
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegioselective microwave synthesis and derivatization of 1,5-diaryl-3-amino-1,2,4-triazoles and a study of their cholinesterase inhibition properties†
Sabrina Neves Santosab,
Gabriela Alves de Souzaab,
Thiago Moreira Pereiraab,
Daiana Portella Francoab,
Catarina de Nigris Del Cistiab,
Carlos Mauricio R. Sant'Annab,
Renata Barbosa Lacerdaab and
Arthur Eugen Kümmerle *ab
*ab
aLaboratório de Diversidade Molecular e Química Medicinal (LaDMol-QM, Molecular Diversity and Medicinal Chemistry Laboratory), Chemistry Institute, Universidade Federal Rural do Rio de Janeiro, Seropédica, Rio de Janeiro, 239897-000, Brazil. E-mail: akummerle@hotmail.com
bPrograma de Pós-Gradução em Química (PPGQ), Universidade Federal Rural do Rio de Janeiro, Seropédica, Rio de Janeiro, 239897-000, Brazil
First published on 1st July 2019
Abstract
Herein we describe the development of an efficient one-pot regioselective synthesis protocol to obtain N-protected or N-deprotected 1,5-diaryl-3-amino-1,2,4-triazoles from N-acyl-N-Boc-carbamidothioates. This improved protocol using microwave irradiation and low reaction times (up to 1 h) furnished desired compounds in yields ranging from 50 to 84%. This chemistry is useful for a variety of aromatic groups with electronically diverse substituents. The design and correct derivation of the amino group led to compounds able to inhibit cholinesterases with good IC50 of up to 1 μM. Also, the mode of action (mixed-type) and SAR analysis for this series of compounds was described by means of kinetic and molecular modelling evaluations, showing potential for this class of compounds as new scaffolds for this biological activity.
Introduction
The five-membered azaheterocyle aminotriazole is widely described in the literature and has received great attention from the scientific community due to its diversity of biological activities. This increased attention to this 1,2,4-triazole nucleus comes from its possible non-classical bioisosteric replacement of a cis-amide bond from bioactive compounds.1 3-amino-1,2,4-triazoles have been described as an anti-inflammatory agent (A),2 as a treatment for bronchial asthma (B),3 as an inhibitor of methionine aminopeptidase (C)4 and a NPY receptor,5 and as a selective positive allosteric modulator of the α7 nicotinic acetylcholine receptor (D)x (Fig. 1).6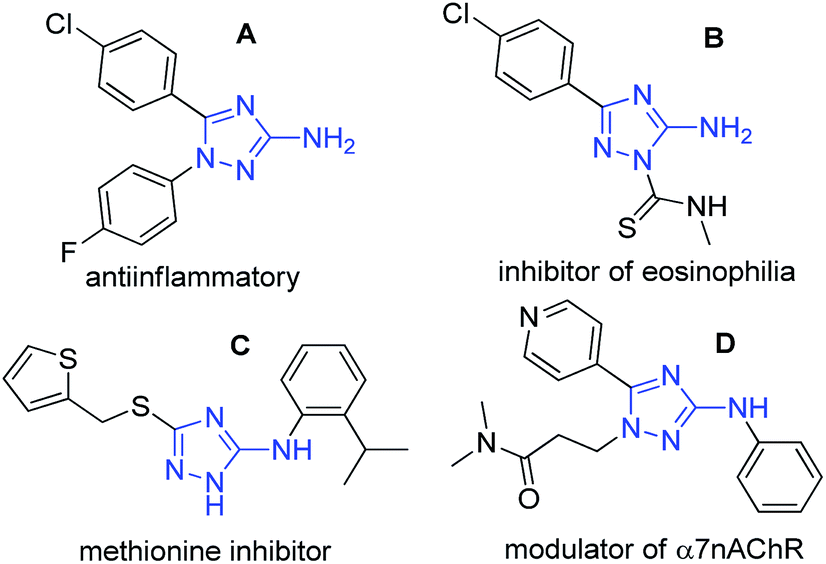 | ||
| Fig. 1 Bioactive compounds with 3-amino-1,2,4-triazole nucleus. | ||
Several procedures have already been described for the synthesis of general 3-amino-1H-1,2,4-triazoles, with great attention to solid phase, microwaves and combinatorial synthesis.7 However, few of them explored the synthesis of 1,5-diaryl-3-amino-1,2,4-triazoles. The first methodology, described by Katritzky8 and also adapted to solid phase synthesis by Houghten,9 employs a cyclization reaction from an electrophilic moiety (N-acylguanidines derived from benzotriazole or N-acyl carbamidothioate) and a nucleophilic one (alkyl or substituted aryl hydrazines) (Fig. 2). Unfortunately, these processes suffer from long reaction times, few examples, and low yields when both substitutions are aryl groups. Furthermore, in many cases they were unable to produce 3-N,N-unsubstituted amino triazole compounds.
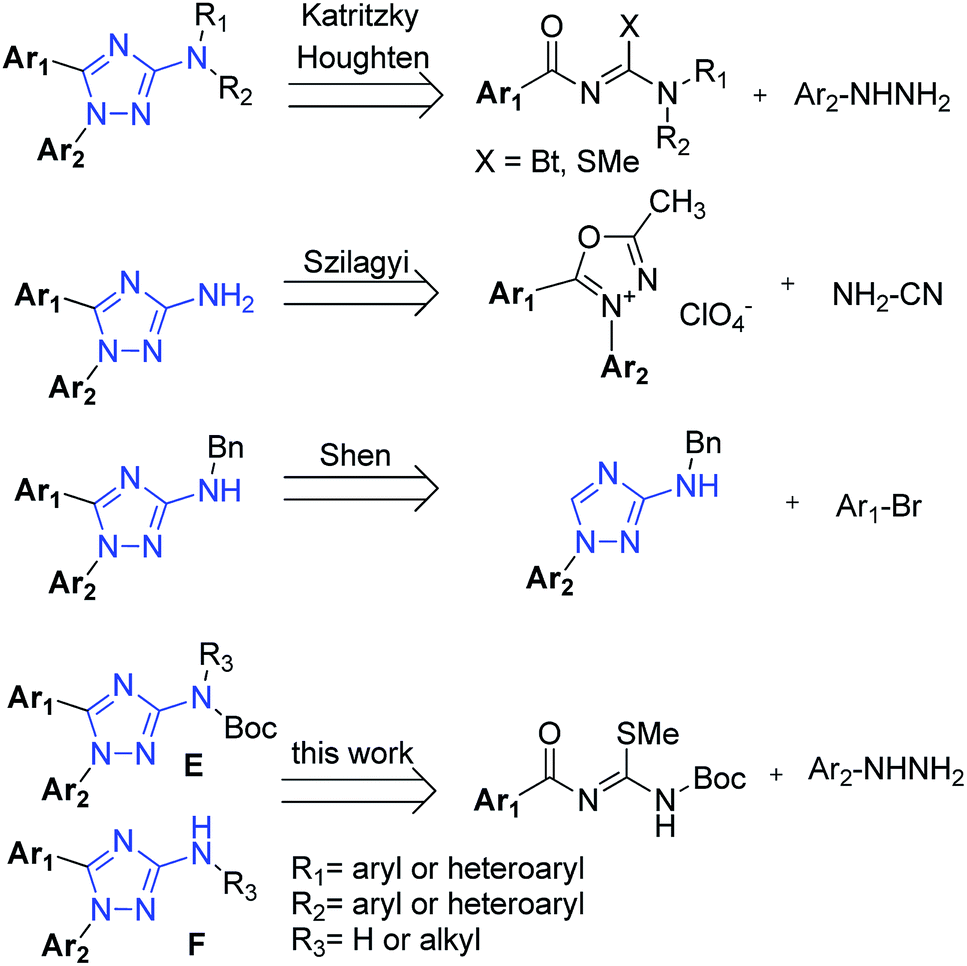 | ||
| Fig. 2 Described synthetic strategies for the synthesis of 1,5-disubstituted 3-amino-1H-1,2,4-triazoles and our strategy for this work. | ||
The second methodology, described by Szilagyi,2 is based on a two steps mechanism: the opening of 1,3,4-oxadiazolium perchlorate nucleus by the nucleophile cyanamide followed by its recyclization (Fig. 2). Despite better in scope and reactional yields compared to the Katritzky method, the explosive nature of perchloric acid and perchlorate salts makes this methodology dangerous and not indicated to scale-up synthesis.10,11
The third procedure was recently described by Shen,12 presenting an interesting strategy for the C-arylation of aminotriazoles at position 1 by means of a Negish coupling (Fig. 2). Despite of good yields and scope demonstrated, the high cost of this reaction associated with its drastic conditions in the final deprotection (6 M H2SO4, 80 °C, 16 h) can make this reaction limited in some cases. Thus, new simpler and more effective methodologies for the synthesis of 1,5-diaryl-3-amino-1,2,4-triazoles are still required.
Our goals in this work were: (1) to develop a simple one-pot regioselective synthesis protocol to obtain N-protected (E) or N-deprotected (F) 1,5-diaryl-3-amino-1,2,4-triazoles (Fig. 2) in low reaction times and suitable for diverse R1 and R2 aromatic groups, through an improvement of Katritzky procedure using N-acyl-N-Boc-carbamidothioates; (2) given the importance of microwave irradiation to heterocyclic chemistry,13 we decided to evaluate its influence in our synthesis. Furthermore, this technique proved to be suitable to furnish different 1,2,3 and 1,2,4 triazole derivatives in good yields and low reaction times;7b (3) derivatization studies of N-protected compounds E to furnish series F, when R3 is an alkyl group; and (4) use of N-derivatizations to correct design and obtain new 1,5-diaryl-3-amino-1,2,4-triazole derivatives as inhibitors of cholinesterases.
Results and discussion
Synthesis of triazoles
Our initial objective was to obtain the 1,5-diaryl-3-amino-1,2,4-triazoles (6) without any protection. For this purpose, we planned a three-steps protocol (Schemes 1 and 2). The first step was the synthesis of S-methylisothiourea (1) as previously described,14 by the alkylation of urea with methyl iodide followed by precipitation and filtration (99% yield). The second step was the synthesis of S-methyl-N-benzoyl-isothiourea (3) in 73% yield from the reaction of S-methylisothiourea with an activated benzotriazole intermediate, previously obtained from the in situ reaction of benzoic acid (2a) with N,N′-diisopropylcarbodiimide (DIC) and 1-hydroxybenzo-triazole (HOBt) (Scheme 1).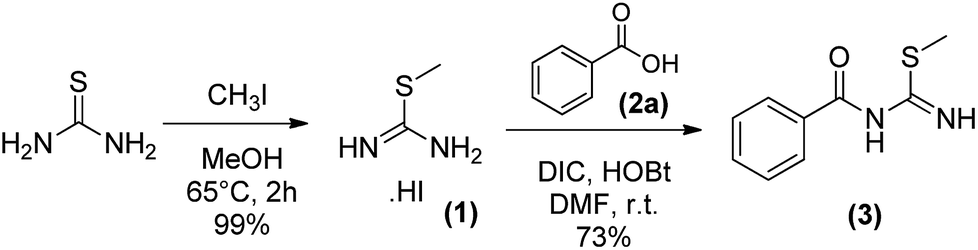 | ||
| Scheme 1 Synthesis of methyl N-acylisothiourea (3). | ||
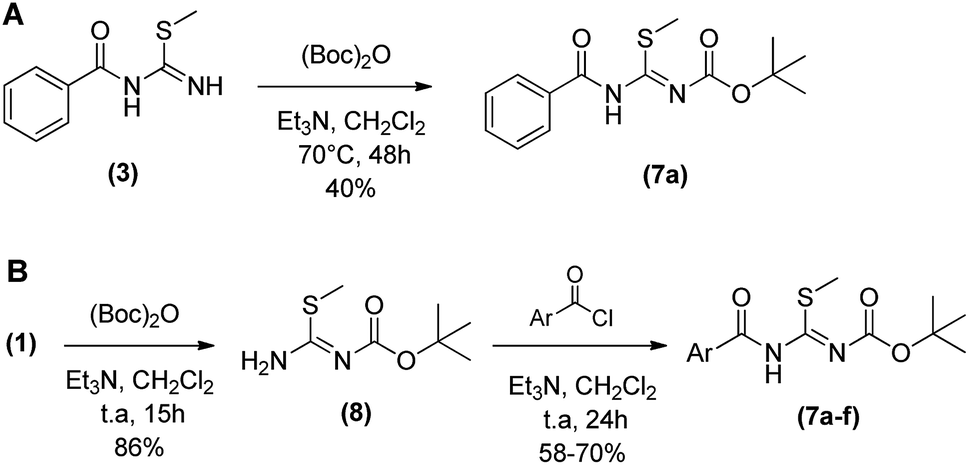 | ||
| Scheme 2 Synthesis of N-Boc-S-methylisothiourea derivatives (7). | ||
The cyclization reaction leading to 1,5-diphenyl-3-amino-1,2,4-triazole (6a) was performed in the last step by reacting N-benzoylisothiourea (3) with phenylhydrazine (4). This reaction was attempted in two different solvents and heating sources (Table 1). The best reactional yield was observed for the conventional heating at 100 °C in acetonitrile.
