
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceActivating peroxymonosulfate by halogenated and methylated quinones: performance and mechanism†
Hong Zhang‡
ab,
Lina Qiao‡b,
Jing Heab,
Na Liab,
Dongmei Zhangab,
Kai Yuab,
Hong Youab and
Jie Jiang *ab
*ab
aSchool of Marine Science and Technology, Harbin Institute of Technology at Weihai, Weihai, Shandong 264209, P. R. China. E-mail: jiejiang@hitwh.edu.cn; Fax: +86-631-5685-359
bState Key Laboratory of Urban Water Resource and Environment, Harbin Institute of Technology, Harbin, Heilongjiang 150090, P. R. China
First published on 30th August 2019
Abstract
Activation of peroxymonosulfate (PMS) by halogenated and methylated quinones for destroying sulfamethoxazole (SMX) was investigated, where 2,6-dimethyl-1,4-benzoquinone (DMBQ), 2,6-dichloro-1,4-benzoquinone (DCBQ), and tetrafluoro-1,4-benzoquinone (TFBQ) were chosen as model quinones. The PMS could be activated by halogenated and methylated quinones efficiently for SMX degradation, and the process showed high pH and quinones dependency. Different from PMS activated by ultraviolet (UV), singlet oxygen (1O2) instead of hydroxyl radical (˙OH) and sulfate radical (SO4˙−) was the primary oxidizing species in the activation process. The formation of 1O2 was confirmed by various quenching studies combined with chemical probes (9,10-diphenylanthracene (DPA)). By sampling in situ and monitoring in real time, droplet spray ionization mass spectrometry (DSI-MS) was applied to capture and identify the intermediates generated in the activation process. A possible mechanism for PMS activation was proposed accordingly. It was found that a series of reactions between PMS and halogenated/methylated quinones formed a dioxirane intermediate, and the subsequent decomposition of this intermediate produced the 1O2. These findings would help to better understand the interactions between PMS and quinones, and provide a novel activator for PMS activation toward environmental contaminants.
1. Introduction
Advanced oxidation processes (AOPs) for the remediation of contaminated soils, groundwater, and sediments have received considerable attention in recent years.1–3 These processes are based mostly on activation of hydrogen peroxide (H2O2),4 titanium dioxide (TiO2),5 persulfate (PDS)6,7 and peroxymonosulfate (PMS)8–10 to generate oxidizing species such as sulfate radical (SO4˙−) (2.5–3.1 V) and hydroxyl radicals (˙OH) (1.9–2.7 V),11 which are highly reactive oxidants toward organic compounds. Among AOPs, PMS has emerged as a popular technique in environmental remediation. Because PMS has high potential of 1.82 V (ref. 12) in generating SO4˙− and ˙OH,13,14 it has been regarded as an alternative of other methods such as H2O2. The application of PMS in destroying refractory organics has been extensively investigated and well-documented.12,15,16To date, various strategies have been developed for activating PMS.12,16 Generally, the activators for PMS activation include homogenous and heterogeneous transition metal catalyst, metal-free heterogeneous catalysts, ultraviolet, ultrasound, conduction electron, and miscellaneous activation. These techniques can efficiently destroy organic pollutants, but they have significant limitations in the application. As an example, ultrasound, UV, and heating are an environmentally friendly and applicable technology. However, they are not cost-effective for water treatment because of the high-energy consumption. Many studies regarding to PMS activation have focused on transition metals, among which Co(II) is found to be very active in initiating sulfate radical generation from PMS.17 But the adverse effect of Co(II) on human health needs to be considered. Meanwhile, concerns about metal leaching result in secondary contamination to water body.18 Therefore, the development of green methods for PMS activation is an area of active research and highly desired.
Quinones such as 1,4-benzoquinone (p-BQ) can efficiently activate PMS with singlet oxygen (1O2) evolution.11 It is notable that quinones are potent redox active compounds and ubiquitous in soils, surface water and even in atmospheric aerosols.19,20 Halogenated quinones can active H2O2 to produce ˙OH by generating a nucleophilic adduct intermediate, but methylated quinones fail to active H2O2.21 Furthermore, halogenated and methylated quinones are also effective in activating PDS with SO4˙− evolution by a semiquinone radical-dependent mechanism.20 However, little is known about the halogenated and methylated quinones on PMS activation so far.
The goals of the present work are (i) to investigate the activation of PMS by halogenated and methylated quinones to catalytically oxidize organic pollutants, (ii) to capture and identify the primary oxidizing species during the activation process, and (iii) to elucidate the mechanism of PMS activated by halogenated and methylated quinones. Herein, activations of PMS by halogenated and methylated quinones for sulfamethoxazole (SMX) removal under various conditions were examined. SMX was chosen as target contaminant, since SMX is very stable and has been frequently detected in soils and water sources. Chemical trapping methods combined with various radical scavengers have been employed to investigate the oxidizing species in the activation process. In situ characterization using droplet spray ionization mass spectrometry (DSI-MS) was applied to analyze the intermediates generated in the catalytic PMS decomposition process. Results indicated that the combination of PMS with halogenated and methylated quinones is an attractive process providing efficient 1O2 generation for SMX degradation.
