
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A comprehensive study of conditions of the biodegradation of a plastic additive 2,6-di-tert-butylphenol and proteomic changes in the degrader Pseudomonas aeruginosa san ai†
Ana Medić a,
Ksenija Stojanovićb,
Lidija Izrael-Živkovića,
Vladimir Beškoskib,
Branka Lončarevićc,
Saša Kazazićd and
Ivanka Karadžić*a
a,
Ksenija Stojanovićb,
Lidija Izrael-Živkovića,
Vladimir Beškoskib,
Branka Lončarevićc,
Saša Kazazićd and
Ivanka Karadžić*a
aDepartment of Chemistry, Faculty of Medicine, University of Belgrade, Višegradska 26, 11000 Belgrade, Serbia. E-mail: ivanka.karadzic@med.bg.ac.rs; Tel: +381113607067
bFaculty of Chemistry, University of Belgrade, Studentski trg 12-16, 11000 Belgrade, Serbia
cInstitute of Chemistry, Technology and Metallurgy, Department of Chemistry, Njegoševa 12, 11000 Belgrade, Serbia
dRuđer Bošković Institute, Bijenička cesta 54, Zagreb, Croatia
First published on 30th July 2019
Abstract
The Pseudomonas aeruginosa san ai strain was investigated for its capability to degrade the 2,6-di-tert-butylphenol (2,6-DTBP) plastic additive, a hazardous and toxic substance for aquatic life. This investigation was performed under different parameter values: 2,6-DTBP concentration, inoculum size, pH, and temperature. The GC-MS study showed that P. aeruginosa efficiently degraded 2,6-DTBP in the pH range of 5–8 at higher temperatures. Under exposure to 2,6-DTBP concentrations of 2, 10, and 100 mg L−1, the strain degraded by 100, 100, and 85%, respectively, for 7 days. Crude enzyme preparation from the biomass of P. aeruginosa san ai showed higher efficiency in 2,6-DTBP removal than that shown by whole microbial cells. Gene encoding for the enzymes involved in the degradation of aromatic compounds in P. aeruginosa san ai was identified. To complement the genomic data, a comparative proteomic study of P. aeruginosa san ai grown on 2,6-DTBP or sunflower oil was conducted by means of nanoLC-MS/MS. The presence of aromatic substances resulted in the upregulation of aromatic ring cleavage enzymes, whose activity was confirmed by enzymatic tests; therefore, it could be concluded that 2,6-DTBP might be degraded by ortho-ring cleavage. A comparative proteomics study of P. aeruginosa san ai indicated that the core molecular responses to aromatic substances can be summarized as the upregulation of proteins responsible for amino acid metabolism with emphasized glutamate metabolism and energy production with upregulated enzymes of glyoxylate bypass. P. aeruginosa san ai has a high capacity to efficiently degrade aromatic compounds, and therefore its whole cells or enzymes could be used in the treatment of contaminated areas.
1. Introduction
The plastic additive 2,6-di-tert-butylphenol (2,6-DTBP) is an industrially important chemical that has been used as an antioxidant in plastics, as well as an oxidation inhibitor and stabilizer for fuel, oil, and gasoline.1,2 Antioxidants slow down the oxidation cycle and prevent the loss of strength, breakdown, or discoloration of plastics. Moreover, 2,6-DTBP is used as a synthetic intermediate for the production of higher-molecular-weight phenolic compounds. Due to the extensive use of 2,6-DTBP, the possible routes for its release into the natural environment, particularly in surface water, are manufacturing processes and leaching from the final products. Because of concerns regarding the widespread use of microplastics and their effects on the aquatic environments and their biota,3 2,6-DTBP could become a serious issue for aquatic animals; this is because 2,6-DTBP has shown highly acute toxicity to crustacean (Gammarus fasciatus), fathead minnow (Pimephales promelas), water flea (Daphnia magna), and zebrafish (Brachydanio rerio) with LC50 values of 0.80, 1.1, 0.45, and 10 mg L−1, respectively.1 In the case of human exposure, skin and eye irritations have been reported.1As chemical decomposition is usually complex and uneconomical, biodegradation is considered to be a promising alternative to remove 2,6-DTBP from polluted areas. The degradation of environmental pollutants—particularly organic compounds (hydrocarbons, including polycyclic aromatic hydrocarbons (PAHs)), phenols, and its derivatives—by certain naturally occurring microorganisms have been considered to be a safe, efficient, cost-effective, and environmentally friendly technology for treating hazardous substances.4,5 However, the biological remediation of aromatic substances is particularly challenging as aromatic rings are highly stable against microbial degradation. Although microorganisms that can degrade, transform, or accumulate toxic organic compounds are not widely distributed in nature, a certain number of bacterial species (e.g., Bacillus stearothermophilus, Bacillus laterosporus, Alcaligenes eutrophus, Rhodococcus erythropolis, and Rhodococcus sp., particularly genus Pseudomonas) have been described to degrade a wide variety of aromatic compounds, although at lower pollutant concentrations in most cases.6 The natural abundance of microorganisms with multidegradative capacity, which may play an important role in the removal of heterogeneous contamination, is well reviewed.7 The Pseudomonas genus is known for its ability to utilize diverse aromatic compounds as the sole C source.8,9 The degradation and utilization of alkylphenols have been reported for several Pseudomonas strains, including Pseudomonas sp. KL28,10 Pseudomonas putida MT4,11 and Pseudomonas veronii INA06.12 Only two Pseudomonas strains, namely, Pseudomonas azelaica13 and Pseudomonas sp. MS-1, have been reported to degrade ortho-monosubstituted alkylphenols,14 whereas Pseudomonas putida TX2 and Pseudomonas sp. TX-1 can degrade para-monosubstituted alkylphenols.15 There is only one publication on the biodegradation of ortho-disubstituted alkylphenols.16
Recent advances in high-throughput genomics and proteomics techniques have opened up new opportunities for understanding the molecular mechanism of biodegradation.17 A specific cellular response during growth on different C sources, including organic pollutants, affects the proteome of the microorganism. An insight into such mechanisms associated with organic pollutant removal, as well as the dynamics of synthesis of certain proteins, can address the gap involving the fundamental understanding of the molecular mechanisms of the adaptation of microorganism, strategies to better control production of its worthwhile metabolites, and apply them toward environmental protection. Simultaneously, differential up- or downregulation of proteins/enzymes under the distinct conditions of growth could serve toward the environmental biomonitoring of stress induced by pollutants in the natural microbial communities and for biomarker measurement. Until now, published data have revealed considerable changes in microbial proteomes during aromatics degradation in comparison to those in natural C sources.18–25 Recently, a global proteomics approach was successfully used to explore phenanthrene catabolic pathways in Arthrobacter phenanthrenovorans exposed to phenanthrene,20 implying an overproduction of enzymes of degradation and their strong downregulation when the culture is grown on aromatic and natural substrates, respectively. Further, global, ge1-based 1-DE in combination with tandem MS proteomics approach was considered to be a sensitive method for mapping the enzymes involved in the phenol degradation of pseudomonad, an indigenous soil.25 No proteomics studies of microbial response toward alkylphenols have been conducted so far.
The β-ketoadipate pathway is an aryl-ring degradation pathway for the conversion of hazardous aromatic pollutants into nontoxic metabolites, which are widely distributed in bacteria (including Pseudomonas).5 This pathway mediates the ortho-cleavage of dihydroxy-substituted arenes through two distinct branches—catechol and protocatechuate. Most of the published strains degrade alkylphenols via alkylcatechols using the ortho- or meta-cleavage of the aromatic ring.14,15 The biodegradation of the ortho-disubstituted derivatives of alkylphenols has not been studied in detail yet.
Pseudomonas aeruginosa, well known for its ability to grow in diverse environments and with different C sources, has shown promising potential in the remediation of sites polluted by hardly biodegradable organic chemicals.26 The main objective of this study involves the estimation of the capability of P. aeruginosa san ai, a culturable environmental isolate from mineral cutting oil,27 to degrade the ortho-disubstituted alkylphenol—2,6-DTBP—under different parameters, namely, 2,6-DTBP concentration, inoculum size, pH, and temperature. The successful identification of catabolic pathways using the proteomic approach20,25 has encouraged us to perform comparative proteomics to explore the biodegradation pathway along with the metabolic adaptation of the microorganism grown on 2,6-DTBP and sunflower oil as the sole C source. The comparative proteomics approach based on nanoliquid chromatography and tandem mass spectrometry (nanoLC-MS/MS) coupled with bioinformatics was applied to identify proteins. The proteomic results of the biodegradation of aromatic compound by P. aeruginosa san ai were validated by the enzymic approach.
2. Material and methods
Chemicals
2,6-DTBP and the organic solvents were purchased from Sigma Aldrich (St. Louis, MO, USA) with purity above 99% and of the HPLC grade. All the other chemicals used in this study were of the pro-analysis grade purchased from Merck (Darmstadt, Germany).Media and growth conditions
The microorganisms used for 2,6-DTBP degradation were Pseudomonas aeruginosa san ai, a strain isolated from industrial mineral metal-cutting oil.27 The strain was deposited in National Collection of Agricultural and Industrial Microorganisms (NCAIM), Faculty of Food Sciences, Corvinus University of Budapest, Budapest, Hungary, labeled as NCAIM B.001380 and ISS WDCM 375 (Institute of Soil Science, Belgrade, Serbia) in the public collection with accession number ISS 619.P. aeruginosa san ai was activated on nutrient agar (Torlak, Belgrade, Serbia) at 30 °C for 24 h and transferred to a 500 mL Erlenmeyer flask, containing 100 mL of minimal salt medium (MSM) supplemented with different sources of carbon and energy, to achieve an initial colony-forming unit (CFU) value of approximately 4 × 107. 2,6-DTBP, sodium benzoate, and sunflower oil were used as the C sources. Moreover, the strain was grown in a complex Luria–Bertani (LB) broth. MSM comprised the following components (per liter): 5 mL phosphate-buffered solution (PBS; KH2PO4 8.5 g L−1, K2HPO4 19.71 g L−1, Na2HPO4·2H2O 16.61 g L−1, and NH4Cl 5 g L−1); 3 mL MgSO4·7H2O solution (46.125 g L−1); 1 mL FeCl3 solution (0.25 g L−1); 1 mL CaCl2·2H2O solution (48.21 g L−1); 1 mL trace element solution (MnSO4·4H2O 0.05 g L−1, (NH4)6Mo7O24·4H2O 0.0347 g L−1, and ZnSO4·7H2O 0.07 g L−1)28 was sterilized by a Millipore filtration system (Merck Millipore, USA) using a membrane filter (0.45 μm). The pH of this medium was maintained between 7.2 and 7.4. An appropriate volume of the stock solution of 2,6-DTBP in n-hexane was injected into sterile 500 mL Erlenmeyer flasks to obtain the desired final concentration and then hexane was allowed to evaporate under the airflow. After forming a thin film of 2,6-DTBP at the bottom of the flask, 100 mL sterilized MSM was added to each flask.29 Sodium benzoate as the C source was dissolved in water and added to MSM. The flasks were incubated at 30 °C for 7 days and shaken at 150 cycles per min using a horizontal shaker (Kuhner, Basel, Switzerland). MSM with appropriate concentrations of C sources without P. aeruginosa san ai was used as the control for nonbiological degradation.
