
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCytotoxic and antimicrobial indole alkaloids from an endophytic fungus Chaetomium sp. SYP-F7950 of Panax notoginseng†
Fei Pengab,
Shao-Yang Houa,
Tian-Yuan Zhanga,
Ying-Ying Wua,
Meng-Yue Zhanga,
Xi-Meng Yana,
Ming-Yu Xiaa and
Yi-Xuan Zhang *a
*a
aSchool of Life Science and Biopharmaceutics, Shenyang Pharmaceutical University, Shenyang 110016, PR China. E-mail: zhangyxzsh@163.com
bQuanzhou Medical College, Quanzhou, PR China
First published on 12th September 2019
Abstract
Two new compounds chetoseminudin F (1) and G (2) together with eleven known compounds were isolated from the solid fermentation products of the endophytic fungus Chaetomium sp. SYP-F7950. The structures of the isolated compounds were elucidated by extensive spectroscopic analyses, including 1D and 2D NMR, and HRFABMS experiments. The absolute configurations of chetoseminudin F (1) and G (2) were determined by comparing the electronic circular dichroism (ECD) spectrum with those of the reported references. A plausible biogenetic pathway for compounds 1–6 and 9–13 was proposed. These isolates were also evaluated for their antimicrobial and antitumor activity, revealing that chetoseminudin F (1) displayed more potent cytotoxicity against MDA-MB-231 cells with an IC50 value of 26.49 μmol L−1 more than the common chemotherapeutic agent (paclitaxel). In antimicrobial assay, compounds 6, 9, 11 and 12 had strong antibacterial activity against Staphylococcus aureus, Bacillus subtilis, Enterococcus faecium and antifungal activity against Candida albicans with minimum inhibitory concentration (MIC) values ranging from 0.12 to 9.6 μg mL−1; meanwhile compounds 6, 8, 9 and 12 exhibited strong cytotoxicity with IC50 values of 2.75–8.68 μmol L−1 against tumor cell lines A549 and MDA-MB-231. In addition, morphological observation showed that treatment with compounds 6, 9 and 12 increased the mean length of B. subtilis by 1.6 to 1.8-fold. In silico molecular docking was applied to study the binding interactions between the compounds and the active sites of filamentous temperature-sensitive protein Z (FtsZ) from B. subtilis. Compounds 6, 9 and 12 displayed the low binding energies, strong H-bond interactions with FtsZ. On the basis of the antimicrobial activities, cellular phenotype observation and docking studies, compounds 6, 9 and 12 are considered to be a promising antimicrobial inhibitor of FtsZ.
Introduction
Chaetomium, which includes about 400 species in the Index Fungorum (http://www.indexfungorum.org/Names/Names.asp) since Kunze first established this genus in 1817,1 is one of the largest genera in Chaetomiaceae family.1 It is widely distributed in different biotopes, such as soil, marine, animal dung, hair, textile, plant seeds and some other substrates being rich in cellulose. Thus, Chaetomium spp. are often used to produce cellulose in industry, and are also used as biocontrol agents in agriculture. Since chaetomin was firstly reported from Chaetomium sp. in 1944,2 a wide variety of compounds have been reported in this genus such as armochaetoglobins A–J,3 xanthenone,4 chaetoglobins,5 and orsellides,6 some of which showed interesting biological activities including cytotoxic, antibacterial, antifungal and anti-inflammatory activities.7–9Panax notoginseng F. H. Chen (Araliaceae) is a valued traditional Chinese medicinal herb,10 which as a perennial plant should be grown in the field for at least three years to obtain high-quality raw roots.11 P. notoginseng is vulnerable to be attacked by soil-borne pathogens including fungi, bacteria, and nematodes due to the required long-term continuous cultivation.12,13 Endophytic fungi play a key role in plant defense and could be used as a promising source for biocontrol agents.14 There is little doubt that secondary metabolites of endophytic fungi play important roles in the antagonism against many pathogens.15,16 As part of our ongoing researches to discovery for new bioactive metabolites from endophytic fungi from P. notoginseng,17–19 a fungus, Chaetomium sp. SYP-F7950, was isolated from the stem of Panax notoginseng collected in Wenshan district of Yunnan province of China. The large-scale fermentation followed by combinatorial chromatography led to isolation of two new compounds, chetoseminudin F (1) and chetoseminudin G (2), together with eleven known compounds (3–13) (Fig. 1). Meanwhile, a plausible biogenetic pathway for compounds 1–6 and 9–13 was proposed. Therefore, the aims of this study are to discover the diversity of endophytic fungi of P. notoginseng and to obtain antimicrobial secondary metabolites from this strain.