In a DSI-MS experiment, a corner of cover slip placed in front of the MS inlet functions as the spray corner and solution reservoir. By applying a high voltage to the solution, a charged-droplet spray is formed between the corner and the MS inlet.22,23 DSI-MS addresses the detection limitation in the sample transfer capillary of electrospray ionization and can monitor chemical reactions in real time. Applications of DSI-MS have included real-time monitoring of photolysis/photocatalysis reactions,24,25 catalytic reactions,22,26 and electrochemical reactions27,28 where the intermediates are fished out and characterized by MS. DSI is particularly suitable for fast reaction kinetics and short-lived reactive intermediates involved reaction.
2. Materials and methods
2.1 Materials
Methanol and acetonitrile at MS grade were purchased from Sigma-Aldrich (Darmstadt, Germany). Benzoic acid (BA), 9,10-diphenylanthracene (DPA), and sodium azide (NaN3) were purchased from Sigma-Aldrich (St. Louis, MO). Ethanol, sodium borate (Na2B4O7·10H2O), and sodium thiosulfate (Na2S2O3) were purchased from Aike Co. (Chengdu, Chian). Sulfamethoxazole (SMX), 2,6-dimethyl-1,4-benzoquinone (DMBQ), 2,6-dichloro-1,4-benzoquinone (DCBQ), and tetrafluoro-1,4-benzoquinone (TFBQ) were obtained from J&K Scientific Ltd. (Beijing, China). The structures of the quinones investigated in this study were given in Table S1.† Ultrapure water was obtained from a Milli-Q water purification system. All the individual solutions were freshly prepared before use.2.2 Experimental procedure and analysis
The removal of SMX by PMS in the presence of DMBQ, DCBQ and TFBQ was conducted in a brown flask on a reciprocating shaker at 25 ± 1 °C in the dark. This aimed to avoid decomposition of quinones and PMS by light irradiation. The experiments were carried out by simultaneously adding different quinones (1–20 μM) and PMS (0.5 mM) into pH-buffered solutions (20 mM sodium borate; pH 7–10) containing SMX (10 μM). All samples were collected at regular time points, quenched with sodium thiosulfate, and stored at 4 °C before being analyzed by LC-MS/MS.A Thermo Finnigan Surveyor LC coupled to a Thermo LTQ mass spectrometer equipped with an electrospray ionization source was used for LC-MS/MS analysis. An Agilent ZORBAX Eclipse plus C18 column (150 × 4.6 mm, 3.5 μm) was used for analytes separation. The isocratic mobile phase consisted of 70% acetonitrile and 30% water containing 0.1% formic acid, and the flow rate was 1 mL min−1. To avoid contamination of the mass spectrometer, a switching valve was used to divert the LC fluid to the waste bottle during the first and last few minutes. The MS parameters were set as follows: spray voltage, 4.5 kV; capillary voltage, 50 V; tube lens, 110 V; capillary temperature, 275 °C; ion maximum injection time, 20 ms; auxiliary gas, 35 arbitrary units; sheath gas, 4 arbitrary units; ion pair for SMX, 254/156. Each experiment was conducted in triplicate, and the averaged data and standard deviation were presented.
2.3 Chemical detection of singlet oxygen
DPA was used to trap the 1O2 formed in PMS activated by different quinones (DMBQ, TCBQ, and TFBQ). The corresponding DPA endoperoxide (DPAO2) can be detected by MS.29 Experiments were conducted in a mixed solution of 0.5 mM PMS, 20 μM different quinones (DMBQ, TCBQ, and TFBQ), and 20 μM DPA in 20 mM sodium borate buffer at pH 10. The reaction was allowed to react for 60 min. After dilution and filtration, the solution was analyzed by LC-MS/MS operated in the positive ion mode.2.4 Intermediate identification
To identify the intermediates in PMS activated by DMBQ, DCBQ and TFBQ, in situ analysis and real-time monitoring of the reaction of PMS and quinones were carried out by droplet spray ionization mass spectrometry (DSI-MS). A detailed description of DSI-MS is given elsewhere.22,24 A photograph of the DSI-MS is shown in Fig. S1.† The protocol for in situ analysis and real-time monitoring of PMS activated by different quinones involved three steps (Fig. S2†). Firstly, 10 μL of methanol/water (v/v: 7/3) was loaded onto the corner at 0 s. When a high voltage of −4 kV was applied to the solution, an electrospray was formed between the corner and the MS inlet. Then, 10 μL of different quinones (DMBQ, TCBQ, and TFBQ) (5 × 10−5 mol L−1) was loaded onto the corner at 10 s. Finally, 30 μL of PMS (5 × 10−4 mol L−1) was loaded onto the corner at 14 s. Data were recorded continuously during these additions of reagents. The sample solutions were adjusted at pH 10 before pipetting to the corner. These experiments were implemented on a Thermo LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). The operation parameters were set as follows: resolution, 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000; capillary temperature, 275 °C; ion maximum injection time, 500 ms; tube lens voltage, −110 V; capillary voltage, −35 V.
000; capillary temperature, 275 °C; ion maximum injection time, 500 ms; tube lens voltage, −110 V; capillary voltage, −35 V.