Bacterial growth was monitored as a change in the optical density (OD) at 580 nm, measured on an ultraviolet-visible spectrophotometer (UV-2600, Shimadzu Kyoto, Japan) using a sterile non-inoculated medium as the reference.30 The bacterial concentration was estimated to be 3 × 108 cells per mL, which corresponded to an absorbance value of 0.47 at 580 nm.
Effects of initial concentration of 2,6-DTBP, pH, temperature, and inoculum size on biodegradation
These effects were examined using MSM (pH 7.2) supplemented with 100 mg L−1 2,6-DTBP inoculated with 4 × 107 CFU per mL and shaken at 150 cycles per min using a horizontal shaker at 30 °C for 7 days, unless otherwise indicated. A sterile medium with 2,6-DTBP as the control of chemical decomposition and inoculated sterile MSM without 2,6-DTBP as the control of microbial growth were designed. All the experiments were performed in three independent replicates and the average values are given in this study. The differences in the results between the replicates in all the experiments did not exceed 1.5%.Several concentrations of 2,6-DTBP, namely, 2, 10, 100, and 400 mg L−1, were added to MSM to investigate the effects of initial concentration.
The effect of pH on the biodegradation efficiency was assessed by individually modifying the pH of MSM to 5, 6, 7, and 8. The pH was adjusted before membrane filtration using HCl or NaOH solutions at a concentration of 0.1 mol L−1.
The influence of temperature on 2,6-DTBP biodegradation was investigated at four different values: 22, 30, 37, and 43 °C.
To study the effect of inoculum size, cell suspension of P. aeruginosa san ai was added to the medium to achieve initial CFU per mL values of 2.6 × 107, 8.3 × 107, and 1.4 × 108.
Microbial adherence to n-hexadecane (MATH) test
Cell hydrophobicity was evaluated via the MATH test.31 Two milliliters of bacterial suspension in distilled water at OD580 = 0.5 was overlaid with 0.5, 1, and 1.5% (v/v) of n-hexadecane. After vortexing for 1 min, the phases were permitted to separate for 15 min. The absorbance at 580 nm of the aqueous phase was then measured. The results were calculated using the following equation: [(A0 − A)/A0] × 100, where A0 and A represent the initial and final ODs of the aqueous phase, respectively.Gas chromatography-mass spectrometry (GC-MS) analysis of 2,6-DTBP and its degradation products
The entire flask culture was extracted by n-hexane for three times. The extracts were dehydrated with anhydrous Na2SO4 and the solvents were evaporated under reduced pressure by a rotary evaporator (Senco GG17, Shanghai, China). The extraction residues were dissolved in n-hexane and analyzed by GC-MS. The gas chromatograph Agilent 7890A GC (HP5-MS capillary column; 30 m × 0.25 mm; film thickness: 0.25 μm; He carrier gas: 1.5 cm3 min−1) coupled to an Agilent 5975C mass selective detector (70 eV) was used. The column was heated from 80 to 300 °C at a rate of 2 °C min−1 and maintained at 300 °C for 20 min; thereafter, it was heated from 300 to 310 °C at a rate of 10 °C min−1 and maintained at 310 °C for 1 min. The injector temperature was 250 °C. The spectrometer was operated in the electrospray ionization (EI) mode over a scan range from m/z 45 to 550. The individual peaks of 2,6-DTBP and its degradation products were identified on the basis of the mass spectra (library: NIST11). The quantification of the compounds in the standard series, controls (sterile medium with 2,6-DTBP), and inoculated samples (sterile medium supplemented with 2,6-DTBP and P. aeruginosa san ai) were performed by an integration of the peak areas (software: GCMS Data Analysis). For the calculations, a standard series of commercial 2,6-DTBP solutions in n-hexane covering the range of 2,6-DTBP concentrations from 2 to 400 mg L−1 (2, 5, 10, 50, 100, 200, and 400 mg L−1), as used in the study, were prepared and analyzed under the same GC-MS conditions as those for the samples. The ratio of the peak areas and the corresponding concentrations were linearly dependent with very high correlation (r2 = 0.994). Moreover, in order to verify the stability of 2,6-DTBP, two identical standard series, as mentioned above, were prepared and analyzed by GC-MS. The first standard series was analyzed after preparation, and the second one after 7 days from preparation (the time of experiments); they were exposed to the same temperature (30 °C) during this period. The obtained results from the GC-MS analysis showed no change in 2,6-DTBP concentrations in the solutions of the standard series after 7 days. Furthermore, a comparison of the GC-MS results of the standard series and control samples, having the same concentration of 2,6-DTBP, indicated that the losses of the target compound in the controls were less than 2%, confirming the accuracy of the experiments. The efficiency of degradation is determined based on the concentrations of the target compound in the control and inoculated samples, and it is expressed in percentage.Crude enzyme extract preparation
Total proteins were isolated from the biomass grown in MSM supplemented with different C sources in the early stationary phase. Biomass was collected by centrifugation, frozen, and homogenized in two volumes of buffer A (50 mM Tris buffer at pH 7.5 supplemented with 0.1 mM phenylmethylsulfonyl fluoride (PMSF) and 0.5 mM 1,4-dithiothreitol (DTT)) in a glass Teflon homogenizer at 4 °C. The resulting homogenates were ultracentrifuged for 2 h at 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g at 4 °C using Beckman Coulter L5-65 with SW 28 rotor (Beckman, Indianapolis, USA). The supernatants were used for proteomics and enzyme assays after the determination of protein content by the Bradford's method.32
000g at 4 °C using Beckman Coulter L5-65 with SW 28 rotor (Beckman, Indianapolis, USA). The supernatants were used for proteomics and enzyme assays after the determination of protein content by the Bradford's method.32
Proteomic analysis
Crude protein extracts obtained from the biomass grown on MSM supplemented with 100 mg L−1 2,6-DTBP or 100 μL L−1 sunflower oil were used for proteomics analysis. The crude protein extract was separated by sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis (PAGE) using 4–15% gradient acrylamide gels (Criterion™ TGX™ Precast Gels, Bio-Rad) according to the manufacturer's instructions. Staining was carried out with Coomassie Brilliant Blue R-250. Twelve successive bands from the gel were cut and washed with 50 mM ammonium bicarbonate and 100% acetonitrile (ACN) for 15 min on the thermomixer (Digital Shaking Drybath, Thermo Fisher Scientific, USA). After volume reduction by a vacuum concentrator (Eppendorf Concentrator 5301, Hamburg, Germany), proteins were digested by trypsin (Promega, Madison, USA) dissolved in 50 mM ammonium bicarbonate to the final concentration of 0.02 μg μL−1. Digestion was performed overnight at room temperature on the thermomixer. The eluate was saved and gel-washed with 5% ACN/0.1% formic acid (FA) and 50% ACN/0.1% FA for 15 min on the thermomixer. The washes were combined and concentrated to near dryness in the vacuum concentrator.Peptide digests were analyzed by EI in the positive mode on an ion-trap instrument called Amazon Speed (Bruker, Bremen, Germany) using a captive spray source. Two analytical replicates of every sample were conducted. Peptides were separated by nanoflow HPLC (NanoAdvance, Bruker, Bremen, Germany). UHPLC Nanotrap (i.d.: 100 μm; length: 25 mm) packed with 200 A C18 stationary phase (5 μm, C18AQ, Michrom) was used for peptide trapping. Analytical columns (100 μm × 100 mm long) packed with 130 A C18 stationary phase (1.7 μm, ACQUITY, UPLC, M-Class, Waters) were coupled to the mass spectrometer (MS). Peptide mixtures obtained after tryptic digestion were applied to the precolumn at a flow rate of 5 μL min−1 in 2% (v/v) ACN with 0.1% (v/v) FA. The peptides were eluted by a linear gradient of A (water, 0.1% FA) and B (ACN, 0.1% FA) as follows. 0 min: A (98%), B (2%); 50 min: A (5%), B (95%); 50–55 min: A (5%) and B (95%) at a flow rate of 400 nL min−1. The ion source conditions were optimized with a calibration solution according to the instrument provider's instructions. All the MS survey scans were performed from m/z 400 to 1400 with enhanced resolution. The data analysis was undertaken by the selection of the five most abundant precursors rejecting singly charged ions.
The tandem mass spectral data were automatically acquired and processed using Hystar 3.2 and Data Analysis 4.2 software (Bruker, Germany). The deconvoluted MGF spectra were searched by using the Mascot database search tool (version 2.3.02). The mascot search parameters were set as follows. Taxonomy: other proteobacteria; enzyme: trypsin; missed cleavages: 2; modifications, cysteine carbamidomethylation and methionine oxidation (variable); precursor mass tolerance: 1.2 Da; MS/MS mass tolerance: 0.6 Da; ions with monoisotopic m/z value and +2 and +3 charge states. Only the top hits in the compounds' list with a significance threshold of p < 0.01 were considered.
Enzyme assays
The activities of catechol 1,2-dioxygenase (C12O) and catechol 2,3-dioxygenase (C23O) were spectrophotometrically measured at 260 and 375 nm, respectively, using UV-2600. The formation of 2-hydroxymuconic semialdehyde, cis,cis-muconic acid, and their derivatives was measured by slightly modifying the procedures proposed by Briganti et al.33 and Mahiudddin et al.34 The reaction mixture for C12O activity determination comprised the following: 1 mL of 50 mM PBS at pH 8.0, 0.1 mL of 100 mM DTT, and 0.1 mL of cell homogenate obtained from biomass grown on 2,6-DTBP or sodium benzoate. The reaction was started by the addition of 0.1 mL of 1 mM substrate (catechol, 2,6-DTBP, and sodium benzoate). The reaction mixture for C23O activity determination comprised the following: 1 mL of 100 mM PBS at pH 8.0, 0.1 mL of cell homogenate, and 0.1 mL of 100 mM substrate (catechol, 2,6-DTBP, and sodium benzoate). One unit of activity was defined as the amount of enzymes producing 1 μmol each of cis,cis-muconate (ε260 = 1.6 × 104 mol−1 cm−1),34 2-hydroxymuconic acid (ε290 = 1.2 × 104 mol−1 cm−1),35 and 2-hydroxymuconic semialdehyde (ε375 = 4.4 × 104 mol−1 cm−1)25 per min at 25 °C. The specific activity is calculated as the enzyme activity per mg of protein.The oxygenolytic activity of the homogenate was measured by amperometrically recording the O2 consumption by a Clark electrode. The experiment was performed at 25 °C in a 5 mL chamber containing 4 mL of 100 mM PBS at pH 8.0, 0.4 mL substrate (2,6-DTBP), and 0.4 mL crude protein extract.36,37
The activity of catalase (CAT) was assayed by the measurement of H2O2 substrate remaining after the action of CAT.38 One unit (U) of CAT activity was defined as 1 μmol of H2O2 degraded into O2 and H2O per min of reaction. The specific activity is calculated as the enzyme activity per mg of protein.