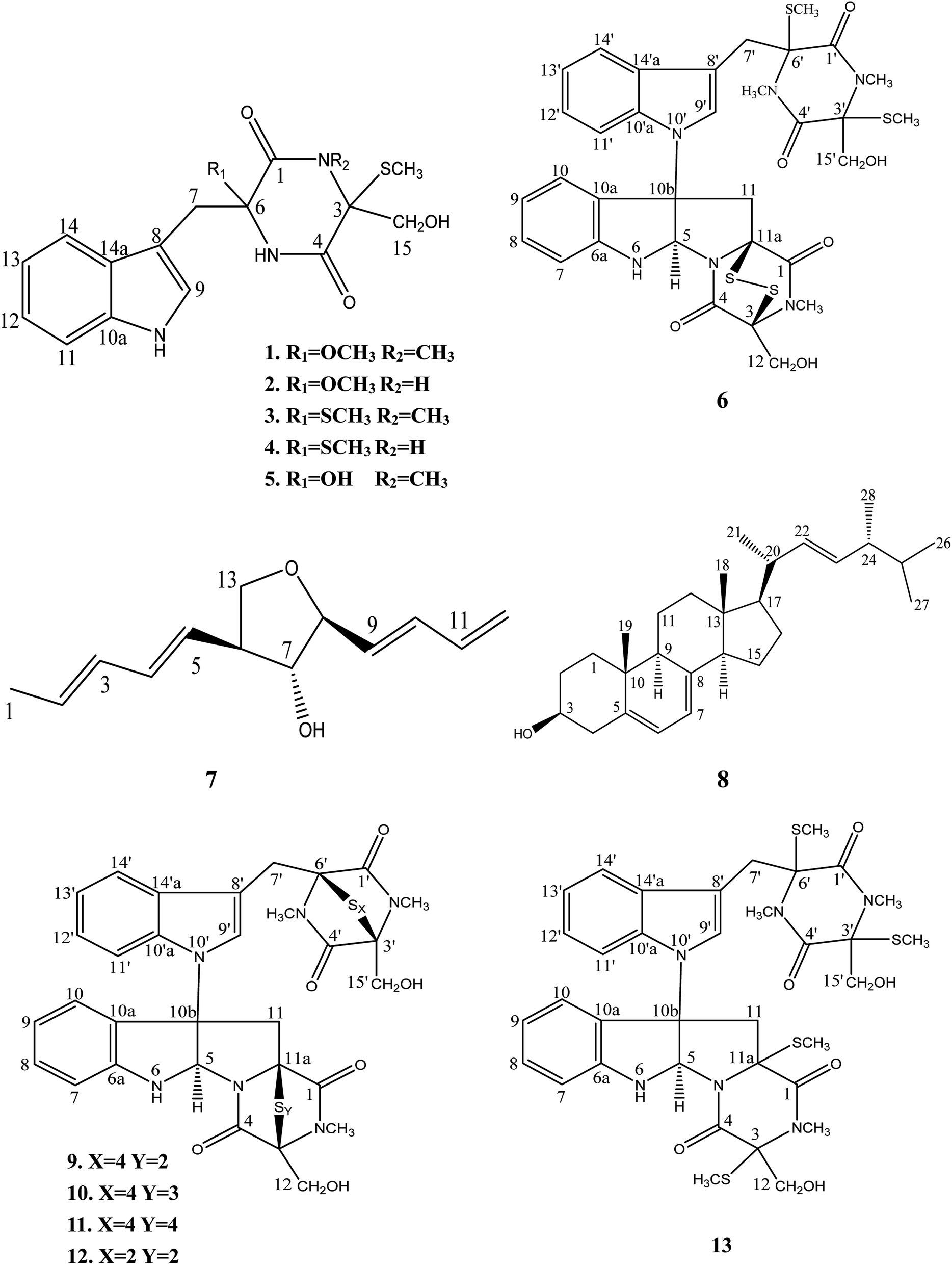 | ||
| Fig. 1 Chemical structure of compounds 1–13 isolated from Chaetomium sp. SYP-F7950. | ||
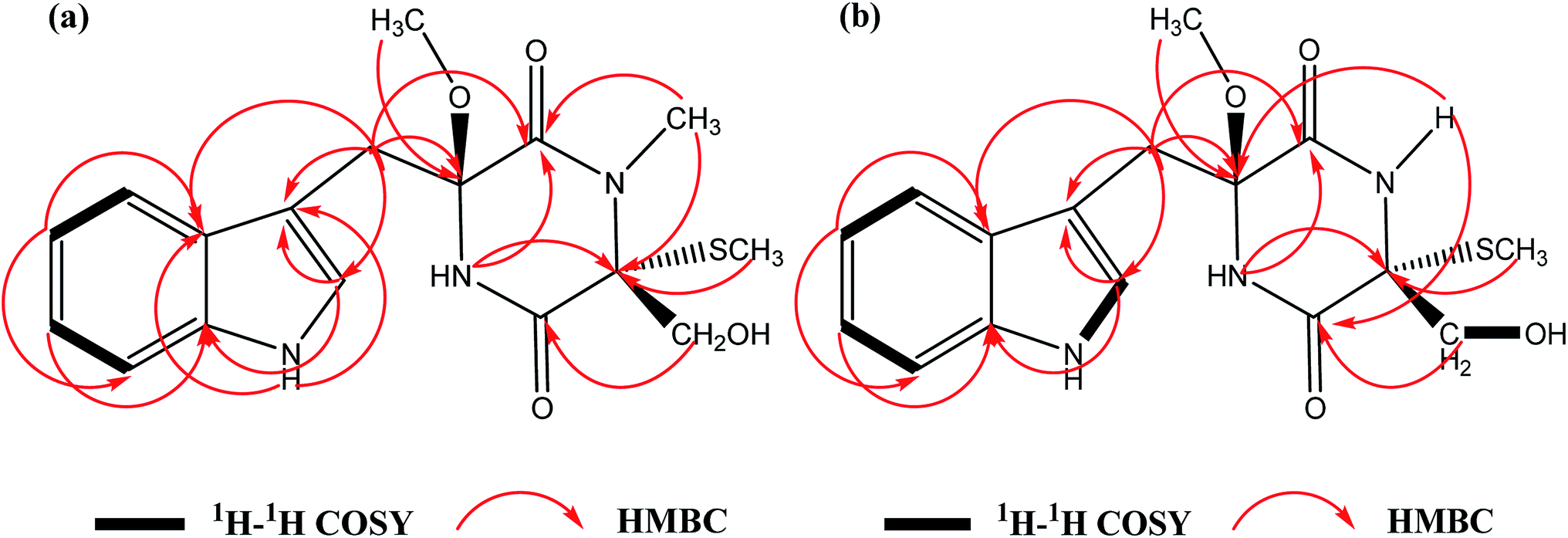 | ||
| Fig. 2 Key 2D correlations of chetoseminudin F (1, a) and G (2, b). | ||
The rise and rapid spread of antibiotic resistance renders it necessary to discovery novel targets and new drug classes. In recent years, bacterial cell division has attracted considerable attention as a potential target pathway for antibiotic treatment. FtsZ is considered to be the major protein of the bacterial cell division machinery (divisome).20,21 FtsZ is an appealing target for new antibacterial drug discovery due to several key reasons. First, it is an essential protein for bacterial viability.22 Second, FtsZ protein is a potentially broad-spectrum antibacterial target because it is highly conserved and has been identified in most bacteria. Third, FtsZ is not present in higher eukaryotes,23 which suggests that FtsZ inhibitors should not be toxic to human cells. All of these suggests that FtsZ may be amenable to inhibitor development. In our study, three indole alkaloids were identified as potential inhibitors of FtsZ by morphological observation and molecular docking, which explore the possible binding mechanism between the lead compounds and FtsZ.
Results and discussion
Compound 1 was obtained as a yellow powder. The UV spectrum of compound 1 gave the absorption of an indole system (274, 281, and 290 nm) (Fig. S10†), and the IR spectrum showed the absorption of OH and NH (3380 and 3225 cm−1), CH3 (2923 cm−1) and amide![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) C
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups (1641 cm−1) (Fig. S12†). The molecular formula of 1 was calculated to be C17H21N3O4S with the molecular weight of m/z 386.1148 [M + Na]+ (calcd for 386.1145) by HRFABMS with 8 degrees of unsaturation. The 1H NMR and 13C NMR spectral data of 1 (Table 1) revealed 17 carbons, including 3 methyl groups, 2 methylenes, 5 methines and 7 quaternary carbons. A comparison of the spectroscopic data between 1 and chetoseminudin E24 indicated their similarities except that compound 1 possessed a CH3–O group (δH 3.17; δC 50.9) at 6 position instead of –OH group in chetoseminudin E, which was further confirmed by the long-range HMBC correlations from CH3–O (δH 3.17) to C6 (δC 87.6). In the NMR spectra of 1, three CH3 signals at δH/δC 2.80/28.3, 0.43/8.3 and 3.17/50.9 were assigned to CH3–N, CH3–S and CH3–O, respectively, and the two CH2 signals at δH/δC 3.02, 3.49/35.2 and 3.42, 3.73/62.8 were assigned to CH2-7 and CH2-15, respectively, by HMBC correlations (Fig. 2a) Examination of the 1H and 13C NMR spectra of 1 showed the presence of a 3-substituted indolyl group [δH 7.04 (1H, d, J = 6 Hz, H-9), 7.26 (1H, d, J = 6 Hz, H-11), 7.01 (1H, t, J = 6 Hz, H-12), 6.94 (1H, t, J = 6 Hz, H-13) and 7.67 (1H, d, J = 6 Hz, H-14); δC 107.3 (C, C-8), 125.4 (CH, C-9), 135.9 (C, C-10a), 111.1 (CH, C-11), 120.8 (CH, C-12), 118.4 (CH, C-13), 118.9 (CH, C-14) and 128.2 (C, C-14a)]. The long-range HMBC correlations from H-2 (NCH3) to C-1 and C-3, H-3 (SCH3) to C-3, H-15 to C-4, H-5 to C-1 and C-3, H-6 (OCH3) to C6 revealed the presence of a monothiodimethyldioxopiperazine moiety, as shown in Fig. 1. No NOESY correlations were observed to determine the relative configuration of 1. As shown in Fig. S11,† the absolute configuration of 1 was determined by comparing ECD spectrum with those of the reported references, revealing that the cotton effects in the experimental ECD spectrum of 1 were consistent with chetoseminudin E.24 Thus, its absolute configuration was determined as 3R,6S. These data confirmed the chemical structure of compound 1, named as chetoseminudin F, as a new chetoseminudin alkaloid characterized by a 3-substituted indolone integrated with a monothiodimethyldioxopiperazine ring with methoxy-substituted at 6 position.