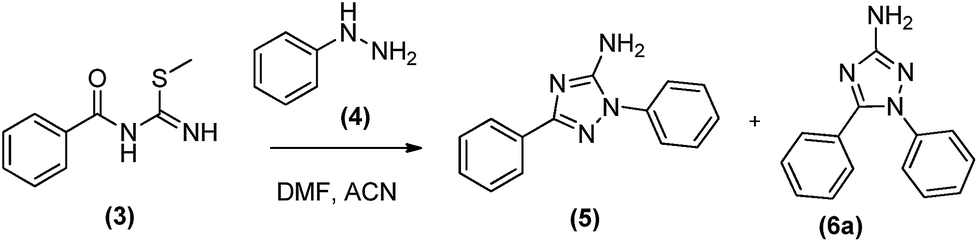
However, long reaction time (48 h) was required to observe the consumption of reactants (Table 1, entry 3) and, although 75% of 6a could be obtained, 13% of its regioisomer (5) was also formed, complicating their chromatographic separation.
Bearing in mind the possible low reactivity and the lack of selectivity demonstrated by 3, we decided to introduce an electron withdrawing group at –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH(S–CH3) moiety by means of a labile tert-butiloxycarbonyl group (–BOC).
NH(S–CH3) moiety by means of a labile tert-butiloxycarbonyl group (–BOC).
The synthesis of N-protected N-acyl-S-methylisothiourea (7) was evaluated reacting 3 with di-tert-butyl dicarbonate in dichloromethane at 70 °C in a sealed borosilicate tube. However only 40% of product 7 was furnished after 48 h (Scheme 2A). Aiming at to optimize the reaction process to obtain 7, a new synthetic route was evaluated (Scheme 2B).
Instead of protecting the low reactivity N-acyl-S-methylisothiourea (3) in the least step, the new process started with the protection of S-methylisothiourea (1) in a similar condition as above described, furnishing the N-Boc-S-methylisothiourea (8) as a white solid in 86% yield after 15 h. Subsequently, 8 was reacted with correspondent acid chlorides in dichloromethane at room temperature for 24 hours, leading to the desired intermediates (7a–f) in yields ranging from 58 to 80%. This new synthetic route was more suitable for obtaining the N-acyl-S-methylisothiourea derivatives (7a–f) with higher yields and lower reaction times. In order to evaluate the reactivity and selectivity of the N-protected N-acyl-S-methylisothiourea (7a), we started the cyclocondensations maintaining the same temperature and solvents described before (i.e. DMF or acetonitrile as solvent and 100 °C) and the Table 2 shows the main results obtained.
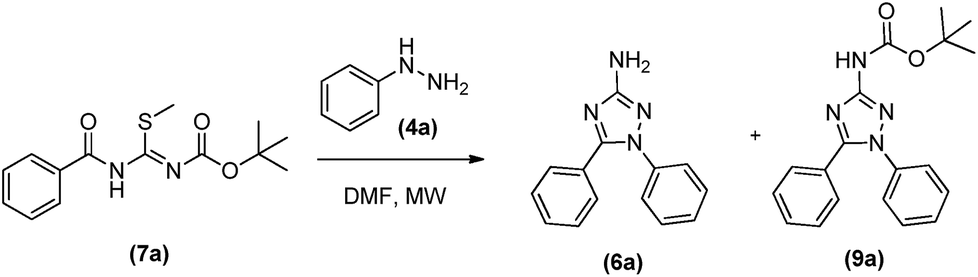
Initially the reactions were done without microwave assistance (Table 2, entries 1–4). Depending on the reaction time it was possible to observe that the cyclocondensation has occurred and the 1,5-diphenyl-3-amino-1,2,4-triazole could be isolated in its both protected (with BOC group) (9a), after reaction of up to 40 min (Table 2, entries 1 and 3), and deprotected form (6a), in the case when DMF was used as solvent in 48 h of reaction (Table 2, entry 2). Curiously, longer reaction times in acetonitrile led to a by-product that we were not able to identify, without any trace of targeted 1,5-diphenyl-3-amino-1,2,4-triazoles (6a and 9a) by the end of reaction (Table 2, entry 4).
In fact, as previous related for others amino-triazoles,7b the use of microwave irradiation proved to be efficacious in increasing reactional yields, leading to the protected product 9a in yields of up to 90% in 40 minutes (Table 2, entry 7), as well as being very efficient in obtaining the final deprotected 1,5-diphenyl-3-amino-1,2,4-triazole (6a), in shorter reaction time (1 h) and higher yield (82%) (Table 2, entry 6) compared to conventional heating. Once more, the reaction in acetonitrile at 150 °C led to the formation of the unknown by-product as previous described for the conventional heating (Table 2, entry 8). Therefore, DMF was chosen as the best solvent to synthesise the desired products, allowing the selective formation of the protected and deprotected 1,5-diphenyl-3-amino-1,2,4-triazoles, depending only on the temperature and reaction time.
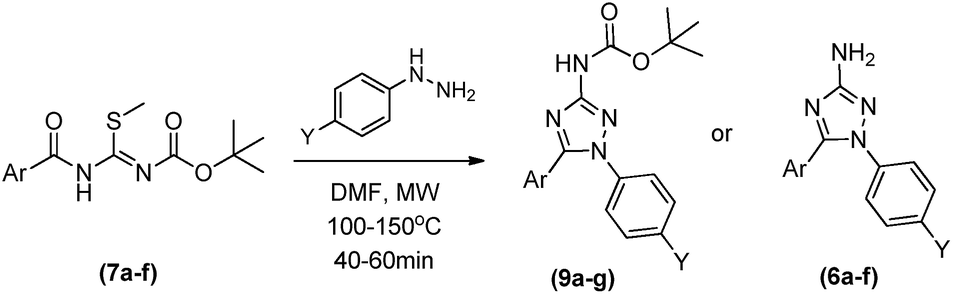
With this set of conditions, the scope of reaction was evaluated varying the aromatic rings coming from both benzoic acid and phenylhydrazine reactants (Table 3). The cyclocondensations furnished the functionalized 1,5-diaryl-3-amino-1,2,4-triazoles in theirs protected and deprotected forms (9a–h and 6a–f) in reasonable to good yields ranging from 47 to 90% in reactions of up to 1 hour. For all compounds, the deprotection has been well controlled, based on the temperature as standardized for 6a and 9a (Table 3). Also, analysing the reactional yields we could not identify a clear influence of electronic effects of substituents on the cyclocondensations, except for the 4-metoxy group that furnished the lowest yields for both 6b and 9b (Table 3).
|
|||||
|---|---|---|---|---|---|
| Prod. | Ar | Y | Timea | Temp.b | Yieldc |
| a Time in minutes.b Temperature in oC.c Isolated yields in %. | |||||
| 9a | Ph | H | 40 | 100 | 87 |
| 9b | 4-OCH3–Ph | H | 40 | 100 | 62 |
| 9c | 4-Cl–Ph | H | 40 | 100 | 71 |
| 9d | 4-NO2–Ph | H | 40 | 100 | 80 |
| 9e | Ph | NO2 | 40 | 100 | 61 |
| 9f | Ph | SO2CH3 | 40 | 100 | 84 |
| 9g | 4,5-DiBr-2-pyrrol | H | 40 | 100 | 90 |
| 9h | 2-Pyrrol | H | 40 | 100 | 79 |
| 6a | Ph | H | 60 | 150 | 82 |
| 6b | 4-OCH3–Ph | H | 60 | 150 | 47 |
| 6c | 4-Cl–Ph | H | 60 | 150 | 68 |
| 6d | 4-NO2–Ph | H | 60 | 150 | 52 |
| 6e | Ph | NO2 | 60 | 150 | 73 |
| 6f | Ph | SO2CH3 | 60 | 150 | 69 |
Finally, as a goal of our medicinal chemistry development program, we decided to explore N-alkylations on the 3-amino group. First, we tried the methylation of the deprotected 1,5-diphenyl-3-amine-1,2,4 triazole (6a) in some conditions, varying the base, temperature, and molar proportion. Curiously, none of these conditions led to the desired alkylated product (results not shown). As we could selective synthetize the protected 1,5-diphenyl-3-amine-1,2,4 triazole (9a), this compound was reacted with methyl iodide, NaH and DMF in a borosilicate sealed tube during around 1 h at 45 °C. The consumption of start material was observed and after chromatographic isolation we could obtained the protected N-metil-1,5-diphenyl-3-amine-1,2,4 triazole (10a) in 62% (Table 4).
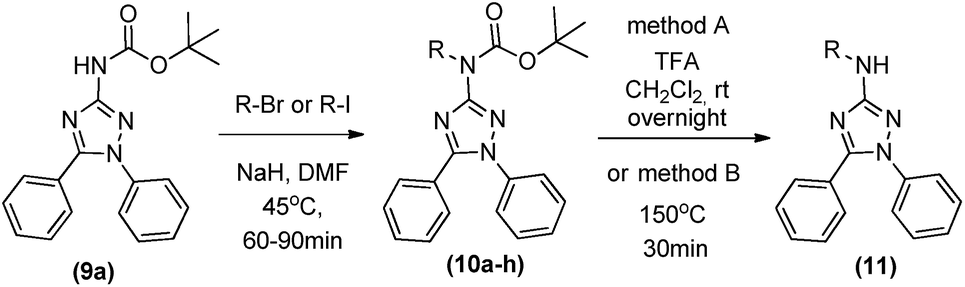
This methodology was applied to other alkylating agents, furnishing the N-alkylated products (10b–h) in reasonable to good yields (41–82%) (Table 4). Bulky alkylating groups as dibromoethane did not work in this synthesis. For bioactivity analysis, it is not so interesting to use compounds with protecting groups. Thus, we first tried a deprotection of Boc group with TFA in dichloromethane at room temperature, leading to 11a in 92% yield. After, we proceeded with a one-pot process of methylation and deprotection from the starting material (9a). After the total methylation of 9a, observed by TLC, we heated the reaction to 150 °C for 30 min and we could observe the deprotection of 10a, furnishing 11 in 59% global yield (2 steps one pot procedure) after chromatographic isolation (Table 4).