The activity of superoxide dismutase (SOD) was determined according to the method based on the oxidation of adrenalin previously described by Sun and Zigman.39 The change in absorbance was determined at 340 nm. One unit (U) of SOD activity is the amount of enzyme that causes a change in absorbance (ΔA/min) of 0.001 under the test conditions. The specific activity was calculated as the enzyme activity per mg of protein.
Enzymatic degradation
Crude protein extracts obtained from biomass grown in MSM with 2,6-DTBP for 48 h were used to investigate the enzymatic degradation of 2,6-DTBP. The experiment was performed in lightproof 500 mL bottles (Duran, Wertheim, Germany) containing 50 mL of 100 mM PBS at pH 8.0, supplemented with 4 mL substrate (2,6-DTBP) at a concentration of 10 mg L−1, and 4 mL of the crude protein extracts; the mixture was constantly stirred (150 rpm) with a magnetic stirrer (Heidolph, Schwabach, Germany) at 29 + 2 °C for 4 days. The remaining amount of 2,6-DTBP was estimated by the GC-MS analysis, as described. Two controls were designed: crude extracts without substance and only substance without cell extracts in PBS at pH 8.0.Respiration analysis
The respiration activity of P. aeruginosa san ai exposed to 2,6-DTBP in MSM was measured using a twelve-channel Micro-Oxymax® respirometer (Columbus Instruments, Columbus, USA) connected to a PC. The experiments were performed in Micro-Oxymax lightproof 500 mL bottles (Duran, Wertheim, Germany) containing 100 mL MSM supplemented with different concentrations of 2,6-DTBP and constantly stirred (150 rpm) with a magnetic stirrer (Heidolph, Schwabach, Germany) at different temperatures for 4 days. The respiration rates (μL min−1) as well as the cumulative O2 consumed and CO2 produced (μL) were determined. Cell respiration was measured every 300 min for 4 days. A sterile medium with 2,6-DTBP and inoculated sterile MSM without 2,6-DTBP were used as the controls. All the experiments were performed in triplicate. The obtained data were evaluated by Micro-Oxymax software.3. Results and discussion
As 2,6-DTBP is particularly harmful to aquatic life, all the experiments of biodegradation by P. aeruginosa san ai were done in MSM, which mimics an aquatic environment, as the growth medium. The direct correlation between the microbial surface hydrophobicity and its potential to degrade nonpolar organic polymers has already been reported;40 the cell surface of P. aeruginosa san ai was preliminarily determined. The degree of cell hydrophobicity of 29% was obtained by the MATH assay, which indicated moderate hydrophobicity41 of P. aeruginosa san ai and suggested its potential toward the biodegradation of different organic compounds. Following this idea, the degradation effects of several aliphatic and aromatic hydrocarbons (n-hexadecane, n-nonadecane, fluorene, phenanthrene, and pyrene) by P. aeruginosa san ai were tested. Interestingly, in chromatograms of the investigated substances, a peak corresponding to 2,6-DTBP, originating from the plastic used in this experiment, was noticed; surprisingly, this peak disappeared relatively rapidly during microbial growth (Fig. 1), which led us to the idea that the microorganism preferred to use 2,6-DTBP as the C source. Indeed, our preliminary experiments clearly demonstrated that P. aeruginosa san ai could remove 2 mg L−1 of 2,6-DTBP within 7 days. The effects of various conditions including initial 2,6-DTBP concentration, pH, and temperature on the degradative capabilities of P. aeruginosa san ai were further investigated.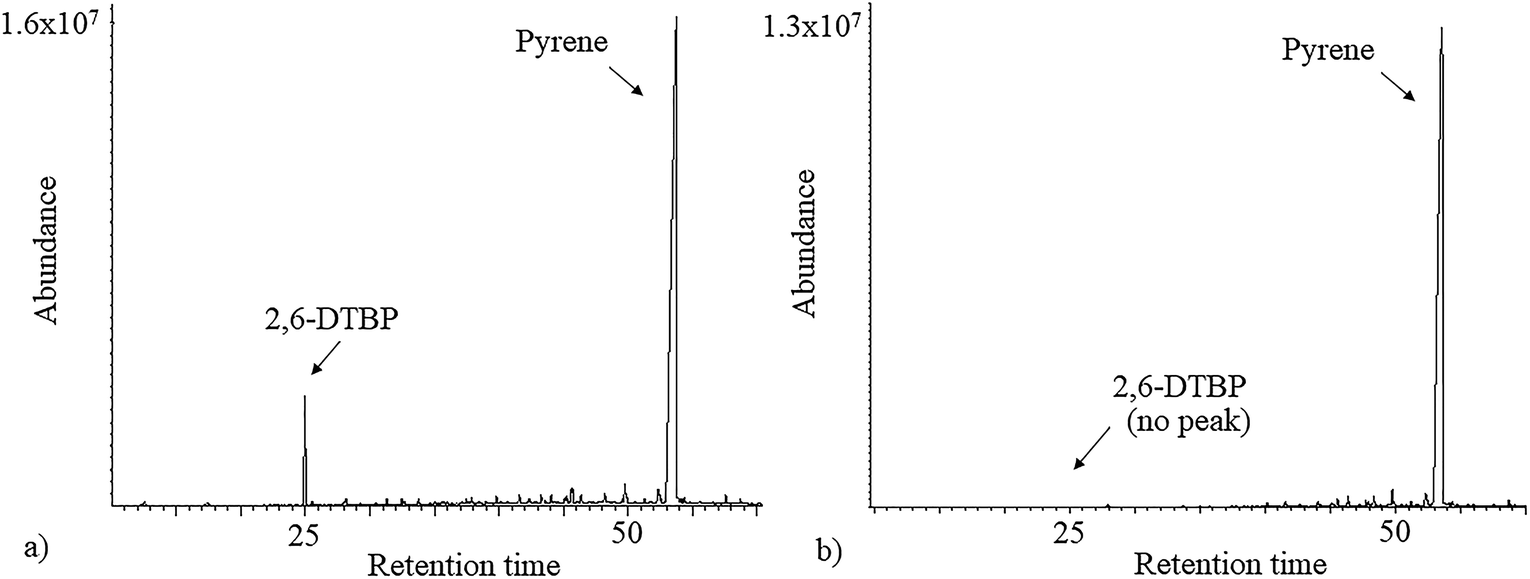 | ||
| Fig. 1 Total ion chromatogram (TIC) of n-hexane extracts of (a) control and (b) inoculated culture broth supplemented with pyrene. | ||
Biodegradation efficiency of P. aeruginosa san ai
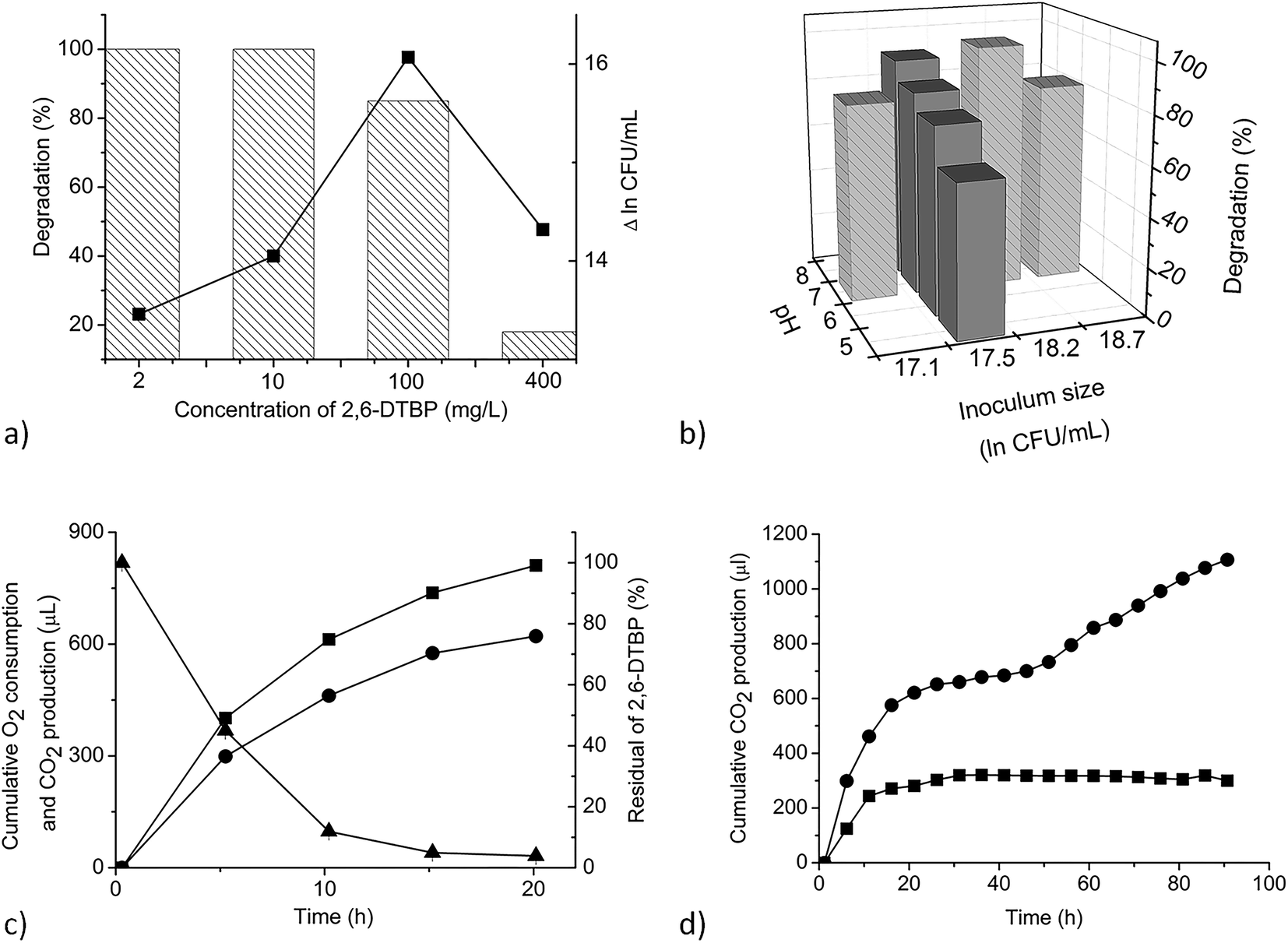 | ||
| Fig. 2 Effects of different factors and dynamics of 2,6-DTBP degradation. (a) Effect of initial substrate and intensity of growth of P. aeruginosa san ai. Efficiency of degradation was measured after 7 days. Biomass increase in the early stationary phase when the culture reached the maximum growth is shown (square) (Δ lnCFU per mL represents the difference between CFU per mL in the early stationary phase and that at time zero). (b) Effects of inoculum size and pH (C2,6-DTBP = 100 mg L−1). Dark gray histograms indicate the effect of pH on degradation efficiency. Light gray histograms with textures show the effect of inoculum size on degradation efficiency. (c) Dynamics of the degradation of 2,6-DTBP (concentration: 4 mg L−1) (triangle), cumulative O2 consumption (square), and cumulative CO2 production (circle) of P. aeruginosa san ai grown at 30 °C (controls were subtracted). (d) Cumulative CO2 production during the growth of P. aeruginosa san ai on 2,6-DTBP (concentration: 10 mg L−1) at 25 °C (square) and 35 °C (circle) (controls were subtracted). Each data point represents the mean of three replicate samples. Some error bars are not visible because they are shorter than the symbol size. | ||
Dynamic of 2,6-DTBP biodegradation
The dynamics of 2,6-DTBP biodegradation was monitored during P. aeruginosa san ai growth on 4 mg L−1 2,6-DTBP, a concentration that is completely soluble in water.43 2,6-DTBP was rapidly removed from the medium, intensively consuming O2 and releasing CO2, as shown in Fig. 2c. The most intensive respiration occurred in the first 5 h, with cumulative O2 consumption of 402 μL and cumulative CO2 production of 293 μL. At the same time, 50% 2,6-DTBP as the sole C source was mineralized. Within the first 10 h, more than 90% 2,6-DTBP was degraded. The cumulative production of CO2 reached the maximum after 20 h when complete 2,6-DTBP was exhausted. P. aeruginosa san ai quickly, within 20 h, removed 2,6-DTBP from the solution (concentration: 4 mg L−1), clearly demonstrating promising potential for the aerobic biodegradation of 2,6-DTBP (Fig. 2c). In fact, biodegradation by P. aeruginosa san ai was incomparably more efficient than anaerobic degradation by an activated sludge, where a conversion of 3.5% at the starting concentration of 0.05 mg L−1 2,6-DTBP to CO2 was reported over 5 days.47As biodegradation under different conditions of initial concentrations, pH, inoculums size, and temperature revealed the promising potential of P. aeruginosa san ai to degrade 2,6-DTBP, we further endeavored to study the genome and changes in the proteome of the strain, as well as to elucidate the metabolic pathways that contribute toward the survival under exposure to 2,6-DTBP.