O groups (1641 cm−1) (Fig. S12†). The molecular formula of 1 was calculated to be C17H21N3O4S with the molecular weight of m/z 386.1148 [M + Na]+ (calcd for 386.1145) by HRFABMS with 8 degrees of unsaturation. The 1H NMR and 13C NMR spectral data of 1 (Table 1) revealed 17 carbons, including 3 methyl groups, 2 methylenes, 5 methines and 7 quaternary carbons. A comparison of the spectroscopic data between 1 and chetoseminudin E24 indicated their similarities except that compound 1 possessed a CH3–O group (δH 3.17; δC 50.9) at 6 position instead of –OH group in chetoseminudin E, which was further confirmed by the long-range HMBC correlations from CH3–O (δH 3.17) to C6 (δC 87.6). In the NMR spectra of 1, three CH3 signals at δH/δC 2.80/28.3, 0.43/8.3 and 3.17/50.9 were assigned to CH3–N, CH3–S and CH3–O, respectively, and the two CH2 signals at δH/δC 3.02, 3.49/35.2 and 3.42, 3.73/62.8 were assigned to CH2-7 and CH2-15, respectively, by HMBC correlations (Fig. 2a) Examination of the 1H and 13C NMR spectra of 1 showed the presence of a 3-substituted indolyl group [δH 7.04 (1H, d, J = 6 Hz, H-9), 7.26 (1H, d, J = 6 Hz, H-11), 7.01 (1H, t, J = 6 Hz, H-12), 6.94 (1H, t, J = 6 Hz, H-13) and 7.67 (1H, d, J = 6 Hz, H-14); δC 107.3 (C, C-8), 125.4 (CH, C-9), 135.9 (C, C-10a), 111.1 (CH, C-11), 120.8 (CH, C-12), 118.4 (CH, C-13), 118.9 (CH, C-14) and 128.2 (C, C-14a)]. The long-range HMBC correlations from H-2 (NCH3) to C-1 and C-3, H-3 (SCH3) to C-3, H-15 to C-4, H-5 to C-1 and C-3, H-6 (OCH3) to C6 revealed the presence of a monothiodimethyldioxopiperazine moiety, as shown in Fig. 1. No NOESY correlations were observed to determine the relative configuration of 1. As shown in Fig. S11,† the absolute configuration of 1 was determined by comparing ECD spectrum with those of the reported references, revealing that the cotton effects in the experimental ECD spectrum of 1 were consistent with chetoseminudin E.24 Thus, its absolute configuration was determined as 3R,6S. These data confirmed the chemical structure of compound 1, named as chetoseminudin F, as a new chetoseminudin alkaloid characterized by a 3-substituted indolone integrated with a monothiodimethyldioxopiperazine ring with methoxy-substituted at 6 position.
| Position | Compound 1 | Position | Compound 2 | ||
|---|---|---|---|---|---|
| δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | ||
| 1 | 165.0 C | — | 1 | 165.3 C | — |
| 2 NCH3 | 28.3 CH3 | 2.80, s, 3H | 2 NH | — | 8.68, s, 1H |
| 3 | 75.2 C | — | 3 | 67.8 C | — |
| 3 SCH3 | 8.3 CH3 | 0.43, s, 3H | 3 SCH3 | 9.7 CH3 | 0.87, s, 3H |
| 4 | 165.5 C | — | 4 | 165.9 C | — |
| 5 NH | — | 9.00, s, 1H | 5 NH | — | 8.72, s, 1H |
| 6 | 87.6 C | — | 6 | 87.7 C | — |
| 6 OCH3 | 50.9 CH3 | 3.17, s, 3H | 6 OCH3 | 50.8 CH3 | 3.18, s, 3H |
| 7 | 35.2 CH2 | 3.02, d, 1H, 12 | 7 | 34.6 CH2 | 2.99, d, 1H, 12 |
| 3.49, d, 1H, 12 | 3.43, d, 1H, 12 | ||||
| 8 | 107.3 C | — | 8 | 107.0 C | — |
| 9 | 125.4 CH | 7.04, d, 1H, 6 | 9 | 125.6 CH | 7.06, d, 1H, 6 |
| 10 NH | — | 10.92, s, 1H | 10 NH | — | 10.91, s, 1H |
| 10a | 135.9 C | — | 10a | 135.9 C | — |
| 11 | 111.1 CH | 7.26, d, 1H, 6 | 11 | 111.1 CH | 7.26, d, 1H, 6 |
| 12 | 120.8 CH | 7.01, t, 1H, 6 | 12 | 120.8 CH | 7.00, t, 1H, 6 |
| 13 | 118.4 CH | 6.94, t, 1H, 6 | 13 | 118.8 CH | 6.94, t, 1H, 6 |
| 14 | 118.9 CH | 7.67, d, 1H, 6 | 14 | 119.0 CH | 7.67, d, 1H, 6 |
| 14a | 128.2 C | — | 14a | 128.2 C | — |
| 15 | 62.8 CH2 | 3.42, d, 1H, 12 | 15 | 65.3 CH2 | 3 28, d, 1H, 12 |
| 3.73, d, 1H, 12 | 3.69, d, 1H, 12 | ||||
Compound 2 was obtained as a yellow powder. The molecular formula of compound 2 was determined to be C16H19N3O4S with the molecular weight of m/z 372.0982 [M + Na]+ (calcd for 372.1096) by HRFABMS, indicating 8 degrees of unsaturation. The UV spectrum of 2 gave the absorption of an indole system (274, 281, and 290 nm) (Fig. S22†), and the IR spectrum of 2 showed the absorption of OH and NH (3419 cm−1), CH3 (2921 cm−1) and amide CO groups (1677 cm−1) (Fig. S24†). The 1H NMR and 13C NMR spectral data of 2 (Table 1) showed that 2 was built up by a combination of a 3-methylindole and monothiodioxopiperazine moieties, and revealed 16 carbons, including 2 methyl groups, 2 methylenes, 5 methines and 7 quaternary carbons. Comparison of NMR spectra of 2 (Table 1) with those of 1 suggested that the CH3–N group at position 2 (δH/δC 2.80/28.3) in 1 was replaced with an H–N group (δH 8.68) in 2, which was confirmed by the long-range HMBC correlations from H–N (δH 8.68) to C4 (δC 165.9) and C6 (δC 87.7). In the NMR spectra of 2, two CH3 signals at δH/δC 0.87/9.7 and 3.18/50.8 were assigned to CH3–S and CH3–O, respectively, and the two CH2 signals at δH/δC 2.99, 3.43/34.6 and 3.28, 3.69/65.3 were assigned to CH2-7 and CH2-15, respectively, by HMBC correlations (Fig. 2b). No NOESY correlations were observed to determine the relative configuration of 2. As shown in Fig. S20,† the absolute configuration of 2 was determined by comparing the electronic circular dichroism (ECD) spectrum with 1, revealing that the cotton effects in the experimental ECD spectrum of 2 were consistent with 1. Thus, its absolute configuration was determined as 3R,6S. All above data confirmed the chemical structure of compound 2 belonging to a new indole alkaloid, named as chetoseminudin G.
Eleven known compounds were identified to be chetoseminudin B (3)25 (Table S1 and Fig. S25–S27†), chetoseminudin C (4)26 (Table S5 and Fig. S28–S30†), chetoseminudin E (5)24 (Table S1 and Fig. S31–S33†), chaetocochin C (6)27 (Table S2 and Fig. S34–S36†), 3-epi-aureonitol (7)28 (Table S3 and Fig. S37–S39†), ergosterol (8)29 (Table S4 and Fig. S40–S42†), chetomin A (9)30 (Table S6 and Fig. S43–S45†), chetomin B30 (10) (Table S6 and Fig. S46–S48†), chetomin C30 (11) (Table S6 and Fig. S49–S51†), chetomin (12)25 (Table S6 and Fig. S52–S54†) and dethiotetra-(methylthio)-chetomin (13)27 (Table S6 and Fig. S55–S57†) by comparing their NMR data and ESI-MS with those reported (see in the ESI†).
The postulated biosynthetic pathway for compounds 1–6 and 9–13 is demonstrated in Fig. 3. For compounds 1–5, all of them began with the condensation of serine and tryptophan by a nonribosomal peptide synthetase,31 followed by C-hydroxylation,32 water elimination,33 sulfurization33 and O- or S-methylation34 to afford compounds 1–5. For compounds 6 and 9–13, they were derived from tryptophan-derived heterodimer via a radical dimerization,35 followed by the peptide coupling reactions with serine and subsequent dioxopiperazine cyclization by a nonribosomal peptide synthetase, N-methylation, water elimination, sulfurization, S-methylation and di or polythiol-oxidation to afford compounds 6 and 9–13 which has been reported previously.31
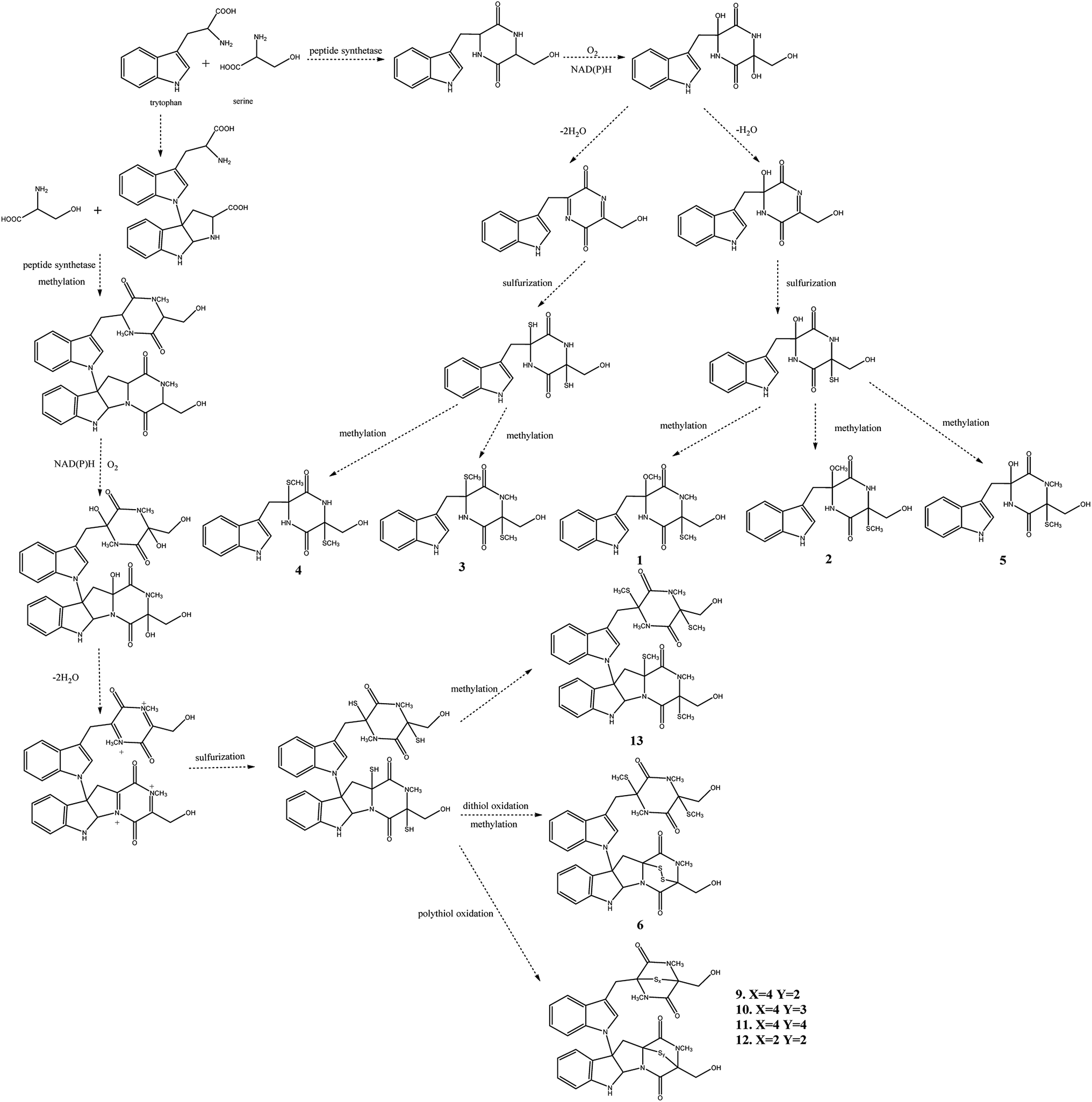 | ||
| Fig. 3 Proposed biosynthetic pathway for compounds 1–6 and 9–13. | ||
Seven test bacteria (Staphylococcus aureus, Bacillus subtilis, Escherichia coli, Enterococcus faecium, Proteus mirabilis, Acinetobacter baumannii and Achromobacter marplatensis) and one fungus (Candida albicans) were selected for antibacterial and antifungal assays (Table 3). In antimicrobial assay, compounds 1 and 2 showed weak inhibitory activity against A. marplatensis and C. albicans with MICs values of 117.7, 76.7, 103.3 and 124.1 μg mL−1 (vancomycin, MIC = 50 μg mL−1 and geneticin, MIC = 6.0 μg mL−1), respectively. However, the above compounds were inactive against another six test microorganisms. At the same time, compounds 6, 9 and 12 exhibited strong inhibitor activities against S. aureus with MIC values of 0.5, 0.12, and 4.3 μg mL−1, respectively. The above compounds also exhibited strong inhibitor activities against B. subtilis with MIC values of 0.25, 0.2 and 2.4 μg mL−1, respectively. Compounds 9, 11 and 12 also displayed strong antagonistic activities against E. faecium with MIC values of 3.6, 4.1 and 3.3 μg mL−1 respectively. In antifungal assay, compounds 11 and 12 significantly inhibited the growth of C. albicans with MIC values of 8.3 and 9.6 μg mL−1 respectively. In anticancer assay, compound 2 exhibited more potent cytotoxic effect against MDA-MB-231 cell with IC50 value of 26.49 μmol L−1 than paclitaxel (Table 2). Meanwhile, compounds 6 and 12 showed obvious cytotoxic activity against MDA-MB-231 with IC50 values of 7.20 and 2.75 μmol L−1. Compounds 6, 8 and 9 displayed obvious cytotoxic activity against A549 with IC50 values of 4.58, 4.84 and 8.68 μmol L−1.
