
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ fabrication of hierarchical biomass carbon-supported Cu@CuO–Al2O3 composite materials: synthesis, properties and adsorption applications†
Hongling Dai,
Min Wang,
Wendong Luo,
Cheng Pan,
Fengping Hu* and
Xiaoming Peng *
*
School of Civil Engineering and Architecture, East China Jiaotong University, Nanchang, 330013, Jiangxi Province, China. E-mail: hufengping1968@126.com; pengxiaoming70@gmail.com
First published on 16th October 2019
Abstract
Hierarchical Cu–Al2O3/biomass-activated carbon composites were successfully prepared by entrapping a biomass-activated carbon powder derived from green algae in the Cu–Al2O3 frame (H–Cu–Al/BC) for the removal of ammonium nitrogen (NH4+-N) from aqueous solutions. The as-synthesized samples were characterized via XRD, SEM, BET and FTIR spectroscopy. The BET specific surface area of the synthesized H–Cu–Al/BC increased from 175.4 m2 g−1 to 302.3 m2 g−1 upon the incorporation of the Cu–Al oxide nanoparticles in the BC surface channels. The experimental data indicated that the adsorption isotherms were well described by the Langmuir equilibrium isotherm equation and the adsorption kinetics of NH4+-N obeyed the pseudo-second-order kinetic model. The static maximum adsorption capacity of NH4+-N on H–Cu–Al/BC was 81.54 mg g−1, which was significantly higher than those of raw BC and H–Al/BC. In addition, the presence of K+, Na+, Ca2+, and Mg2+ ions had no significant impact on the NH4+-N adsorption, but the presence of Al3+ and humic acid (NOM) obviously affected and inhibited the NH4+-N adsorption. The thermodynamic analyses indicated that the adsorption process was endothermic and spontaneous in nature. H–Cu–Al/BC exhibited removal efficiency of more than 80% even after five consecutive cycles according to the recycle studies. These findings suggest that H–Cu–Al/BC can serve as a promising adsorbent for the removal of NH4+-N from aqueous solutions.
1. Introduction
Nutrient eutrophication is a significant environmental issue globally, posing a risk to aquatic ecosystems, which is generally associated with the trigger for water eutrophication due to the discharge of many nutrients, particularly nitrogen (N) and phosphorus (P) compounds, into the ecosystems.1,2 Thus, nutrient removal during wastewater treatment is vital to minimize the impact of nitrogen and phosphorus pollutants on the aquatic ecosystems. Among them, ammonium nitrogen (NH4+-N) is the dominant nitrogen-containing pollutant in the aquatic environment and if its concentration is higher than the permissible level, it is a severe hazard to aquatic ecosystems and contributes significantly to the eutrophication process.3 To meet the strict quality standard requirements, it is imperative to take effective measures to remove effluents containing ammonia nitrogen (NH4+-N) and other nutrients from the aqueous environment.In recent decades, as far as the removal of NH4+-N is concerned, a considerable number of water treatment technologies have been extensively studied, which include precipitation, crystallization, biological removal, and ion exchange.4,5 Adsorption is considered as a reliable, clean and the most frequently adopted technique over other conventional treatment processes due to its low cost, removal efficiency, mild operating conditions and good performance; thus, it is widely used for the removal of ammonium nitrogen from aqueous solutions.6
Various types of adsorbents have been used for the removal of ammonium nitrogen from aqueous solutions, but the main defect in the application of the adsorbents such as polymeric materials is their preparation cost, especially for widespread usage, in comparison with cheap natural biomass materials.7 Biomass recently has been used to prepare and produce bio-adsorbents for removing different pollutants from water due to its low cost, environmentally friendly characteristics and renewable advantages, which has become a research hotspot.8–10 Up to now, some natural biomass materials such as bamboo, straw, bagasse, and corn cob have been applied to prepare carbon adsorbent materials for environmental applications.11,12 Algae biomass are considered to be one of the most abundant plant wastes and it is readily available; its total world production is approximately 1.2 × 104 t dry basis (about 1.6 × 104 t wet basis) annually, which has continuously increased by an average rate of 10%. Furthermore, algae biomass grows widely in the oceans, seas and lakes, and its excessive growth can eventually lead to the significant deterioration of water bodies. It is very difficult to deal with algal biomass due to its extremely high water content and possibility of secondary pollution. Also, its disposal by burning or deposition in landfills involves significant manpower and material resources. However, it is worth noting that algae biomass can be used as a low-cost biomass carbon precursor to prepare cost-effective biosorbents because it is a renewable and abundance natural resource, which can not only help resolve the global environmental issue of eutrophication by removing excessive nutrients, but also have high uptake capacities for the removal of organic or inorganic pollutants from various types of water bodies.13,14 Numerous studies on the utilization of algal biomass as an adsorbent for adsorption have been widely reported. Hence, algae biomass-based activated carbon has been considered as an economical, ecofriendly and promising adsorbent for removing various pollutants from aqueous solutions.15–17
In this study, ammonium nitrogen (NH4+-N) was chosen as a model pollutant to evaluate the removal efficiency of the as-prepared composite because it is the main cause of nutrient eutrophication. The aim of the present study was to prepare a new hierarchical Cu–Al2O3/biomass carbon (H–Cu–Al/BC) composite using green algae as the biomass raw material through the in situ growth technology via the co-precipitation method and to subsequently use it as an adsorbent to remove NH4+-N from aqueous solutions. To get a better understanding of the adsorption behavior, the influence of some adsorption experimental factors including pH, adsorbent dosage, contact time, temperature, salt, and natural organic matter (NOM) and regeneration on its adsorption capacity was investigated. Furthermore, adsorption isotherms and kinetic models were also studied to discuss the mechanisms that control the adsorption process.
2. Experimental
2.1. Materials
All chemicals and reagents used including hydrochloric acid, sodium hydroxide (NaOH), potassium iodide (KI), methyl alcohol, Seignette salt (KNaC4H4O6·4H2O), ammonium chloride (NH4Cl), humic acid (HAc, a major constituent of NOM, used as NOM), acetic acid (CH3COOH), aluminium nitrate (Al(NO3)3·9H2O), sodium hypochlorite (NaClO2), ammonium hydroxide (NH3·H2O) and copper nitrate (Cu(NO3)2·3H2O) were of analytical grade. All solutions were prepared with deionized water (DW).2.2. Preparation of materials
Green algae were chosen as the precursor for the biomass carbon (BC). The green algae samples were collected from Taihu Lake and were ground into a powder (<0.5 mm) through a mesh sieve; they were washed several times with hot DW to get rid of the impurities and ash from the surface and then dried at 40 °C in an oven. Subsequently, the samples were pyrolysed in a muffle furnace under a nitrogen gas atmosphere at 600 °C and maintained for 3 h. Then, the obtained sample was decolorized using acidified NaClO2 and its pH maintained at about 4. Finally, the prepared white sample was immersed in 300 mL of deionized distilled water and dried at 60 °C for 10 h.The hierarchical Cu–Al2O3/biomass carbon material was synthesized via the co-precipitation method. In a typical experiment, 15 g Al(NO3)3·9H2O was dissolved in 200 mL deionized water to obtain an alumina sol solution. Ammonium hydroxide was then added to the above solution to adjust the pH to around 9 with continuous and vigorous stirring to form an aluminum hydroxide colloid at room temperature. Then, the colloid was filtered and subsequently dissolved in 400 mL DW, and nitric acid solution was added dropwise to the mixture solution under vigorous magnetic stirring until the pH reached 4. Then, the sample was aged under static conditions for 3 h at 85 °C. Subsequently, the obtained white biomass was washed with DW water and ethanol three times, respectively, and further dried at 60 °C for 10 h. Thereafter, the resulting mixture was calcined at 450 °C for 2 h under a nitrogen atmosphere. Finally, a black powder was obtained (the prepared material was denoted as Al2O3/BC). Additionally, pure biomass carbon was prepared via the same method without the impregnation of Al2O3, which was denoted as BC.