Genomic and proteomic insights into aromatic biodegradation
To explore the 2,6-DTBP catabolic pathways, genomic and proteomic analyses were applied. A collection of genes encoding to the proteins involved in the catabolism of aromatic compounds were identified in P. aeruginosa san ai (Genbank: BioProject PRJNA19571; accession number: JMKR00000000).48The biodegradation of alkylphenols by several Pseudomonas strains indicated that both C12O and C23O had wide substrate specificity for a number of alkylcatechols, which are the initial products of alkylphenol oxidation.15 With respect to these results, the genome of P. aeruginosa san ai was analyzed by KEGG and the presence of gene encoding to proteins involved in the catabolism of aromatic compounds (Fig. 3a) was detected in the K260DRAFT_scf7180000000062_quiver.15 and K260DRAFT_scf7180000000064_quiver.13 scaffolds. In particular, all the gene coding for proteins involved in the β-ketoadipate ortho-degradation pathway—catABC of catechol branch and pcaBCDG of protocatechuate branch—have been identified (Fig. 3a). Genomic analysis clearly indicated the potential of P. aeruginosa san ai for the degradation of aromatics through the ortho-pathway. To validate the genomic data, we used comparative proteomics to identify the proteins involved in the catabolism of 2,6-DTBP by P. aeruginosa san ai when grown on 2,6-DTBP versus sunflower oil as the C source.
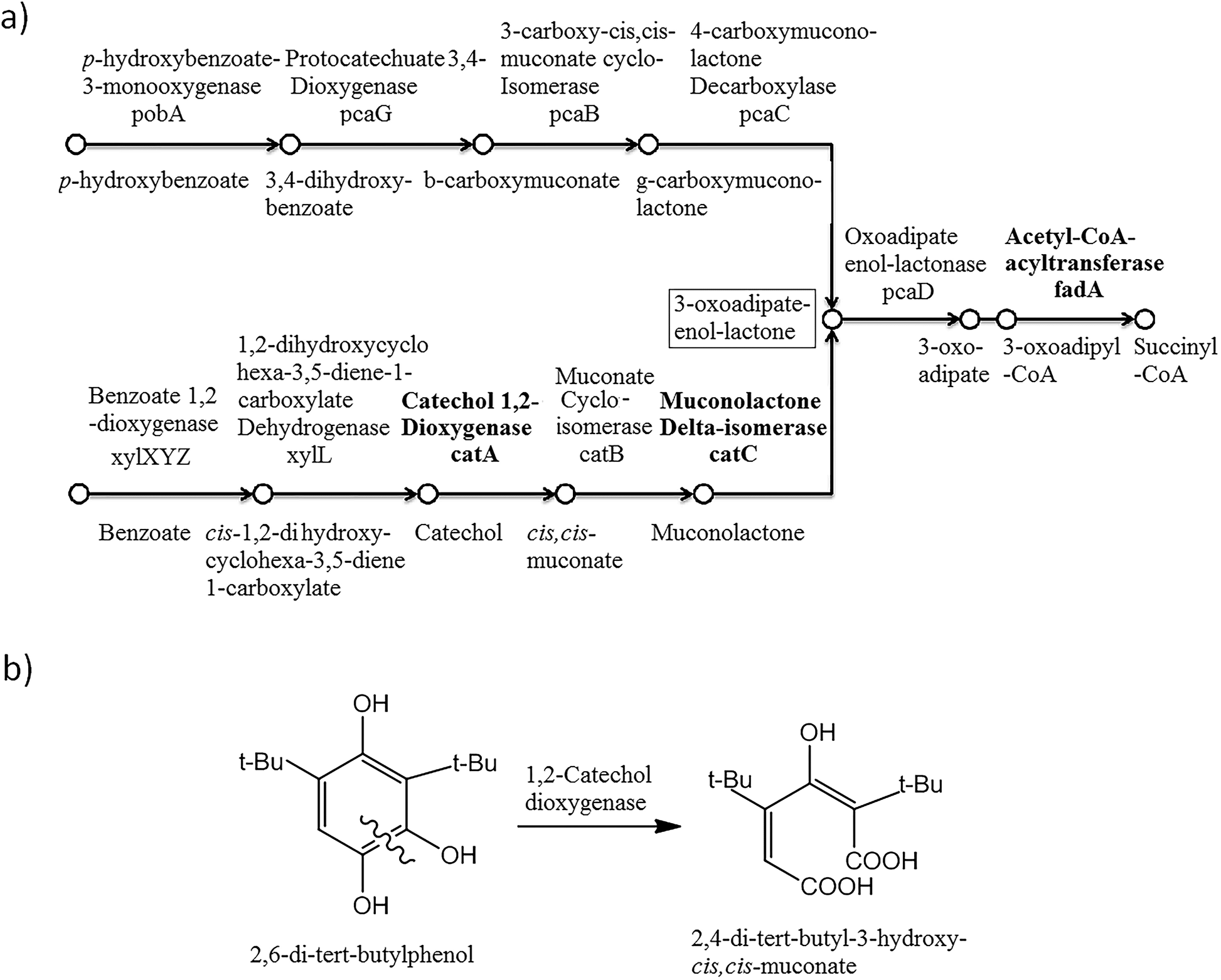 | ||
| Fig. 3 β-ketoadipate pathway in P. aeruginosa san ai. Compounds, genes, and enzymes are indicated. (a) Gene and coding protein, respectively, are as follows. pobA: p-hydroxybenzoate-3-monooxygenase; pcaG: protocatechuate 3,4-dioxygenase; pcaB: 3-carboxy-cis,cis-muconate cycloisomerase; pcaC: 4-carboxymuconolactone-decarboxylase; pcaD: oxoadipate-enol-lactonase; catA: catechol 1,2-dioxygenase; catC: muconolactone δ-isomerase; catB: muconate cycloisomerase; xylL: 1,2-dihydroxycyclohexa-3,5-diene-1-carboxylate dehydrogenase; xylXYZ: benzoate/toluate 1,2-dioxygenase. (b) Proposed ortho-degradation of 2,6-DTBP with 2,4-di-tert-butyl-3-hydroxy-cis,cis-muconic acid as the degradation product. | ||
Proteins from cultures grown on two different substrates, namely, 2,6-DTBP versus sunflower oil, were separated by SDS PAGE (Fig. 4a), digested by trypsin, and analyzed using nanoLC-MS/MS. A list of differentially regulated proteins identified in 2,6-DTBP- versus oil-supplemented media during the growth of P. aeruginosa san ai is provided in Table 1. A total of 86 proteins were identified, out of which 43 and 29 were uniquely identified in 2,6-DTBP- and oil-amended cultures, respectively.
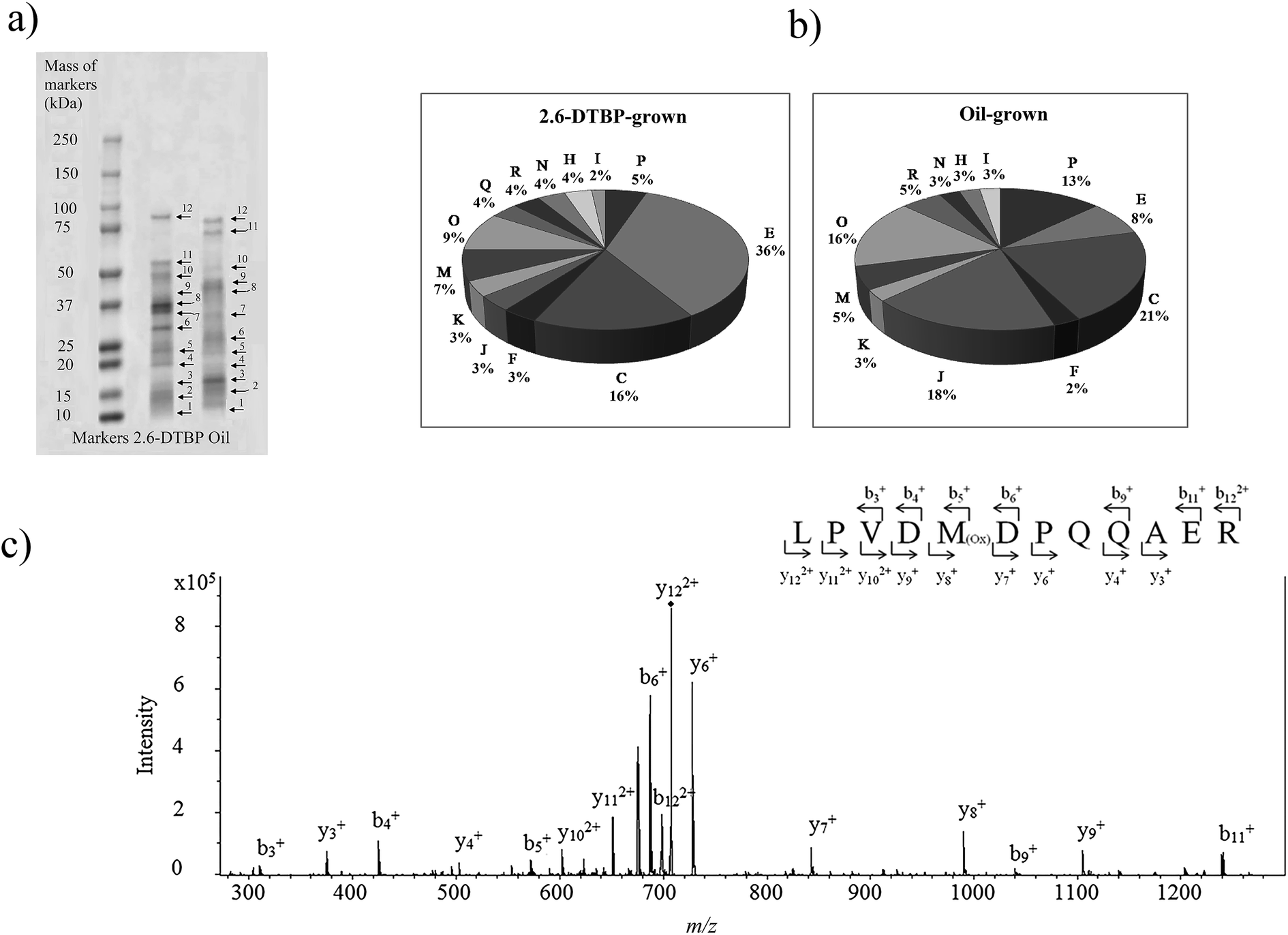 | ||
| Fig. 4 (a) SDS-PAGE electrophoresis of P. aeruginosa san ai proteins with the corresponding bonds (spot no. 1–12 for nanoLC-MS/MS analysis). (b) Functional classification of the proteins identified from P. aeruginosa san ai grown in MSM supplemented with 2,6-DTBP and oil. Metabolic categories. COG: C energy production; E: amino acid metabolism; I: lipid metabolism; J: translation; H: coenzyme metabolism; K: transcription; M: cell wall biogenesis; N: cell motility; O: PTM chaperon function; P: inorganic ion transport; R: general function prediction. COG categories: https://www.ncbi.nlm.nih.gov/COG/. (c) Tandem mass spectrum of LPVDMDPQQAER peptide from muconolactone δ-isomerase (m/z = 707.80). Proteins were analyzed by the proteomic method. Muconolactone δ-isomerase was identified by MASCOT search with a score of 90. Specific b and y ions were observed within the measured mass accuracy of 44 ppm. More than 30% of the fragment ions were identified to belong to the assigned peptide. | ||
| Identified protein | Entry name | Mascot score | Peptide matched | Coverage (%) | Molecular mass (kDa) | Spot no | COG | Fold changea |
|---|---|---|---|---|---|---|---|---|
| a Fold change in relative abundance is the ratio of the abundance of proteins between DTBP-grown versus oil-grown cultures from three biological replicates. + are upregulated and – are downregulated proteins. The absence of proteins is identified in oil-grown cultures, but not in DTBP-grown cultures.b The same bond number in both the gels (proteins from oil- and DTBP-grown cells). | ||||||||
| Azurin | AZUR_PSEAE | 1191 | 10 | 60 | 16 | 2b | C | +1.1 |
| Azurin | AZUR_PSEAI | 696 | 12 | 76 | 13.9 | 1 | C | Absence |
| ATP synthase subunit alpha | ATPA_PSEAB | 81 | 4 | 8 | 55.5 | 11 | C | Presence |
| ATP synthase subunit beta | ATPB_PSEAB | 78 | 3 | 9 | 49.5 | 10 | C | −1.2 |
| ATP synthase gamma chain | ATPG_PSEAB | 85 | 2 | 6 | 31.5 | 6 | C | Presence |
| Isocitrate lyase | ACEA_PSEAE | 76 | 2 | 5 | 58.9 | 11 | C | Presence |
| Isocitrate dehydrogenase [NADP] | IDH_PSEAB | 118 | 4 | 10 | 45.6 | 9 | C | Absence |
| Electron transfer flavoprotein subunit alpha | ETFA_PSEAE | 46 | 1 | 3 | 31.4 | 7 | C | Absence |
| Electron transfer flavoprotein subunit beta | ETFB_PSEAB | 65 | 2 | 11 | 26.4 | 6 | C | Absence |
| C4-dicarboxylate-binding periplasmic protein DctP | DCTP_PSEAE | 176 | 6 | 22 | 37 | 9 | C | Presence |
| Succinate-CoA ligase [ADP-forming] subunit alpha | SUCD_PSEAE | 57 | 1 | 4 | 30.2 | 6 | C | Absence |
| Dihydrolipoyl dehydrogenase | DLDH1_PSEAE | 190 | 6 | 17 | 48.6 | 10 | C | −1.2 |
| Glycerol kinase 2 | GLPK2_PSEAE | 185 | 6 | 14 | 55.9 | 11 | C | Presence |
| Cytochrome c4 | CYC4_PSEAE | 132 | 2 | 15 | 20.7 | 4 | C | Presence |
| Glutarate-semialdehyde dehydrogenase DavD | DAVD_PSEAE | 76 | 2 | 6 | 51.6 | 10 | E | Presence |
| Glycine dehydrogenase | GCSP1_PSEAE | 702 | 11 | 12 | 103.9 | 12 | E | Presence |
| Glycine cleavage system H protein 1 | GCSH1_PSEAE | 48 | 1 | 7 | 13.8 | 1 | E | Absence |
| Glutaminase-asparaginase | ASPQ_PSEAE | 177 | 6 | 18 | 38.6 | 8 | E | Presence |
| Putrescine-binding periplasmic protein SpuD | SPUD_PSEAB | 132 | 5 | 16 | 40.6 | 9 | E | +8.1 |
| Spermidine-binding periplasmic protein SpuE | SPUE_PSEAE | 54 | 1 | 5 | 40 | 9 | E | Presence |
| Aromatic-amino-acid aminotransferase | PHHC_PSEAE | 142 | 7 | 22 | 43.2 | 9 | E | Presence |
| Leucine-, isoleucine-, valine-, threonine-, and alanine-binding protein | BRAC_PSEAE | 128 | 2 | 6 | 39.7 | 9 | E | +8.1 |
| Aliphatic amidase | AMIE_PSEAB | 70 | 3 | 10 | 38.5 | 8 | E | Presence |
| Histidine ammonia-lyase | HUTH_PSEAB | 56 | 1 | 2 | 53.7 | 10 | E | Presence |
| Urocanate hydratase | HUTU_PSEA7 | 241 | 3 | 5 | 61.2 | 11 | E | Presence |
| Aspartate aminotransferase | AAT_PSEAE | 80 | 1 | 3 | 43.3 | 9 | E | Presence |
| Branched-chain-amino-acid aminotransferase | ILVE_PSEAE | 161 | 3 | 11 | 34.1 | 7 | E | Presence |
| Ketol-acid reductoisomerase (NADP(+)) | ILVC_PSEAB | 164 | 4 | 17 | 36.4 | 7 | E/H | Presence |
| Arginine deiminase | ARCA_PSEAE | 215 | 5 | 15 | 46.4 | 9 | E | Presence |
| Ornithine carbamoyltransferase, catabolic | OTCC_PSEAE | 420 | 13 | 35 | 38.1 | 8 | E | Presence |
| Argininosuccinate lyase | ARLY_PSEAB | 99 | 4 | 11 | 51.6 | 10 | E | Presence |
| N-Acetyl-gamma-glutamyl-phosphate reductase | ARGC_PSEAB | 92 | 3 | 8 | 36.6 | 8 | E | Presence |
| Methylmalonate-semialdehyde dehydrogenase [acylating] | MMSA_PSEAE | 339 | 7 | 18 | 53.6 | 11 | E | Presence |
| 5-Aminovalerate aminotransferase DavT | DAVT_PSEAE | 129 | 5 | 15 | 45.2 | 9 | E | −1.2 |
| S-Adenosylmethionine synthase | METK_PSEAB | 78 | 2 | 6 | 42.7 | 9 | E | Presence |
| Formate-dependent phosphoribosylglycinamide formyltransferase | PURT_PSEAB | 47 | 2 | 6 | 42.3 | 9 | F | Presence |
| Nucleoside diphosphate kinase | NDK_PSEAB | 46 | 1 | 6 | 15.4 | 2 | F | Absence |
| Orotate phosphoribosyltransferase | PYRE_PSEAB | 83 | 1 | 8 | 23.3 | 4 | F | Presence |
| 6,7-Dimethyl-8-ribityllumazine synthase | RISB_PSEAB | 85 | 2 | 19 | 16.4 | 2 | H | Presence |
| Delta-aminolevulinic acid dehydratase | HEM2_PSEAE | 167 | 6 | 21 | 37 | 8 | H | Presence |
| Acyl carrier protein 1 | ACP1_PSEAE | 78 | 3 | 32 | 8.7 | 1 | I | Absence |
| Acetyl-CoA acetyltransferase | ATOB_PSEAE | 135 | 4 | 11 | 40.4 | 9 | I | Presence |
| Lipid A deacylase PagL | PAGL_PSEAE | 48 | 1 | 5 | 18.4 | 3 | I | Absence |
| Isoleucine-tRNA ligase | SYI_PSEAB | 60 | 1 | 1 | 105.4 | 12 | J | Presence |
| Glutamyl-tRNA(Gln) amidotransferase subunit A | GATA_PSEAE | 164 | 2 | 5 | 51.8 | 10 | J | Presence |
| Ribosome-recycling factor | RRF_PSEAB | 56 | 1 | 5 | 20.5 | 4 | J | Absence |
| 30S ribosomal protein S7 | RS7_PSE14 | 57 | 2 | 15 | 17.6 | 3 | J | Absence |
| 50S ribosomal protein L21 | RL21_PSEA8 | 44 | 1 | 10 | 11.7 | 1 | J | Absence |
| 30S ribosomal protein S16 | RS16_PSEA7 | 61 | 2 | 33 | 9.2 | 1 | J | Absence |
| 30S ribosomal protein S9 | RS9_PSEA7 | 44 | 1 | 6 | 14.6 | 2 | J | Absence |
| 50S ribosomal protein L20 | RL20_PSEA7 | 44 | 1 | 8 | 13.3 | 1 | J | Absence |
| 50S ribosomal protein L25 | RL25_PSEA8 | 164 | 4 | 27 | 21.9 | 4 | J | Absence |
| Elongation factor Ts | EFTS_PSEAB | 77 | 3 | 16 | 30.6 | 6 | K | Presence |
| Elongation factor Tu | EFTU_PSEAB | 86 | 2 | 5 | 43.3 | 9 | K | Presence |
| DNA-binding protein HU-beta | DBHB_PSEAE | 122 | 2 | 33 | 9.1 | 1 | K | Absence |
| Outer membrane porin F | PORF_PSEAE | 161 | 5 | 16 | 37.6 | 8 | M | Absence |
| Outer membrane protein OprJ | OPRJ_PSEAE | 85 | 2 | 5 | 51.9 | 10 | M | Presence |
| Outer membrane protein OprM | OPRM_PSEAE | 48 | 1 | 2 | 52.6 | 10 | M | Presence |
| Outer-membrane lipoprotein carrier protein | LOLA_PSEAB | 74 | 1 | 6 | 23.1 | 5 | M | Presence |
| Protein TolB | TOLB_PSEAE | 80 | 1 | 2 | 47.7 | 10 | M | Presence |
| Porin D | PORD_PSEAE | 114 | 5 | 10 | 48.4 | 9 | M | Absence |
| Porin B | PORB_PSEAE | 123 | 7 | 16 | 50.8 | 10 | M | Absence |
| A-type flagellin | FLICA_PSEAI | 622 | 8 | 23 | 40 | 8,12 | N | +6.5 |
| Glutathione hydrolase proenzyme | GGT_PSEAE | 72 | 2 | 4 | 58.9 | 11 | O | Presence |
| 60 kDa chaperonin | CH60_PSEA7 | 183 | 4 | 10 | 57 | 11 | O | −9.1 |
| Chaperone SurA | SURA_PSEAE | 48 | 1 | 3 | 46.9 | 10 | O | Presence |
| Thiol:disulfide interchange protein DsbA |
DSBA_PSEAB | 894 | 7 | 34 | 23.4 | 5 | O | +1.22 |
| Thiol peroxidase | TPX_PSEAE | 46 | 1 | 6 | 17.2 | 2 | O | Absence |
| Thioredoxin | THIO_PSEAE | 50 | 1 | 11 | 11.9 | 1 | O | Absence |
| Alkyl hydroperoxide reductase C | AHPC_PSEAB | 78 | 3 | 17 | 20.5 | 4 | O | Presence |
| Chaperone protein DnaK | DNAK_PSEAB | 64 | 2 | 3 | 68.4 | 10 | O | Absence |
| 10 kDa chaperonin | CH10_PSEU5 | 64 | 2 | 18 | 10.3 | 1 | O | Absence |
| Bacterioferritin | BFR_PSEAE | 66 | 1 | 5 | 17.9 | 2 | P | Absence |
| Catalase | CATA_PSEAE | 76 | 1 | 2 | 55.6 | 11 | P | Presence |
| Fe(3+)-pyochelin receptor | FPTA_PSEAE | 82 | 3 | 4 | 79.9 | 11 | P | +1.1 |
| Ferric uptake regulation protein | FUR_PSEAE | 59 | 1 | 9 | 15.2 | 2 | P | Absence |
| Mercuric transport protein periplasmic component | MERP_PSEFL | 76 | 2 | 19 | 9.5 | 1 | P | Absence |
| Superoxide dismutase [Fe] | SODF_PSEAE | 50 | 1 | 5 | 21.3 | 4 | P | 1.0 |
| Phosphate-binding protein PstS | PSTS_PSEAB | 98 | 2 | 8 | 34.5 | 7 | P | Absence |
| Muconolactone delta-isomerase | CATC_PSEAE | 90 | 1 | 8 | 11.3 | 1 | Q | Presence |
| Toluene-4-monooxygenase | TMOA_PSEME | 61 | 2 | 9 | 58.1 | 11 | Q | Presence |
| N5-carboxyaminoimidazole ribonucleotide mutase | PURE_PSEAE | 138 | 2 | 19 | 16.9 | 3 | R | Presence |
| Ecotin | ECOT_PSEAB | 55 | 1 | 7 | 17.3 | 3 | R | +1.6 |
| UPF0312 protein PLES_04211 | Y421_PSEA8 | 2659 | 9 | 52 | 20.8 | 4 | S | 1.0 |
| Uncharacterized protein PA3922 | Y3922_PSEAE | 101 | 3 | 8 | 51.2 | 10 | S | Presence |
| Uncharacterized protein PA1579 | Y1579_PSEAE | 60 | 1 | 6 | 22.1 | 4 | S | +1.1 |
Muconolactone δ-isomerase (CATC_PSEAE) (Fig. 4a and c), a unique enzyme that contributes toward the aromatic ortho-degradation via catechol, and acetyl-CoA-acyltransferase, which is responsible for β-ketoadipate transformation into succinyl, were differentially upregulated (Fig. 3a). CATC was not found in the oil-grown culture, implying it's strong downregulation. In accordance with the upregulation of the ortho-pathway, which was determined by the proteomic analysis, ring cleavage reactions were analyzed by the enzymatic tests. As ring fission is oxygen-consuming, the enzyme activity of crude protein extracts on 2,6-DTBP was measured by an oxygen electrode. The enzyme extract of P. aeruginosa san ai grown on LB and 2,6-DTBP exhibited specific activities of 0.02 and 0.23 U mg−1, respectively, indicating the aromatic ring processing reaction, as well as implying the inducibility of enzyme(s) responsible for degradation. As suggested in the earlier studies regarding the biodegradation of alkylphenols using ortho- or meta-aromatic pathways via alkylcatechols,14,15 we investigated the ring cleavage enzyme activities on several aromatic compounds, namely, benzoate, catechol, and 2,6-DTBP, using the biomass of P. aeruginosa san ai grown on different C sources (Table 2). Catechol ortho-cleavage as a result of C12O activity was monitored as an increase in absorbance at 260 nm (indicating the accumulation of cis,cis-muconic acid),34 at 290 nm (implying the presence of 2,4-dialkyl-3-hydroxy-cis,cis-muconic acid, which was proposed to be the product of 2,6-DTBP degradation (Fig. 3b) (bathochromic shift of OH– group is approximately 30 nm)),49 and meta-cleavage was measured at 375 nm (accumulation of 2-hydroxymuconic semialdehydes).33,34 As shown in Table 2, our results revealed that all the tested substrates were preferentially ortho-cleaved. The catechol branch of the β-ketoadipate pathway was found to be preferred for several molecules: nitroaromatics, phenol, toluene, and benzene, while protocatechuate was derived from lignin monomers and chlorinated aromatics.5 Our results are in agreement with the ortho-cleavage of catechol and sodium benzoate by P. putida50 and P. aeruginosa 142.51–53 Furthermore, the ortho-degradation of catechol by crude enzyme preparation from biomass grown on 2,6-DTBP implies that 2,6-DTBP could be ortho-converted by the same enzyme to the derivative of cis,cis-muconic acid. The upregulation of CATC_PSEAE along with the enzyme activity of C12O supports the hypothesis that the biodegradation of 2,6-DTPB might be passing through the ortho-cleavage of the ring. Large absorbance enhancement at 290 nm could imply the existence of 2,4-dialkyl-3-hydroxy-cis,cis-muconic acid as the product of the ortho-cleavage of 2,6-DTBP assuming that the previous hydroxylation was available at positions 3 and 4, a step that was not revealed in this study. The hydroxylation reaction can be a result of the monooxygenase or hydroxylating dioxygenase action that added hydroxyl groups,54 yielding an intermediate that is further cleaved by a dioxygenase.55,56 Our proteomic study revealed the upregulation of toluene-4-monooxygenase (TMOA), the enzyme responsible for the hydroxylation of the aromatic substrate. The relatively broad substrate specificity of TMOA has been reported;28 therefore, it can be considered to be a good candidate for the hydroxylation of aromatic 2,6-DTBP.
| C-source | Substrate | Specific activity, U mg−1 | |
|---|---|---|---|
| A260 | A375 | ||
| a Absorbance determined at 290 nm only for 2,6-DTBP. | |||
| Sodium benzoate | Sodium benzoate | 0.160 | 0.002 |
| Catechol | 0.150 | 0.010 | |
| 2,6-DTBP | 0.110; 0.150a | 0.008 | |
| 2,6-DTBP | 2,6-DTBP | 0.100; 0.160a | 0.004 |
| Catechol | 0.080 | <0.001 | |
| Peptone | Sodium benzoate | 0.020 | <0.001 |
| 2,6-DTBP | 0.030 | <0.001 | |
| Catechol | 0.009 | 0.007 | |
Mechanisms of P. aeruginosa san ai adaptation to the aromatic substrate
The proteomics analysis of P. aeruginosa san ai indicated that the core molecular responses to 2,6-DTBP can be summarized as the upregulation of proteins responsible for amino acid metabolism and energy production, while the translation, energy production, and inorganic ion transport and metabolism dominate in oil-amended cultures (Fig. 4b and 5).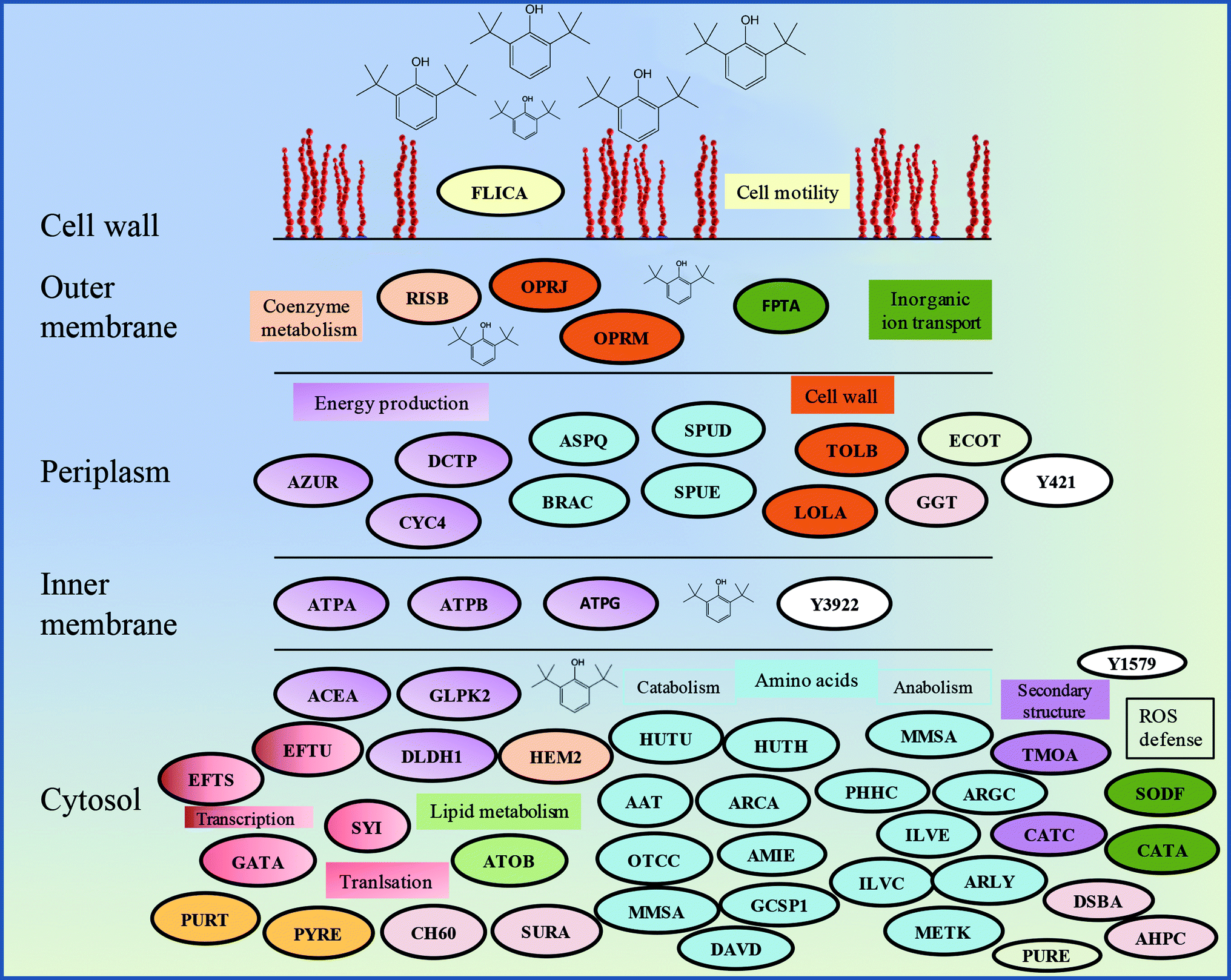 | ||
| Fig. 5 Schematic model of the response of P. aeruginosa san ai to the 2,6-DTBP plastic additive. Protein abbreviations are the same as that in Table 1. Proteins that belong to the same COG are labeled by the same color. | ||
Cell membrane proteins
It seems that the most massive changes occurred in the membrane when P. aeruginosa san ai was grown in the presence of DTBP versus oil as the C sources. In the DTBP-grown culture, the outer membrane OPRJ and OPRM proteins are differentially expressed, while porines D and B (PORD and PORB, respectively) are found in the oil-grown culture (Fig. 5). The overexpression of the channel-forming component of a multidrug resistance efflux pump OPRJ and OPRM— the major efflux pump for n-hexane and p-xylene—indicates the active response of the cell toward exposure to aromatic hydrocarbons; this is in good agreement with the fact that P. aeruginosa san ai can resist and grow when exposed to 2,6-DTBP. The increased content of the protein subunits of the solvent efflux pump systems and decreased contents of porins was registered in P. putida KT2440 exposed to phenol.22 An energized inner membrane is required for efflux that is in accordance with the upregulation of ATP synthase; this implies an increased production and consumption of ATP that can be partially used for efflux. Moreover, the identified protein Tol B (TOLB) and outer-membrane lipoprotein carrier protein (LOLA) are important for membrane integrity. TOLB from the Tol Pal system links the inner and outer membranes and the peptidoglycan layer, while LOLA acts in the translocation of lipoproteins from the inner membrane to the outer membrane. In accordance with the already mentioned chaperone SurA (SURA), which is involved in the correct assembly of the outer membrane proteins, OPRJ, OPRM, and TOLB show an adjustment of the membrane structure for the purpose of cell survival when exposed to 2,6-DTBP. At the same time, PORD and PORB reveal that an intensive diffusion of nutritive compounds, such as glycerol (PORB) and amino acids (PORD), occurs in the oil-grown culture. The enhanced glycerol amount can be attributed to the action of extracellular lipase from P. aeruginosa san ai that hydrolyzes oil to glycerol and fatty acids.45Posttranslational modifications are mainly related to protein redox processing: glutathione hydrolase proenzyme (GGT), thiol:disulfide interchange protein DsbA (DSBA), and alkyl hydroperoxide reductase C (AHPC) in DTBP cultures and thiol peroxidase (TPX) and thioredoxin (THIO) in oil-amended cultures (Table 1). As the mechanism of toxicity of phenol derivatives involves the production of reactive oxygen species (ROS) (e.g., superoxide, hydroxyl radicals, hydrogen peroxide, and free radicals), enzymes that can neutralize ROS were identified as differentially expressed in the current study, such as catalase (catA) and AHPC, which are kinetically more potent H2O2 scavengers than CAT, as well as the fact that they are probably the primary scavengers of endogenous H2O2 (Fig. 5). Proteomic data were validated in the enzymatic study, revealing a slight increase in the SOD and even six times higher CAT-specific activity in P. aeruginosa san ai grown on 2,6-DTBP versus oil, confirming the oxidative stress caused by 2,6-DTPB (Table 3). Furthermore, the upregulation of DSBA responsible for SH-group processing revealed that the emphasized thiol–disulfide exchange was impacted by DTBP, too. In oil-amended cultures, thioredoxin that catalyzes dithiol–disulfide exchange reactions and thiol-specific peroxidase that catalyzes the reduction of hydrogen peroxide and organic hydroperoxides to water and alcohols were identified. Our results imply an efficient ROS defense and redox homeostasis maintenance in P. aeruginosa san ai cells exposed to both DTBP and oil.
| C-source | Enzyme activities | |
|---|---|---|
| SOD, U mg−1 | Catalase, U mg−1 | |
| 2,6-DTBP | 18.7 | 2.68 |
| Oil | 9.51 | 0.40 |
Degradation of 2,6-DTBP by crude enzyme preparation
Crude enzyme preparation obtained from P. aeruginosa san ai was investigated to degrade 2,6-DTBP (concentration: 10 mg mL−1). The crude enzyme preparation obtained from P. aeruginosa san ai grown on 2,6-DTBP, quickly within four days, degraded 98% of 2,6-DTBP (concentration: 10 mg L−1). A similar strategy used toward hydrocarbon removal was successfully applied by Kadri et al.62 They reported that crude enzymes extracted from Alcanivorax borkumensis showed high efficiency in terms of hydrocarbon removal. This approach demonstrated the better capacity of crude enzyme preparation in the bioremediation of aromatic pollutants as compared to using whole microbial cells. Accordingly, our results show that crude enzymes from P. aeruginosa san ai degraded 2,6-DTBP (concentration: 10 mg L−1) really rapidly in a shorter time (4 days) at almost the same efficiency as that obtained when using microorganisms for biodegradation (7 days), namely, 98 and 100%, respectively.4. Conclusion
The effects of plastics, particularly microplastics, on aquatic environments and their biota have become a growing problem on our planet. Therefore, 2,6-di-tert-butylphenol as a plastic additive has been considered to be a serious issue for aquatic animals. This study was undertaken to estimate the capacity for 2,6-DTBP biodegradation by using the widespread Pseudomonas bacteria and the effects that it causes on these microorganisms. P. aeruginosa san ai rapidly degraded 2,6-DTBP during the growth in a mineral medium that mimics the aquatic environment. A high degradation capacity of 85% out of 100 mg L−1 of 2,6-DTBP for 7 days was determined. The removal of 2,6-DTBP completely dissolved in water (4 mg L−1) by P. aeruginosa san ai was almost accomplished within 10 h, causing intensive respiration. Further, it has been shown that crude enzymes isolated from P. aeruginosa san ai grown on 2,6-DTBP as the sole C source degraded 2,6-DTBP rapidly in a shorter time with higher efficiency than those obtained when using whole cells.A simple strategy, based on one-dimension electrophoresis of the total protein extracts followed by proteomic analysis (nanoLC-MS/MS), revealed that the proteins that were upregulated in the presence of 2,6-DTBP could be assigned to the functional groups of energy production and amino acid metabolism. As a response to the exposure to 2,6-DTBP, the culture shifts into a glyoxylate shunt to save energy and carbon. Further, at the cell membrane level, the content of solvent efflux pump systems increases and that of porines decreases, implying a large change in the membrane structure. Proteins responsible for redox homeostasis are upregulated in both DTBP- and oil-amended cultures, but enzymatic tests have shown more intensive oxidative stress in DTBP-amended cultures. Genomic, enzymatic, and proteomic data accordingly revealed that 2,6-DTBP could be ortho-cleaved. All these mechanistic insights related to catabolism and microbial adaptation to 2,6-DTBP may have an impact on the biodegradation, bioremediation, and biocatalysis by P. aeruginosa san ai. Finally, although ortho-disubstituted phenols are complex molecules for biodegradation, the P. aeruginosa san ai strain holds promising potential for the biotransformation of 2,6-DTBP; as such, the strain could be a promising agent in the bioremediation of the environment, particularly water, contaminated with organic pollutants.
The multiomics approach based on a combination of genomics and proteomics has provided insights into the global metabolic and regulatory networks of P. aeruginosa san ai that are exposed to 2,6-DTBP. While the genome of P. aeruginosa san ai revealed the existence of genes responsible for proteins that could serve for microorganism adaptation, a comparative proteomic analysis enabled the profiling of up- and downregulated proteins involved in the metabolic control of microorganisms grown on different C sources. As a result, the proteomic study gave an overall understanding of the response mechanisms toward pollutants and an opportunity to selectively use induced core proteins for environmental biomonitoring in natural microbial communities exposed to organic pollutants. The sets of up- or downregulated proteins obtained by proteomics might be used as multi-markers toward environmental contamination. Ultimately, P. aeruginosa san ai can be used for designing new strategies for environmental protection, as well as for the engineering of novel strains and novel proteins with improved characteristics for more effective bioremediation of contaminated areas.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financed by the Ministry of Education, Science and Technological Development, Republic of Serbia, Projects III 43004 and 176006.References
- OECD/SIDS, Screening Information Data Set (SIDS) of OECD High Production Volume Chemicals Programme, 1994 Search PubMed.
- Catalog Plastic Additive Standards Guide, New Haven, USA, 2018, http://https://www.accustandard.com/plastic-additive-catalog-2nd-edition Search PubMed.
- S. Rezania, J. Park, M. Md Din, S. Mat Taib, A. Talaiekhozani, K. Yadav and H. Kamya, Mar. Pollut. Bull., 2018, 133, 191–208 CrossRef CAS PubMed.
- M. Vidali, Pure Appl. Chem., 2001, 73, 1163–1172 CAS.
- T. Wells and J. Ragauskas, Trends Biotechnol., 2012, 30, 627–637 CrossRef CAS PubMed.
- A. Krastanov, Z. Alexieva and H. Yemendzhiev, Eng. Life Sci., 2013, 13, 76–87 CrossRef CAS.
- J. Brzeszcz and P. Kaszycki, Biodegradation, 2018, 29, 359–407 CrossRef PubMed.
- K. H. Kwon and S. H. Yeom, Bioprocess Biosyst. Eng., 2009, 32, 435–442 CrossRef CAS PubMed.
- V. Sridevi, M. V. V. Chandana Lakshmi, M. Manasa and M. Sravani, IJESAT, 2012, 3, 695–705 Search PubMed.
- J. J. Jeong, J. H. Kim, C. Kim, I. Hwang and K. Lee, Microbiology, 2013, 149, 3265–3277 CrossRef PubMed.
- M. Takeo, S. K. Prabu, C. Kitamura, M. Hirai, H. Takahashi, D. Kato and S. Negoro, J. Biosci. Bioeng., 2006, 102, 352–361 CrossRef CAS PubMed.
- B. Ajithkumar, V. Ajithkumar and R. Iriye, Res. Microbiol., 2003, 154, 17–23 CrossRef CAS PubMed.
- H. P. Kohler, M. Maarel and D. Kohler-Staub, Appl. Environ. Microbiol., 1993, 59, 860–866 CAS.
- T. Toyama, N. Maeda, M. Murashita, Y. Chang and S. Kikuchi, Biodegradation, 2010, 21, 157–165 CrossRef CAS PubMed.
- N. N. Tuan, H. Ch. Hsieh, Y. W. Lin and Sh. L. Huang, Bioresour. Technol., 2011, 102, 4232–4240 CrossRef PubMed.
- Y. Zhang, Z. Fang, D. Xu, Y. Xiao, J. Zhao and Z. Qiang, J. Environ. Sci., 2005, 17, 271–275 CAS.
- A. Chauhan and R. K. Jain, Biodegradation, 2010, 21, 861–879 CrossRef CAS PubMed.
- L. Kurbatov, D. Albrecht, H. Herrmann and L. Petruschka, Environ. Microbiol., 2006, 8, 466–478 CrossRef CAS PubMed.
- F. Li, W. Song, J. Wei, C. Liu and C. Yu, Int. Biodeterior. Biodegradation, 2016, 110, 189–198 CrossRef CAS.
- E. Vandera, M. Samiotaki, M. Parapouli, G. Panayotou and A. I. Koukkou, J. Proteomics, 2015, 113, 73–89 CrossRef CAS PubMed.
- S. Verhoef, H. Ballerstedt, J. M. R. Volkers, H. J. de Winde and H. J. Ruijssenaars, Appl. Microbiol. Biotechnol., 2010, 87, 679–690 CrossRef CAS PubMed.
- C. Roma-Rodrigues, P. M. Santos, D. Benndorf, E. Rapp and I. Sá-Correia, J. Proteomics, 2010, 73, 1461–1478 CrossRef CAS PubMed.
- M. P. Santos, D. Benndorf and I. Sá-Correi, Proteomics, 2004, 4, 2640–2652 CrossRef PubMed.
- J. S. Seo, Y. S. Keum and Q. X. Li, Int. J. Environ. Res. Public Health, 2009, 6, 278–309 CrossRef CAS PubMed.
- I. Tsirogianni, M. Aivaliotis, M. Karas and G. Tsiotis, Biochim. Biophys. Acta, 2004, 1700, 117–123 CrossRef CAS PubMed.
- R. S. Kahlon, Biodegradation and bioremediation of organic chemical pollutants by Pseudomonas, in Pseudomonas: Molecular and applied biology, ed. R. Kahlon, Springer International Publishing Switzerland, 2016. pp. 343–417 Search PubMed.
- I. Karadzic, A. Masui and N. Fujiwara, J. Biosci. Bioeng., 2004, 98, 145–152 CrossRef CAS PubMed.
- X. Tao, G. Lu, J. Liu, T. Li and L. Yang, Int. J. Environ. Res. Public. Health, 2009, 6, 2470–2480 CrossRef CAS PubMed.
- M. Lin, X. Hu, W. Chen, H. Wang and C. Wang, Int. Biodeterior. Biodegradation, 2014, 94, 176–181 CrossRef CAS.
- M. Heyd, A. Kohnert, T. H. Tan, M. Nusser, F. Kirschhöfer, G. Brenner-Weiss, M. Franzreb and S. Berensmeier, Anal. Bioanal. Chem., 2008, 391, 1579–1590 CrossRef CAS PubMed.
- M. Rosenberg, D. Gutnick and E. Rosenberg, FEMS Microbiol. Lett., 1980, 9, 29–33 CrossRef CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- F. Briganti, E. Pessione, C. Giunta and A. Scozzafava, FEBS Lett., 1997, 416, 61–64 CrossRef CAS PubMed.
- Md. Mahiudddin, A. N. M. Fakhruddin and A. A. Mahin, ISRN Microbiol., 2012, 741820 Search PubMed.
- A. Mars, J. Kingma, S. Kaschabek, W. Reineke and D. Janssen, J. Bacteriol., 1999, 181, 1309–1318 CAS.
- S. Mancini, H. K. Abicht, Y. Gonskikh and M. Solioz, Mol. Microbiol., 2015, 95, 645–659 CrossRef CAS PubMed.
- J. M. Sala-Trepat and W. C. Evans, Eur. J. Biochem., 1971, 20, 400–413 CrossRef CAS PubMed.
- F. B. Roland and W. S. Irwin, J. Biol. Chem., 1952, 195, 133–140 Search PubMed.
- M. Sun and S. Zigman, Anal. Biochem., 1987, 90, 81–89 CrossRef.
- P. Tribedi and A. K. Sil, J. Appl. Microbiol., 2013, 116, 295–303 CrossRef PubMed.
- T. A. Kadam, L. Rupa, D. K. Balhal, N. D. Totewad and G. Gyananath, Asian J. Exp. Sci., 2009, 23, 135–139 Search PubMed.
- J. D. Walker, Testing decisions of the TSCA Interagency Testing Committee for chemicals on the Canadian Environmental Protection Act domestic substances list and priority substances list: Di-tert-butylphenol, ethyl benzene, brominated flame retardants, phthalate esters, chloroparaffins, chlorinated benzenes and anilines, Environmental toxicity and risk assessment, ASTM, Philadelphia, 1996 Search PubMed.
- C. Zhang, G. Zeng, L. Yuan, J. Yu, J. Li, G. Huang, B. Xi and H. Liu, Chemosphere, 2007, 68, 181–190 CrossRef CAS PubMed.
- M. Sasaki, J. Maki, K. Oshiman, Y. Matsumura and T. Tsuchido, Biodegradation, 2005, 16, 449–459 CrossRef CAS PubMed.
- I. Karadzic, A. Masui, L. Izrael-Zivkovic and N. Fujiwara, J. Biosci. Bioeng., 2006, 102, 82–89 CrossRef CAS PubMed.
- K. M. Basha, A. Rajendran and V. Thangavelu, Asian J. Exp. Biol. Sci., 2010, 1, 219–234 CAS.
- D. Freitag, L. Ballhorn, H. Geyer and F. Korte, Chemosphere, 1985, 14, 1589–1616 CrossRef CAS.
- L. Izrael-Živković, V. Beškoski, M. Rikalović, S. Kazazić, N. Shapiro, T. Woyke, G. Gojgić-Cvijović, M. Vrvić, N. Maksimović and I. Karadžić, Extremophiles, 2019, 23, 399–405 CrossRef PubMed.
- R. Silverstain and G. Bassler, Spectrometry Identification of Organic Compounds, John Wiley&Sons, Inc., New York, 1967 Search PubMed.
- K. Loh and S. Chua, Enzyme Microb. Technol., 2002, 30, 620–626 CrossRef CAS.
- C. F. Feisti and G. D. Hegeman, J. Bacteriol., 1969, 100, 869–877 Search PubMed.
- B. E. Haigler, C. A. Pettigrew and J. C. Spain, Appl. Environ. Microbiol., 1992, 58, 2237–2244 CAS.
- V. Romanov and R. P. Hausinger, J. Bacteriol., 1994, 176, 3368–3374 CrossRef CAS PubMed.
- C. S. Karigar and S. S. Rao, Enzyme Res., 2011, 805187 Search PubMed.
- S. Harayama, M. Kok and E. L. Neidle, Annu. Rev. Microbiol., 1992, 46, 565–601 CrossRef CAS PubMed.
- F. H. Vaillancourt, J. T. Bolin and L. D. Eltis, Crit. Rev. Biochem. Mol. Biol., 2006, 41, 241–267 CrossRef CAS PubMed.
- D. White, The Physiology and Biochemistry of Procariotes, Oxford University Press, New York, 2nd edn, 2000 Search PubMed.
- M. Unell, P. E. Abraham, M. Shah, B. Zhang, C. Rückert and N. C. VerBerkmoes, J. Proteome Res., 2009, 8, 1953–1964 CrossRef CAS PubMed.
- A. C. Badejo, C. W. Choi, A. O. Badejo, K. H. Shin, J. H. Hyun and Y. G. Lee, Biodegradation, 2013, 24, 741–752 CrossRef CAS PubMed.
- M. Eschbach, K. Schreiber, K. Trunk, J. Buer, D. Jahn and M. Schobert, J. Bacteriol., 2004, 186, 4596–4604 CrossRef CAS PubMed.
- L. Izrael-Živković, M. Rikalović, G. Gojgić-Cvijović, S. Kazazić, M. Vrvić, I. Brčeski, V. Beškoski, B. Lončarević, K. Gopčević and I. Karadžić, RSC Adv., 2018, 8, 10549–10560 RSC.
- T. Kadri, S. Magdouli, T. Rouissi and S. Kaur Brar, Biochem. Eng. J., 2018, 132, 279–287 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04298a |
| This journal is © The Royal Society of Chemistry 2019 |