Design and synthesis of triazole cholinesterase inhibitors
The knowledge of neurotransmitter disorders in Alzheimer Disease (AD) has led to the approval of drugs with symptomatic effects.15The cholinergic hypothesis of AD states that the degeneration of cholinergic neurons in basal forebrain nuclei causes disorders in the presynaptic cholinergic terminals in the hippocampus and neocortex, regions of extreme importance for memory disorders and other cognitive symptoms.16 Because of neurodegeneration, the activity of cholinergic neurons and levels of neurotransmitter ACh are reduced and the main approach to improve cholinergic neurotransmission is to increase the availability of ACh by inhibition of cholinesterases.17
Bear in mind the total control in the synthesis and derivatization of our N-alkyl-1,5-diphenyl-3-amine-1,2,4 triazoles and the description of cholinesterase inhibition and neuroprotection of triazine derivative (12),18 we proposed the synthesis of triazoles (13). The design was carried out based on the description that multipotent activity of diaryltriazines (12) comes from the diaryltriazine moiety and not from the piperazine-ethyl side chain.18 Thus, we imagined that our 1,5-diphenyl-3-amine-1,2,4 triazoles could be envisaged from a non-classic bioisosterism19 of ring contraction from diaryltriazines (12). And finally, a molecular hybridization20 with the classical drug donepezil (14) could furnished the desired triazoles as cholinesterase inhibitors (Fig. 3).
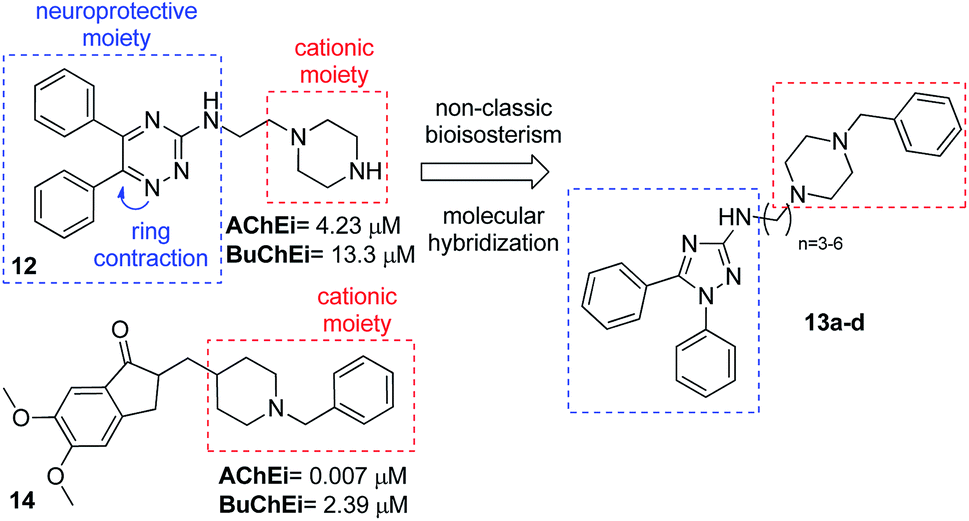 | ||
| Fig. 3 Structural design of N-alkyl-1,5-diphenyl-3-amine-1,2,4 triazoles (13a–d) as cholinesterase inhibitors. | ||
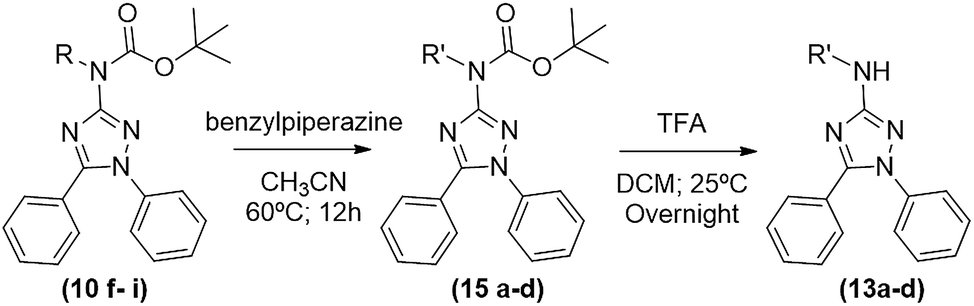
The desired 1,5-diphenyl-3-amine-1,2,4 triazoles (13a–d) were obtained in a two-step synthesis (Table 5): (1) a reaction of compounds 10f–i with 3 equivalents of benzylpiperazine in acetonitrile for 12 h at 60 °C, furnishing products 15a–d in yields ranging from 66 to 93% after chromatographic purification; and (2) a deprotection of Boc group with TFA in dichloromethane, as above described, leading to desired products 13a–d in yields ranging from 88 to 94%. In these cases, the thermal deprotection at 150 °C, despite of being faster (around 30–60 min) did not work very well originating some impurities.
Biological profile
The cholinesterase (AChE and BuChE) inhibition activities of the designed triazole derivatives (13a–d) and the non-substituted triazole 6a were determined by the Ellman's method21 using donepezil as reference compound.As depicted in Table 6, compounds (13a–d) presented good inhibitory activities against AChE with IC50 values varying from 1.14 to 11.00 μM for compounds 13a and 13c respectively.
| Compound | AChE IC50a,b (μM) | BuChE IC50a,c (μM) |
|---|---|---|
| a Concentration required for 50% inhibition of ChEs, data were shown in mean ± SD of triplicate independent experiments.b AChE from electric eel.c BuChE from horse serum.d NA – not active, inhibition < 20% at 30 μM. | ||
| 13a | 1.14 ± 0.031 | 1.79 ± 0.060 |
| 13b | 3.79 ± 0.061 | 1.63 ± 0.111 |
| 13c | 11.00 ± 0.251 | 1.65 ± 0.059 |
| 13d | 5.22 ± 0.188 | 2.21 ± 0.068 |
| 6a | NAd | NAd |
| Donepezil | 0.007 ± 0.0002 | 2.39 ± 0.105 |
The inhibition profile of triazoles (13a–d) against AChE was quite similar of that related in the literature with a regular decreasing of activity with the increasing of the number of methylene spacers. Compound 13a, which has the smallest methylene chain, was the most potent compound. Curiously, compound 13c (IC50 = 11.00 μM), which has five methylenes in the spacer chain, was an exception, being considerably less potent than its upper homolog (13d, IC50 = 5.22 μM). This result may represent a tendency of an alternating, or serrated, curve as the carbon spacer chain increases. Sometimes this curve can occur in a homologous series when variations in activity are observed according to whether the number of carbons is even or odd, because of the different 3D disposition of attached functional groups. This difference in activity when plotted versus the methylene spacer number originate this “serrated” profile. Interestingly serrated curve variations for AChE inhibitory activity were also observed in the literature for donepezil analogues.22
Additionally, all the tested triazoles (13a–d) were also capable of inhibiting BuChE (Table 6) with equipotent IC50 values, ranging from 1.63 to 2.21 μM (compounds 13b and 13d respectively). These potency values were comparable to the reference compound donepezil (IC50 = 2.39 μM). Additional results obtained by kinetic study with derivative 13a, revealed a mixed-type inhibition with a very similar non-competitive constant 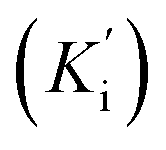 for AChE
for AChE 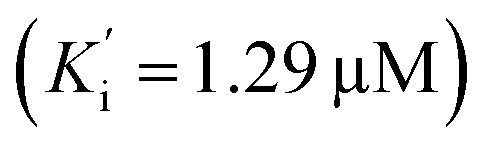 and BuChE
and BuChE 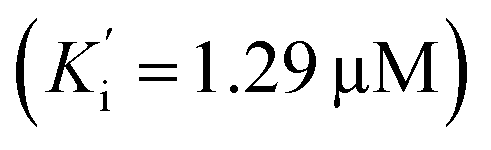 . Additionally, the competitive inhibition constant (Ki) for this compound was only 1.7 times higher for BuChE (Ki = 0.62 μM) compared to acetylcholinesterase (Ki = 0.36 μM) (Fig. 4). The obtained results suggest that the assayed triazoles (13a–d) presented good mixed-type inhibition potency, as the prototypal triazine (12), but without showing selectivity between AChE and BuChE enzymes. Nevertheless, with the molecular understanding of how triazoles interact with cholinesterases, further modifications could lead to a better selective profile.
. Additionally, the competitive inhibition constant (Ki) for this compound was only 1.7 times higher for BuChE (Ki = 0.62 μM) compared to acetylcholinesterase (Ki = 0.36 μM) (Fig. 4). The obtained results suggest that the assayed triazoles (13a–d) presented good mixed-type inhibition potency, as the prototypal triazine (12), but without showing selectivity between AChE and BuChE enzymes. Nevertheless, with the molecular understanding of how triazoles interact with cholinesterases, further modifications could lead to a better selective profile.
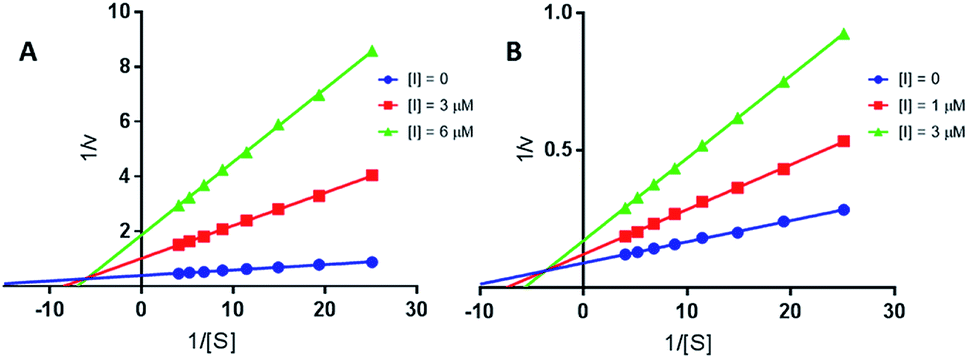 | ||
| Fig. 4 Lineweaver–Burk plots of EeAChE (A) and EcBuChE (B) inhibition kinetics of compound 13a. Inset: concentrations used for 13a are depicted with [I] graphic symbol. | ||
Furthermore, the planned derivatizations were correct and important for the activity since 6a had no significant cholinesterase inhibition at 30 μM.
Molecular modeling
All four scoring functions available in GOLD 5.6 were able to produce docking poses in the redocking of N,N′-di-1,2,3,4-tetrahydroacridin-9-yl-pentane-1,5-diamine with RMSD values lower than 1.00 Å. GoldScore23 was the only function for which the pose with the lowest RMSD value (0.62 Å) was also the pose with the highest docking score, so it was chosen for the subsequent docking procedure.In the GOLD program, the docking functions yield the “fitness scores”, which are dimensionless values. The score values are a guide of how good the docking pose is, with a higher score indicating a better interaction between the ligand and the binding site. All compounds were predicted to interact effectively with EeAChE and EcBuChE using the GoldScore fitness score function.
Comparing compounds 13a and 13c, which presented the greater difference in activity as EeAChE inhibitors, it was obtained a better fitness score for 13a using the 1C2O EeAChE structure and the ligands protonated at the N atom attached to the benzyl group (Table 7). The fitness score was higher for compound 13c but closer to that of compound 13a (both with the same protonation mode used for the EaAChE docking) when they were docked in the EcBuChE model. All these results are in qualitative accordance with the experimental activity data.
| Ligand | EeAChE | EcBuChE |
|---|---|---|
| 13a | 83.7 | 69.7 |
| 13c | 79.7 | 71.5 |
Similarly to what was observed in the complex between N,N′-di-1,2,3,4-tetrahydroacridin-9-yl-pentane-1,5-diamine in T. californica AChE by Rydberg and collaborators24 and also in accordance with our previous results,25 it could be observed that the present compounds were able to spread from the peripheral site to the active site inside the EeAChE structure, completely occupying the gorge connecting them. This is in accordance with the experimental results that showed a mixed-type inhibition for these compounds.
In Fig. 5, it is presented a superposition of the docking poses of the strongest (13a) and the weakest (13c) EeAChE inhibitors of the series. It can be observed that in the active site, the phenyl substituents of the triazole ring interact by means of T-stacking interactions with Trp86 for both inhibitors, in a similar way as described for the prototype 12.18 Most of interactions involve the side chains of aromatic rings, including some located at the gorge, which is lined with aromatic residues. However, conversely from 12, one of these residues (Tyr124) is donating a hydrogen bond to one of the N atoms of the triazole ring in our compounds.