| Substance no. | IC50 (μmol L−1) | |
|---|---|---|
| Cell line | ||
| A549 | MDA-MB-231 | |
| a The cells were cultured with various concentrations of different samples for 48 h. | ||
| 1 | >30 | 26.49 |
| 2 | >30 | >30 |
| 3 | >30 | >30 |
| 4 | >30 | >30 |
| 5 | 20.81 | >30 |
| 6 | 4.58 | 7.20 |
| 7 | >30 | >30 |
| 8 | 4.84 | >30 |
| 9 | 8.68 | >30 |
| 10 | >30 | >30 |
| 11 | >30 | >30 |
| 12 | >30 | 2.75 |
| 13 | 15.66 | 22.30 |
| Paclitaxel | <0.3 | 30.00 |
| Compound (μg mL−1) | MICs (μg mL−1) | |||||||
|---|---|---|---|---|---|---|---|---|
| Staphylococcus aureus | Bacillus subtilis | E. coli | Enterococcus faecium | Proteus mirabilis | Acinetobacter baumannii | Achromobacter marplatensis | Candida albicans | |
| 1 | >200 | >200 | >200 | >200 | >200 | >200 | 76.7 | 124.1 |
| 2 | >200 | >200 | >200 | >200 | >200 | >200 | 117.7 | 103.3 |
| 3 | >200 | >200 | >200 | >200 | >200 | >200 | >200 | >200 |
| 4 | >200 | >200 | >200 | >200 | >200 | >200 | >200 | >200 |
| 5 | >200 | >200 | >200 | >200 | >200 | >200 | >200 | >200 |
| 6 | 0.5 | 0.25 | >200 | 19.3 | >200 | >200 | 29.0 | 12.4 |
| 7 | >200 | >200 | >200 | >200 | >200 | >200 | >200 | >200 |
| 8 | >200 | >200 | >200 | >200 | >200 | >200 | >200 | >200 |
| 9 | 0.12 | 0.2 | >200 | 3.6 | >200 | >200 | 165.8 | 20.4 |
| 10 | 65.2 | 30.5 | >200 | 12.4 | >200 | >200 | 69.1 | 18.8 |
| 11 | 44.3 | 50.0 | >200 | 4.1 | >200 | >200 | 41.8 | 8.3 |
| 12 | 4.3 | 2.4 | >200 | 3.3 | >200 | >200 | 42.6 | 9.6 |
| 13 | 23.4 | 15.0 | >200 | 37.3 | >200 | >200 | 43.3 | 12.6 |
| Vancomycin | 2.5 | 1.5 | 50.0 | 10.0 | 50.0 | 30.0 | 50.0 | — |
| Geneticin | — | — | — | — | — | — | — | 6.0 |
The cells of B. subtilis treated with the compounds 6, 9 and 12 were observed to study the possible antibacterial mechanism in more detail. Interestingly, as shown in Fig. 4, the rod-shaped cells of B. subtilis grew into longer filaments under the treatment of compounds 6, 9 and 12, which reached 1.6, 1.8 and 1.7-fold length of the untreated cells, respectively. To explain this interesting result, the filamentous temperature-sensitive protein Z (FtsZ), the key protein of cell division,36 was applied to illustrate the mechanism of cells that became filamentous. Thus, a molecular docking study was conducted to validate the deduction. The docking results are shown in Table 4 and Fig. 5. The FtsZ from B. subtilis (PDB: ID 2VXY) displayed the docking score (Table 4) of 6, 9 and 12 that were all lower than its ligand, thus indicating better binding abilities. Compound 6 displayed four conventional hydrogen bonds and one carbon hydrogen bonds with neighbouring amino acid residues. Compound 9 also displayed four conventional hydrogen bonds and one carbon hydrogen bonds with neighbouring amino acid residues. The docking scores −158 kcal mol−1 (6) and −156 kcal mol−1 (9), indicate that two compounds may both form lower binding energy and stable binding sites with the target protein. Compound 12 formed four conventional hydrogen bonds with the ALA 71, ALA 73, ASN 25 and GLY 72 residues as listed in Table 4 and Fig. 5. In total, the docking results revealed that compounds 6, 9 and 12 exhibited low binding energies, strong H-bond interactions with the FtsZ from B. subtilis to validate the phenotypic consequence and antimicrobial activity. Therefore, the morphological changes and in silico docking simulations provided strong proof that compounds 6, 9 and 12 are potential inhibitors of FtsZ from B. subtilis.
 | ||
| Fig. 4 Electron micrographs of B. subtilis in the absence or presence of compounds 6, 9 and 12. (A) Untreated control cells of B. subtilis with average length of 4.60 μm. (B–D) Filamentous cells of B. subtilis treated with compounds 6 (B), 9 (C) and 12 (D) showed average length of 7.35, 8.30 and 7.72 μm, respectively. | ||
| Compound | PDB (ID) | Docking score (kcal mol−1) | Residues | |
|---|---|---|---|---|
| Conventional hydrogen bond | Carbon hydrogen bond | |||
| 6 | 2VXY | −158 | ARG143, GLY108, GLY110, THR109 | GLY107 |
| 9 | 2VXY | −156 | ALA71, ALA73, ASN44, THR109 | ASP46 |
| 12 | 2VXY | −154 | ALA71, ALA73, ASN25, GLY72 | |
| Ligand | 2VXY | −83 | ALA71, ALA73, GLY21, GLY108, GLY110, THR109 | GLY107 |
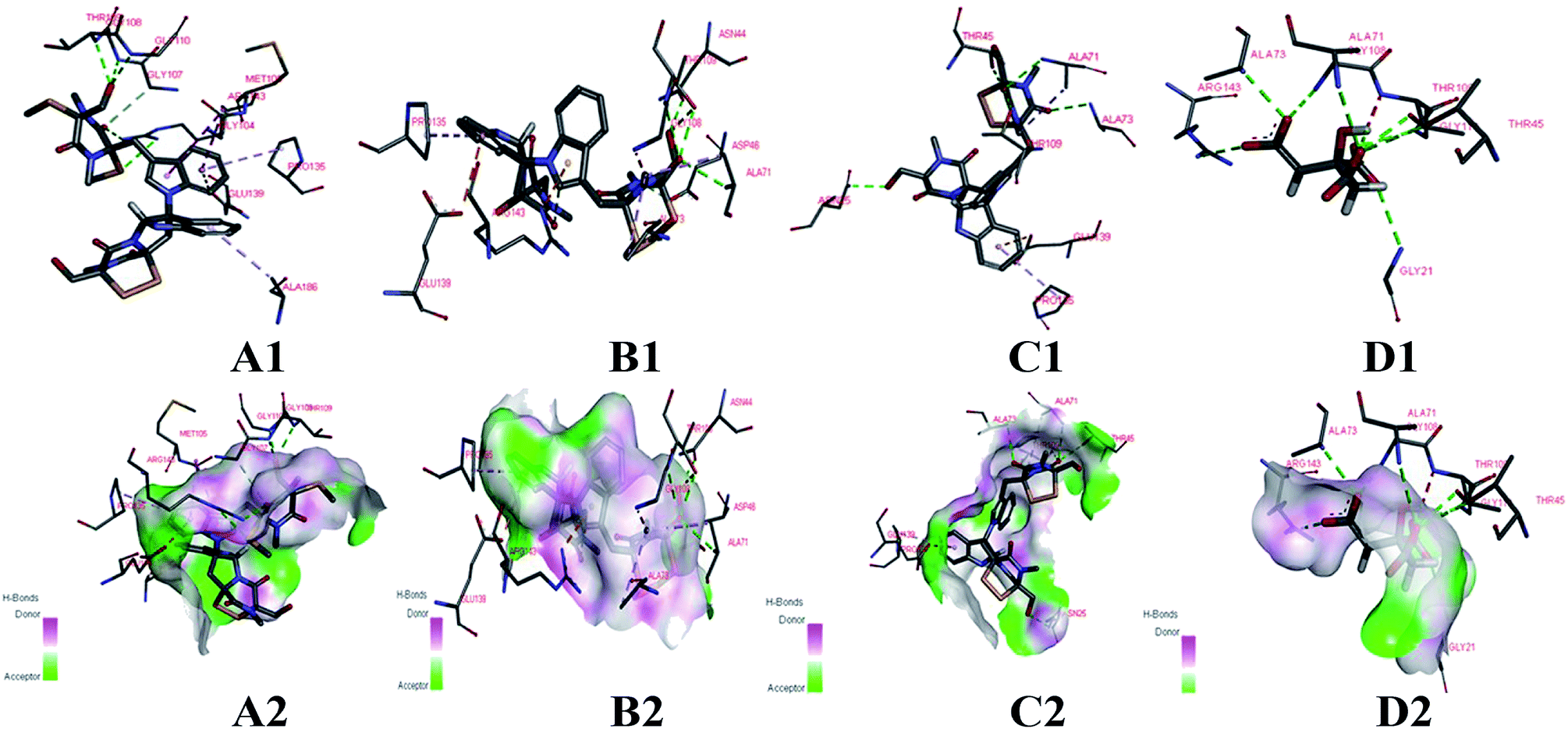 | ||
| Fig. 5 Docking models of compounds 6, 9, 12 and ligand with FtsZ of B. subtilis (PDB ID: 2VXY). (A1, B1, C1 and D1) The binding sites of H-bond or hydrophobic between 6, 9, 12, ligand and FtsZ. (A2, B2, C2 and D2) The 3D docking model with H-bond pocket between 6, 9, 12, ligand and FtsZ. | ||
Conclusion
This study resulted in isolation and identification of two new indole alkaloids, chetoseminudin F (1) and G (2), and eleven known compounds from Chaetomium globosum on the basis of 1D, 2D NMR, HRESIMS. The absolute configuration of chetoseminudin F (1) and G (2) were determined as 3R,6S by comparing the electronic circular dichroism (ECD) spectrum with chetoseminudin E.24 In antimicrobial assays, compounds 6, 9, 11 and 12 had strong antibacterial activity against Staphylococcus aureus, B. subtilis, Enterococcus faecium and antifungal activity against Candida albicans with minimum inhibitory concentration (MIC) values ranging from 0.12 to 9.6 μg mL−1. In antitumor assays, chetoseminudin G (2) displayed more potent cytotoxicity in MDA-MB-231 cells with IC50 value of 26.49 μmol L−1 than the common chemotherapeutic agent (paclitaxel). Meanwhile, compounds 6, 8, 9 and 12 also exhibited strong cytotoxicity with IC50 values of 2.75–8.68 μmol L−1 against cell lines A549 and MDA-MB-231. TEM images indicated the mean length of B. subtilis increased under treatment with compounds 6, 9 and 12. In addition, these compounds showed low binding energies and strong H-bond interactions with the FtsZ from B. subtilis. These results revealed that the antimicrobial mechanism of these compounds against B. subtilis could be achieved by combination with FtsZ, resulting in the inhibition of cell division. On the basis of the antimicrobial activities, phenotypic consequences and docking studies, these indole alkaloids were identified to be a promising antimicrobial lead molecule. Our current findings would greatly expand the chemical and pharmacological diversities of the metabolites of Chaetomium species.Experimental method
General experimental procedures
UV spectra were recorded on a Shimadzu UV 2201 spectrophotometer. ECD spectra were recorded on a Bio-Logic Science MOS-450 spectrometer. Bruker AV-600 spectrometer was used in the NMR experiments. Optical rotations were obtained on PerkinElmer 241. HRESIMS data were acquired on a Bruke TOF mass spectrometer. Preparative HPLC was performed on SHIMADZU 20A-DAD HPLC using Welch Materials Ultimate XB-C18 (5 μm) 10 × 250 mm column. Silica gel (200–300 mesh) was purchased from Qingdao Marine Chemical Factory (Qingdao, China). Size exclusion chromatography was done on Sephadex LH-20 (Amersham, GE in USA).Fungal material
The fungal strain in this research was isolated from the root of Panax notoginseng, collected from Wenshan, Yunnan, China, and identified based on the DNA sequence (18S-ITSI-5.8S-ITS2-28S), which has been submitted to GenBank with accession number MH819653. BLAST search result revealed that the isolate belonged to the genus Chaetomium sp. with high identity (99%) to the species of C. globosum, and the fungus was deposited at the School of Life Science and Biopharmaceutics, Shenyang Pharmaceutical University.Fermentation and extraction
Fermentation was carried out in Erlenmeyer flask (250 mL) each containing sterile water (53 mL) and rice (40 g) before sterilization at 121 °C for 30 min. The spore of Chaetomium sp. (2.0 mL) was inoculated in each Erlenmeyer flask (250 mL), then cultivated at 28 °C for 40 days. The fermented material was soaked with ethyl acetate (12 L) and the organic solvent was removed under reduced pressure to yield the crude extract (16.8 g), which was dissolved using 90% MeOH–H2O (1 L) and extracted by cyclohexane (1 L) to obtain the residue (10.5 g).Isolation and purification
The extract (10.5 g) was separated by silica gel for column chromatography (CC) eluted with petroleum ether–EtOAc (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:1) and CH2Cl2–MeOH (100:1 to 1:1), yielding eleven fractions (1–11). Fraction 5 (675.5 mg) was separated by C18-reversed phase silica gel CC using MeOH–H2O gradient elution to afford three subfractions. Subfraction 5-2 (28.5 mg) was separated by preparative HPLC (CH3CN–H2O, 50%) to afford compound 7 (34.3 mg). Subfraction 5-3 (62.6 mg) was repeatedly crystalized with MeOH–H2O at room temperature to yield compound 8 (2.0 mg). Fraction 6 (360.5 mg) was charged on Sephadex LH-20 column (200 mL) with MeOH as eluent to afford six subfractions. Subfraction 6-6 (150 mg) was separated by preparative HPLC with 60% CH3CN–H2O to yield compounds 9 (4.5 mg), 10 (7.2 mg) and 11 (5.1 mg). Compounds 6 (11.9 mg) and 12 (4.5 mg) were afforded from fraction 7 (430.2 mg) through C18-reversed phase silica gel CC with MeOH–H2O gradient elution and preparative HPLC (CH3CN–H2O, 51%). Fraction 8 (477.8 mg) was crystalized with MeOH–H2O at room temperature to afford compound 13 (36.5 mg). Fraction 9 (106.7 mg) was separated by Sephadex LH-20 gel filtration chromatography eluted with MeOH to five subfractions. Purification of subfraction 9-4 (70.0 mg) by preparative HPLC (CH3CN–H2O, 36%) led to the isolation of compound 3 (13.0 mg). Separation of fraction 10 (979.6 mg) by C18-reversed phase silica gel CC with MeOH–H2O gradient elution afforded two subfractions. Finally, subfraction 10-1 (106.0 mg) was subject to preparative HPLC (CH3CN–H2O, 25%), which led to the isolation of compounds 1 (12 mg), 2 (7.9 mg), 4 (8.3 mg) and 5 (8.4 mg).
ε) 274, 281, and 290 nm; IR (KBr) νmax 3380, 3225, 2923, 2851, 1677, 1641, 1448, 1434, 1386, 1287, 1265, 1242, 1207, 1155, 1099, 1025, 1011, 982, 923, 896, 877, 835, 772, 742, 725, 637, 626, 594, 566, 485, 429 cm−1; 1H (600 MHz, DMSO-d6) and 13C NMR (150 MHz, DMSO-d6) data, see Table 1, Fig. S3 and S4;† HRESIMS m/z 386.1148 [M + Na]+ (calcd for C17H21N3NaO4S, 471.1145).ε) 274, 281, and 290 nm; IR (KBr) νmax 3419, 2921, 2851, 1677, 1457, 1422, 1384, 1323, 1274, 1236, 1196, 1160, 1100, 1068, 1012, 955, 891, 745, 715, 647, 603, 557, 431 cm−1; 1H (600 MHz, DMSO-d6) and 13C NMR (150 MHz, DMSO-d6) data, see Table 1, Fig. S15 and S16;† HRESIMS m/z 372.0982 [M + Na]+ (calcd for C16H19N3NaO4S, 349.1096).
:1 to 1:1) and CH2Cl2–MeOH (100:1 to 1:1), yielding eleven fractions (1–11). Fraction 5 (675.5 mg) was separated by C18-reversed phase silica gel CC using MeOH–H2O gradient elution to afford three subfractions. Subfraction 5-2 (28.5 mg) was separated by preparative HPLC (CH3CN–H2O, 50%) to afford compound 7 (34.3 mg). Subfraction 5-3 (62.6 mg) was repeatedly crystalized with MeOH–H2O at room temperature to yield compound 8 (2.0 mg). Fraction 6 (360.5 mg) was charged on Sephadex LH-20 column (200 mL) with MeOH as eluent to afford six subfractions. Subfraction 6-6 (150 mg) was separated by preparative HPLC with 60% CH3CN–H2O to yield compounds 9 (4.5 mg), 10 (7.2 mg) and 11 (5.1 mg). Compounds 6 (11.9 mg) and 12 (4.5 mg) were afforded from fraction 7 (430.2 mg) through C18-reversed phase silica gel CC with MeOH–H2O gradient elution and preparative HPLC (CH3CN–H2O, 51%). Fraction 8 (477.8 mg) was crystalized with MeOH–H2O at room temperature to afford compound 13 (36.5 mg). Fraction 9 (106.7 mg) was separated by Sephadex LH-20 gel filtration chromatography eluted with MeOH to five subfractions. Purification of subfraction 9-4 (70.0 mg) by preparative HPLC (CH3CN–H2O, 36%) led to the isolation of compound 3 (13.0 mg). Separation of fraction 10 (979.6 mg) by C18-reversed phase silica gel CC with MeOH–H2O gradient elution afforded two subfractions. Finally, subfraction 10-1 (106.0 mg) was subject to preparative HPLC (CH3CN–H2O, 25%), which led to the isolation of compounds 1 (12 mg), 2 (7.9 mg), 4 (8.3 mg) and 5 (8.4 mg).
ε) 274, 281, and 290 nm; IR (KBr) νmax 3380, 3225, 2923, 2851, 1677, 1641, 1448, 1434, 1386, 1287, 1265, 1242, 1207, 1155, 1099, 1025, 1011, 982, 923, 896, 877, 835, 772, 742, 725, 637, 626, 594, 566, 485, 429 cm−1; 1H (600 MHz, DMSO-d6) and 13C NMR (150 MHz, DMSO-d6) data, see Table 1, Fig. S3 and S4;† HRESIMS m/z 386.1148 [M + Na]+ (calcd for C17H21N3NaO4S, 471.1145).ε) 274, 281, and 290 nm; IR (KBr) νmax 3419, 2921, 2851, 1677, 1457, 1422, 1384, 1323, 1274, 1236, 1196, 1160, 1100, 1068, 1012, 955, 891, 745, 715, 647, 603, 557, 431 cm−1; 1H (600 MHz, DMSO-d6) and 13C NMR (150 MHz, DMSO-d6) data, see Table 1, Fig. S15 and S16;† HRESIMS m/z 372.0982 [M + Na]+ (calcd for C16H19N3NaO4S, 349.1096).Antimicrobial assays
Compounds 1–13 were assayed for their antibacterial activities against Staphylococcus aureus, Bacillus subtilis, Escherichia coli, Enterococcus faecium, Proteus mirabilis, Acinetobacter baumannii, Achromobacter marplatensis and antifungal activity against Candida albicans according to our previous method.19 Compounds 1–13 were dissolved in DMSO at an initial concentration at 10 mg mL−1. Ampicillin, was used as a positive control. The S. aureus, B. subtilis, E. coli, E. faecium, P. mirabilis, A. baumannii, and A. marplatensis were preincubated at 37 °C for 24 h in Luria–Bertani (LB) media, respectively. Candida albicans was incubated at 28 °C for 48 h in PDA media. The bacterial suspension was diluted to the predetermined starting concentration OD600 = 0.2. Then 1.0 mL bacterial solution was added into 24.0 mL LB media. The bacterial solution was transferred into a 96-well plate (198 μL per well) and the 2 μL of compounds 1–13. After 24 h cultivated, absorbance was determined at 600 nm by a microplate reader. Every experiment was performed in three biological replicates in triplicate to validate the screening method.Antitumor assays
Human lung carcinoma cell A549 and human breast cancer cell MDA-MB-231, purchased from American Type Culture Collection (ATCC, Rockville, MD, USA), were cultured in DMEM medium, supplemented with 10% fetal bovine serum (Yuanheng Shengma Biology Technology Research Institute, Beijing, People's Republic of China), 100 U mL−1 penicillin, and 100 mg mL−1 streptomycin. Cultures were maintained in an incubator (NuAir, USA) with 5% CO2 at 37 °C.The viability of cells was performed using an MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (Sigma, USA) method in 96-well microplates, as reported previously,37 with slight modification. Cells were plated in 96-well culture plates (93103 per well) and allowed to adhere for 24 h before treated with fungal metabolites at a series of concentration (0, 0.5, 1, 5, 10, and 20 mg mL−1) for 48 hours. Add 20 mL MTT (5 mg mL−1) per well was added prior to 4 h incubation at 37 °C. Upon medium removal, formazans depositing on the plate were dissolved in 150 mL DMSO, then, the cell viability was detected by a Tecan microplate reader (Austria) at λ = 492 nm, and the IC50 values were obtained from the MTT viability curves using GraphPad Prism 4.0.
Morphological observation of B. subtilis in the absence or presence of active compounds
To further study the possible antibacterial mechanism of the active compounds in more detail, the cells of B. subtilis were treated with the purified compounds, and observed using transmission electron microscopy (TEM, HT7700, Japan). B. subtilis cells were grown separately at 37 °C on agar plates and diluted with LB broth to an OD600 value of 0.2. Compounds 6, 9 and 12 at the working concentrations of 0.5, 0.4 and 4.8 μg mL−1 (twice the MIC), respectively, were used to treat the B. subtilis cells. Samples of the treated cells and controls were cultivated at 37 °C for an additional 3 h. The bacterial suspensions were dyed with 2% phosphotungstic acid (1:1, v/v, pH 6.5) for 3–5 min, and a transmission scan was performed as previously described.38
Molecular docking
FtsZ is a prokaryotic homologue to the eukaryotic tubulin. Thus, molecular docking was used to investigate the interaction of FtsZ and the active compounds. The crystal structure of FtsZ from B. subtilis was obtained from the RCSB Protein Data Bank using the PDB Code 2VXY.19 The protein structure was prepared using Discovery Studio 4.3 (Accelrys Inc., San Diego, USA). Water molecules were removed, and hydrogen atoms were added. The prepared structure was saved in the PDB format for docking studies. The 3D structures of the compounds were prepared and Gasteiger–Hückel charges were added using Sybyl software (Tripos, America). The ligand was subjected to energy minimization with Tripos force filed parameters.39 Blind docking was conducted using the Molegro Virtual Docker 4.0 (Molegro ApS, Aarhus, Denmark) programme. The 3D docking grid was sufficiently large to cover the protein.Conflicts of interest
The authors have no conflicts of interest with any parties or individuals.Acknowledgements
This research program is financially supported by the National Natural Science Foundation of China (No. 81703397, 31770545), the Fujian Natural Science Foundation (No. 2016J01175), the Science and Technique Programs of Yunnan Province (2016ZF001-001, 2017IB038), the National Science and Technology Major Project (2018ZX09735001-002-002), Key Research and Development Program of Liaoning Province (2017107013), Basic Scientific Research Program in Higher Education of Liaoning Province (2017LQN17).References
- L. Keyang, Z. Yisheng, L. Li, W. Xuewei and D. Gang, Molecules, 2013, 18, 10944–10952 CrossRef PubMed.
- W. B. Geiger, J. E. Conn and S. A. Waksman, J. Bacteriol., 1944, 48, 531–536 CAS.
- C. Chen, J. Wang, J. Liu, H. Zhu, B. Sun, J. Wang, J. Zhang, Z. Luo, G. Yao and Y. Xue, J. Nat. Prod., 2015, 78, 1193–1201 CrossRef CAS PubMed.
- A. Pontius, A. Krick, S. Kehraus, R. Brun and G. M. König, J. Nat. Prod., 2008, 71, 1579–1584 CrossRef CAS PubMed.
- M. G. Hui, Y. Z. Wei, D. Gang, P. Saparpakorn, C. S. Yong, S. Hannongbua and X. T. Ren, Chem. Commun., 2008, 40, 5978–5980 Search PubMed.
- Q. L. Xu, Y. S. Xiao, Y. Shen, H. M. Wu, X. Zhang, X. Z. Deng, T. T. Wang, W. Li, R. X. Tan and R. H. Jiao, J. Asian Nat. Prod. Res., 2017, 20, 1–8 Search PubMed.
- J. Zhang, H. M. Ge, R. H. Jiao, J. Li, H. Peng, Y. R. Wang, J. H. Wu, Y. C. Song and R. X. Tan, Planta Med., 2010, 76, 1910–1914 CrossRef CAS PubMed.
- L. Xiang, T. Ye, Y. Sheng-Xiang, Z. Ya-Mei and Q. Jian-Chun, Bioorg. Med. Chem. Lett., 2013, 23, 2945–2947 CrossRef PubMed.
- W. Li, X. Yang, Y. Yang, R. Duang, G. Chen, X. Li, Q. Li, S. Qin, S. Li and L. Zhao, Nat. Prod. Res., 2016, 30, 2616–2619 CrossRef CAS PubMed.
- T. X. D. Tina, X. M. Cui, Z. H. Song, J. Z. Kui, Z. N. Ji, C. K. Lo and W. K. T. Karl, J. Agric. Food Chem., 2003, 51, 4617–4623 CrossRef PubMed.
- H. B. Guo, X. M. Cui, N. An and G. P. Cai, Genet. Resour. Crop Evol., 2010, 57, 453–460 CrossRef.
- L. Ma, Y. H. Cao, M. H. Cheng, Y. Huang, M. H. Mo, Y. Wang, J. Z. Yang and F. X. Yang, Antonie van Leeuwenhoek, 2013, 103, 299–312 CrossRef PubMed.
- J. H. Lee, Y. H. Yu, Y. H. Kim, S. H. Ohh and W. M. Park, Plant Pathol. J., 1990, 6, 13–20 Search PubMed.
- Z. Y. Fan, C. P. Miao, X. G. Qiao, Y. K. Zheng, H. H. Chen, Y. W. Chen, L. H. Xu, L. X. Zhao and H. L. Guan, J. Ginseng Res., 2016, 40, 97–104 CrossRef PubMed.
- A. A. Gunatilaka, J. Nat. Prod., 2006, 69, 509–526 CrossRef CAS PubMed.
- P. Chomcheon, S. Wiyakrutta, T. Aree, N. Sriubolmas, N. Ngamrojanavanich, C. Mahidol, S. Ruchirawat and P. Kittakoop, Chem. Eur J., 2010, 16, 11178–11185 CrossRef CAS PubMed.
- Y. N. An, X. Zhang, T. Y. Zhang, M. Y. Zhang, Q. Zhang, X. Y. Deng, F. Zhao, L. J. Zhu, G. Wang and J. Zhang, Sci. Rep., 2016, 6, 27396 CrossRef CAS PubMed.
- F. Wang, W. Zhao, C. Zhang, S. Chang, R. Shao, J. Xing, M. Chen, Y. Zhang and S. Si, RSC Adv., 2019, 9, 16035–16039 RSC.
- J. Xie, Y. Y. Wu, T. Y. Zhang, M. Y. Zhang, F. Peng, B. Lin and Y. X. Zhang, Fitoterapia, 2018, 131, 35–43 CrossRef CAS PubMed.
- D. W. Adams and E. Jeff, Nat. Rev. Microbiol., 2009, 7, 642–653 CrossRef CAS PubMed.
- J. Errington, R. A. Daniel and D. J. Scheffers, Microbiol. Mol. Biol. Rev., 2003, 67, 52–65 CrossRef CAS PubMed.
- M. G. Pinho and J. Errington, Mol. Microbiol., 2010, 50, 871–881 CrossRef PubMed.
- N. R. Stokes, N. Baker, J. M. Bennett, J. Berry, I. Collins, L. G. Czaplewski, A. Logan, R. Macdonald, L. Macleod and H. Peasley, Antimicrob. Agents Chemother., 2013, 57, 317–325 CrossRef CAS PubMed.
- F. Q. Wang, Q. Y. Tong, H. R. Ma, H. F. Xu, S. Hu, W. Ma, Y. B. Xue, J. J. Liu, J. P. Wang and H. P. Song, Sci. Rep., 2015, 5, 9294 CrossRef CAS PubMed.
- H. Fujimoto, M. Sumino, E. Okuyama and M. Ishibashi, J. Nat. Prod., 2004, 67, 98–102 CrossRef CAS PubMed.
- I. Marmouzi, M. E. A. Faouzi, N. Saidi, Y. Cherrah, N. Rehberg, S. S. Ebada, W. Ebrahim, R. Kalscheuer and P. Proksch, Chem. Nat. Compd., 2017, 6, 1208–1211 CrossRef.
- G. Y. Li, B. G. Li, T. Yang, J. F. Yan, A. G. Y. Liu and G. L. Zhang, J. Nat. Prod., 2006, 69, 1374–1376 CrossRef CAS PubMed.
- R. G. Marwah, M. O. Fatope, M. L. Deadman, Y. M. Al-Maqbali and J. Husband, ChemInform, 2007, 63, 8174–8180 CAS.
- H. C. Kwon, S. D. Zee, S. Y. Cho, S. U. Choi and K. R. Lee, Arch. Pharmacal Res., 2002, 25, 851–855 CrossRef CAS.
- M. H. Wang, Y. C. Hu, B. D. Sun, M. Yu, S. B. Niu, Z. Guo, X. Y. Zhang, T. Zhang, G. Ding and Z. M. Zou, Org. Lett., 2018, 20, 1806–1809 CrossRef CAS PubMed.
- T. R. Welch and R. M. Williams, Tetrahedron, 2013, 69, 770–773 CrossRef CAS PubMed.
- D. H. Scharf, T. Heinekamp, N. Remme, P. Hortschansky, A. A. Brakhage and C. Hertweck, Appl. Microbiol. Biotechnol., 2012, 93, 467–472 CrossRef CAS PubMed.
- C. Davis, S. Carberry, M. Schrettl, I. Singh, J. Stephens, S. Barry, K. Kavanagh, G. Challis, D. Brougham and S. Doyle, Chem. Biol., 2011, 18, 542–552 CrossRef CAS PubMed.
- B. Li, R. R. Forseth, A. A. Bowers, F. C. Schroeder and C. T. Walsh, ChemBioChem, 2012, 13, 2521–2526 CrossRef CAS PubMed.
- N. Timothy, C. A. Lewis, K. J. Eastman and P. S. Baran, J. Am. Chem. Soc., 2010, 132, 7119–7137 CrossRef PubMed.
- R. L. Lock and E. J. Harry, Nat. Rev. Drug Discovery, 2008, 7, 324–338 CrossRef CAS PubMed.
- Z. Luo, F. Wang, J. Zhang, X. Li, M. Zhang, X. Hao, Y. Xue, Y. Li, F. D. Horgen and G. Yao, J. Nat. Prod., 2012, 75, 2113–2120 CrossRef CAS PubMed.
- P. Sass, M. Josten, K. Famulla, G. Schiffer, H. G. Sahl, L. Hamoen and H. Brötzoesterhelt, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 17474–17479 CrossRef CAS PubMed.
- S. Ballu, R. Itteboina, S. K. Sivan and V. Manga, J. Recept. Signal Transduction Res., 2018, 38, 61–70 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04747f |
| This journal is © The Royal Society of Chemistry 2019 |