3. Results and discussion
3.1 Effect of pH on SMX degradation
The degradations of SMX by PMS in the presence of DMBQ, DCBQ and TFBQ over a pH range of 7–10 were conducted, and the results are shown in Fig. 1. In the absence of quinones, the degradation of SMX by PMS was negligible within the pH investigated. Comparatively, SMX degradation gradually increased from pH 7 to 10 by PMS in the presence of quinones. For instance, when the PMS was in the presence of DMBQ, the degradation of SMX in 9 min increased from 7% to 70% with pH from 7 to 10. In the case of PMS/DCBQ in 9 min, with the increase of pH from 7 to 10, the degradation of SMX increased from 12% to 100%. As for PMS/TFBQ in 9 min, the degradation of SMX at pH 7 was only 16%, while that of SMX at pH 10 reached 100%. In addition, the first-order rate constants derived from Fig. 1 were listed in Table S1.† These rate constants also suggest that pH has a significant effect on the SMX degradation. For instance, in the case of PMS/TFBQ, with increasing pH from 8 to 10, the degradation rate increased from 0.0702 to 0.6323 min−1. Moreover, the pH in experiment of pH 10 showed negligible change (Fig. S3†).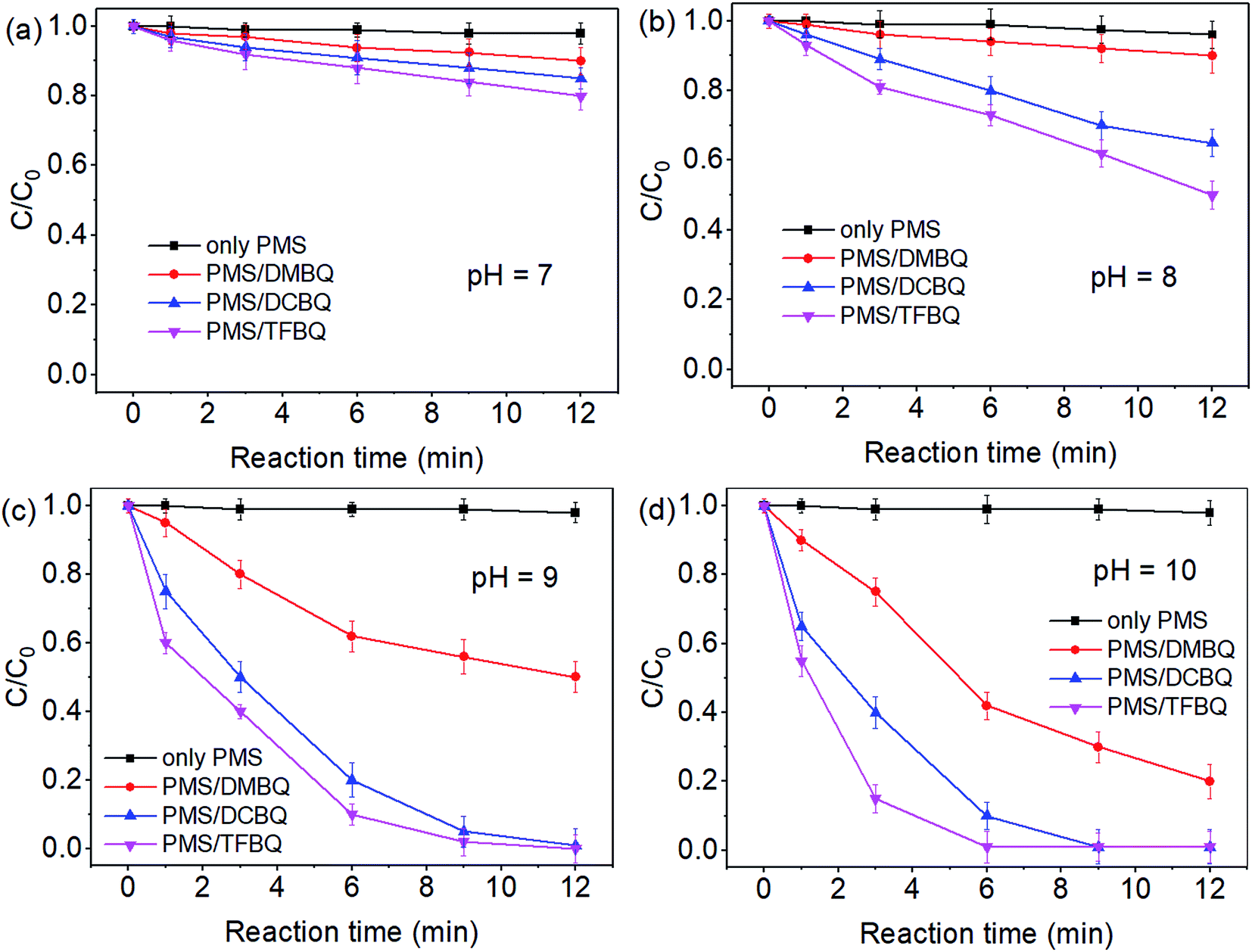 | ||
| Fig. 1 Effect of pH on SMX degradation. (a) pH 7; (b) pH 8; (c) pH 9; (d) pH 10. Experimental conditions: [PMS]0 = 0.50 mM, [SMX]0 = 10 μM, [BQ]0 = 20 μM, 20 mM borate buffer, and T = 25 °C. | ||
After the reactions, residual contents of PMS over pH 7–10 were also determined (Fig. S4†). In control experiments with PMS alone, the decomposition of PMS was negligible within the pH investigated. However, when the PMS was in the presence of quinones, the degradation of PMS significantly enhanced with the increase of pH from 7 to 10. This pH dependent decomposition of PMS was consistent with SMX degradation (Fig. S4† and 1). Therefore, pH adjustment may be a good option to enhance reaction rates if necessary.