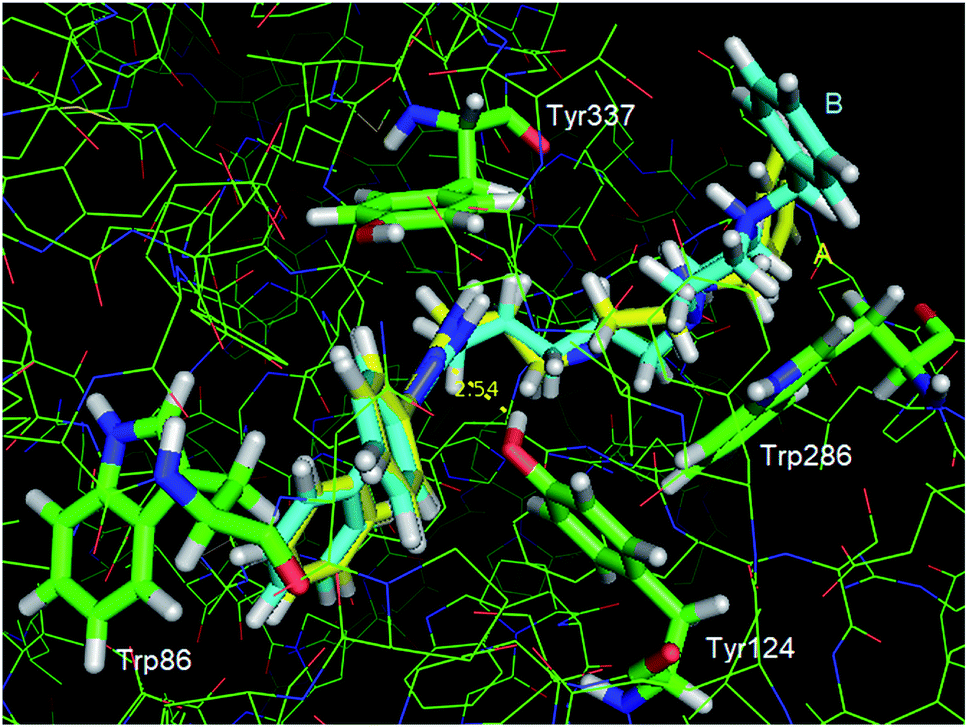 | ||
| Fig. 5 Superposition of the interaction poses of compounds 13a (A, carbon atoms in yellow) and 13b (B, carbon atoms cyan) with EeAChE obtained by molecular docking (Goldscore function). Color code: oxygen atoms, red; nitrogen atoms, blue; hydrogen atoms, white; carbon atoms, green. Dashed yellow line: hydrogen bond distance (Å). Figure generated with PyMol 0.99 (DeLano Scientific LLC). | ||
The main difference between the interaction modes of these compounds (13a and 13c) occur at the peripheral binding site (PAS). Because of its smaller spacer chain, the protonated piperazine ring of compound 13a is located exactly over the Trp286 side chain, which enables the formation of a strong cation-π interaction. However, because compound 13c side chain is two methylenes longer, a strong interaction between its protonated piperazine ring and Trp286 is impaired, which explains its weaker interaction with the enzyme. Worth of note, this interaction differs from others cholinesterase inhibitors bearing cationic groups like piperazine and piperidine, once these groups normally interact with the catalytic site (CAS) instead of PAS like ours.
To better quantify the effect of the difference in the interaction pattern with the PAS, we implemented PM6 calculations for 13a and 13c at the docking geometries of the inhibitor/Trp286 pairs.
From the resulting enthalpies of formation (ΔHf), the ΔHf of the corresponding inhibitor and of the Trp286 residue were subtracted, resulting in the interaction enthalpy (ΔHint) between each inhibitor and the Trp286 residue. For the 13c/Trp286 pair, the calculated ΔHint was −41.5 kJ mol−1, whereas for the 13a/Trp286 pair, ΔHint was −59.5 kJ mol−1. With these values, it is clear how the differences in the interaction geometries favors the interaction with the ligand with a shorter spacer, 13a.
Predicted ADMET profile analysis
To accede the drugability of tested triazoles, we proceeded with in silico evaluations that showed a good ADMET profile for these compounds. Parameters as topological polar surface area (TPSA), consensus log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P, logS, human intestinal absorption (HIA), blood brain barrier permeation (BBB), and P-glycoprotein (P-gP) substrate and drug-likeness profile were analyzed (ESI†).26 TPSA value was calculated as 49.22 Å2 and consensus logP ranged from 4.27 to 5.18. The moderate polarity and relative lipophilicic characteristics put our compounds in the yellow compartment of BOILED-Egg model (Fig. 6), having a high probability to gastrointestinal absorption and access to the CNS,27 which is fundamental for the distribution of central-acting molecules. Additionally, the most potent compound 13a presented a good drug-likeness profile according to Lipinski,28 Veber29 and Egan30 rules.
P, logS, human intestinal absorption (HIA), blood brain barrier permeation (BBB), and P-glycoprotein (P-gP) substrate and drug-likeness profile were analyzed (ESI†).26 TPSA value was calculated as 49.22 Å2 and consensus logP ranged from 4.27 to 5.18. The moderate polarity and relative lipophilicic characteristics put our compounds in the yellow compartment of BOILED-Egg model (Fig. 6), having a high probability to gastrointestinal absorption and access to the CNS,27 which is fundamental for the distribution of central-acting molecules. Additionally, the most potent compound 13a presented a good drug-likeness profile according to Lipinski,28 Veber29 and Egan30 rules.
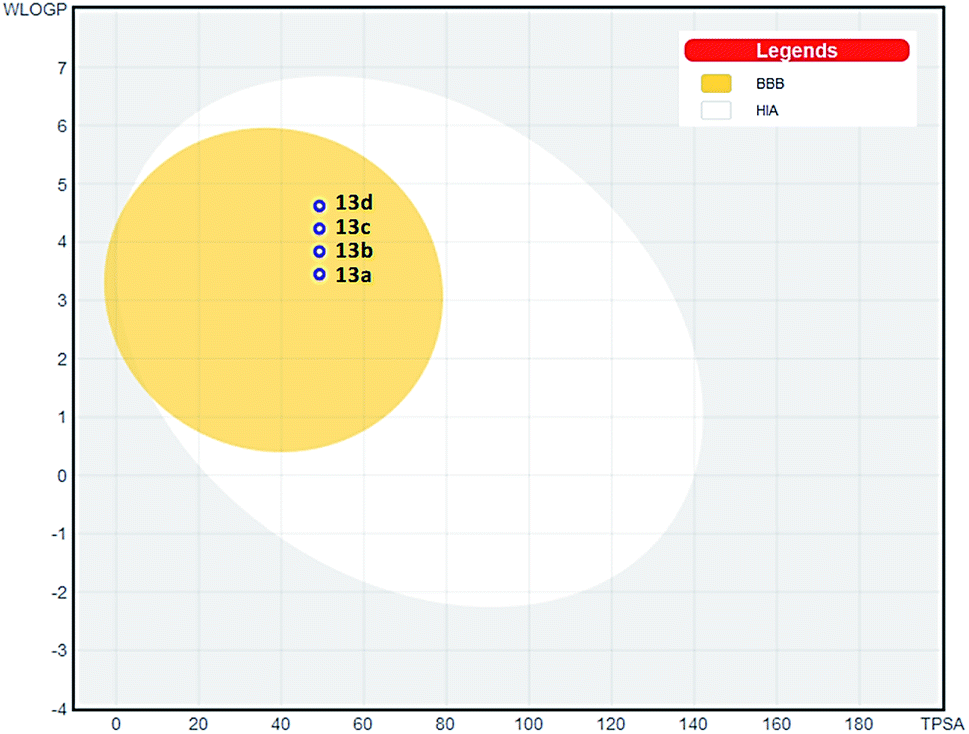 | ||
| Fig. 6 BOILED-Egg ADMET model27 for triazole compounds 13a–d. (HIA) gastrointestinal absorption; (BBB) brain penetration. | ||
Conclusions
We could develop an efficient and regioselective one-pot synthesis protocol for obtaining N-protected or N-deprotected functionalized 1,5-diaryl-3-amino-1,2,4-triazoles starting from N-acyl-N-Boc-carbamidothioates. This protocol demonstrated that microwave irradiation was very useful reducing reaction times to up to 1 h and presenting yields ranging from 50 to 84%. This chemistry summed to bioisosteric and molecular hybridization techniques allowed us to discover a new series of compounds able to inhibit cholinesterases (without selectivity) with good IC50 of up to 1 μM acting by a mixed-type way. Molecular modelling evaluations demonstrated the importance of phenyl substituents as well as the triazole ring to the interaction with CAS site of AChE, showing potential for this class of compounds as new scaffolds for this biological activity.Experimental
Materials and instruments
All chemicals were purchased from commercial suppliers and used without further purification. The1 H and 13C NMR spectra were recorded using a 400 MHz or 500 MHz Brucker Avance spectrometer (AC-400 or AC-500), using DMSO-d6 or CDCl3 and TMS as an internal standard. The melting points recorded are uncorrected. Elemental analyzes were carried out on a Thermo Scientific Flash EA 1112 Series CHN-Analyzer. A Biotage Initiator 2 Reactor was used for microwave reactions using an internal IR temperature probe.All commercial reagents were used without prior purification. Reactions were monitored by thin layer chromatography (TLC) using silica gel plates and visualized in UV light (254 nm and 365 nm). The multiplicities were called sl (singlet), d (doublet), t (triplet), q (quartet), dd (double doublet) and m (multiplet).