Then, 0.408 g of the above prepared Al2O3/BC and 0.56 g of hexamethylenetetramine were dissolved in 75 mL deionized water under vigorous magnetic stirring; this solution was hydrothermally treated at 120 °C for 10 h to obtain AlOOH/BC. The obtained as-prepared product was filtered and washed with DW, dried at 60 °C for 12 h, and then calcined under a nitrogen atmosphere by increasing the temperature from ambient temperature at a rate of 5 °C min−1 to 450 °C and maintaining that temperature for 2 h to acquire hierarchical Al2O3/BC (denoted as H–Al/BC).
The H–Al/BC powder was added into a solution with Cu(NO3)2·3H2O and 50 mL DW; the mixture was then ultrasonicated in an ultrasonic bath and ultrasonically dispersed for 30 min, and the obtained product was denoted as H–Cu–Al/BC (a schematic illustration of the preparation of H–Cu–Al/BC is given in Scheme S1†).
2.3. Characterization of the prepared materials
The elemental composition and surface morphologies of the prepared materials were characterized using Hitachi S-4800 with a 15 kV accelerating voltage and a field-emission scanning electron microscope (SEM, S-4800, Hitachi Corp., Tokyo, Japan) and energy dispersive X-ray spectroscopy (EDS). Fourier transform infrared spectroscopy (FT-IR) was performed from 4000 cm−1 to 400 cm−1 with KBr pellets using an AVATAR 360 spectrophotometer (Nicolet, USA). The X-ray diffraction (XRD) patterns of the prepared materials were observed using a Bruker X-ray diffractometer with Cu-Kα radiation in the 2θ range from 10° to 80°. N2 adsorption–desorption was employed at 77 K on an ASAP 2020 apparatus for the analysis of specific surface area and pore-size distributions. Transmission electron microscopy (TEM) (Philips CM120, Netherlands) was performed at 200 kV and used to study the morphologies and crystalline characteristics of the prepared materials.2.4. Batch adsorption experiments
A batch of ammonium nitrogen (NH4+-N) adsorption experiments was carried out to investigate the NH4+-N removal efficiency, adsorption kinetics and isotherms for the prepared series of biomass carbon. A fixed amount of sample and 100 mL of NH4+-N solution with different initial concentrations were placed in conical flasks and continuously shaken at 120 rpm using a temperature-controlled thermostat shaking bath for 24 h. The influence of the experimental parameters was investigated including temperature (20, 40 and 60 °C), pH (2–10) and ionic strength (K+, Na+, Ca2+, Mg2+, Al3+ and humic acid) to optimize the adsorption experimental conditions. The influence of various parameters on the adsorption was investigated by varying one parameter at a time and keeping the other parameters constant; the pH of the solution was adjusted using 0.1 M of HCl or NaOH. After adsorption, the samples were passed through a 0.45 μm filter and the concentration of NH4+-N was analyzed by the spectrophotometry method using a UV spectrophotometer. All experiments were performed in triplicate. The removal percentage of NH4+-N was calculated from the following expression:
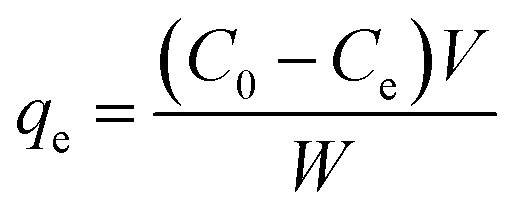 | (1) |
Langmuir
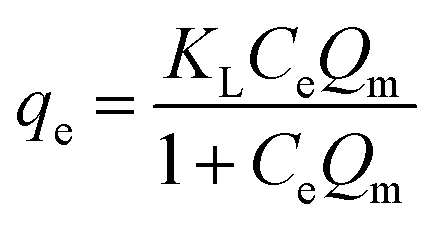 | (2) |
Freundlich
| qe = KFCe1/n | (3) |
Temkin
qe = B1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(ACe) ln(ACe)
| (4) |
Pseudo-first-order
|
ln(qe − qt) = lnqe − k1t
| (5) |
Pseudo-second-order
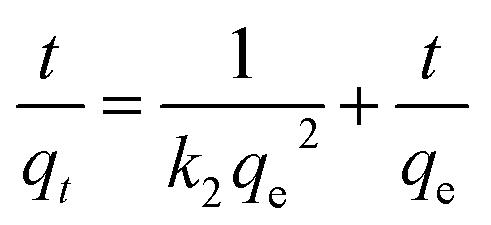 | (6) |
Intra-particle diffusion
| qt = kpt1/2 + C | (7) |
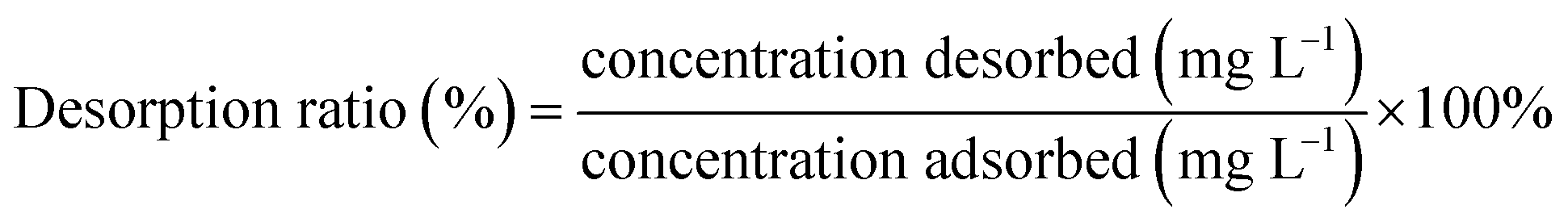 | (8) |
3. Results and discussion
3.1. Characterization of the prepared materials
Fig. S1† presents the wide-angle XRD patterns of all the prepared materials in the large-angle regions. The powder XRD patterns depicted a distinct difference between BC and the H–Cu–Al/BC nanocomposite because aluminum and copper oxide were present in the BC framework. As shown in Fig. S1,† intense well-resolved diffraction peaks were observed for H–Al/BC at 2θ = 29.3°, 45.4° and 66.3°, which can be indexed to the typical Al2O3 structure (JCPDS card no. 04-0878). Furthermore, for H–Cu–Al/BC, three new sharp and intense symmetric peaks appeared at 2θ = 43.4°, 50.4° and 74.1°, which corresponded to the (111), (200) and (220) planes of the Cu harmonic reflections (JCPDS card no. 04-0836). In addition, the dominant diffraction peaks of H–Cu–Al/BC were observed at 2θ = 35.5°, 38.7°, 58.3° and 66.2°, which could be indexed to the (11−1), (111), (202) and (31−1) planes of CuO (JCPDS card no. 48-1548), respectively, confirming that the metal copper oxide nanoparticles were uniformly dispersed and anchored on H–Al/BC.21 The new peak of CuAl2O4 (JCPDS card no. 33-0448) appeared for H–Cu–Al/BC. It is noteworthy that some other compounds possibly copper- and aluminum-containing oxide compounds were present in the composites, which can be explained by the fact that small diffraction peaks appeared in the XRD pattern. In conclusion, the XRD results indicate that the Cu–Al nanoparticles are favorable for the formation of the hierarchical biomass carbon structure, suggesting that these as-prepared samples are highly crystalline, dispersed and stable materials.22The N2 adsorption–desorption isotherms and pore size distributions for the three prepared materials were obtained using BET analysis (Fig. S2a†). It can be observed that the curves of the three materials clearly show typical type-IV characteristics according to the IUPAC classification with a hysteresis loop of type H2 at high relative pressures (P/P0 = 0.4–1.0) due to multilayer adsorption; this corresponds to pore filling by capillary condensation, indicating the existence of a well-developed mesoporous structure of the nanocomposites. Correspondingly, the analysis revealed that a larger amount of mesopores and some small-sized micropores were present in the three materials. The average pore diameter of the three materials was mainly around 4.0–5.0 nm (Fig. S2b†).