3.2 Effect of quinones concentrations on SMX degradation
The degradations of SMX by PMS under different concentrations of quinones were also investigated. From Fig. 2, the degradation of SMX significantly enhanced with the increase of quinones concentrations. For instance, in the case of PMS/DCBQ in 9 min, SMX degradation increased from 42% to 98% with the DCBQ concentrations from 10 to 20 μM. As for PMS/TFBQ in 6 min, the SMX degradation was only 40% when the concentration of TFBQ was 10 μM; whereas the SMX was completely degraded at the concentration of 20 μM.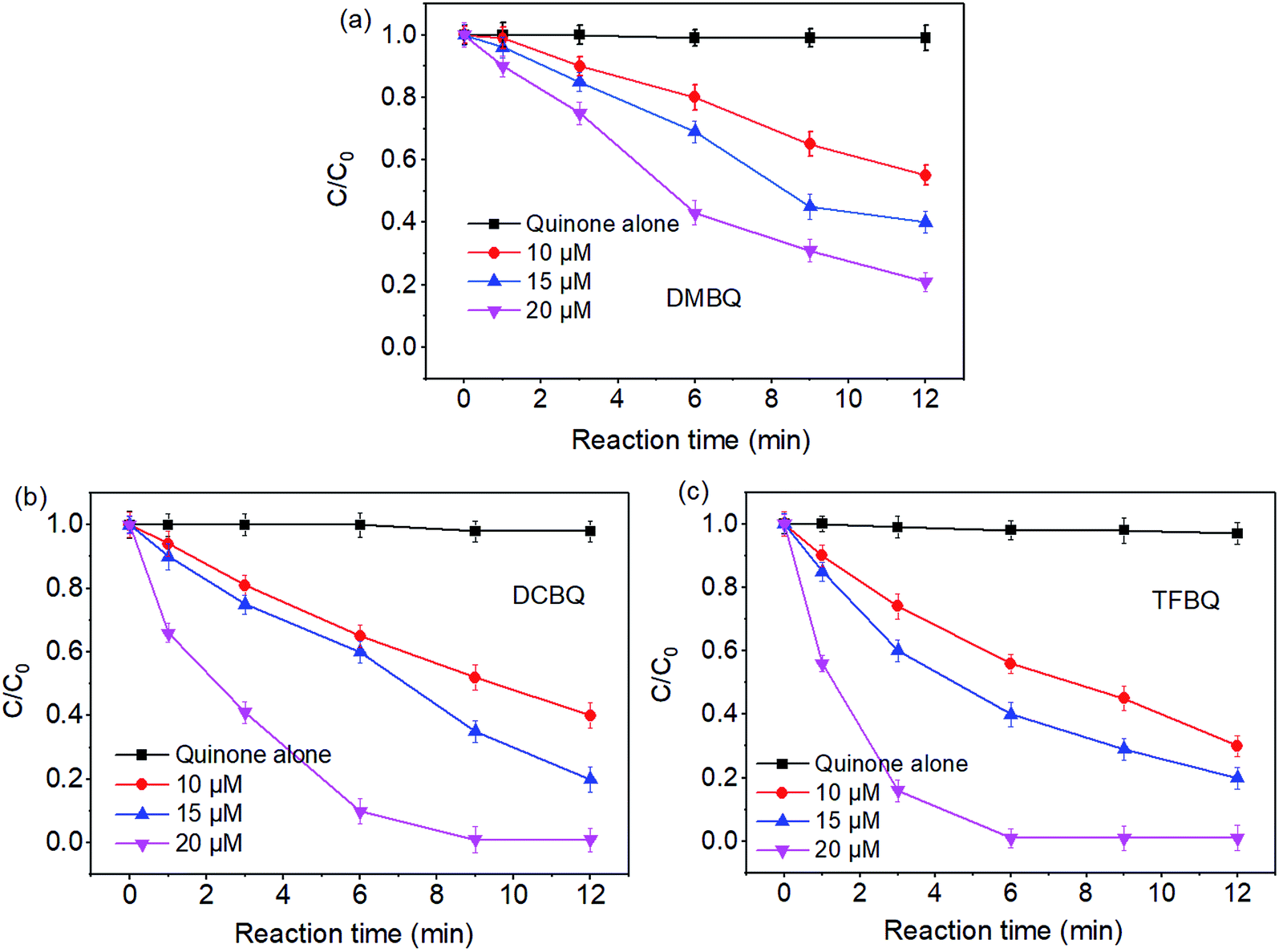 | ||
| Fig. 2 Effect of quinones concentrations on SMX degradation. (a) DMBQ; (b) DCBQ; (c) TFBQ. Experimental conditions: [PMS]0 = 0.50 mM, [SMX]0 = 10 μM, 20 mM borate buffer, pH = 10, and T = 25 °C. | ||
As shown in Fig. 2, in control experiments with quinones alone, the SMX degradation was negligible. This observation indicates that the relatively strong oxidant semiquinone radicals appeared in quinones solution20 could not directly degrade SMX. Similar results have also been reported in previous studies.11 These results suggest that halogenated and methylated quinones can significantly enhance the degradation of SMX by PMS. From Fig. 1 and 2, it seems that SMX degraded by PMS showed both pH and quinones dependency.