Synthesis of S-methyl-urea (1)
Methyl iodide (0.82 mL, 13.13 mmol) was added in a sealed tube containing thiourea (0.50 g, 6.56 mmol) in methanol (5 mL). The mixture was stirred at 65 °C for 2 hours. At the end of the reaction the mixture was concentrated on a rotary evaporator to give 1.42 g of a brown solid in 99% yield. 1H NMR (500 MHz, DMSO) (δ ppm): 2.58 (s, 3H); DEPT-Q (125 MHz, DMSO) (δ ppm): 171.09, 13.55.Synthesis of 2,5-dioxopyrrolidine-1-benzoate
Benzoic acid (0.10 g, 0.82 mmol), N,N′-dicyclohexylcarbodi-imide (DCC) (0.22 g, 0.82 mmol) and N-hydroxysuccinimide (0.12 g, 0.82 mmol) were solubilized in THF (10 mL). The resulted solution was kept under stirring at room temperature for 20 hours. Reaction mixture was filtered and evaporated on a rotary evaporator to give 216 mg of a beige solid in 71% yield that was used without any further purification.Synthesis of S-methyl-N-benzoyl-isothiourea (3)
To a solution of methylisothiourea (0.06 g, 0.28 mmol) in saturated sodium bicarbonate (2 mL) was slowly added under stirring a solution of 2,5-dioxopyrrolidine-1-benzoate (0.06 g, 0.28 mmol) in dichloromethane (10 mL). The reaction mixture was stirred at room temperature for 18 hours. After its completion, the reaction was diluted in dichloromethane (20 mL) and washed with water (3 × 10 mL). The organic phase was dried over anhydrous sodium sulphate and concentrated on a rotary evaporator to give a white amorphous solid in 72% yield.1H NMR (400 MHz, CDCl3) (δ ppm): 8.28–8.26 (d, 2H), 7.53–7.51 (m, 1H), 7.45–7.41 (m, 2H), 2.63 (sl, 3H); DEPT-Q (100 MHz, CDCl3) (δ ppm): 137.27, 132.03 (2x), 129.68 (2x), 128.04, 13.84.General procedure for 3-amine-1,2,4 triazoles preparation (5 and 6a) from 3
In a sealed borosilicate tube, methyl N′-benzoylcarbamido-thioate (3) (1 eq.) and phenylhydrazine (4) (1.1 eq.) were solubilized in the correspondent solvent (2 mL). The reaction mixture was stirred at temperature and time determined in Table 1. After completion of reaction the slurry was diluted with ethyl acetate (20 mL) and partitioned with water (10 mL). The organic phase was dried over anhydrous sodium sulphate and concentrated on a rotary evaporator. The products were purified by silica gel column chromatography (FLASH Chromatography System – Biotage), using an ultra-10 g snap column (n-hexane: ethyl acetate, 5–75% gradient elution).1,3-diphenyl-1,2,4-triazol-5-amine (5)
Brown solid; 13% yield; mp = 159–160 °C; 1H NMR (500 MHz, CDCl3) (δ ppm): 8.06–8.06 (d, 2H), 7.64–7.62 (d, 2H), 7.56–7.54 (t, 2H), 7.44–7.40 (m, 5H), 4.80 (s, 2H); DEPT-Q (125 MHz, CDCl3) (δ ppm): 159.28, 153.92, 136.81, 133.35, 130.76, 129.88, 129.21, 128.50, 128.23, 127.39, 126.16 (2X), 123.45 (2X). Elemental analysis calcd (%) for C14H12N4: C, 71.17; H, 5.12; N, 23.71; found: C 71.05, H 5.22, N 23.63.1,5-Diphenyl-1,2,4-triazol-3-amine (6a)
Brown solid; 75% yield, mp = 150–152 °C; 1H NMR (400 MHz, CDCl3) (δ ppm): 7.47–7.45 (d, 2H), 7.39–7.32 (m, 8H), 4.27 (sl, 2H); DEPT-Q (100 MHz, CDCl3) (δ ppm): 162.67, 153.09, 138.28, 129.85, 129.22 (2x), 128.74 (2x), 128.46 (2x), 128.28, 128.04, 125.18 (2x). Elemental analysis calcd (%) for C14H12N4: C, 71.17; H, 5.12; N, 23.71; found: C 71.21, H 5.35, N 23.48.General procedure for synthesis of 3-amine-1,2,4-triazoles (6b–6f) from 7a–d
In a sealed borosilicate tube containing Boc-S-methyl-N-benzoyl-isothiourea intermediates 7a–d (1 eq.) in DMF (2 mL) it was added the corresponding hydrazine (1.1 eq.). The reaction was carried out in microwave irradiation at 150 °C for 60 minutes. After the end of the reaction, the slurry was partitioned between ethyl acetate (20 mL) and water (10 mL). The organic phase was dried over anhydrous sodium sulphate and concentrated on a rotary evaporator. Products were separated by FLASH Chromatography System (Biotage), using an ultra-10 g snap column (n-hexane: ethyl acetate, 5–75% gradient elution).1,5-Diphenyl-1,2,4-triazol-3-amine (6a)
Brown solid; 82% yield; spectral data described before.5-(4-methoxyphenyl)-1-phenyl-1,2,4-triazol-3-amine (6b)
Brown solid; 47% yield, mp = 198–200 °C; 1H NMR (500 MHz, CDCl3) (δ ppm): 7.44–7.37 (m, 7H), 6.94–6.93 (d, 2H), 6.86–6.84 (d, 2H), 3.82 (sl, 3H); DEPT-Q (125 MHz, CDCl3) (δ ppm): 161. 23, 153.04, 130.42, 129.48, 129.20, 129.06, 125.50, 114.06, 113.35, 55.34. Elemental analysis calcd (%) for C15H16N4O: C, 67.65; H, 5.30; N, 21.04; found: C 67.62, H 5.21, N 21.28.5-(4-chlorophenyl)-1-phenyl-1,2,4-triazol-3-amine (6c)
Beige solid; 68% yield, mp = 151–153 °C; 1H NMR (400 MHz, CDCl3) (δ ppm): 7.43–7.42 (m, 5H), 7.33–7.31 (m, 4H); DEPT-Q (100 MHz, CDCl3) (δ ppm): 161.76, 151.26, 137.66, 136.53, 130.01 (2X), 128.98 (2X), 128.90, 125.43, 125.20 (2X), 101.30. Elemental analysis calcd (%) for C14H11N4Cl: C, 62.11; H, 4.10; N, 20.70; found: C 62.00, H 4.31, N 20.63.5-(4-nitrophenyl)-1-phenyl-1,2,4-triazol-3-amine (6d)
Yellow solid; 52% yield, mp = 202–204 °C; 1H NMR (500 MHz, CDCl3) (δ ppm): 8.19–8.17 (d, 2H), 7.65–7.64 (d, 2H), 7.45 (sl, 3H), 7.32 (m, 2H), 4.33 (sl, 2H); DEPT-Q (125 MHz, CDCl3) (δ ppm): 162.97, 150.72, 148.25, 137.64, 133.77, 129.68 (2X), 129.55 (2X), 129.17, 125.32 (2X), 123.73 (2X). Elemental analysis calcd (%) for C14H11N5O2: C, 59.78; H, 3.94; N, 24.90; found: C 59.55, H 4.02, N 24.95.1-(4-nitrophenyl)-5-phenyl-1,2,4-triazol-3-amine (6e)
Brown solid; 73% yield, mp = 173–176 °C; 1H NMR (500 MHz, CDCl3) (δ ppm): 8.27–8.26 (d, 2H), 7.54–7.49 (m, 5H), 7.45–7.43 (m, 2H), 4.99 (sl, 2H); DEPT-Q (125 MHz, CDCl3) (δ ppm): 160.70, 146.97, 142.07, 131.72, 129.35 (2X), 128.95 (2X), 125.25, 125.00 (2X), 124.93 (2X). Elemental analysis calcd (%) for C14H11N5O2: C, 59.78; H, 3.94; N, 24.90; found: C 59.43, H 4.14, N 24.78.1-(4-(methylsulfonyl)phenyl)-5-phenyl-1,2,4-triazol-3-amine (6f)
Brown solid; 69% yield, mp = 213–215 °C; 1H NMR (500 MHz) CDCl3 (δ ppm): 7.94–7.92 (d, 2H), 7.53 (d, 2H), 7.45 (m, 3H), 7.42–7.39 (m, 2H), 4.38 (sl, 2H), 3.08 (sl, 3H); DEPT-Q (125 MHz, CDCl3) (δ ppm): 163.04, 153.84, 142.25, 139.25, 130.60 (2X), 128.94, 128.80 (2X), 128.60 (2X), 127.56, 124.78 (2X), 44.46. Elemental analysis calcd (%) for C15H14N4O2S: C, 57.31; H, 4.49; N, 17.82; found: C 57.32, H 4.54, N 17.66.General synthesis of compounds (7a–7d)
In a flask containing benzoic acid (1 eq.) and dichloromethane (5 mL) it was added oxalyl chloride (4 eq.) and catalytic amount of dimethylformamide (DMF). After total consumption of the correspondent carboxylic acid, the reaction was concentrated on a rotary evaporator and diluted with dichloromethane (5 mL). This solution was added dropwise to another solution containing the protected S-metil-isothiurea (8) (1 eq.) and triethylamine (2 eq.) in dichloromethane (10 mL) and kept under stirring overnight. Then, mixture was diluted with dichloromethane (20 mL) and washed with aqueous sodium bicarbonate 10% (3 × 10 mL). Organic phase was dried over sodium sulphate and evaporated on a rotary evaporator.tert-Butyl(benzamido(methylthio)methylene)carbamate (7a)
White solid; 70% yield; 1H NMR (500 MHz, CDCl3) (δ ppm): 8.29–8.28 (d, 2H), 7.56–7.46 (dd, 3H), 2.59 (sl, 3H), 1.54 (sl, 9H); DEPT-Q (125 MHz, CDCl3) (δ ppm): 172.03, 132.81, 130.12 (2X), 128.25 (2X), 27.98 (3X), 14.92. Elemental analysis calcd (%) for C14H18N2O3S: C, 57.12; H, 6.16; N, 9.52; found: C 56.89, H 6.38, N 9.33.tert-Butyl((4-methoxybenzamido)(methylthio)methylene)-carbamate (7b)
White solid; 68% yield; 1H NMR (400 MHz, CDCl3) (δ ppm): 8.11–8.09 (d, 2H), 7.06–7.04 (d, 2H), 3.85 (sl, 3H), 2.49 (sl, 3H), 1.46 (sl, 9H). DEPT-Q (100 MHz, CDCl3) (δ ppm): 171.00, 163.44, 151.12, 132.32 (2X), 129.31, 113.49 (2X), 83.26, 55.43, 27.98 (3X), 14.86. Elemental analysis calcd (%) for C15H20N2O4S: C, 55.54; H, 6.21; N, 8.64; found: C 55.72, H 6.29, N 8.77.tert-Butyl(4-chlorobenzamido(methylthio)methylene)-carbamate (7c)
White solid; 63% yield; 1H NMR (500 MHz, CDCl3) (δ ppm): 12.54 (sl, 1H), 8.22–8.20 (d, 2H), 7.43–7.41 (d, 2H), 2.58 (sl, 3H), 1.53 (sl, 9H); DEPT-Q (125 MHz, CDCl3) (δ ppm): 174.93, 172.63, 150.95, 139.17, 135.14, 131.48 (2X), 128.55 (2X), 83.64, 27.98 (3X), 14.97. Elemental analysis calcd (%) for C14H17ClN2O3S: C, 51.14; H, 5.21; N, 8.52; found: C 51.00, H 5.39, N 8.41.tert-Butyl((4-nitrobenzamido)(methylthio)methylene)carbamate (7d)
White solid; 58% yield; 1H NMR (500 MHz, CDCl3) (δ ppm): 7.04 (s, 1H); 2.51 (s, 3H); 1.52 ppm (s, 9H); DEPT-Q (125 MHz, CDCl3) (δ ppm): 174.28, 173.68, 150.76, 150.21, 142.03, 130.95 (2X), 123.27 (2X), 84.06, 27.95 (3X), 15.12. Elemental analysis calcd (%) for C14H17N3O5S: C, 49.55; H, 5.05; N, 12.38; found: C 49.21, H 5.24, N 12.02.tert-Butyl(1H-pyrrole-2-carboxamido(methylthio)-methylene)carbamate (7e)
White solid; 80% yield; 1H NMR (500 MHz, CDCl3) (δ ppm): 12.49 (s, 1H), 9.55 (s, 1H), 7.10 (s, 1H), 7.02 (s, 1H), 6.30 ppm (s, 1H), 2.52 (s, 3H), 1.51 (s, 9H). DEPT-Q (125 MHz, CDCl3) (δ ppm): 170.5, 151.1, 130.5, 116.8, 110.9, 83.5, 27.9, 14.6. Elemental analysis calcd (%) for C12H17N3O3S: C, 50.87; H, 6.05; N, 14.83; found: C 50.71, H 5.98, N 14.96.tert-Butyl(4,5-dibromo-1H-pyrrole-2-carboxamido(methylthio)-methylene)carbamate (7f)
White solid; 69% yield; 1H NMR (500 MHz, CDCl3) (δ ppm): 12.53 (sl, 1H), 8.43–8.42 (d, 2H), 8.30–8.28 (d, 2H), 2.61 (sl, 3H), 1.55 (sl, 9H); DEPT-Q (125 MHz, CDCl3) (δ ppm): 119.3; 84.0; 28.0; 15.1. Elemental analysis calcd (%) for C12H15Br2N3O3S: C, 32.67; H, 3.43; N, 9.53; found: C 32.38, H 3.64, N 9.48.Synthesis of tert-butyl [amine(methylthio)methylene] carbamate (8)
In a flask containing methyl isothiourea (1 g, 4.58 mmol), dichloromethane (10 mL) and triethylamine (639 μL, 4.58 mmol), in an ice-bath, it was slowly added a solution of di-tert-butyl dicarbonate (Boc–O–Boc) (0.5 g, 2.29 mmol) in dichloromethane (5 mL). Then, ice bath was removed and reaction was left under stirring overnight at room temperature. After consumption of reactants, the reaction mixture was diluted with dichloromethane (15 mL), washed with water (2 × 10 mL), dried over sodium sulphate and concentrated on a rotary evaporator. A white solid product was obtained in 86% yield and used without any further purification.1H NMR (500 MHz, DMSO) (δ ppm): 8.58 (sl, 2H), 2.32 (sl, 3H), 1.41 (sl, 9H); DEPT-Q (125 MHz, DMSO) (δ ppm): 171.87, 160.98, 78.11, 26.17 (3X), 13.13.