The textural parameters calculated from the adsorption isotherms for the three materials using BET analysis are summarized in Table S1.† As shown in Table S1,† the BET specific surface area and the total volume of BC are lower than those of H–Al/BC and H–Cu–Al/BC, suggesting that the materials have not developed porosity. Comparatively, higher values were obtained after modification, where the introduction of aluminum metal oxide nanoparticles into the BC matrix affected both the micropore and mesopore volumes. It was found that porosity increased when the aluminum metal oxide nanoparticles were successfully incorporated into the BC surface channels; the total volume of H–Cu–Al/BC increased from 0.15 to 0.27 m3 g−1 and the BET surface area increased from 175.4 to 226.7 m2 g−1. However, it is noteworthy that the total volume and mesopore volume of H–Cu–Al/BC showed a certain degree of decrease as the copper metal oxide nanoparticles were introduced into the material matrix. This phenomenon can be ascribed to the incorporated copper species possibly covering some pores. Additionally, the hierarchy factor (HF) increased when the aluminum and copper metal oxide nanoparticles were introduced in the surface frame, suggesting that the mesopore surface was enhanced with the simultaneous preservation of microporosity.23 The total pore volume and mesopore volume also increased with the conversion of BC to H–Al/BC and H–Cu–Al/BC; however, it is worth noting that the average pore size (Dp) of BC decreased compared with those of H–Al/BC and H–Cu–Al/BC. The BET surface area increased by almost 2 times after grafting with Cu and Al oxide nanoparticles. This result may be due to the fact that the adsorbent matrix structure shrank during high-temperature carbonization and Cu–Al oxide could act as a catalyst in the development of a high surface area; this would help create a more porous structure by opening the previously inaccessible pores and generating some new pores, resulting in a higher BET surface area and total pore volume. In general, for an adsorbent, a large surface area and pore volume can result in a high contact area and sufficient active sites for NH4+-N removal. Therefore, H–Cu–Al/BC may result in higher adsorption capacity for NH4+-N. In addition, according to the analysis results, the average pore size of all the as-prepared samples exceeded the micropore range (<2.0 nm). The average pore diameter of the as-prepared samples was in the range of 3–5 nm and thus, they can be considered mesoporous materials. This mesoporous feature could play an important role in the adsorption properties of the carbon materials since mesoporosity of the as-prepared materials allowed ions to penetrate more easily into their pores; thus, the samples possess suitable properties as adsorbents for removing NH4+-N from aqueous solutions because the molecular dimensions of NH4+-N in water are 0.321 × 0.123 × 0.217 nm.24
As we all know, the adsorption capacity of an adsorbent is commonly related to its own porosity and surface functional groups. As shown in Fig. S3,† it is worth noting that H–Cu–Al/BC shows higher SBET values and higher adsorption capacity for NH4+-N compared to BC and H–Al/BC under two different water quality conditions. The NH4+-N uptake increased from 12.36 to 60.33 mg g−1 under deionized water when the Cu and Al oxides were introduced into the BC surface channels. Compared to the adsorption capacity under deionized water, the adsorption capacity increased from 7.32 to 25.56 mg g−1 under simulated actual water. It could thus be inferred that the adsorption capacity of the three samples was positively related to their BET surface area under different water quality conditions.
The morphologies of the three samples were observed at 100×, 150×, 1500× and 2000× magnifications. The SEM images are presented in Fig. S4.† The surface morphology of BC exhibited micrometer-sized massive structures (Fig. S4a†). Fig. S4c† shows that a certain number of nanorod bundles are aggregated and well-dispersed in the BC carbon matrix; the average diameter of these nanorods is 30–50 nm, corresponding to an average length of 5 μm. The figures show that the surface morphological feature of raw BC is much smoother than those of modified H–Al/BC and H–Cu–Al/BC. As shown in Fig. S4e, f, h, and i,† clearly, the BC surface is surrounded by numerous small nanocrystals with about 50 nm thickness. These nanocrystals with a random orientation are in contact with each other, resulting in an irregular pore arrangement, which endows the as-prepared modified materials with a high specific surface area and total volume in comparison with that for pure BC.13 It is worth noting that the introduction of the compounds of aluminum and copper in the BC matrix did not seem to affect its structure since the morphology of BC showed no obvious changes. Furthermore, this observation was in good agreement with the results of XRD, as described above.
The TEM images provide detailed information on the morphological structure changes in the samples before and after modification. The unloaded sample before adsorption exhibited some compact and irregular elongated crystals (Fig. S5A†), whereas after loading, the surface became fragmented and some Cu or Al metal and metal oxide particles were distributed on the surface wall (Fig. S5B and C†); this was consistent with the XRD patterns and SEM images.
To better understand the adsorption process, the FTIR spectrum of H–Cu–Al/BC is displayed in Fig. S6.† In the FTIR spectrum, a broad and strong stretching band at approximately 3450 cm−1 is detected for the as-prepared samples, which is mainly ascribed to the O–H valence vibration of the interlayer adsorbed water molecules and is also probably partially related to the O–H vibration of the –COOH groups in the adjacent layers. The new peak for H–Cu–Al/BC that appeared at approximately 900 cm−1 could be assigned to the Al or Cu oxide lattice vibrations in the layers.25 In addition, the absorption band at 1431 cm−1 in the spectrum was assigned to the asymmetric stretching modes (CO32− ion), which was probably caused by the exposure of the CO32− ions in the interlayer galleries.26 Thus, the hierarchical Cu–Al/biomass carbon could be successfully synthesized based on the above observations.
The forms of the elements in the H–Cu–Al/BC composite before and after adsorption were analyzed by XPS. As shown in Fig. S7a,† the sample is mainly composed of the C, O, Al and Cu elements. The XPS spectrum of C 1s (Fig. S7b†) can be decomposed into four peaks with the binding energies of 283.9 eV, 284.7 eV, 285.7 and 288.6 eV. The peaks at 283.9 and 284.7 eV correspond to C–C and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, and the peaks centered at 285.7 and 288.6 correspond to C–O and C–OO. As shown in Fig. S7c,† the O 1s line in the spectra of the used H–Cu–Al/BC has one peak at 531.1 eV, which can be attributed to the OH groups adsorbed on the sample surface. In addition, the Al 2p binding energy peak of H–Cu–Al/BC is located at 74.5 eV (Fig. S7d†), which indicates that Al was in the form of Al(III) species. In Fig. S7e,† the XPS spectra of Cu 2p can be divided into six peaks at around 932.7 eV, 934.8 eV, 941.1 eV, 943.3 eV, 954.4 eV and 962.6 eV. According to a previous result, the fresh H–Cu–Al/BC particles did not have Cu(I) and only Cu(II) was detected on the surface of BC. The peak at the binding energy of 934.8 eV was attributed to the Cu(II) oxide species, and the new peak at 932.7 eV suggested that Cu(II) was transformed into Cu(I) partially.
C, and the peaks centered at 285.7 and 288.6 correspond to C–O and C–OO. As shown in Fig. S7c,† the O 1s line in the spectra of the used H–Cu–Al/BC has one peak at 531.1 eV, which can be attributed to the OH groups adsorbed on the sample surface. In addition, the Al 2p binding energy peak of H–Cu–Al/BC is located at 74.5 eV (Fig. S7d†), which indicates that Al was in the form of Al(III) species. In Fig. S7e,† the XPS spectra of Cu 2p can be divided into six peaks at around 932.7 eV, 934.8 eV, 941.1 eV, 943.3 eV, 954.4 eV and 962.6 eV. According to a previous result, the fresh H–Cu–Al/BC particles did not have Cu(I) and only Cu(II) was detected on the surface of BC. The peak at the binding energy of 934.8 eV was attributed to the Cu(II) oxide species, and the new peak at 932.7 eV suggested that Cu(II) was transformed into Cu(I) partially.