3.3 Identification of oxidizing species
Comparatively, the degradation of SMX markedly slowed down when NaN3 was added, as shown in Fig. 3. For instance, SMX was completely degraded in 20 min without NaN3; whereas with 0.1 M NaN3 added, only 20%, 23% and 29% SMX were degraded by PMS in the presence of DMBQ, DCBQ and TFBQ, respectively. These results indicated that neither ˙OH nor SO4˙− were formed, and that 1O2 may be the primary oxidizing species that lead to the degradation of SMX.
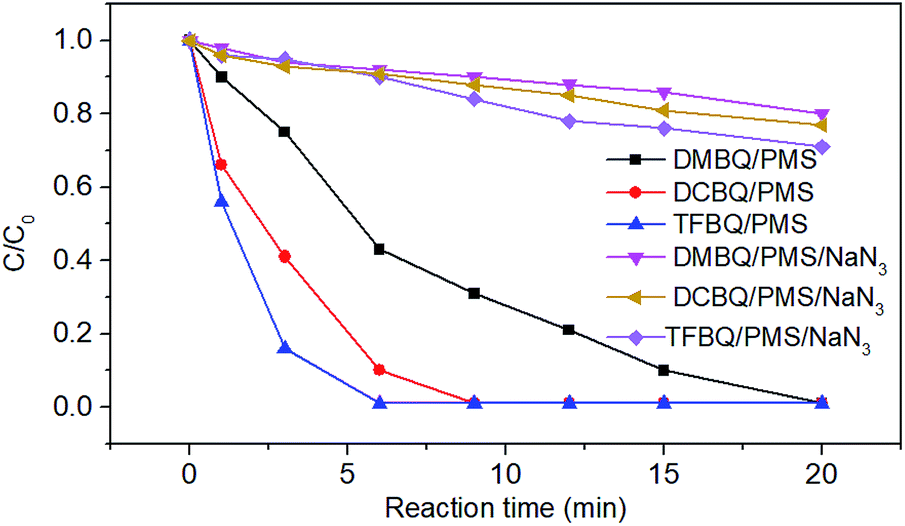 | ||
| Fig. 3 Effect of scavengers on SMX degradation in PMS activated by DMBQ, DCBQ and TFBQ. Experimental conditions: [PMS]0 = 0.50 mM, [Quinone]0 = 20 μM, [SMX]0 = 10 μM, 20 mM borate buffer, pH = 10, and T = 25 °C. | ||
 | ||
| Fig. 4 (a) MS/MS spectra of DPAO2 generated in PMS/different quinones/DPA; (b) effect of PMS concentrations on the generation of DAPO2. | ||
3.4 Activation mechanism
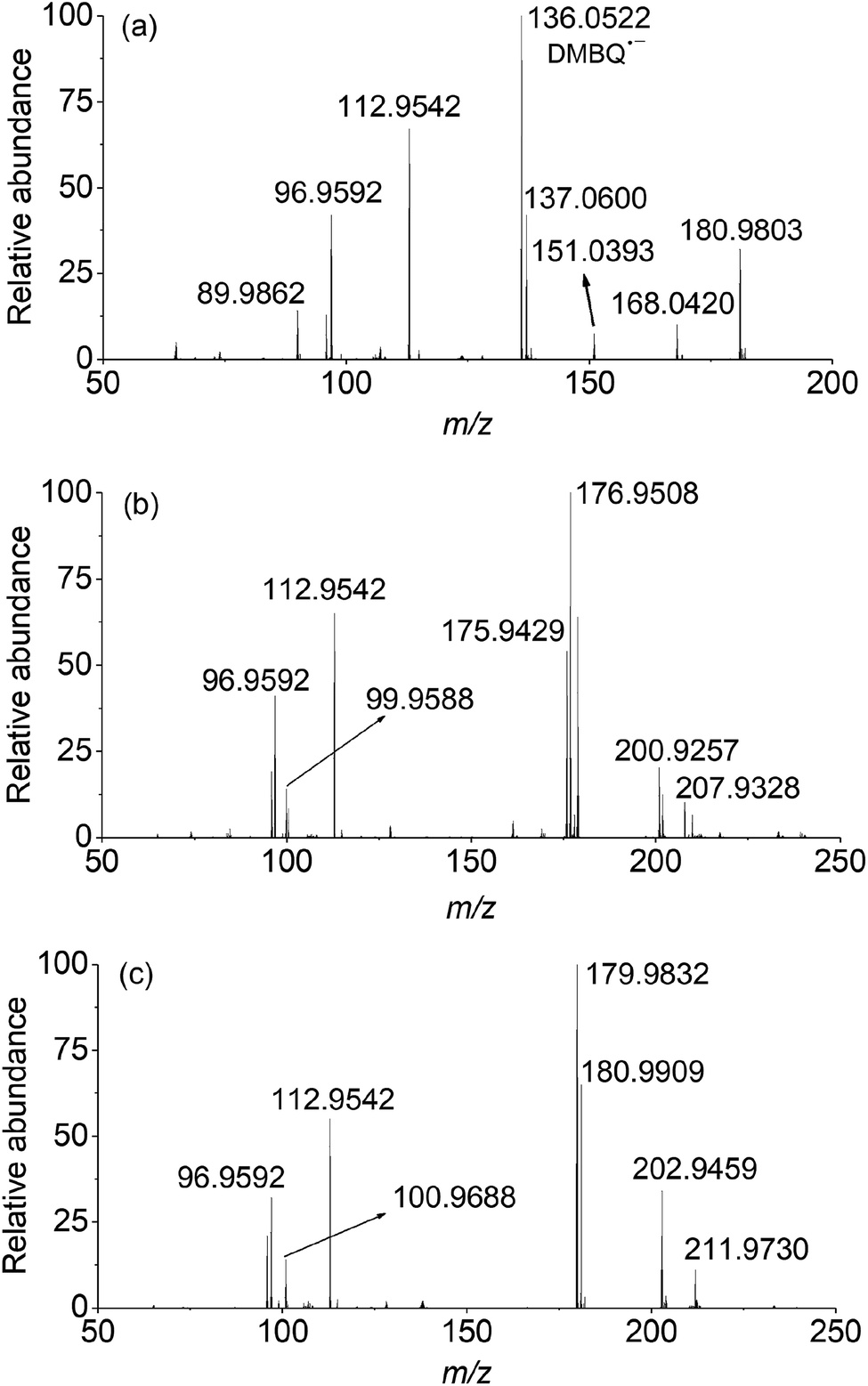 | ||
| Fig. 5 In situ and real-time characterization of intermediates in PMS/different quinones using DSI-MS. (a) PMS/DMBQ, (b) PMS/DCBQ, and (c) PMS/TFBQ. Mass spectra displayed the reaction time of 40 s for each case. | ||
In the case of PMS activated by DBMQ (Fig. 5a), the signal at m/z 136.0522 and 151.0393 corresponded to DMBQ˙− and [DMBQ + O − H]− respectively. Peak at m/z 137.0600 was assigned as [DMBQ + H]−. This is due to that DMBQ underwent an electrochemical reduction at the spraying tip forming hydro-DMBQ. It is similar to what has been reported in previous studies.35 The signals at m/z 112.9542 and 96.9592 were assigned as HSO5− and HSO4−. In addition, two signals at m/z 180.9803 and 89.9862 were evident. Peak of m/z 180.9803 was tentatively assigned as a peroxide adduct intermediate, which is due to nucleophilic addition of two molecules of HSO5− to the carbonyl group