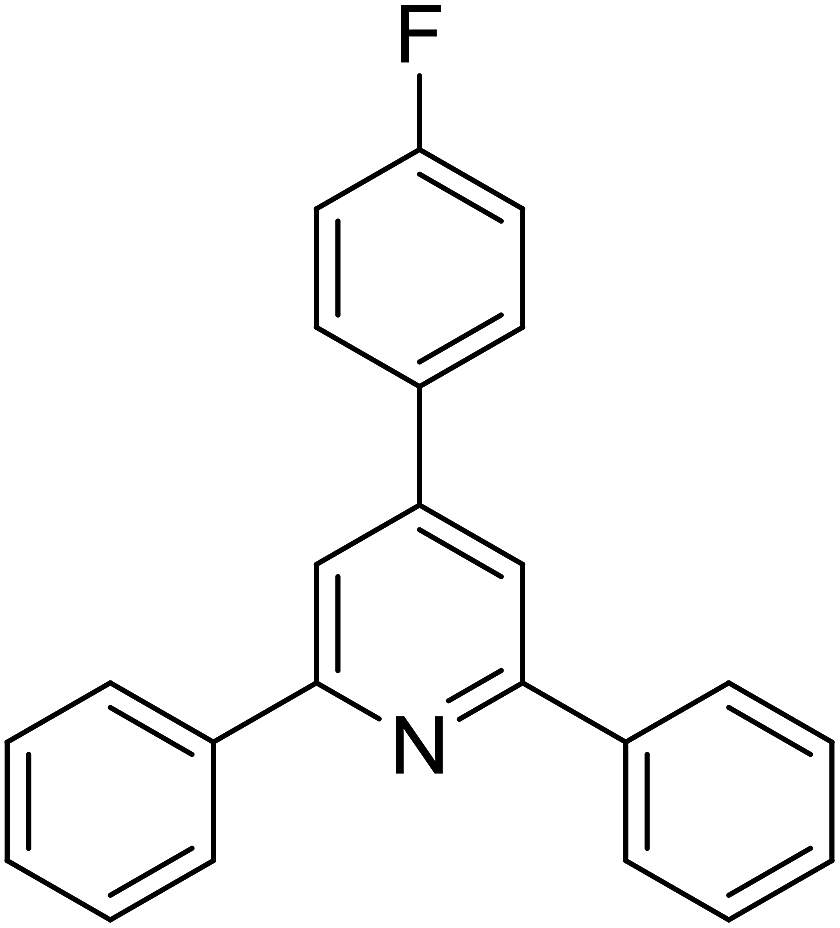
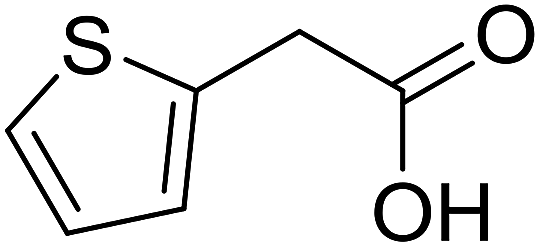
With these achievements, we consequently extended the scope of this work to the synthesis of several 2,4,6-triphenylpyridines via oxidative cyclization reactions between ketoximes and phenylacetic acids utilizing the La0.6Sr0.4CoO3 perovskite catalyst (Table 3). The reaction was conducted in toluene at 140 °C for 8 h using 3 equivalents of ketoxime and 3 equivalents of DTBP, in the presence of 5 mol% catalyst. At the end of the experiment, the triphenylpyridine product was purified by column chromatography. First, different acetophenone oxime acetates were employed for the oxidative cyclization with phenylacetic acid. Under these conditions, 2,4,6-triphenylpyridine was obtained in 79% yield (entry 1). Acetophenone oxime acetates possessing