3.2. Effect of temperature on the adsorption process
The effects of temperature on the adsorption capacity of the as-prepared adsorbents for AN at 20 °C, 40 °C and 60 °C were investigated using different initial concentrations of NH4+-N. As illustrated in Fig. 1, the NH4+-N removal capacity is maximum at 60 °C; the adsorption capacity of NH4+-N remarkably increased from 35.31 mg g−1 to 54.74 mg g−1 as the temperature increased from 20 °C to 60 °C, respectively, indicating that the adsorption process was endothermic in nature. This can be explained by the fact that the adsorption capacity of NH4+-N under a high temperature is favorable for Brownian movement. Therefore, there is a tendency for the NH4+-N ions to adsorb from the solution into the solid phase due to their improved mobility with an increase in temperature. Another reason that can explain this phenomenon is that the kinetic energy of the NH4+-N ions in the adsorption process increased with an increase in temperature. With an increase in temperature, a larger quantity of NH4+-N ions could be adsorbed on the adsorbent since more NH4+-N ions acquired enough energy to be bound to the available active sites of the adsorbent, resulting in an increase in the NH4+-N adsorption capacity.27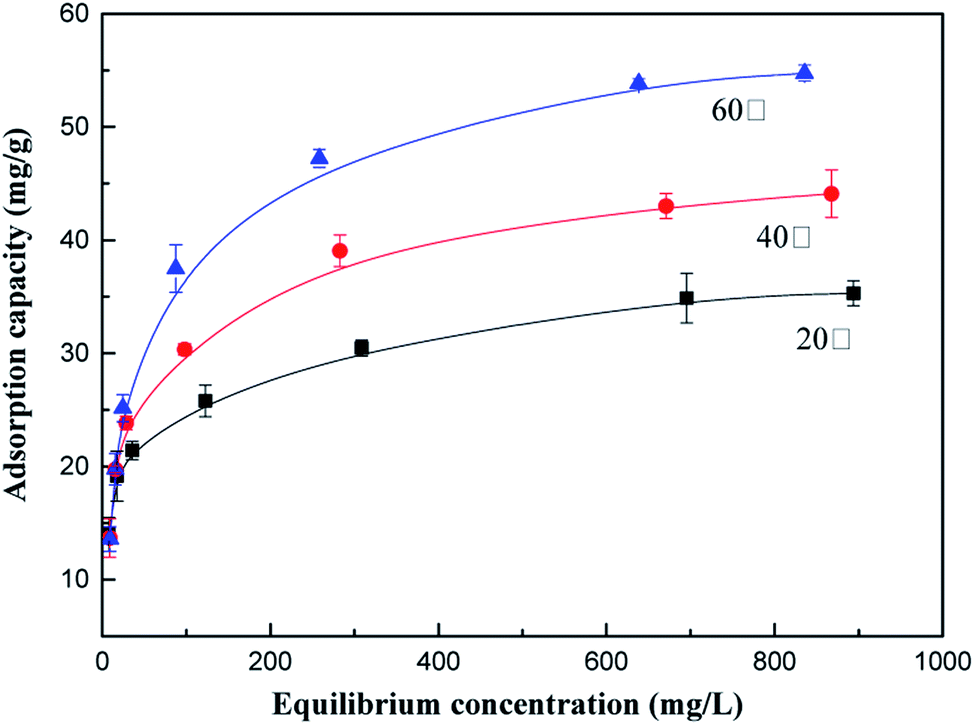 | ||
| Fig. 1 Effect of different temperatures on the adsorption process. | ||
3.3. Effect of initial pH on the adsorption process
The variation in solution pH plays a fundamental role in efficient pollutant adsorption from an aqueous solution, which determines the dissociation or association of protons in NH4+-N and the characteristics of the adsorbent. In an aqueous solution, depending on the value of pH, NH4+-N is predominantly present in the form of NH4+ or NH3(aq). This form can be described under different pH values by eqn (10) and (11).|
NH3(aq) + H2O ⇄ NH4+ + OH− (atacidicpH)
| (10) |
|
NH4+ ⇄ H+ + NH3(aq) (atalkalinepH, pKa = 9.3)
| (11) |
Fig. 2 shows the effect of the solution pH (2.0–12.0) on the adsorption of NH4+-N under equilibrium conditions, and the final pH was measured and presented in this figure. Fig. 2 shows that the pH values affect the adsorption of NH4+-N significantly. As shown in eqn (10), NH4+-N presents a dissociation ionic state under acidic conditions; the interaction of the adsorbent surface and ionic solution is facilitated, which leads to the positive surface charge of the adsorbent. The experimental results showed that the adsorption capacity of the as-prepared adsorbents for NH4+-N significantly increased with an increase in the solution pH from 2 to 8, and the maximum removal efficiency was obtained around pH 8. Subsequently, the adsorption capacity of the material decreased with an increase in pH from 8 to 12. This is observed because NH4+-N mostly exists in the form of NH4+ at a low solution pH; the hydrogen ions (H+) are in excess and strongly compete with the NH4+ ions for the active sites on the as-prepared adsorbents in the solution. As the pH increased, the adsorption efficiency of NH4+-N increased because of the electrostatic attraction interaction between the negatively charged surface of the as-prepared adsorbents and the positively charged surface of NH4+-N under basic conditions, leading to enhanced NH4+-N adsorption. Subsequently, most NH4+ ions could be transformed into aqueous NH3 at a very high pH, resulting in a reduction in the amount of NH4+-N removed.28 In addition, this finding is notable since the final solution pH almost did not change in comparison with the initial pH after the adsorption process, which indicated that the adsorbents were pH-independent; this can help avoid secondary pollution on account of the adsorbent materials and also promote the practical applications in actual water bodies.29
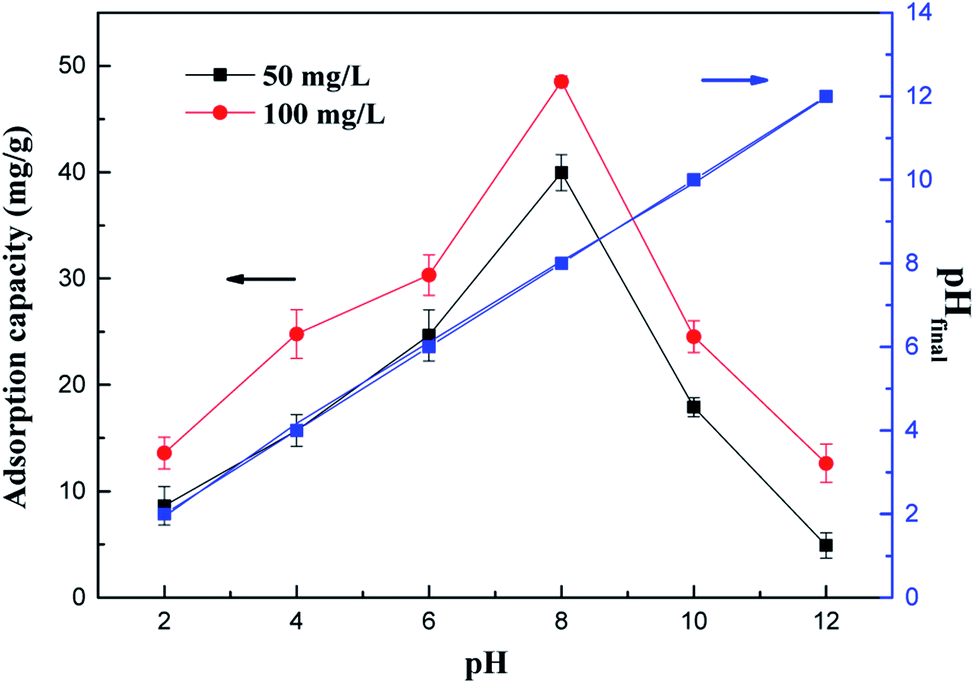 | ||
| Fig. 2 The effect of pH on the removal efficiency and pHfinal of the adsorbent. | ||
As a result, the optimum pH for NH4+-N adsorption was found to be around 8 and the subsequent adsorption experiments were performed at this pH value.
3.4. Effect of contact time on the adsorption process
Fig. 3 shows the adsorption capacity for NH4+-N on the three prepared materials. At the initial stage, it was observed that the rapid increase in the solution phase concentration was followed by a slow increase beyond 60 min in the as-prepared adsorbents, and adsorption equilibrium was almost achieved within the initial 60 min. This above result may be explained due to the saturation of active sites present on the surface of the adsorbent as the adsorption process within a relatively short period of time.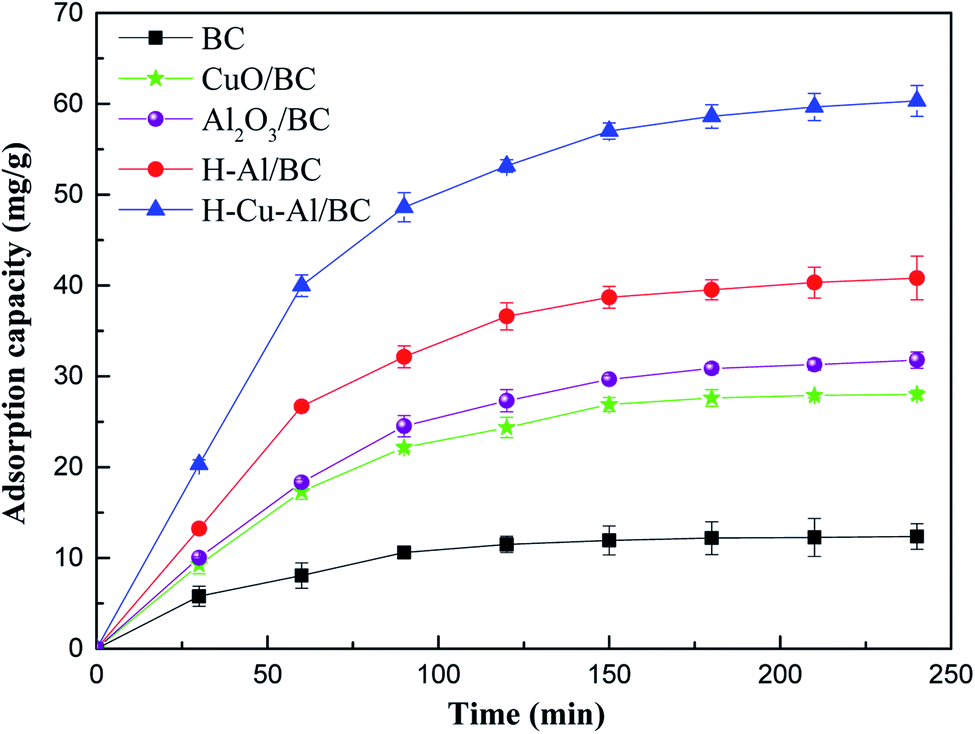 | ||
| Fig. 3 The removal capacity of NH4+-N on the as-prepared materials (initial concentration of 100 mg L−1). | ||
Besides, it is obvious that the adsorptive properties of the samples increased significantly with the incorporation of Cu–Al oxides into the BC frame, and the equilibrium adsorption capacity of H–Cu–Al/BC reached 62.3 mg g−1, which was significantly higher than those of raw BC and H–Al/BC. For comparison, the equilibrium adsorption capacity followed the order H–Cu–Al/BC > H–Al/BC > Al2O3/BC > CuO/BC. This improvement in the adsorption amount of NH4+-N can be explained as follows: the structure property of the absorbents was related to the large surface area and high total pore volume, and the synergistic effect of Cu–Al was also a key factor. When the aluminum and copper metal oxides loaded on the BC surface aggregated, new surfaces and channels were formed, leading to higher porosity and larger specific surface for both H–Al/BC and H–Cu–Al/BC in comparison with that for BC.
Additionally, H–Cu–Al/BC showed a faster adsorption rate and higher adsorption capacity for NH4+-N than those of H–Al/BC and BC. The adsorption capacity of the as-prepared materials for NH4+-N followed the order H–Cu–Al/BC > H–Al/BC > BC. There are two main reasons for this phenomenon. The first may be because the aluminum and copper nanocrystals and BC carbon nanorods are simultaneously constructed by the dispersion and structure-directing effects of H–Cu–Al/BC, which can provide abundant adsorption sites and effectively facilitate the adsorption of NH4+-N. The other reason may be mainly due to the synergistic effect and other adsorption mechanisms between the Cu–Al metal oxides and BC carbon in the NH4+-N pollutant removal process.30
3.5. Adsorption equilibrium isotherms
The three adsorption isotherms of the as-prepared materials for NH4+-N were investigated. As shown in Table S2,† the highest correlation coefficients for NH4+-N on the three materials indicate that the adsorption data are best described by the Langmuir isotherm model in comparison with the Freundlich and Temkin models; this implies that the energy distribution on the three materials is homogeneous and that the uptake of NH4+-N is likely to be achieved by chemical adsorption. This result also demonstrates that the adsorption of NH4+-N takes place on homogeneous sites and monolayer adsorption occurs on the surface of the as-prepared materials. The Langmuir constant (KL), which is related to the affinity between the active sites of the adsorbent and adsorbate, indicated that H–Cu–Al/BC has higher affinity in comparison to H–Al/BC and pure BC.Additionally, H–Cu–Al/BC showed the best adsorption capacities and fastest removal rates, and its maximum adsorption capacity for NH4+-N could be up to around 81.54 mg g−1; this value was approximately 2 times greater than that of H–Al/BC and 7 times higher than that of BC due to its more abundant pores, specific surface area and some other interaction effects. These results further confirm the favorable effect of the copper ions and copper oxides decorated on H–Cu–Al/BC to improve the sorption capacity for phosphorus compared to that for H–Al/BC and pure BC. A comparison of the maximum monolayer adsorption capacity of NH4+-N with those of various adsorbents is shown in Table 1. The adsorption capacity of H–Cu–Al/BC for NH4+-N was relatively higher than those reported in some previous works.