of DMBQ. Peak of m/z 89.9862 was attributed to the conjugate base of the adduct intermediate. In this sense, peaks of m/z 180.9803 and 89.9862 were detected with two and four charges, respectively. The comparison of isotopic envelope and theoretical simulation (Thermo Xcalibur Qual Browser 2.0) clearly confirmed this assumption (Fig. S7 and S8†). To further confirm these assignments, tandem mass spectrometry (MS/MS) was performed for structural characterization. These two ions showed similar dissociation behaviors where both of them gave daughter ions of m/z 113 corresponding to HSO5− (Fig. S9†).
In addition, a signal at m/z 168.0420, tentatively assigned as a dioxirane intermediate ([C8H8O4]˙−) was unambiguously observed. The abundance of this ion during mass selection and isolation was absence, even when the isolation window increased from 1 to 10 Th. This suggested that this ion could not be effectively subjected to MS/MS experiments. However, the isotopic envelope of m/z 168.0420 compared well with theoretical simulation (Fig. S10†). The high accurate mass listed in Table S4† and isotopic envelope of this intermediate sufficiently confirmed the assignment of this ion. Fig. 5b and c depict the mass spectra of PMS activated by DCBQ and TFBQ, respectively. The assignments and high accurate mass of these intermediates/products were listed in Tables S5 and S6.† Furthermore, the dissociation behaviors of these intermediates showed similar characteristic peaks (m/z 113, HSO5−) (Fig. S11 and S12†), which is consistent with PMS activated by DMBQ (Fig. S9†). These results show the same behavior where dissociation of intermediates in activation of PMS by DMBQ, DCBQ and TFBQ generates HSO5− and reproduced the corresponding quinones. This also supports the fact that PMS activated by DMBQ, DCBQ and TFBQ followed the same activation pathway.