an electron-donating substituent on the benzene ring were also reactive towards the reaction. Under the standard conditions, 4-phenyl-2,6-di-p-tolylpyridine (entry 2), 2,6-bis(3-methoxyphenyl)-4-phenylpyridine (entry 3), and 2,6-bis(2-methoxyphenyl)-4-phenylpyridine (entry 4) were generated in 62%, 74%, and 67% yields, respectively. Similarly, acetophenone oxime acetates containing an electron-withdrawing substituent on the benzene ring were utilized, producing 2,6-bis(4-chlorophenyl)-4-phenylpyridine (entry 5), 2,6-bis(3-chlorophenyl)-4-phenylpyridine (entry 6), and 2,6-bis(4-bromophenyl)-4-phenylpyridine (entry 7) in 55%, 68%, and 73% yields, respectively. (E)-3,4-Dihydronaphthalen-1(2H)-one O-acetyl oxime was reactive, and the reaction afforded the corresponding product in 69% yield (entry 8). Using (E)-1-(thiophen-2-yl)ethan-1-one O-acetyl oxime led to 72% yield of the desired product (entry 9). Moving to phenylacetic acids containing a substituent, the oxidative cyclization reactions with acetophenone oxime acetate afforded the corresponding triphenylpyridine products in reasonable yields (entries 10–12). Heterocyclic acetic acids were also competent substrates, affording the products in moderate yields (entries 13–15).
|
||||
|---|---|---|---|---|
| Entry | Reactant 1 | Reactant 2 | Product | Yieldb (%) |
| a Reaction conditions: phenylacetic acids (0.2 mmol), ketoximes (0.6 mmol), DTBP (0.6 mmol), toluene (2 mL), La0.6Sr0.4CoO3 catalyst (5 mol%), 8 h at 140 °C under air for 2 h and under argon for 6 h.b Isolated yield. | ||||
| 1 | 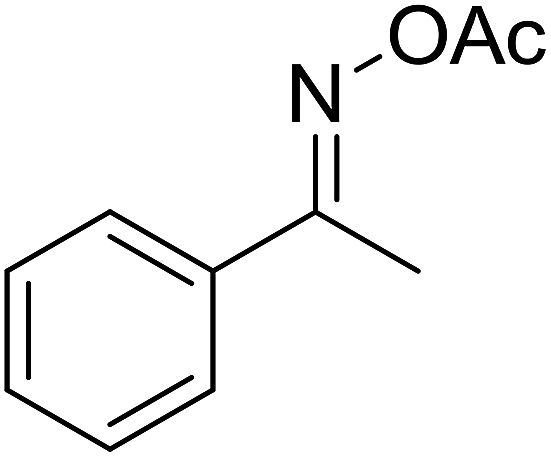 |
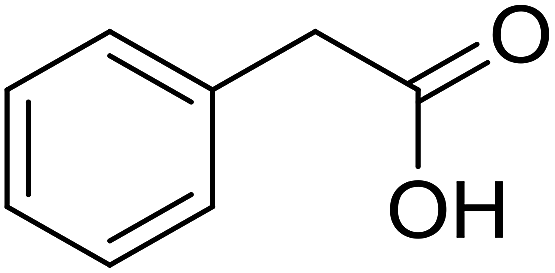 |