3.6. Adsorption kinetics
The experimental data are shown in Fig. S8† and the kinetic model parameter results are summarized in Table S3.† The experimental results demonstrate that the kinetics may slightly affect the performance of the NH4+-N adsorption process, which may include both surface sorption and intra-particle diffusion processes, and it might be that the surface sorption along with intra-particle diffusion occurs simultaneously; nevertheless, the adsorption process is accomplished within a short period of contact time. The plots of the kinetics data are multilinear and consist of three linear segments for NH4+-N during the adsorption process, indicating that some other mechanism may also play a significant role in the adsorption process.The first sharp phase can be attributed to boundary layer diffusion due to the mass transfer from NH4+-N in solution to the external surface of the as-prepared adsorbents, where the NH4+-N molecules can easily occupy most available adsorption active sites on the adsorbent surface; thus, the rate of the process is very fast. In addition, the initial rapid adsorption could be due to electrostatic attraction, which led to the fast transportation of the NH4+-N molecules from the solution onto the surface of the adsorbent. The second part showed a gradual adsorption step, and this phenomenon can be ascribed to the adsorption on the exterior surface reaching saturation; the NH4+-N molecules slowly diffused from the surface sites and entered the inner pores of the as-prepared adsorbents by intraparticle diffusion through the pores. The plots of qt versus t1/2 do not pass through the origin and have an intercept value C, which indicates the thickness of the boundary layer.33
Furthermore, as shown from Table S3,† the NH4+-N sorption on the as-prepared adsorbents follows the pseudo-second-order model with the highest R2 value compared to the pseudo-first-order and intra-particle diffusion models; the calculated qe,cal value from the pseudo-second-order model is close to the experimental values (qe,exp). This indicates that the pseudo-second-order model is the most suitable in describing the kinetics of NH4+-N on the as-prepared adsorbents, representing monolayer adsorption; thus, the rate of sorption is dominated by chemisorption in the adsorption process. For the pseudo-second-order model, it was found that the initial adsorption rate (k2qe2) showed a tendency to increase with the initial concentration. According to the results of the non-linear relation between the rate constants and the initial concentrations, intraparticle diffusion is not the rate-limiting step of the adsorption process. This may involve some mechanisms in the whole process, including ion exchange and physical adsorption.34
3.7. Recycle studies
Recyclability is one of the most fundamental issues to consider when determining the adsorption mechanism and evaluating the reusability of a spent adsorbent after recovering the pollutant extracted from the aqueous solution.35 As seen from Fig. 4, high-efficiency regeneration of the spent adsorbent was achieved using an HCl solution as the eluent for desorption. The main reason for this phenomenon is that the HCl solution plays an important role in the adsorption of NH4+-N, which can be reused. In addition, it was observed that the desorption efficiency of the spent H–Cu–Al/BC decreased, and NH4+-N desorbed from the untreated H–Cu–Al/BC was 5 to 15.0% of NH4+-N measured in the desorption experiments. In addition, the results showed that the adsorption capacity of NH4+-N decreased gradually with an increase in the number of regeneration cycles. After the 2nd cycle, the efficiency of H–Cu–Al/BC in the removal of NH4+-N was found to be above 89%. With further cycling, this value decreased to 82.7% after the 4th cycle. However, the adsorption capacity of the regenerated adsorbent was still found to be 79.54% without obvious efficiency loss after five consecutive cycles under similar conditions. Thus, it was concluded from the results that the H–Cu–Al/BC sorbent can still maintain a good reusability adsorption performance, especially for the subsequent five cycles, nearly without a dramatic decline in efficiency, indicating that H–Cu–Al/BC exhibits excellent reusability and chemical stability.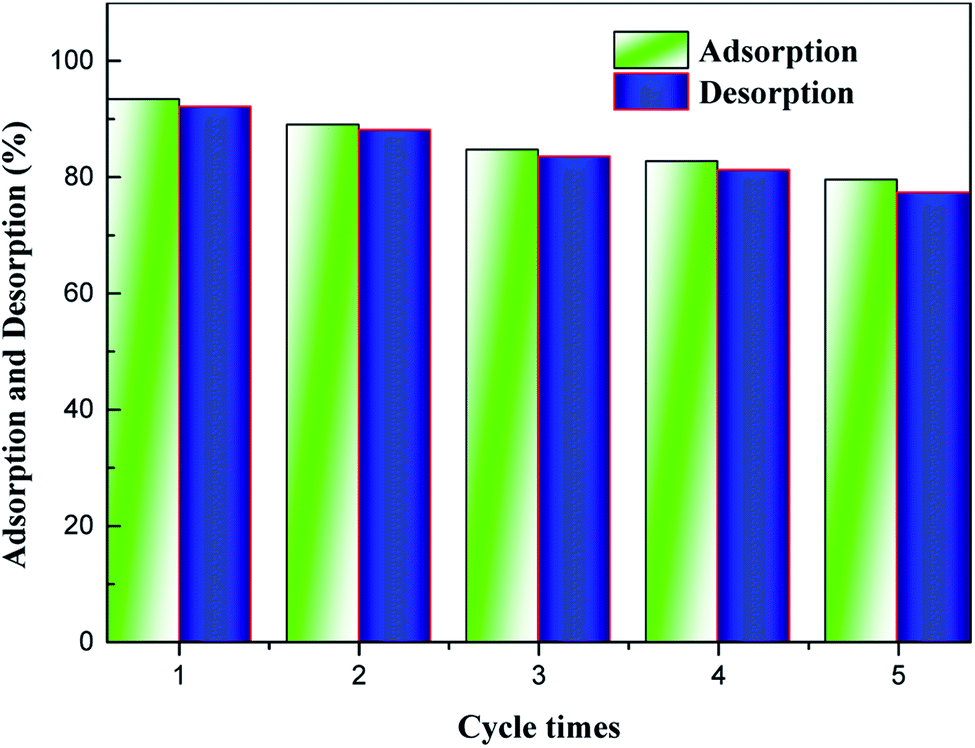 | ||
| Fig. 4 Effect of regeneration cycles on the uptake of NH4+-N by H–Cu–Al/BC. | ||
3.8. Application in simulated actual water samples
Actual groundwater and wastewater contain a combination of different substances; the presence of different substances including various salts or co-ions can affect the adsorption properties of NH4+-N, which is non-negligible in aqueous solutions. Also, humic acid (used as NOM) is ubiquitous in water bodies, which can act as a radical scavenger by competing for NH4+-N. Considering that K+, Na+, Ca2+, Mg2+ and Al3+ are the common coexisting ions present in real water, the as-prepared adsorbents were also used in real wastewater samples containing NH4+-N in order to evaluate their practical applications.36 First, 0.1 g of the as-prepared adsorbent was added to 100 mL simulated wastewater at 25 °C for 24 h for the adsorption test, which contained different salts or actual wastewater (the presence of both co-ions and NOM). The components of the simulated wastewater are presented in Table S4;† the results for the removal of NH4+-N from the wastewater (containing a mixture of NOM, K+, Na+, Ca2+, Mg2+ and Al3+) and DI water are displayed in Fig. S9.†As shown in Fig. S9,† it can be clearly seen that the presence of salts or actual wastewater (the presence of both co-ions and NOM) has varying degrees of influence on the adsorption of NH4+-N. The ions can affect the electrostatic interactions since electrolytes can compete with adsorbates for electrostatic sites. The presence of the ions or NOM had a negative influence on the removal of NH4+-N. Among them, the presence of equivalent amounts of Al3+ ions and NOM obviously inhibited the NH4+-N adsorption, and cations such as K+, Na+, Ca2+ and Mg2+ showed no obvious effect on the NH4+-N adsorption. On the other hand, the influence of NOM was much more significant than that of the ions. The decrease in adsorption capacity may be because both NOM and cations in solution could fiercely compete with NH4+-N for certain active sorption sites on the adsorbent surface, leading to remarkable decrease in the NH4+-N removal efficiency. It is worth noting that the adverse effects of the NH4+-N adsorption were obtained for a mixture of multivalent cation solution compared with individual ions or NOM, which was mainly due to the strong interaction between the as-prepared nanocomposite material surface and multivalent anions. In addition, anion exchange can easily occur at sorption active sites, which results from the H-bonding interactions between the salt anion and the nanocomposite material, leading to lower NH4+-N adsorption efficiency.37
It is clearly evident that the performance of the NH4+-N removal by H–Cu–Al/BC was much better than those of H–Al/BC and pure BC. Particularly, it is noteworthy that both high and low NH4+-N effluent concentrations of wastewater could be effectively treated by H–Cu–Al/BC and H–Al/BC; the effluent with a low NH4+-N concentration met the Chinese Standard for the permissible limit of discharge concentration and was further far below the level of the WHO standards (0.5–1.0 ppm), indicating that the as-prepared adsorbents could be recommended as potential adsorbents to effectively remove NH4+-N in practical water treatment.
3.9. Mechanisms of NH4+-N adsorption on the prepared materials
The amount of NH4+-N adsorbed by an adsorbent in the adsorption process is related to its own unique structure and in principle is proportional to its own surface physical and chemical properties.38 The BET analysis result showed that an increase in the BET surface area and total pore volume of the as-prepared adsorbents led to an increase in the number of adsorption sites available to adsorb the NH4+-N molecules, which increased the ability to adsorb ammonia nitrogen. Besides, the molecular dimensions of NH4+-N are 0.321 × 0.123 × 0.217 nm; it can easily reach and enter the channels or inter-pores of the as-prepared materials with the average pore sizes of around 4–5 nm.Moreover, according to the above discussion, it can be concluded that cation exchange is the main mechanism for the adsorption of NH4+-N. The adsorption process can be expressed as follows:5,31
| Cu2+,Al(adsorbent)3+ + NH4(solution)+ → NH4(adsorbent)+ + Cu2+,Al(solution)3+ | (12) |
The above result indicates that the NH4+-N adsorption is not only governed by the cation exchange process but also probably controlled by other processes; these include electrostatic interactions, planar aromatic structures, chemical bonding, hydrogen bonds, π–π electron donor–acceptor interactions, complexation and ion synergistic interactions and they are the most likely forces that contribute to the binding, which could be the adsorption mechanism interactions between the as-prepared materials and NH4+-N that also contribute to controlling the NH4+-N adsorption on the as-prepared adsorbents from aqueous solutions.12 The above four interactions synergistically contributed to the high adsorption capacity and removal efficiency for the NH4+-N adsorption on H–Cu–Al/BC (Scheme 1). As can be seen from Table S1,† H–Cu–Al/BC has abundant pores, which are composed of macro-, meso- and micro-pores to form a hierarchical porous structure. The pore structure including the BET specific surface area and the total volume increased with the successful incorporation of aluminum and copper oxides, which could provide more active sites for the NH4+-N molecules.39 The above trends may be explained by the rich void structure of the as-prepared materials, which allowed the NH4+-N molecules to move easily from the exterior and interior layers to the active sites. H–Cu–Al/BC and H–Al/BC possess Al–O−, which presents a negative charge surface character, and NH4+-N shows a strongly positive charge; thus, the mechanism of the electrostatic interactions between both the NH4+-N molecules and the two adsorbents might contribute to the adsorption process. Furthermore, the presence of a non-negligible portion of copper and aluminum oxides can also be involved in the synergetic effect with NH4+-N during the adsorption process. In addition, the Cu content loaded on the adsorbent is important for the removal of NH4+-N via complexation.32
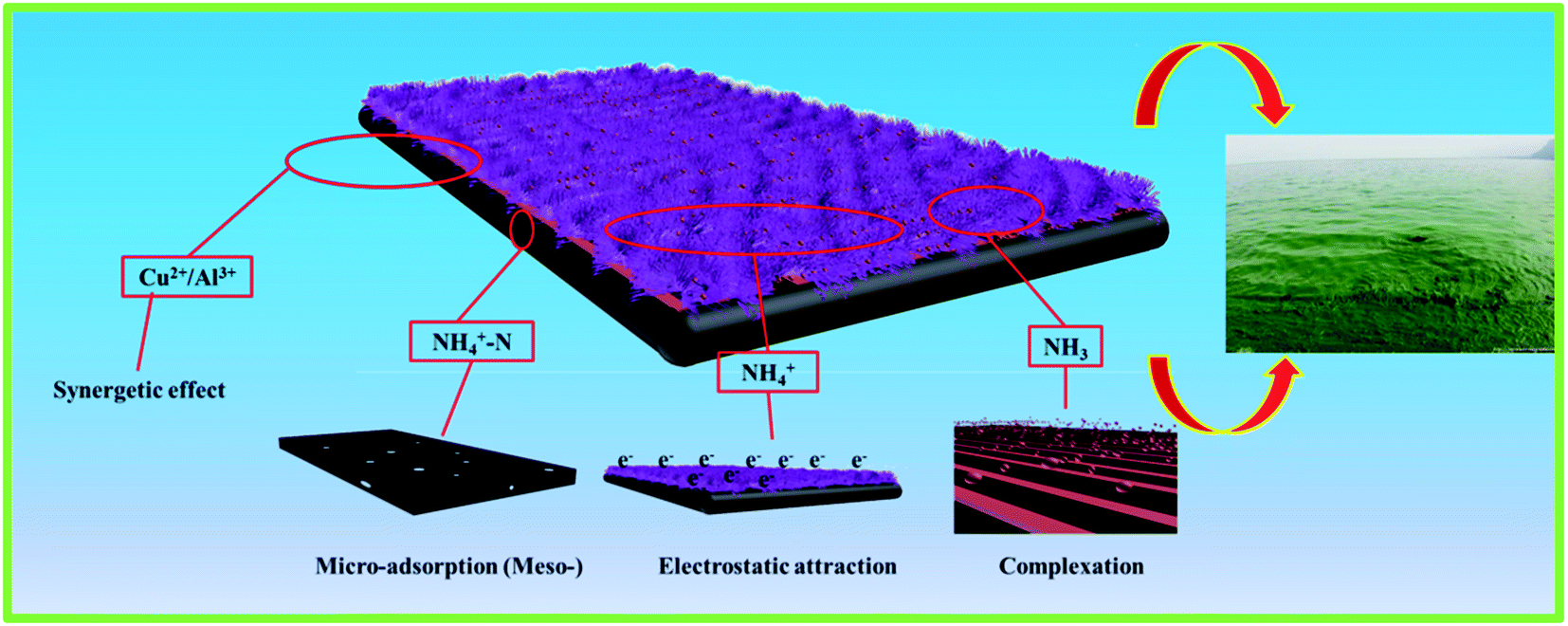 | ||
| Scheme 1 The possible major adsorption mechanism of H–Cu–Al/BC. | ||
According to the above discussion, this can be further confirmed by the fact that the ion exchange, complexation and synergetic effects are the dominant mechanisms for the adsorption of the NH4+-N molecules onto the adsorbent.
3.10. Future work
In the future, it is necessary to evaluate the cycle recovery and availability of the captured recovered nitrogen compounds directly or indirectly for applications in agriculture. Thus, the application of environment-friendly biomass carbon adsorption materials in the removal of ammonia nitrogen shows a good prospect, and it can effectively ameliorate environmental eutrophication issues such as in water bodies and improve the quality of real water. The wastewater emission standard in China has become increasingly strict, especially for environmentally sensitive areas. Thus, to satisfy this strict discharge standard, as discussed above, further studies will focus on the integration of safe drinking water supply and conventional wastewater treatment with ammonium recovery to increase the economic feasibility of the wastewater treatment and improve the sustainability of the wastewater treatment facilities.Besides, this research work can serve as a good reference point and precedence to further research interest on nutritive substance adsorption and the mechanism behavior by different groups of biomass carbon. In addition, it is still a big challenge to remove nutritive substances from low concentration wastewater safely, economically and eco-friendly.
4. Conclusion
In this study, a series of nanocomposites were successfully prepared via the co-precipitation and hydrothermal methods; subsequently, the performance of the nanocomposites for NH4+-N sorption from aqueous solutions was also investigated in batch experiments. The aluminum and copper oxides developed and created a porous structure by catalyzing the dehydration reaction and promoting the depolymerization reaction. The synthesized H–Cu–Al/BC was found to have a certain number of mesopores, with a narrow average pore size distribution of 3.94 nm and a BET surface area of 302.3 m2 g−1. All the as-prepared materials exhibited excellent adsorption properties and high efficiency for the removal of NH4+-N from aqueous solutions. However, H–Cu–Al/BC exhibited better adsorption capacity and adsorption affinity towards NH4+-N than pure BC and H–Al/BC under the same experimental conditions. The adsorption capacity of H–Cu–Al/BC was 7 times higher than that of pure BC. Furthermore, it had a good fit with the Langmuir model and the maximum adsorption capacity followed the order H–Cu–Al/BC > H–Al/BC > pure BC. These results imply that the as-prepared adsorbents can have a potential application prospect in the field of wastewater treatment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by The Natural Science Foundation of Jiangxi Province (No. 20192BAB206038, 20192BAB216029); Science and Technology Support Program of Jiangxi Province (No. 20171BAB206047); Provincial Key Laboratory of Jiangxi Province (No. 20192BCD40013).References
- D. C. Gooddy, D. J. Lapworth, S. A. Bennett, T. H. E. Heaton, P. J. Williams and B. W. J. Surridge, Water Res., 2016, 88, 623–633 CrossRef.
- Z. Tian, Y. Qiu, J. Zhou, X. Zhao and J. Cai, Mater. Lett., 2016, 180, 162–165 CrossRef CAS.
- X. Zhang, Y. Zhou, B. Yu, N. Zhang, L. Wang, H. Fu and J. Zhang, Chem. Eng. J., 2017, 328, 152–158 CrossRef CAS.
- H. Huo, H. Lin, Y. Dong, H. Cheng, H. Wang and L. Cao, J. Hazard. Mater., 2012, 229–230, 292–297 CrossRef CAS.
- X. Peng, M. Wang, F. Hu, F. Qiu, H. Dai and Z. Cao, J. Alloys Compd., 2019, 770, 1055–1063 CrossRef CAS.
- P. Duan, T. Ma, Y. Yue, Y. Li, X. Zhang, Y. Shang, B. Gao, Q. Zhang, Q. Yue and X. Xu, Environ. Sci.: Nano, 2019, 6, 1799–1811 RSC.
- B. Wang, J. Lehmann, K. Hanley, R. Hestrin and A. Enders, Chemosphere, 2015, 138, 120–126 CrossRef CAS.
- W. Abou Saoud, A. A. Assadi, M. Guiza, A. Bouzaza, W. Aboussaoud, I. Soutrel, A. Ouederni, D. Wolbert and S. Rtimi, Chem. Eng. J., 2018, 344, 165–172 CrossRef CAS.
- Y. Q. Huang, C. P. Yang, Z. C. Sun, G. M. Zeng and H. J. He, RSC Adv., 2015, 5, 11475–11484 RSC.
- C. Y. Zhu, W. L. Yang, H. J. He, C. P. Yang, J. P. Yu, X. Wu, G. M. Zeng, S. Tarre and M. Green, Chemosphere, 2018, 200, 380–387 CrossRef CAS.
- L. Zhang, Z. Wang, X. Xu, C. Chen, B. Gao and X. Xiao, Sci. Total Environ., 2019, 653, 897–907 CrossRef CAS PubMed.
- X. Peng, F. Hu, T. Zhang, F. Qiu and H. Dai, Bioresour. Technol., 2018, 249, 924–934 CrossRef CAS.
- Z. S. Birungi and E. M. N. Chirwa, J. Hazard. Mater., 2015, 299, 67–77 CrossRef CAS.
- L. Vafajoo, R. Cheraghi, R. Dabbagh and G. McKay, Chem. Eng. J., 2018, 331, 39–47 CrossRef CAS.
- Y. Xiong, J. Xu, W. Shan, Z. Lou, D. Fang, S. Zang and G. Han, Bioresour. Technol., 2013, 127, 464–472 CrossRef CAS.
- M. J. Wu, H. Y. Liu and C. P. Yang, Int. J. Environ. Res. Public Health, 2019, 16, 205–226 CrossRef.
- Y. Cheng, C. P. Yang, H. J. He, G. M. Zeng, K. Zhao and Z. Yan, J. Environ. Eng., 2016, 142, C4015001 CrossRef.
- Z. Sun, X. Qu, G. Wang, S. Zheng and R. L. Frost, Appl. Clay Sci., 2015, 107, 46–51 CrossRef CAS.
- C. Chen, T. Ma, Y. Shang, B. Gao, B. Jin, H. Dan, Q. Li, Q. Yue, Y. Li, Y. Wang and X. Xu, Appl. Catal., B, 2019, 250, 382–395 CrossRef CAS.
- W. Lee, S. Yoon, J. K. Choe, M. Lee and Y. Choi, Sci. Total Environ., 2018, 639, 1432–1439 CrossRef CAS PubMed.
- J. Li, X. Tang, H. Yi, Q. Yu, F. Gao, R. Zhang, C. Li and C. Chu, Appl. Surf. Sci., 2017, 412, 37–44 CrossRef CAS.
- Y. Shang, C. Chen, P. Zhang, Q. Yue, Y. Li, B. Gao and X. Xu, Chem. Eng. J., 2019, 122004, DOI:10.1016/j.cej.2019.122004.
- J. Perez-Ramirez, D. Verboekend, A. Bonilla and S. Abello, Adv. Funct. Mater., 2009, 19, 3972–3979 CrossRef CAS.
- J. Wu, L.-L. Ma and R. J. Zeng, Chemosphere, 2018, 211, 608–616 CrossRef CAS.
- J. L. Li, S. H. Zhang, Y. Chen, T. M. Liu, C. Y. Liu, X. Zhang, M. T. Yi, Z. Y. Chu and X. X. Han, RSC Adv., 2017, 7, 29051–29057 RSC.
- W. Tian, X. Kong, M. Jiang, X. Lei and X. Duan, Mater. Lett., 2016, 175, 110–113 CrossRef CAS.
- M. T. Vu, H.-P. Chao, T. Van Trinh, T. T. Le, C.-C. Lin and H. N. Tran, J. Cleaner Prod., 2018, 180, 560–570 CrossRef CAS.
- T. M. Vu, V. T. Trinh, D. P. Doan, H. T. Van, T. V. Nguyen, S. Vigneswaran and H. H. Ngo, Sci. Total Environ., 2017, 579, 612–619 CrossRef CAS.
- S. Mallakpour and V. Behranvand, J. Cleaner Prod., 2018, 190, 525–537 CrossRef CAS.
- T. Zhang, X. Yue, L. Gao, F. Qiu, J. Xu, J. Rong and J. Pan, J. Cleaner Prod., 2017, 144, 220–227 CrossRef CAS.
- F. Yang, L. Sun, W. Xie, Q. Jiang, Y. Gao, W. Zhang and Y. Zhang, Sci. Total Environ., 2017, 607–608, 1391–1399 CrossRef CAS.
- S. P. Boeykens, M. N. Piol, L. Samudio Legal, A. B. Saralegui and C. Vázquez, J. Environ. Manage., 2017, 203, 888–895 CrossRef CAS.
- S. Berner, P. Araya, J. Govan and H. Palza, J. Ind. Eng. Chem., 2018, 59, 134–140 CrossRef CAS.
- Z. Wan and K. Li, Chemosphere, 2018, 194, 370–380 CrossRef CAS.
- C. Wan, S. Ding, C. Zhang, X. Tan, W. Zou, X. Liu and X. Yang, Sep. Purif. Technol., 2017, 180, 1–12 CrossRef CAS.
- C. Fang, X. Zhang, Y. Lei, Y. Yuan and Y. Xiang, J. Cleaner Prod., 2018, 181, 618–630 CrossRef CAS.
- M. J. Manto, P. Xie, M. A. Keller, W. E. Liano, T. Pu and C. Wang, Chemosphere, 2018, 198, 501–509 CrossRef CAS PubMed.
- J. Huang, N. R. Kankanamge, C. Chow, D. T. Welsh, T. Li and P. R. Teasdale, J. Environ. Sci., 2018, 63, 174–197 CrossRef CAS.
- Q. Yin, R. Wang and Z. Zhao, J. Cleaner Prod., 2018, 176, 230–240 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04385c |
| This journal is © The Royal Society of Chemistry 2019 |