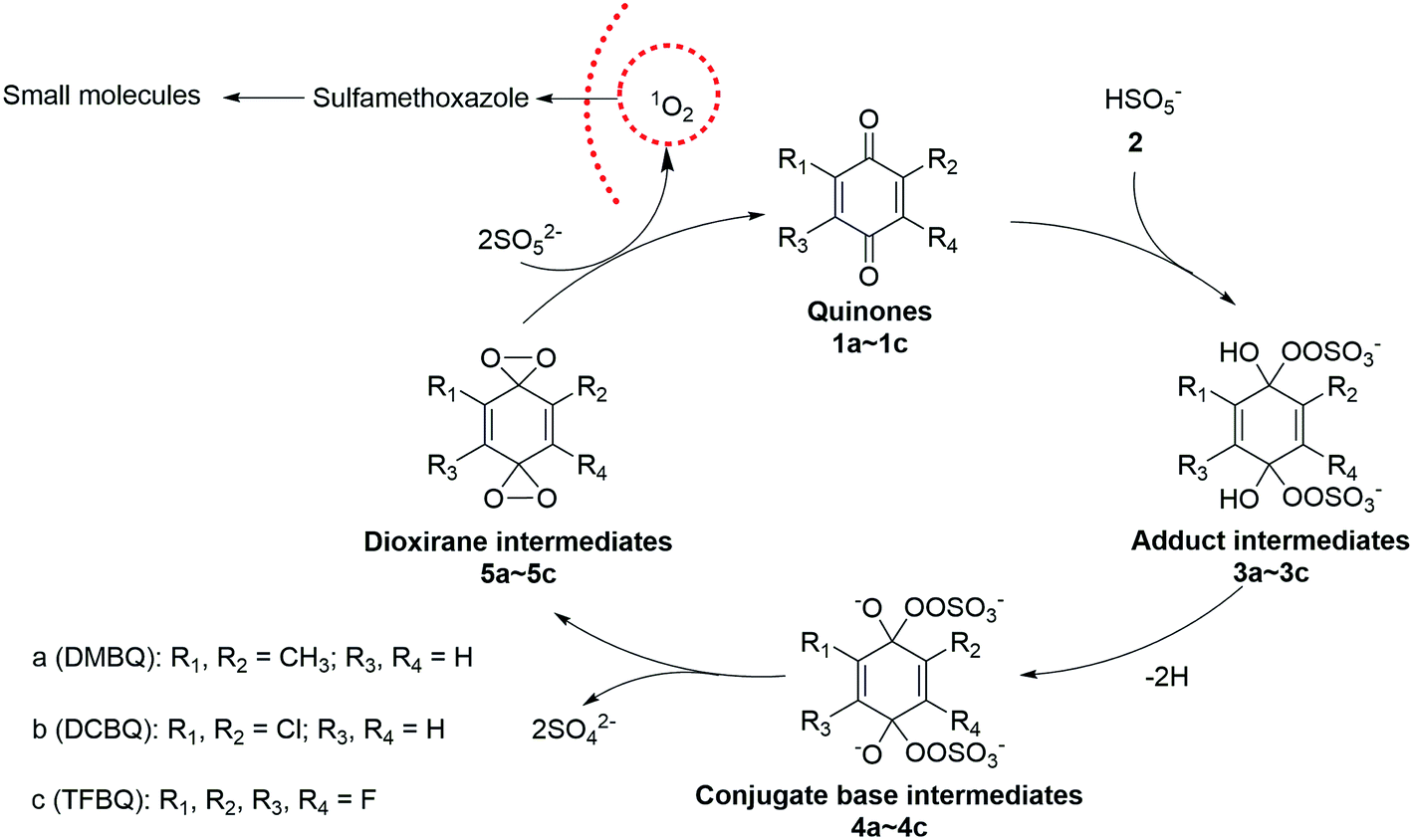 | ||
| Fig. 6 Possible mechanism of PMS activated by halogenated and methylated quinones. | ||
PMS has been activated by various techniques such as heating, UV, p-BQ, and transition metal.12 In the present study, the activation pathway contrasted with PMS activated by UV and transition metal where ˙OH and/or SO4˙− are the primary oxidizing species. Because the halogenated quinones have some toxic, we reiterate that this methodology is suitable for the area where halogenated quinones and pollutants simultaneously exist in wastewater and only PMS is added to the system. This avoids the possibility of contaminate from quinones.
4. Conclusion
In conclusion, the activation of peroxymonosulfate (PMS) by halogenated and methylated quinones (DMBQ, DCBQ, and TFBQ) could degrade SMX efficiently in aqueous solution. The kinetic studies suggested that the activation process was pH and quinones dependency, providing a guidance for reaction rate optimization. Chemical trapping combined with special quencher studies revealed that 1O2 instead of ˙OH and SO4˙− was the primary oxidizing species. This nonradical oxidant was effective in organic oxidation, meanwhile presenting a better selectivity to target contaminants from background organic matters. By sampling in situ analysis and monitoring in real time, DSI-MS successfully capture and identify the intermediates/products in the reaction of PMS with different quinones. A mechanism was proposed involving the formation of a peroxide adduct intermediate, a dioxirane intermediate, and the subsequent generation of 1O2. The success in PMS activated by different quinones advances quinone-mediated PMS activation. The differences in behavior associated with the quinone functional groups help the options of the activating reagent. The findings of this study provide a new pathway of PMS activation and efficient degradation technique for environmental contaminants.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Key R&D Program of China (No. 2016YFF0100302), Natural Science Foundation of China (No. 21705030, 21904029) and the Fundamental Research Funds for the Central Universities (No. HIT.NSRIF.2020087).References
- X. G. Duan, H. Q. Sun, Z. P. Shao and S. B. Wang, Appl. Catal., B, 2018, 224, 973–982 CrossRef CAS.
- Z. P. Xing, J. Q. Zhang, J. Y. Cui, J. W. Yin, T. Y. Zhao, J. Y. Kuang, Z. Y. Xiu, N. Wan and W. Zhou, Appl. Catal., B, 2018, 225, 452–467 CrossRef CAS.
- L. W. Matzek and K. E. Carter, Chemosphere, 2016, 151, 178–188 CrossRef CAS PubMed.
- F. X. Chen, S. L. Xie, X. L. Huang and X. H. Qiu, J. Hazard. Mater., 2017, 322, 152–162 CrossRef CAS PubMed.
- P. Chen, F. L. Wang, Z. F. Chen, Q. X. Zhang, Y. H. Su, L. Z. Shen, K. Yao, Y. Liu, Z. W. Cai, W. Y. Lv and G. G. Liu, Appl. Catal., B, 2017, 204, 250–259 CrossRef CAS.
- R. C. Zhang, P. Z. Sun, T. H. Boyer, L. Zhao and C. H. Huang, Environ. Sci. Technol., 2015, 49, 3056–3066 CrossRef CAS PubMed.