Synthesis of compounds 9a–9e
In a sealed borosilicate tube containing triazole (7a–7f) (1 eq.) in 2 mL of acetonitrile, it was added the corresponding hydrazine (1.1 eq.). Reaction was stirred at 100 °C for 40 minutes as shown in Table 3. After the end of the reaction, the reactional slurry was partitioned between ethyl acetate (20 mL) and water (10 mL). The organic phase was dried over anhydrous sodium sulphate and concentrated on a rotary evaporator. The products were separated with a FLASH Chromatography System (Biotage), using an ultra-10 g snap column (n-hexane: ethyl acetate, 5–75% gradient elution).tert-Butyl(1,5-diphenyl-1,2,4-triazol-3-yl)carbamate (9a)
Brown solid; 87% yield, mp = 147–149 °C; 1H NMR (400 MHz, CDCl3) (δ ppm): 7.49 (m, 3H), 7.39 (sl, 6H), 7.34–7.32 (d, 2H), 1.52 (s, 9H); DEPT-Q (100 MHz, CDCl3) (δ ppm): 156.33, 153.05, 151.32, 138.08, 130.11, 129.23 (2X), 128.93 (2X), 128.50 (2X), 125.75 (2X), 81.34, 28.21 (3X). Elemental analysis calcd (%) for C19H20N4O2: C, 67.84; H, 5.99; N, 16.66; found: C 68.02, H 6.07, N 16.62.tert-Butyl(5-(4-methoxyphenyl)-1-phenyl-1,2,4-triazol-3-yl)carbamate (9b)
Brown solid; 62% yield, mp = 164–167 °C; 1H NMR (400 MHz, CDCl3) (δ ppm): 7.59 (sl, 1H), 7.43–7.39 (m, 7H), 6.84–6.82 (d, 2H), 3.81 (s, 3H), 1.51 (s, 9H); DEPT-Q (100 MHz, CDCl3) (δ ppm): 161.01, 155.94, 151.28, 138.11, 130.43 (2X), 129.25 (2X), 128.77, 125.82 (2X), 113.94 (2X), 81.34, 55.31, 28.18 (3X). Elemental analysis calcd (%) for C20H22N4O3: C, 65.56; H, 6.05; N, 15.29; found: C 65.90, H 6.07, N 14.99.tert-Butyl(5-(4-chlorophenyl)-1-phenyl-1,2,4-triazol-3-yl)carbamate (9c)
White solid; 71% yield, mp = 140–142 °C; 1H NMR (400 MHz, CDCl3) (δ ppm): 7.94 (sl, 1H), 7.43–7.41 (m, 4H), 7.38 (d, 2H), 7.31–7.25 (m, 3H), 1.49 (s, 9H); DEPT-Q (100 MHz, CDCl3) (δ ppm): 156.47, 151.93, 151.33, 137.75, 136.37, 130.18 (2X), 129.38 (2X), 129.05 (2X), 128.82 (2X), 125.74, 125.78, 81.37, 28.14 (3X). Elemental analysis calcd (%) for C19H19ClN4O2: C, 61.54; H, 5.16; N, 15.11; found: C 61.23, H 5.34, N 14.82.tert-Butyl(5-(4-nitrophenyl)-1-phenyl-1,2,4-triazol-3-yl)carbamate (9d)
White solid; 80% yield, mp = 166–168 °C; 1H NMR (400 MHz, CDCl3) (δ ppm): 8.19–8.17 (d, 2H), 7.69–7.66 (d, 2H), 7.47–7.45 (m, 3H), 7.39–7.36 (m, 3H), 1.54 (s, 9H); DEPT-Q (100 MHz, CDCl3) (δ-ppm): 156.71, 151.10, 148.41, 137.40, 133.22, 129.78 (2X), 129.67 (2X), 129.62, 125.79 (2X), 123.72 (2X), 81.76, 28.18 (3X). Elemental analysis calcd (%) for C19H19N5O4: C, 59.84; H, 5.02; N, 18.36; found: C 59.78, H 5.12, N 18.21.tert-Butyl (1-(4-nitrophenyl)-5-phenyl-1,2,4-triazol-3-yl)carbamate (9e)
Brown solid; 61% yield, mp = 147–150 °C; 1H NMR (400 MHz, DMSO) (δ ppm): 10.12 (s, 1H), 8.35–8.33 (d, 2H), 7.65 (d, 2H), 7.49–7.47 (m, 5H), 1.49 (s, 9H); DEPT-Q (100 MHz, DMSO) (δ ppm): 157.53, 153.88, 152.38, 147.19, 143.08, 131.21, 129.52 (2X), 127.85, 126.48 (2X), 125.52 (2X), 80.19, 28.61 (3X). Elemental analysis calcd (%) for C19H19N5O4: C, 59.84; H, 5.02; N, 18.36; found: C 59.95, H 5.01, N 18.14.tert-Butyl (1-(4-(methylsulfonyl)phenyl)-5-phenyl-1,2,4-triazol-3-yl)carbamate (9f)
Brown solid; 84% yield, mp = 179–181 °C; 1H NMR (400 MHz, CDCl3) (δ ppm): 7.96–7.94 (d, 2H), 7.90 (s, 1H), 7.61–7.60 (d, 2H), 7.46 (m, 3H), 7.41–7.38 (m, 2H), 3.07 (s, 3H), 1.50 (s, 9H); DEPT-Q (100 MHz, CDCl3) (δ ppm): 156.94, 153.56, 151.12, 142.09, 140.00, 130.85, 128.96 (2X), 128.64 (2X), 126.82, 125.73 (2X), 81.66, 44.46, 28.11 (3X). Elemental analysis calcd (%) for C20H22N4O4S: C, 57.96; H, 5.35; N, 13.52; found: C 57.59, H 5.55, N 13.28.tert-Butyl (1-phenyl-5-(1H-pyrrol-2-yl)-1-phenyl-1H-1,2,4-triazol-3-yl)carbamate (9g)
Brown solid; 90% yield, mp = 170–172 °C; 1H NMR (500 MHz, CDCl3) (δ ppm): 12.49 (s, 1H), 9.55 (s, 1H), 7.10 (s, 1H), 7.02 (s, 1H), 6.30 ppm (s, 1H), 2.52 (s, 3H), 1.51 (s, 9H). DEPT-Q (125 MHz, CDCl3) (δ ppm): 170.5, 151.1, 130.5, 116.8, 110.9, 83.5, 27.9, 14.6. Elemental analysis calcd (%) for C17H19N5O2: C, 62.75; H, 5.89; N, 21.52; found: C 62.79, H 5.95, N 21.38.tert-Butyl (5-(4,5-dibromo-1H-pyrrol-2-yl)-1-phenyl-1H-1,2,4-triazol-3-yl)carbamate (9h)
Brown solid; 79% yield; 1H NMR (500 MHz, CDCl3) (δ ppm): 12.98 (sl, 1H), 9.77 (sl, 1H), 7.59–7.48 (m, 5H), 5.65 (s, 1H), 1.45 (s, 9H). DEPT-Q (125 MHz, CDCl3) (δ ppm): 155.9, 145.9, 137.5, 130.3, 129.8, 126.7, 120.2, 114.0, 105.5, 100.0, 28.5. Elemental analysis calcd (%) for C17H17Br2N5O2: C, 42.26; H, 3.55; N, 14.49; found: C 42.13, H 3.58, N 14.25.General synthesis of compounds 10a–10h
The corresponding alkyl halide (2 eq.) was added in a sealed borosilicate tube containing triazole 9a (1 eq.) and sodium hydride (4 eq.) in anhydrous DMF (2 mL). The reaction was stirred at 45 °C for 60–90 minutes. When the end of reaction was determined by TLC (hexane: ethyl acetate 30%), mixture was diluted in 20 mL of ethyl acetate and washed with water (3 × 10 mL). The organic phase was dried over sodium sulphate, concentrated and purified by an isolation with FLASH Chromatography System (Biotage), using an ultra-10 g snap column (n-hexane: ethyl acetate, 10–50% gradient elution).tert-Butyl (1,5-diphenyl-1,2,4-triazol-3-yl)(methyl) carbamate (10a)
Yellow oil; 62% Yield; 1H NMR (400 MHz, CDCl3) (δ ppm): 7.50–7.48 (d, 2H), 7.40–7.33 (m, 8H), 3.43 (sl, 3H), 1.55 (sl, 9H); DEPT-Q (100 MHz, CDCl3) (δ ppm): 160.11, 153.56, 153.11, 137.97, 130.01, 129.28 (2X), 128.91 (2X), 128.47 (2X), 127.64, 125.46 (2X), 81.29, 35.54, 28.26 (3X). Elemental analysis calcd (%) for C20H22N4O2: C, 68.55; H, 6.33; N, 15.99; found: C 68.32, H 6.51, N 15.88.tert-Butyl (1,5-diphenyl-1,2,4-triazol-3-yl)(ethyl) carbamate (10b)
Beige solid; 65% yield, mp = 98–99 °C; 1H NMR (500 MHz, CDCl3) (δ ppm): 7.50–7.48 (d, 2H), 7.40–7.31 (m, 8H), 3.91–3.86 (m, 2H), 1.53 (sl 9H), 1.33–1.30 (t, 3H); DEPT-Q (125 MHz, CDCl3) (δ ppm): 159.40, 153.34, 137.99, 129.96, 129.24 (2X), 128.91 (2X), 128.44 (2X), 127.70, 125.45 (2X), 80.99, 43.58, 28.28 (2X), 14.07. Elemental analysis calcd (%) for C21H24N4O2: C, 69.21; H, 6.64; N, 15.37; found: C 69.00, H 6.68, N 15.49.tert-Butyl (1,5-diphenyl-1,2,4-triazol-3-yl)(propyl) carbamate (10c)
Beige solid; 65% Yield, mp = 97–99 °C; 1H NMR (500 MHz, CDCl3) (δ-ppm): 7.50–7.48 (d, 2H), 7.41–7.32 (m, 8H), 3.80–3.77 (t, 2H), 1.79–1.72 (m, 2H), 1.53 (sl, 9H), 0.98–0.95 (t, 3H); DEPT-Q (125 MHz, CDCl3) (δ ppm): 159.60, 153.62, 153.18, 130.01, 129.98, 129.26 (2X), 128.94 (2x), 128.46 (2X), 127.73, 125.48 (2X), 80.98, 50.19, 28.29 (3X), 22.09, 11.23. Elemental analysis calcd (%) for C22H26N4O2: C, 69.82; H, 6.92; N, 14.80; found: C 69.99, H 6.95, N 14.58.tert-Butyl allyl (1,5-diphenyl-1,2,4-triazol-3-yl) carbamate (10d)
White solid; 41% yield, mp = 82–84 °C; 1H NMR (500 MHz, CDCl3) (δ ppm): 7.49–7.47 (d, 2H), 7.40–7.7.31 (m, 8H), 6.06–5.98 (m, 1H), 5.32–5.15 (dd, 2H), 4.47–4.46 (d, 2H), 1.53 (sl, 9H); DEPT-Q (125 MHz, CDCl3) (δ ppm): 159.40, 153. 20, 153, 12, 138.00, 133.85, 129.97, 129.24 (2X), 128.94 (2X), 128.44 (2X), 127.69, 125.50 (2X), 116.61, 81.36, 50.79, 28.24 (3X). Elemental analysis calcd (%) for C22H24N4O2: C, 70.19; H, 6.43; N, 14.88; found: C 70.22, H 6.52, N 14.97.tert-Butyl benzyl (1,5-diphenyl-1,2,4-triazol-3-yl) carbamate (10e)
White solid; 79% yield, mp = 110–113 °C; 1H NMR (500 MHz, CDCl3) (δ ppm): 7.48–7.47 (m, 4H), 7.39 (m, 5H), 7.32–7.31 (m, 6H), 7.25–7.23 (m, 1H), 5.10 (sl, 2H), 1.47 (sl, 9H); DEPT-Q (125 MHz, CDCl3) (δ ppm): 159.68, 153.27, 153.07, 138.52, 137.96, 129.95, 129.21 (2X), 128.91 (2X), 128.42 (2X), 128.18 (2X), 127.66, 126.91, 125.49 (2X), 125.14, 118.96, 112.82, 81.54, 56.52, 51.84, 47.84, 28.16 (3X). Elemental analysis calcd (%) for C26H26N4O2: C, 73.22; H, 6.14; N, 13.14; found: C 72.97, H 6.42, N 13.23.tert-Butyl(4-bromobutyl)(1,5-diphenyl-1,2,4-triazol-3-yl)carbamate (10f)
Brown oil; 82% yield; 1H NMR (500 MHz, CDCl3) (δ ppm): 7.50–7.48 (d, 2H), 7.41–7.32 (m, 8H), 3.89–3.87 (t, 2H), 3.48–3.46 (t, 2H), 2.01–1.97 (m, 2H), 1.90–1.87 (m, 2H), 1.54 (sl, 9H); DEPT-Q (125 MHz, CDCl3) (δ ppm): 159.40, 153.50, 153.30, 137.99, 130.08, 129.33 (2X), 128.96 (2X), 128.53 (2X), 127.66, 125.51 (2X), 81.39, 47.41, 33.51, 29.94, 28.31 (3X), 27.47. Elemental analysis calcd (%) for C23H27BrN4O2: C, 58.60; H, 5.77; N, 11.89; found: C 58.55, H 5.84, N 11.68.tert-Butyl(5-bromopentyl)(1,5-diphenyl-1,2,4-triazol-3-yl)carbamate (10g)
Brown oil; 52% yield; 1H NMR (400 MHz, CDCl3) (δ ppm): 7.50–7.48 (d, 2H), 7.41–7.23 (m, 7H), 3.86–3.82 (t, 2H), 3.44–3.40 (t, 2H), 1.94–1.90 (m, 2H), 1.79–1.77 (m, 2H), 1.53 (sl, 9H); DEPT-Q (100 MHz, CDCl3) (δ ppm): 159.48, 153.49, 153.22, 137.96, 130.03, 129.29 (2X), 128.48 (2X), 127.64, 125.46 (2X), 81.21, 48.13, 33.77, 32.38, 28.29 (3X), 27.88, 25.30. Elemental analysis calcd (%) for C24H29BrN4O2: C, 59.38; H, 6.02; N, 11.54; found: C 59.01, H 5.89, N 11.34.tert-Butyl(6-bromohexyl)(1,5-diphenyl-1,2,4-triazol-3-yl)carbamate (10h)
Brown oil; 65% yield; 1H NMR (400 MHz, CDCl3) (δ ppm): 7.50–7.48 (d, 2H), 7.41–7.32 (m, 8H), 3.84–3.81 (t, 2H), 3.42–3.39 (t, 2H), 1.89–1.85 (m, 2H), 1.76–1.75 (m, 2H), 1.53 (sl, 9H); DEPT-Q (100 MHz, CDCl3) (δ ppm): 159.51, 153.55, 153.21, 137.97, 130.01, 129.29 (2X), 128.93 (2X), 128.48 (2X), 127.66, 125.46 (2X), 101.13, 81.11, 48.28, 33.91, 32.70, 28.51, 28.29 (3X), 27.82, 25.80. Elemental analysis calcd (%) for C25H31BrN4O2: C, 60.12; H, 6.26; N, 11.22; found: C 59.86, H 6.43, N 11.17.General synthesis of compounds 15a–15d
Benzylpiperazine (3 eq.) was added in a sealed borosilicate tube containing triazole 10f–h (1 eq.) and acetonitrile (2 mL). Reaction was stirred at 60 °C until the end of the reaction, determined by TLC (DCM:methanol 10%). Then, mixture was concentrated and purified by an isolation with FLASH Chromatography System (Biotage), using an ultra-10 g snap column (DCM:methanol, 1–50% gradient elution).
tert-Butyl (3-(4-benzylpiperazin-1-yl)propyl)(1,5-diphenyl-1H-1,2,4-triazol-3-yl)carbamate (15a)
Brown oil; 66% yield; 1H NMR (500 MHz, CDCl3) (δ ppm): 7.47–7.45 (d, 2H), 7.40–7.30 (m, 13H), 3.92–3.89 (t, 2H), 3.61 (s, 2H), 2.81–2.76 (m, 8H), 2.12 (m, 2H), 1.51 (sl, 9H), 1.25 (sl, 2H); DEPT-Q (125 MHz, CDCl3) (δ ppm): 159.03, 153.43, 153.33, 137.78, 130.09, 129.41 (2X), 129.28 (2X), 128.84 (3X), 128.47 (2X), 128.42 (2X), 127.66, 127.40, 125.39 (2X), 81.59, 62.11, 55.17, 52.07, 50.88, 46.12, 28.21, 24.56. Elemental analysis calcd (%) for C27H35N5O2: C, 70.25; H, 7.64; N, 15.17; found: C 69.99, H 7.86, N 15.01.tert-Butyl (4-(4-benzylpiperazin-1-yl)butyl)(1,5-diphenyl-1H-1,2,4-triazol-3-yl)carbamate (15b)
Brown oil; 86% yield; 1H NMR (500 MHz, CDCl3) (δ ppm): 7.48–7.47 (d, 2H), 7.40–7.31 (m, 12H), 3.83 (m, 2H), 3.56 (s, 2H), 2.69 (m, 7H), 1.78 (m, 4H), 1.51 (sl, 9H), 1.26 (m, 1H), 0.88–0.83 (m, 1H); DEPT-Q (125 MHz, CDCl3) (δ ppm): 163.24, 137.86, 130.10 (2X), 129.26 (3X), 128.89 (2X), 128.51 (3X), 128.37 (2X), 127.44 (2X), 125.43, 81.35, 62.35, 57.47, 52.29, 51.02, 47.71, 28.26, 26.30 (2X). Elemental analysis calcd (%) for C28H37N5O2: C, 70.71; H, 7.84; N, 14.72; found: C 70.56, H 8.01, N 14.58.tert-Butyl (5-(4-benzylpiperazin-1-yl)pentyl)(1,5-diphenyl-1H-1,2,4-triazol-3-yl)carbamate (15c)
Brown oil; 93% yield; 1H NMR (500 MHz, CDCl3) (δ ppm): 7.48–7.46 (d, 2H), 7.40–7.30 (m, 13H), 3.83–3.80 (t, 2H), 3.61 (s, 2H), 2.86 (m, 6H), 1.89 (m, 2H), 1.76–1.72 (m, 2H), 1.51 (sl, 9H), 1.45–1.42 (m, 2H); DEPT-Q (125 MHz, CDCl3) (δ ppm): 159.33, 153.58, 153.28, 137.86, 130.06, 129.30, 128.49 (2X), 127.54 (2X), 125.43, 81.25, 61.88, 57.28, 49.58, 47.77, 28.26, 27.94, 23.81. Elemental analysis calcd (%) for C29H39N5O2: C, 71.13; H, 8.03; N, 14.30; found: C 70.89, H 8.22, N 14.29.tert-Butyl (6-(4-benzylpiperazin-1-yl)hexyl)(1,5-diphenyl-1H-1,2,4-triazol-3-yl)carbamate (15d)
Brown oil; 81% yield; 1H NMR (500 MHz, CDCl3) (δ ppm): 7.48–7.47 (d, 2H), 7.40–7.31 (m, 12H), 3.81–3.78 (t, 2H), 3.65–3.57 (m, 2H), 3.88 (m, 2H), 2.95–2.80 (m, 7H), 1.88 (m, 2H), 1.73–1.70 (m, 2H), 1.51 (sl, 9H), 1.41 (m, 4H), 0.94–0.89 (m, 1H); DEPT-Q (125 MHz, CDCl3) (δ ppm): 159.40, 153.59, 153.26, 137.90, 130.05, 129.30 (2X), 128.90 (2X), 128.80 (2X), 128.56 (2X), 127.59, 125.44 (2X), 81.15, 61.80, 57.21, 48.11, 46.80, 31.50, 29.56, 28.30, 28.27, 26.38, 25.87, 25.27, 22.61, 13.93, 11.80. Elemental analysis calcd (%) for C30H41N5O2: C, 71.54; H, 8.20; N, 13.90; found: C 71.30, H 8.45, N 13.71.General synthesis of compounds (13a–13d)
In a sealed borosilicate tube containing triazole 12a–d (1 eq.) solubilized dichloromethane (4 mL) in an ice-bath, it was added trifluoracetic acid (35 eq.). The ice bath was removed and reaction stirred at room temperature overnight. The end of reaction was determined by TLC and saturated sodium bicarbonate solution was added until pH 7 and then, extracted with dichloromethane, dried over sodium sulphate and concentrated on a rotary evaporator.N-(3-(4-benzylpiperazin-1-yl)propyl)-1,5-diphenyl-1H-1,2,4-triazol-3-amine (13a)
Brown oil; 94% yield; 1H NMR (400 MHz, CDCl3) (δ ppm): 7.45–7.43 (d, 2H), 7.37–7.30 (m, 13H), 3.54 (sl, 2H), 3.44–3.42 (t, 2H), 2.66–2.43 (m, 10H), 1.89–1.84 (m, 2H); DEPT-Q (100 MHz, CDCl3) (δ-ppm): 163.98, 152.82, 138.83, 129.79, 129.23, 128.46, 128.16, 128.11, 127.64, 125.24, 62.21, 57.34, 43.06, 27.53. Elemental analysis calcd (%) for C28H32N6O6: C, 74.30; H, 7.13; N, 18.57; found: C 74.15, H 7.39, N 18.28.N-(4-(4-benzylpiperazin-1-yl)butyl)-1,5-diphenyl-1H-1,2,4-triazol-3-amine (13b)
Brown oil; 95% yield; 1H NMR (400 MHz, CDCl3) (δ ppm): 7.44–7.43 (d, 2H), 7.38–7.30 (m, 13H), 3.60 (sl, 2H), 3.39–3.37 (t, 2H), 2.75 (m, 6H), 1.76–1.70 (m, 10H); DEPT-Q (100 MHz, CDCl3) (δ ppm): 164.12, 152.79, 138.90, 137.90, 129.70, 129.25, 129.19, 128.76, 128.42, 128.06, 125.25, 62.92, 56.61, 53.14, 52.86, 42.62, 29.67, 26.34. Elemental analysis calcd (%) for C29H34N6: C, 74.65; H, 7.34; N, 18.01; found: C 74.48, H 7.51, N 17.82.N-(5-(4-benzylpiperazin-1-yl)pentyl)-1,5-diphenyl-1H-1,2,4-triazol-3-amine (13c)
Brown oil; 88% yield; 1H NMR (400 MHz, CDCl3) (δ ppm): 7.42–7.41 (d, 2H), 7.35–7.29 (m, 13H), 4.82 (m, 1H), 4.69 (m, 1H), 4.32–4.31 (sl, 1H), 3.60 (sl, 2H), 3.37–3.33 (m, 2H), 2.82 (m, 10H), 1.84 (m, 2H), 1.72–1.66 (m, 2H), 1.48–1.42 (m, 2H); DEPT-Q (100 MHz, CDCl3) (δ ppm): 163.95, 152.75, 138.28, 129.74, 129.20, 128.68, 128.41, 128.11, 128.05, 127.71, 125.22, 61.93, 57.30, 49.95, 43.05, 29.63, 29.21, 24.05. Elemental analysis calcd (%) for C30H36N6: C, 74.97; H, 7.55; N, 17.48; found: C 74.66, H 7.68, N 17.27.N-(6-(4-benzylpiperazin-1-yl)hexyl)-1,5-diphenyl-1H-1,2,4-triazol-3-amine (13d)
Brown oil; 90% yield 1H NMR (400 MHz, CDCl3) (δ ppm): 7.45–7.43 (d, 2H), 7.37–7.31 (m, 12H), 4.27 (sl, 1H), 3.54 (sl, 2H), 3.37–3.33 (m, 2H), 2.59–2.50 (m, 10H), 1.67–1.57 (m, 4H), 1.45–1.35 (m, 4H); DEPT-Q (100 MHz, CDCl3) (δ ppm): 164.11, 152.79, 138.42, 129.76, 129.29, 128.77, 128.46, 128.31, 128.20, 128.12, 127.27, 125.31, 62.73, 58.20, 52.73, 52.07, 43.56, 29.77, 27.11, 26.68, 25.91. Elemental analysis calcd (%) for C31H38N6: C, 75.27; H, 7.74; N, 16.99; found: C 74.99, H 7.96, N 16.75.Biological assessment
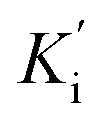 (competitive and non-competitive inhibition constants respectively), Km (Michaelis–Menten constant) and Vmax values were calculated with Graphpad Prism 7.0 using the non-linear regression models for enzyme kinetics – inhibition and enzyme Kinect – substrate vs. velocity.
(competitive and non-competitive inhibition constants respectively), Km (Michaelis–Menten constant) and Vmax values were calculated with Graphpad Prism 7.0 using the non-linear regression models for enzyme kinetics – inhibition and enzyme Kinect – substrate vs. velocity.We implemented redocking experiments as a previous test for the ability of the docking program GOLD 5.6 (CCDC Software Ltd., Cambridge, UK) to find reliable solutions for the docking to cholinesterases. Since all EeAChE structures accessible in the Protein Data Bank (PDB) do not have ligands in their crystallographic structures, the scoring functions available in the program were tested through redocking procedures with the PDB structure 2CMF (Torpedo californica AChE co-crystallized with alkylene-linked bis-tacrine dimer).24 Hydrogen atoms were added to proteins structures based on ionization and tautomeric states defined by GOLD. The bonds of groups capable to donate hydrogen bonds present in serine, threonine, tyrosine and lysine side chains were set free to rotate during the docking procedure to allow the best orientation of the hydrogen bonds. In the course of the searching procedure, 100000 genetic operations (crossover, migration, mutation) were used for each docking run. Radius of binding sites for the enzymes was tested at 15 Å and 20 Å around atoms from adequate amino acids selected based on literature information for each binding site. The same score function, corresponding amino acids and radius were adopted for the docking studies with the EcBChE model.
Three EeAChE structures are available in PDB, with codes 1EEA,32 1C2B33 and 1C2O34 and all of them were evaluated in our docking studies. There is no available crystallographic structure for EcBChE, so it was necessary the construction of a 3D model from a sequence available in the UniProtKB/Swiss-Prot protein sequence database (entry Q9N1N9) with the protein structure homology-modelling server, Swiss-Model,31 as previously described,24 using as a template the human BChE with PDB code 4TPK.35 The resolution of the template is 2.52 Å, the identity between sequences is 90.40%, with a coverage 0.95. The global and per-residue model quality has been assessed using the QMEAN scoring function.
In the GOLD program, the docking functions yield the “fitness scores”, which are dimensionless values. The score of each pose identified is calculated as the negative of the sum of a series of energy terms involved in the protein–ligand interaction process, so that the more positive the score, the better is the interaction. The score values are a guide of how good the docking pose is, with a higher score indicating a better interaction between the ligand and the binding site.
Spartan'14 program [Wavefunction, Inc.] was utilized to construct and optimize the molecules in study, with the PM6 method. Since these compounds have two plausible protonation sites in the piperazine ring, structures protonated at each of them and also with both of them protonated were considered in our studies. Several poses were obtained for each compound in all proteins, and the best-ranked pose for each one was chosen for analysis of the interactions with the amino acid residues.
Conflicts of interest
There is no conflict to declare.Acknowledgements
Fellowship and financial support for this study was provided by Conselho Nacional de Desenvolvimento Científico e Tecnológico - Brasil (CNPq), Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro – Brasil (FAPERJ) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Finance Code 001.Notes and references
- S. Borg, G. Estenne-Bouhtou, K. Luthman, I. Csöregh, W. Hesselink and U. Hacksell, J. Org. Chem., 1995, 60, 3112 CrossRef CAS.
- E. Bozo, G. Szilagyi and J. Janaky, Arch. Pharm., 1989, 322, 583 CrossRef CAS PubMed.
- Y. Naito, F. Akahoshi, S. Takeda, T. Okada, M. Kajii, H. Nishimura, M. Sugiura, C. Fukaya and Y. J. Kagitani, J. Med. Chem., 1996, 39, 3019 CrossRef CAS PubMed.
- J. P. Marino, P. W. Fisher, G. A. Hofmann, R. B. Kirkpatrick, C. A. Janson, R. K. Johnson, C. Ma, M. Mattern, T. D. Meek, M. D. Ryan, C. Schulz, W. W. Smith, D. G. Tew, T. A. Tomazek, D. F. Veber, W. C. Xiong, Y. Yamamoto, K. Yamashita, G. Yang and S. K. Thompson, J. Med. Chem., 2007, 50, 3777 CrossRef CAS PubMed.
- J.-L. Fauchere, J.-C. Ortuno, J. Duhault, J. A. Boutin and N. Levens, European Patent EP 1044970, 2000.
- G. Winterer, J. Gallinat, J. Brinkmeyer, F. Musso, J. Kornhuber, N. Thuerauf, D. Rujescu, R. Favis, Y. Sun, M. A. Franc, S. Ouwerkerk-Mahadevan, L. Janssens, M. Timmers and J. R. Streffer, Neuropharmacology, 2013, 64, 197 CrossRef CAS PubMed.
- (a) S. Maddila, R. Pagadala and S. B. Jonnalagadda, Lett. Org. Chem., 2013, 10, 693 CrossRef CAS; (b) J. Meng and P. P. Kung, Tetrahedron Lett., 2009, 50, 1667 CrossRef CAS; (c) G. M. Makara, Y. Ma and L. Margarida, Org. Lett., 2002, 4, 1751 CrossRef CAS; (d) A. V. Bogolyubsky, O. Savych, A. V. Zhemera, S. E. Pipko, A. V. Grishchenko, A. I. Konovets, R. O. Doroshchuk, D. N. Khomenko, V. S. Brovarets, Y. S. Moroz and M. Vybornyi, ACS Comb. Sci., 2018, 20, 461 CrossRef CAS PubMed.
- A. R. Katritzky, B. V. Rogovoy, V. Y. Vvedensky, K. Kovalenko, P. J. Steel, V. I. Markov and B. Forood, Synthesis, 2001, 6, 897 CrossRef.
- Y. Yu, J. M. Ostresh and R. A. Houghten, Tetrahedron Lett., 2003, 44, 7841 CrossRef CAS.
- D. G. Churchill, J. Chem. Educ., 2006, 83, 1798 CrossRef CAS.
- A. A. Schiltand, L. C. McBride, Perchloric Acid and Perchlorates, ed. G. F. Smith, Chemical Co, Columbus (USA), 2003 Search PubMed.
- J. Shen, B. Wong, C. Gu and H. Zhang, Org. Lett., 2015, 17, 4678 CrossRef CAS PubMed.
- (a) D. Garella, E. Borretto, A. Di Stilo, K. Martina, G. Cravotto and P. Cintas, Med. Chem. Commun., 2013, 4, 1323 RSC; (b) N. M. Nascimento-Júnior, A. E. Kümmerle, E. J. Barreiro and C. A. M. Fraga, Molecules, 2011, 16, 9274 CrossRef PubMed.
- S. M. Hickey, T. D. Ashton, S. K. Khosa, R. N. Robson, J. M. White, J. Li, R. L. Nation, H. Y. Yu, A. G. Elliott, M. S. Butler, J. X. Huang, M. A. Cooperd and F. M. Pfeffer, Org. Biomol. Chem., 2015, 13, 6225 RSC.
- R. Anand, K. D. Gill and A. A. Mahdi, Neuropharmacology, 2014, 76, 27 CrossRef CAS PubMed.
- A. V. Terry and J. J. Buccafusco, J. Pharmacol. Exp. Ther., 2003, 306, 821 CrossRef CAS PubMed.
- C. P. Ferri, M. Prince, C. Brayne, M. Ganguli, K. Hall, K. Hasegawa, H. Hendrie, Y. Huang, A. Jorm, C. Mathers, P. R. Menezes, E. Rimmer and M. Scazufca, Lancet, 2005, 366, 2112 CrossRef.
- A. Sinha, R. S. Tamboli, B. Seth, A. M. Kanhed, S. Kant Tiwari, S. Agarwal, S. Nair, R. Giridhar, R. K. Chaturvedi and M. R. Yadav, Mol. Neurobiol., 2015, 52, 638 CrossRef CAS.
- L. M. Lima and E. J. Barreiro, Curr. Med. Chem., 2005, 12, 23 CrossRef CAS.
- C. Viegas-Junior, A. Danuello, V. d. S. Bolzani, E. J. Barreiro and C. A. Fraga, Curr. Med. Chem., 2007, 14, 1829 CrossRef CAS.
- G. L. Ellman, K. D. Courtney, V. Andres and R. M. Featherstone, Biochem. Pharmacol., 1961, 7, 88 CrossRef CAS.
- H. Sugimoto, Y. Iimura, Y. Yamanishi and K. Yamatsu, J. Med. Chem., 1995, 24, 4821 CrossRef.
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 267, 727 CrossRef CAS PubMed.
- E. H. Rydberg, B. Brumshtein, H. M. Greenblatt, D. M. Wong, D. Shaya, L. D. Williams, P. R. Carlier, Y. P. Pang, I. Silman and J. L. Sussman, J. Med. Chem., 2006, 49, 5491 CrossRef CAS PubMed.
- G. A. de Souza, S. J. da Silva, C. N. Del Cistia, P. Pitasse-Santos, L. O. Pires, Y. M. Passos, Y. Cordeiro, C. M. Cardoso, R. N. Castro, C. M. R. Sant'Anna and A. E. Kümmerle, J. Enzyme Inhib. Med. Chem., 2019, 34, 631 CrossRef PubMed.
- D. Antoine, M. Olivier and Z. Vincent, Sci. Rep., 2017, 7, 42717 CrossRef.
- D. Antoine and Z. Vincent, ChemMedChem, 2016, 11, 1117 CrossRef PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3 CrossRef CAS PubMed.
- D. F. Veber, R. J. Stephen, C. Hung-Yuan, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615 CrossRef CAS PubMed.
- W. J. Egan, K. M. Merz and J. J. Baldwin, J. Med. Chem., 2000, 43, 3867 CrossRef CAS PubMed.
- L. Pezzementi, F. Nachon and A. Chatonnet, PLoS One, 2011, 6, e17396 CrossRef CAS PubMed.
- M. L. Raves, K. Giles, J. D. Schrag, M. F. Schmid, G. N. Phillips Jr, W. Chiu, A. J. Howard, I. Silman and J. L. Sussman, Quaternary Structure of Tetrameric Acetylcholinesterase, in Structure and Function of Cholinesterases and Related Proteins, ed. B. P. Doctor, P. Taylor, D. M. Quinn, R. L. Rotundo and M. K. Gentry, Springer, Boston, MA, 1998 Search PubMed.
- Y. Bourne, J. Grassi, P. E. Bougis and P. Marchot, J. Biol. Chem., 1999, 274, 30370 CrossRef CAS PubMed.
- M. Biasini, S. Bienert, A. Waterhouse, K. Arnold, G. Studer, T. Schmidt, F. Kiefer, T. G. Cassarino, M. Bertoni, L. Bordoli and T. Schwede, Nucleic Acids Res., 2014, 42, W252 CrossRef CAS PubMed.
- B. Brus, U. Kosak, S. Turk, A. Pislar, N. Coquelle, J. Kos, J. Stojan, J. P. Colletier and S. Gobec, J. Med. Chem., 2014, 57, 8167 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04105b |
| This journal is © The Royal Society of Chemistry 2019 |