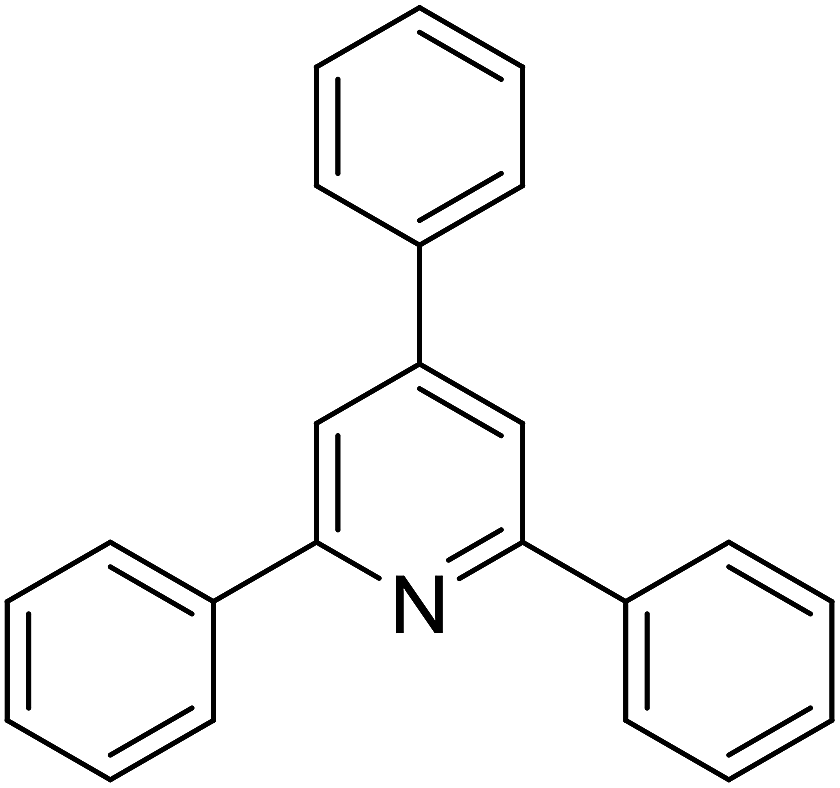 |
79 |
| 2 | 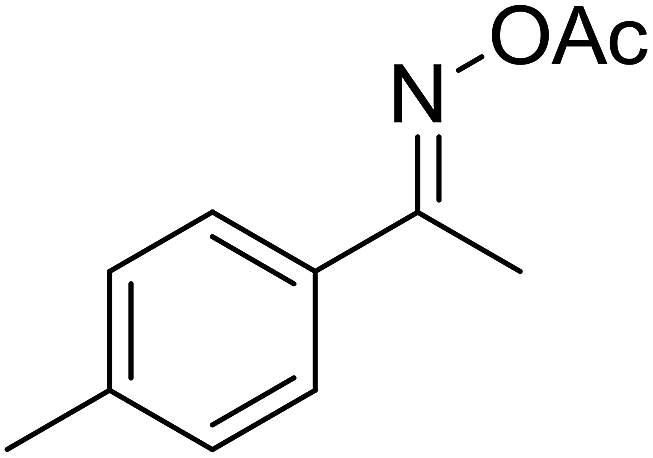 |
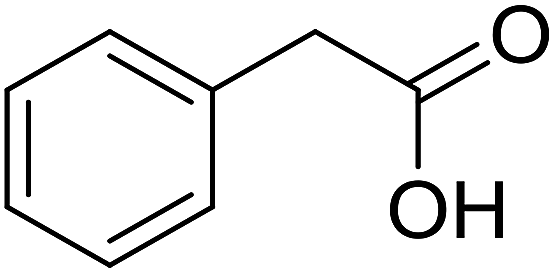 |
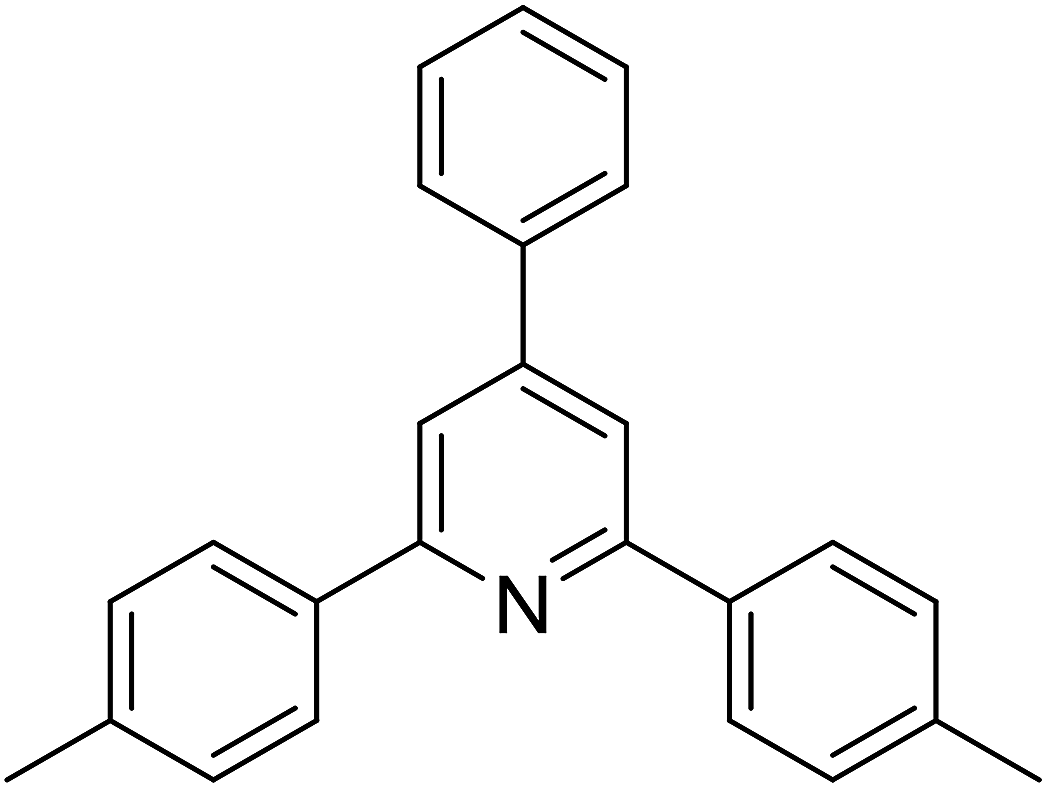 |
62 |
| 3 | 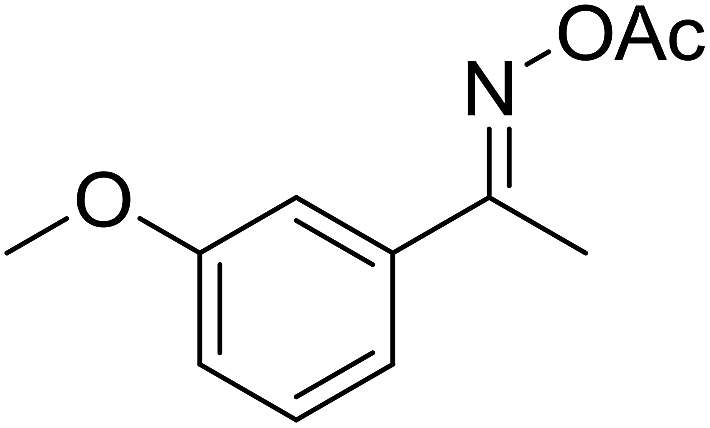 |
 |
 |
74 |
| 4 |  |
 |
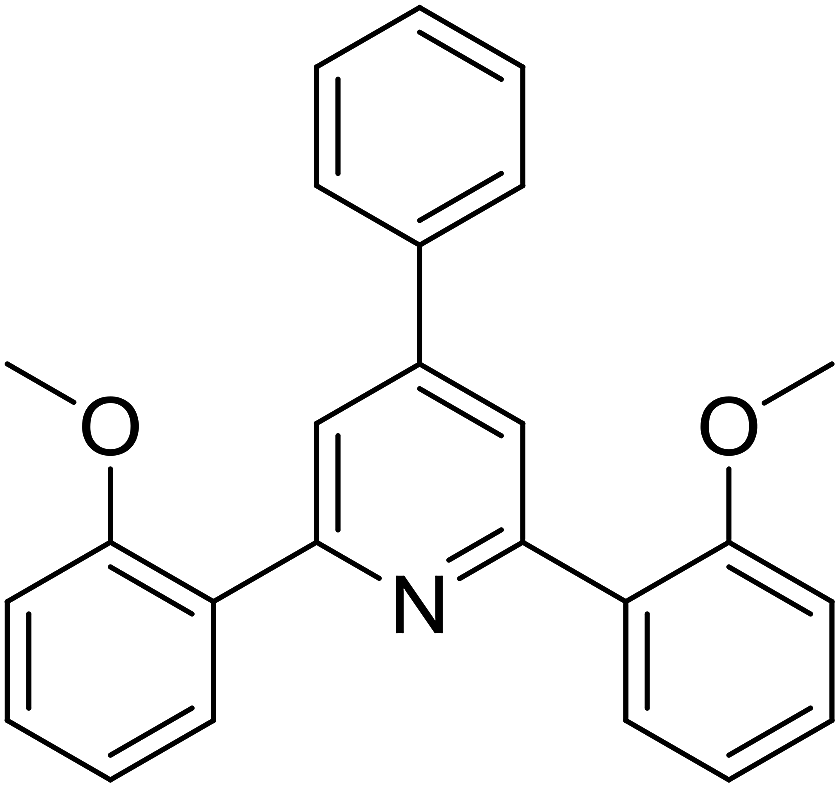 |
67 |
| 5 | 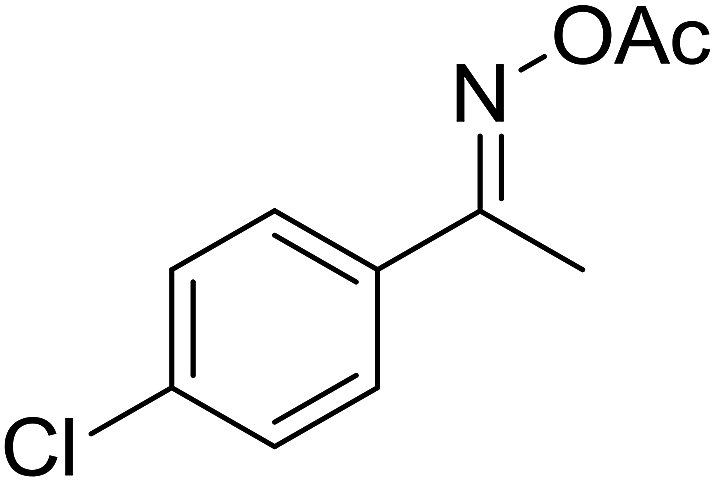 |
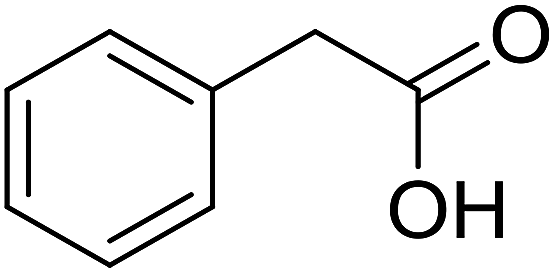 |
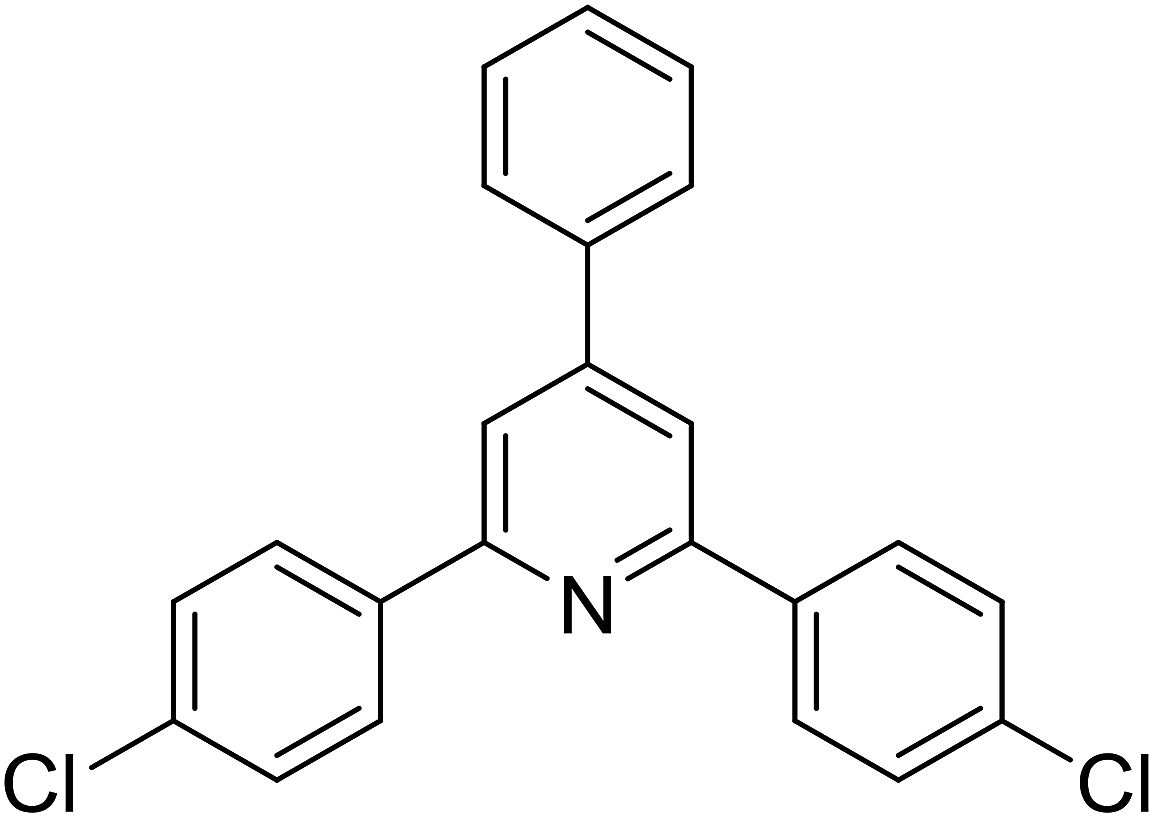 |
55 |
| 6 | 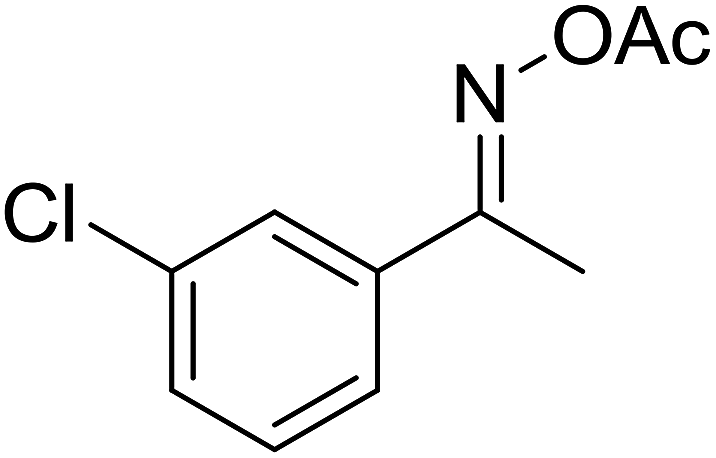 |
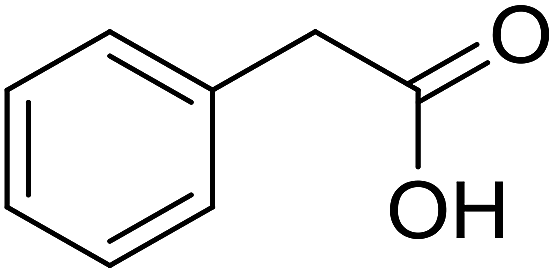 |
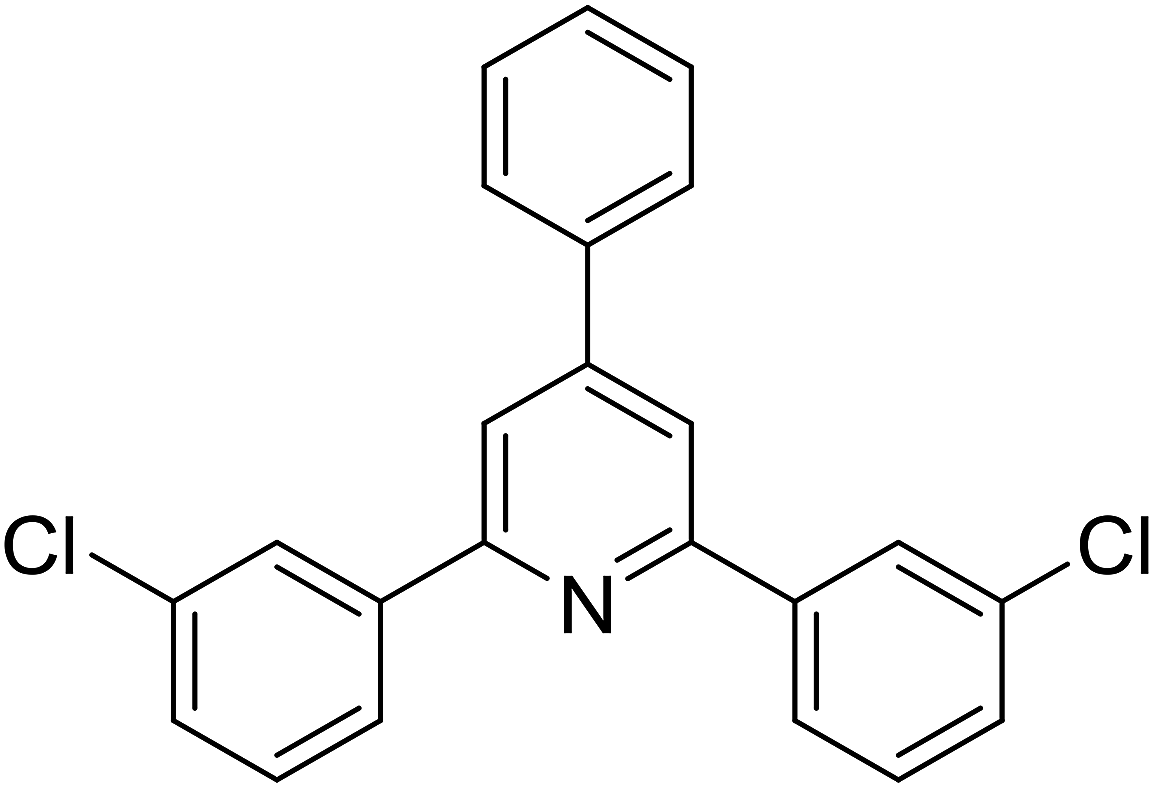 |
68 |
| 7 | 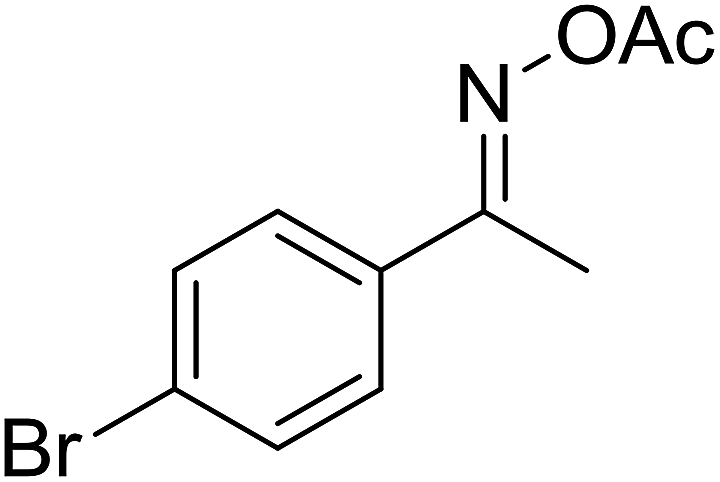 |
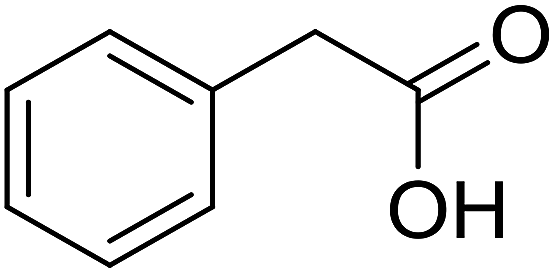 |
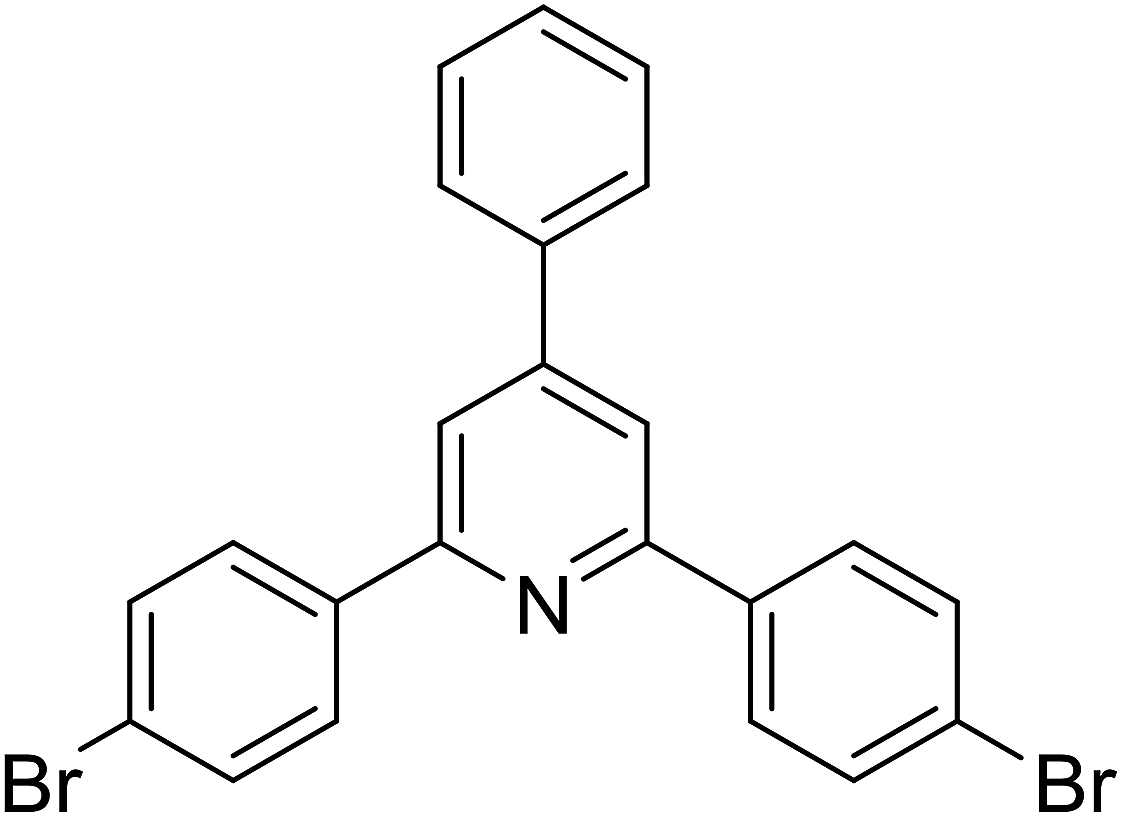 |
73 |
| 8 | 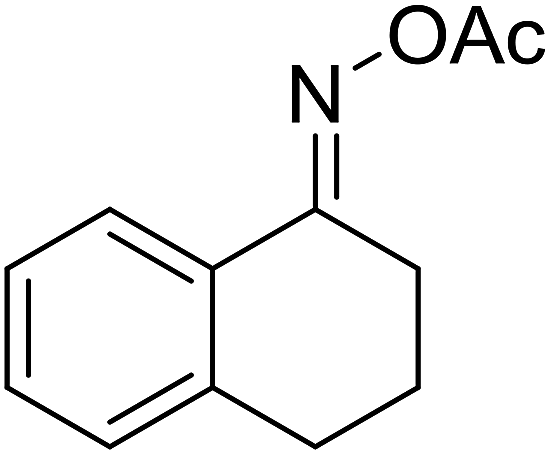 |
 |
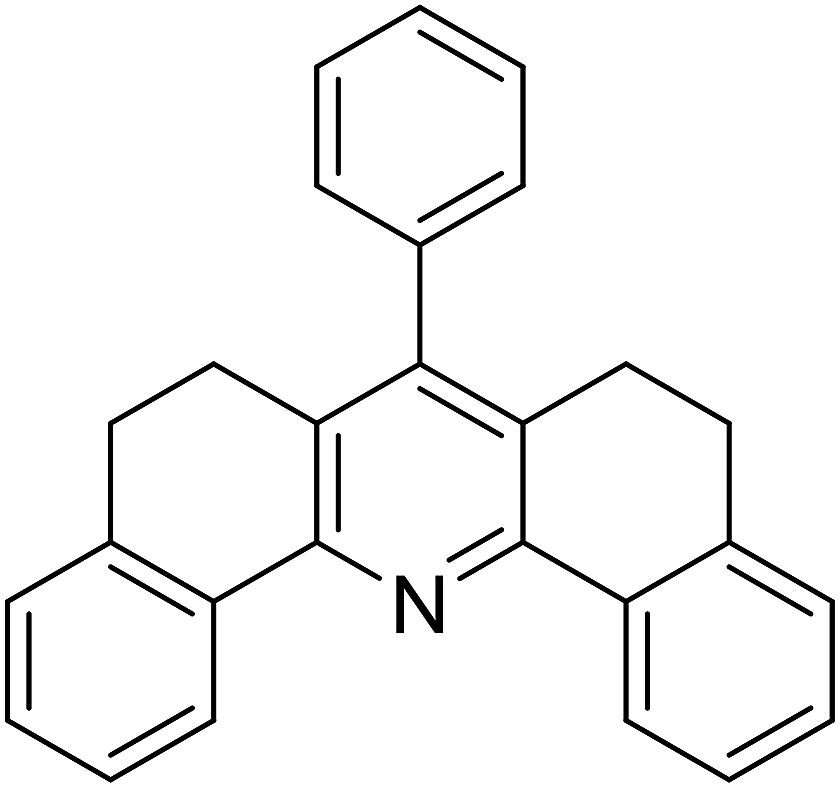 |
69 |
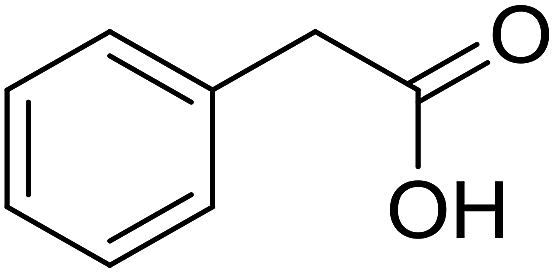
| 9 | 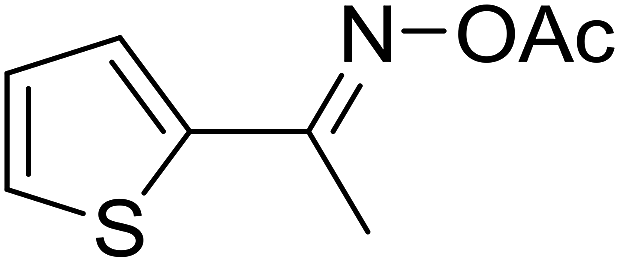 |
 |
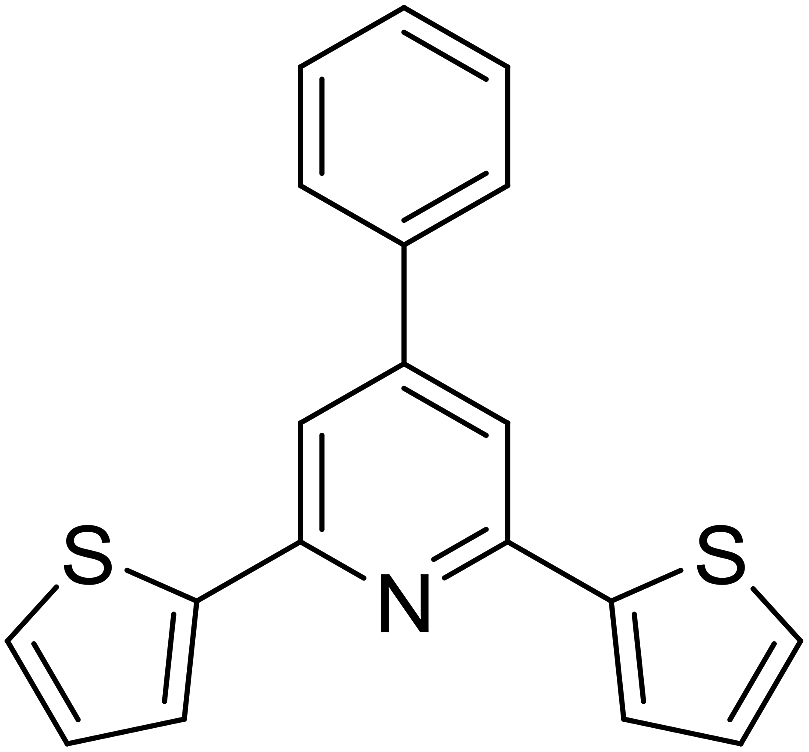 |
72 |
| 10 | 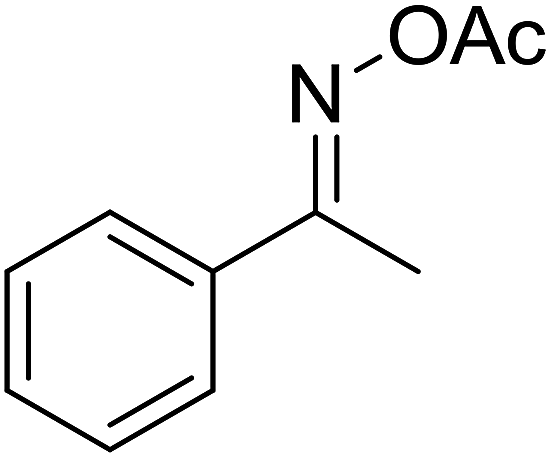 |
 |
 |
54 |
| 11 | 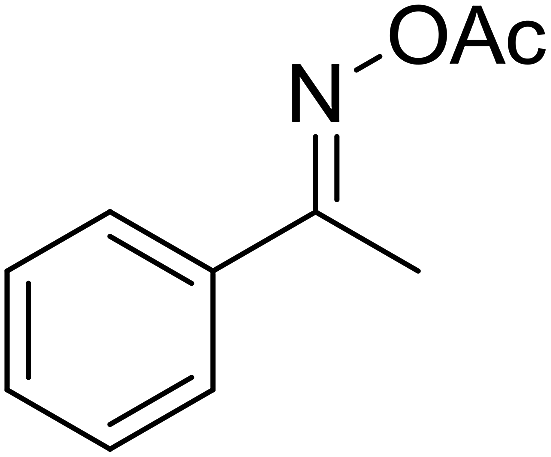 |
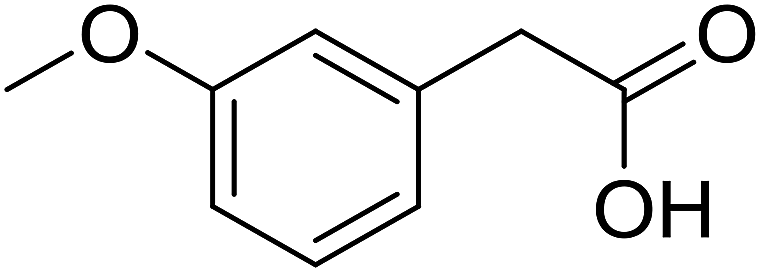 |
 |
53 |
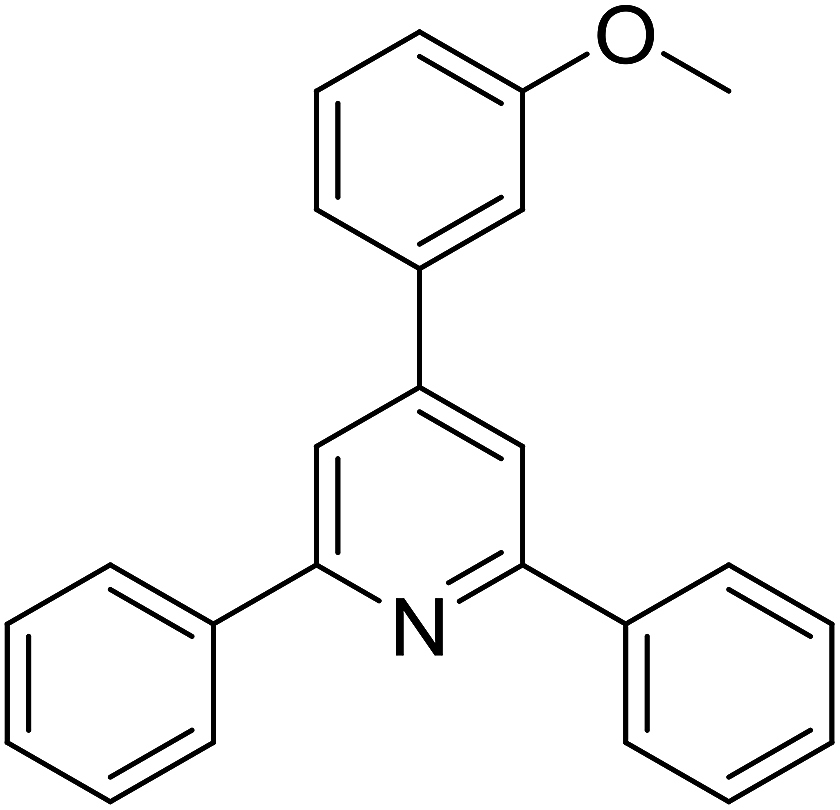
| 12 |  |
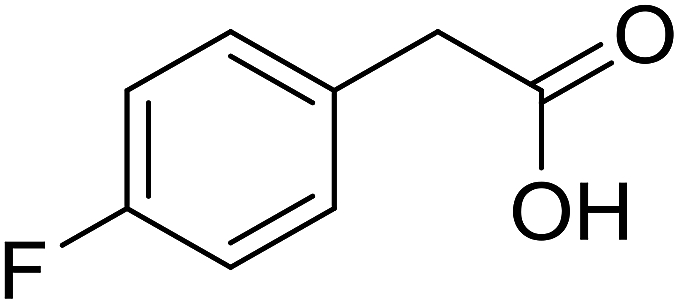 |
 |
72 |
| 13 | 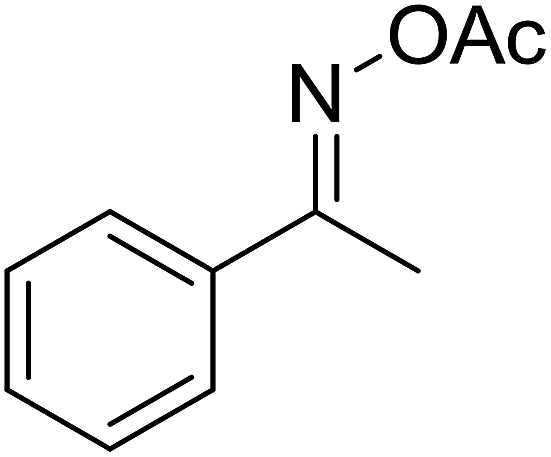 |
 |
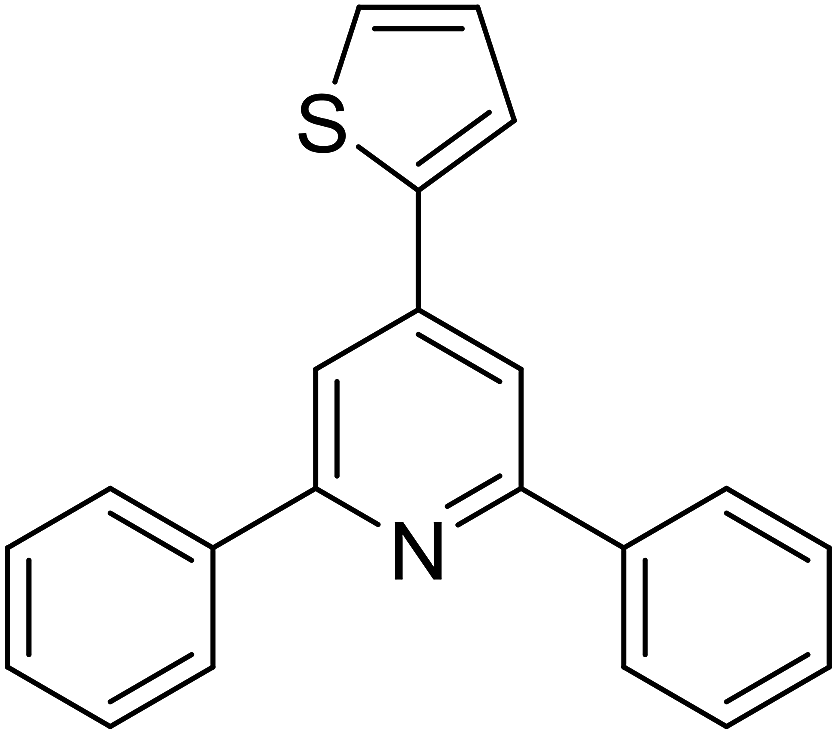 |
53 |
| 14 | 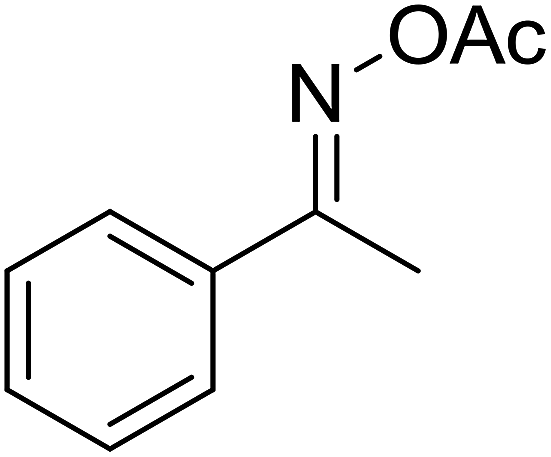 |
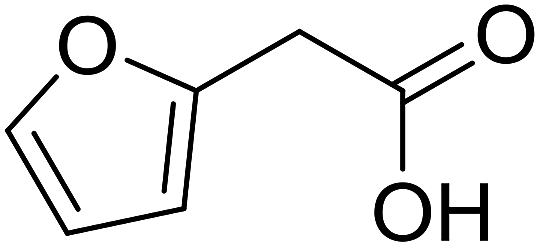 |
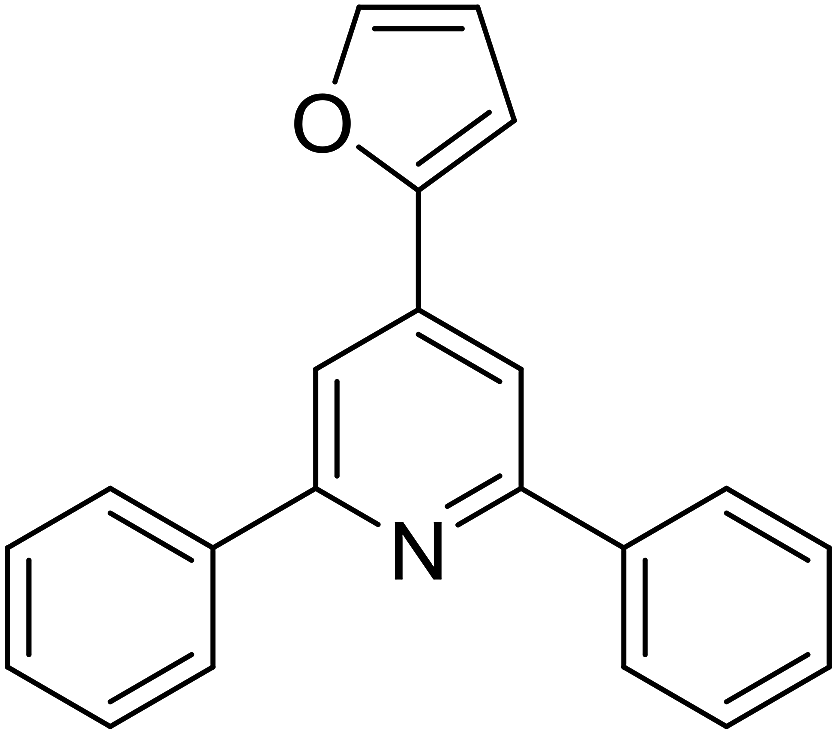 |
59 |
| 15 | 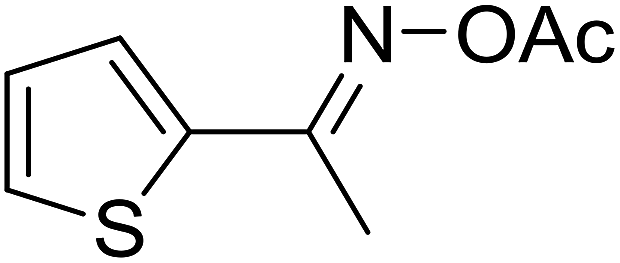 |
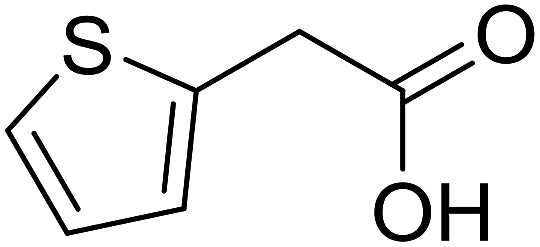 |
 |
50 |
4. Conclusions
In summary, a strontium-doped lanthanum cobaltite perovskite with the formula of La0.6Sr0.4CoO3 was prepared via a gelation and calcination method, and used as a heterogeneous catalyst for the synthesis of triphenylpyridines via a cyclization reaction between ketoximes and phenylacetic acids. The reaction proceeded via the oxidative functionalization of the sp3 C–H bond in phenylacetic acid in the presence of di-tert-butylperoxide as the oxidant. The La0.6Sr0.4CoO3-catalyzed transformation was significantly controlled by the solvent, and toluene was the best option. La0.6Sr0.4CoO3 was more active towards the oxidative cyclization reaction than numerous homogeneous and heterogeneous catalysts. Additionally, the strontium-doped perovskite offered dramatically higher catalytic activity than the pristine LaCoO3. Single oxides or salts of lanthanum, strontium, and cobalt demonstrated low catalytic performance for the transformation, indicating the importance of the synergistic effect in the perovskite catalyst. Furthermore, the La0.6Sr0.4CoO3 catalyst was facilely recovered and reused for the synthesis of triphenylpyridines without a considerable decline in its catalytic efficiency. The new combination of ketoximes with easily available phenylacetic acids via the oxidative functionalization of sp3 C–H bonds under recyclable perovskite-based catalysis conditions will make this method intriguing to the chemical and pharmaceutical industries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Vietnam National University – Ho Chi Minh City (VNU-HCM) is acknowledged for financial support via project no. NV2019-20-02.References
- S. Sowmiah, J. M. S. S. Esperança, L. P. N. Rebelo and C. A. M. Afonso, Org. Chem. Front., 2018, 5, 453–493 RSC.
- Z. Xu, D. S. Wang, X. Yu, Y. Yang and D. Wang, Adv. Synth. Catal., 2017, 359, 3332–3340 CrossRef CAS.
- H. Wu, J. Wu, W. Zhang, Z. Li, J. Fang, X. Lian, T. Qin, J. Hao, Q. Zhou and S. Wu, Bioorg. Med. Chem. Lett., 2019, 29, 870–872 CrossRef CAS PubMed.
- Y. Yan, H. Li, Z. Li, B. Niu, M. Shi and Y. Liu, J. Org. Chem., 2017, 82, 8628–8633 CrossRef CAS PubMed.
- H. Xu, J.-C. Zeng, F.-J. Wang and Z. Zhang, Synthesis, 2017, 49, 1879–1883 CrossRef CAS.
- Z.-H. Ren, Z.-Y. Zhang, B.-Q. Yang, Y.-Y. Wang and Z.-H. Guan, Org. Lett., 2011, 13, 5394–5397 CrossRef CAS PubMed.
- Y. Fu, P. Wang, X. Guo, P. Wu, X. Meng and B. Chen, J. Org. Chem., 2016, 81, 11671–11677 CrossRef CAS PubMed.
- M.-N. Zhao, Z.-H. Ren, L. Yu, Y.-Y. Wang and Z.-H. Guan, Org. Lett., 2016, 18, 1194–1197 CrossRef CAS PubMed.
- Y. Yi, M.-N. Zhao, Z.-H. Ren, Y.-Y. Wang and Z.-H. Guan, Green Chem., 2017, 19, 1023–1027 RSC.
- H. Huang, X. Ji, W. Wu, L. Huang and H. Jiang, J. Org. Chem., 2013, 78, 3774–3782 CrossRef CAS PubMed.
- M. Adib, N. Ayashi and P. Mirzaei, Synlett, 2016, 27, 417–421 CrossRef CAS.
- F. Ling, L. Shen, Z. Pan, L. Fang, D. Song, Z. Xie and W. Zhong, Tetrahedron Lett., 2018, 59, 3678–3682 CrossRef CAS.
- K. Ishihara, T. Shioiri and M. Matsugi, Tetrahedron Lett., 2019, 60, 1295–1298 CrossRef CAS.
- Z. Han, J. Lv and J. Zhang, Tetrahedron, 2019, 75, 2162–2168 CrossRef CAS.
- N. Wang, L. Liu, W. Xu, M. Zhang, Z. Huang, D. Shi and Y. Zhao, Org. Lett., 2019, 21, 365–368 CrossRef CAS PubMed.
- Y. Xia, J. Cai, H. Huang and G.-J. Deng, Org. Biomol. Chem., 2018, 16, 124–129 RSC.
- W. Zheng, W. Yang, D. Luo, L. Min, X. Wang and Y. Hu, Adv. Synth. Catal., 2019, 361, 1995–1999 CrossRef CAS.
- W.-J. Yin, B. Weng, J. Ge, Q. Sun, Z. Li and Y. Yan, Energy Environ. Sci., 2019, 12, 442–462 RSC.
- E. Shi, Y. Gao, B. P. Finkenauer, Akriti, A. H. Coffey and L. Dou, Chem. Soc. Rev., 2018, 47, 6046–6072 RSC.
- T. Qiu, Y. Hu, F. Xu, Z. Yan, F. Bai, G. Jia and S. Zhang, Nanoscale, 2018, 10, 20963–20989 RSC.
- L. Mao, C. C. Stoumpos and M. G. Kanatzidis, J. Am. Chem. Soc., 2019, 141, 1171–1190 CrossRef CAS PubMed.
- F. Polo-Garzon and Z. Wu, J. Mater. Chem. A, 2018, 6, 2877–2894 RSC.
- D. B. Mitzi, Chem. Rev., 2019, 119, 3033–3035 CrossRef CAS PubMed.
- H. Shin, M. Baek and D. H. Kim, Mol. Catal., 2019, 468, 148–153 CrossRef CAS.
- A. Vignesh, M. Prabu and S. Shanmugam, ACS Appl. Mater. Interfaces, 2016, 8, 6019–6031 CrossRef CAS PubMed.
- H. Sun, J. He, Z. Hu, C.-T. Chen, W. Zhou and Z. Shao, Electrochim. Acta, 2019, 299, 926–932 CrossRef CAS.
- K. Kawashima, M. Hojamberdiev, H. Wagata, E. Zahedi, K. Yubuta, K. Domen and K. Teshima, J. Catal., 2016, 344, 29–37 CrossRef CAS.
- D. Wang, Y. Peng, Q. Yang, S. Xiong, J. Li and J. Crittenden, Environ. Sci. Technol., 2018, 52, 7443–7449 CrossRef CAS PubMed.
- R. P. Forslund, J. T. Mefford, W. G. Hardin, C. T. Alexander, K. P. Johnston and K. J. Stevenson, ACS Catal., 2016, 6, 5044–5051 CrossRef CAS.
- Q. Yang, D. Wang, C. Wang, X. Li, K. Li, Y. Peng and J. Li, Catal. Sci. Technol., 2018, 8, 3166–3173 RSC.
- Q. Meng, W. Wang, X. Weng, Y. Liu, H. Wang and Z. Wu, J. Phys. Chem. C, 2016, 120, 3259–3266 CrossRef CAS.
- C. A. Chagas, F. S. Toniolo, R. N. S. H. M. es and M. Schmal, Int. J. Hydrogen Energy, 2012, 37, 5022–5031 CrossRef CAS.
- J. A. Onrubia, B. Pereda-Ayo, U. De-La-Torre and J. R. González-Velasco, Appl. Catal., B, 2017, 213, 198–210 CrossRef CAS.
- M. Ao, G. H. Pham, V. Sage and V. Pareek, J. Mol. Catal. A: Chem., 2016, 416, 96–104 CrossRef CAS.
- M. Ao, G. H. Pham, V. Sage and V. Pareek, Fuel, 2017, 206, 390–400 CrossRef CAS.
- L. Wang, Q. Pang, Q. Song, X. Pan and L. Jia, Fuel, 2015, 140, 267–274 CrossRef CAS.
- X. Chen, T. Chen, F. Ji, Y. Zhou and S.-F. Yin, Catal. Sci. Technol., 2015, 5, 2197–2202 RSC.
- L. Deng, B. Huang and Y. Liu, Org. Biomol. Chem., 2018, 16, 1552–1556 RSC.
- Q. Shi, W. Zhang, Y. Wang, L. Qu and D. Wei, Org. Biomol. Chem., 2018, 16, 2301–2311 RSC.
- Q. Feng and Q. Song, J. Org. Chem., 2014, 79, 1867–1871 CrossRef CAS PubMed.
- M.-N. Zhao, L. Yu, N.-F. Mo, Z.-H. Ren, Y.-Y. Wang and Z.-H. Guan, Org. Chem. Front., 2017, 4, 597–602 RSC.
- C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1999–2070 CrossRef.
- J. Iqbal, B. Bhatia and N. K. Nayyar, Chem. Rev., 1994, 94, 519–564 CrossRef CAS.
- K. Rida, A. Benabbas, F. Bouremmad, M. A. Peña and A. Martínez-Arias, Catal. Commun., 2006, 7, 963–968 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04096j |
| This journal is © The Royal Society of Chemistry 2019 |