- X. X. Pan, L. Q. Yan, R. J. Qu and Z. Y. Wang, Chemosphere, 2018, 196, 95–104 CrossRef CAS PubMed.
- W. Tang, Y. Zhang, H. Guo and Y. Liu, RSC Adv., 2019, 9, 14060–14071 RSC.
- J. Zhu, Z. Zhu, H. Zhang, H. Lu and Y. Qiu, RSC Adv., 2019, 9, 2284–2291 RSC.
- N. Jaafarzadeh, F. Ghanbari and M. Ahmadi, Chemosphere, 2017, 169, 568–576 CrossRef CAS PubMed.
- Y. Zhou, J. Jiang, Y. Gao, J. Ma, S. Y. Pang, J. Li, X. T. Lu and L. P. Yuan, Environ. Sci. Technol., 2015, 49, 12941–12950 CrossRef CAS PubMed.
- F. Ghanbari and M. Moradi, Chem. Eng. J., 2017, 310, 41–62 CrossRef CAS.
- J. Hou, L. Xu, Y. Han, Y. Tang, H. Wan, Z. Xu and S. Zheng, RSC Adv., 2019, 9, 974–983 RSC.
- X. Zhang, J. Yao, W. Peng, W. Xu, Z. Li, C. Zhou and Z. Fang, RSC Adv., 2018, 8, 33681–33687 RSC.
- W. Oh, Z. Dong and T. Lim, Appl. Catal., B, 2016, 194, 169–201 CrossRef CAS.
- P. Hu and M. Long, Appl. Catal., B, 2016, 181, 103–117 CrossRef CAS.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2003, 37, 4790–4797 CrossRef CAS PubMed.
- Y. X. Wang, Z. M. Ao, H. Q. Sun, X. G. Duan and S. B. Wang, Appl. Catal., B, 2016, 198, 295–302 CrossRef CAS.
- M. Fujii, A. Imaoka, C. Yoshimura and T. D. Waite, Environ. Sci. Technol., 2014, 48, 4414–4424 CrossRef CAS PubMed.
- G. D. Fang, J. Gao, D. D. Dionysiou, C. Liu and D. M. Zhou, Environ. Sci. Technol., 2013, 47, 4605–4611 CrossRef CAS PubMed.
- B. Z. Zhu, H. T. Zhao, B. Kalyanaraman and B. Frei, Free Radical Biol. Med., 2002, 32, 465–473 CrossRef CAS PubMed.
- J. Jiang, H. Zhang, M. Li, M. T. Dulay, A. J. Ingram, N. Li, H. You and R. N. Zare, Anal. Chem., 2015, 87, 8057–8062 CrossRef CAS PubMed.
- H. Zhang, N. Li, X. D. Li, J. Jiang, D. D. Zhao and H. You, Rapid Commun. Mass Spectrom., 2016, 30, 51–55 CrossRef CAS PubMed.
- H. Zhang, N. Li, Y. Wang, D. Zhao, J. He, H. You and J. Jiang, Chemosphere, 2017, 184, 932–938 CrossRef CAS PubMed.
- H. Zhang, N. Li, D. Zhao, J. Jiang and H. You, J. Am. Soc. Mass Spectrom., 2017, 28, 1939–1946 CrossRef CAS PubMed.
- H. Zhang, K. Yu, J. He, N. Li, H. You and J. Jiang, Microchem. J., 2018, 139, 437–442 CrossRef CAS.
- K. Yu, H. Zhang, J. He, R. N. Zare, Y. Wang, L. Li, N. Li, D. Zhang and J. Jiang, Anal. Chem., 2018, 90, 7154–7157 CrossRef CAS PubMed.
- K. Y. Hong Zhang, H. Jing, N. Li, H. You and J. Jiang, Analyst, 2018, 143, 4247–4250 RSC.
- S. Miyamoto, G. R. Martinez, A. P. B. Martins, M. H. G. Medeiros and P. Di Mascio, J. Am. Chem. Soc., 2003, 125, 4510–4517 CrossRef CAS PubMed.
- W. D. Oh, Z. L. Dong, G. Ronn and T. T. Lim, J. Hazard. Mater., 2017, 325, 71–81 CrossRef CAS PubMed.
- Y. H. Guan, J. Ma, X. C. Li, J. Y. Fang and L. W. Chen, Environ. Sci. Technol., 2011, 45, 9308–9314 CrossRef CAS PubMed.
- H. E. Gsponer, C. M. Previtali and N. A. Garcia, Toxicol. Environ. Chem., 1987, 16, 23–37 CrossRef CAS.
- S. Miyamoto, G. R. Martinez, M. H. G. Medeiros and P. Di Mascio, J. Am. Chem. Soc., 2003, 125, 6172–6179 CrossRef CAS PubMed.
- J. Jiang, D. Zhao, H. Zhang, J. He and N. Li, Anal. Methods, 2017, 9, 4201–4206 RSC.
- F. Qin, Y. Y. Zhao, Y. L. Zhao, J. M. Boyd, W. J. Zhou and X. F. Li, Angew. Chem., Int. Ed., 2010, 49, 790–792 CrossRef CAS PubMed.
- A. R. Gallopo and J. O. Edwards, J. Org. Chem., 1981, 46, 1684–1688 CrossRef CAS.
- J. O. Edwards, R. H. Pater and R. Curci, Photochem. Photobiol., 1979, 30, 63–70 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04789a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |