
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical degradation of ciprofloxacin with a Sb-doped SnO2 electrode: performance, influencing factors and degradation pathways†
Yanguang Mua,
Cong Huanga,
Haipu Li *ab,
Leilei Chena,
Ding Zhanga and
Zhaoguang Yang*ab
*ab,
Leilei Chena,
Ding Zhanga and
Zhaoguang Yang*ab
aCenter for Environment and Water Resources, College of Chemistry and Chemical Engineering, Central South University, Changsha 410083, PR China. E-mail: lihaipu@csu.edu.cn; zgyang@csu.edu.cn
bKey Laboratory of Hunan Province for Water Environment and Agriculture Product Safety, Changsha 410083, PR China
First published on 20th September 2019
Abstract
Sb-doped SnO2 electrodes were prepared with the practical sol–gel method and were used for the electrocatalytic degradation of ciprofloxacin (CIP) in aqueous solution. Results from the electrochemical characterization (including cyclic voltammetry, linear sweep voltammetry, and electrochemical impedance spectroscopy) showed that the electrode with 16 coating times (SSO-16) had the highest oxygen evolution potential of 2.2 V (vs. SCE) and the highest electrochemically active area of 3.74 cm2. The results of scanning electron microscopy and X-ray diffraction showed that the coating times could affect the surface morphology and crystal structure of the electrodes, and the SSO-16 electrode had a denser surface, higher crystallinity, and smaller grain size (28.6 nm). Moreover, the experimental parameters for CIP degradation with SSO-16 were optimized, and the removal ratio of CIP reached to almost 100% within 60 min. In addition, the possible degradation pathways of CIP were proposed. And the stability and reusability of the SSO-16 electrode were also studied. These results are valuable for the preparation of high electrocatalytic performance electrodes by a sol–gel coating method for electrochemical degradation of antibiotics.
1. Introduction
In recent years, increasing amounts of antibiotics have been produced and consumed. In 2011, the estimated consumption of antibiotics was 89![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 tons in China, while it had doubled in 2016.1,2 Meanwhile, antibiotics have been frequently detected in surface water,3 effluent and excess sludge of municipal wastewater treatment plants,4,5 and even ground water.6–8 The traditional wastewater treatment technology cannot completely remove these antibiotics, because the physical process can not degrade the contaminants and the biological treatment process will be restricted by the antibacterial properties of the antibiotics.9,10 Considering the potential threat of antibiotic residues to the global environment and human health, the effective degradation of antibiotics is becoming a matter of concern for environmental protection.
700 tons in China, while it had doubled in 2016.1,2 Meanwhile, antibiotics have been frequently detected in surface water,3 effluent and excess sludge of municipal wastewater treatment plants,4,5 and even ground water.6–8 The traditional wastewater treatment technology cannot completely remove these antibiotics, because the physical process can not degrade the contaminants and the biological treatment process will be restricted by the antibacterial properties of the antibiotics.9,10 Considering the potential threat of antibiotic residues to the global environment and human health, the effective degradation of antibiotics is becoming a matter of concern for environmental protection.
Advanced oxidation processes, including Fenton oxidation, electrochemical oxidation and photocatalysis, have always been praised as viable methods on wastewater treatment.11–16 By comparison, electrochemical oxidation has great advantages in the treatment of refractory organic wastewater because of its high processing efficiency, simple equipment, convenient operation, and environmental friendliness.17 The anode materials are vital for electrochemical oxidation because they determine the formation of hydroxyl radicals.18 The electrode materials, including SnO2,19 PbO2,20 and boron doped diamond,21 had been widely used in wastewater treatment. Among them, the Sb-doped SnO2 electrode has a wonderful application prospect due to its excellent electrochemical activity, chemical stability, and electrical conductivity.22 Currently, the common preparation methods include sol–gel coating method,23–25 electrodeposition,26,27 spray pyrolysis,28 and hydrothermal synthesis.29 Since the sol–gel coating method is simple in operation and easy to mass produce, the Sb-doped SnO2 electrode is usually prepared by this method.25 About the sol–gel method, the influences of the doping amount of Sb element and calcination temperature on the performance of the electrode had been studied.22,30,31 The results showed that the electrode would possess good catalytic activity when the doping amount of Sb was 6–9% and the calcination temperature was 550 °C. However, little research has investigated the electrocatalytic degradation efficiency of the electrodes prepared with different numbers of coating operations.
As an effective broad-spectrum antibiotic of the third generation fluoroquinolones, ciprofloxacin (CIP) has strong antibacterial activity against many bacteria, such as Escherichia coli, Pseudomonas aeruginosa, and Staphylococcus aureus. However, it is quite stable in the environment. Herein, CIP was selected as the target to evaluate the electrocatalytic activities of the investigated Sb-doped SnO2 electrodes, which were fabricated by the sol–gel method with different coating times. In order to understand the effects of the coating times on the structure and composition of the electrodes, the electrodes were characterized by scanning electron microscopy (SEM) and X-ray diffraction (XRD). And the electrochemical properties of these electrodes were also investigated by cyclic voltammetry (CV), linear sweep voltammetry (LSV), and electrochemical impedance spectroscopy (EIS). Moreover, the effects of current density, electrolyte concentration, initial concentration and initial pH of the substrate solutions on the removal of CIP were also explored so that it could provide valuable information for the removal of CIP in practical applications. Meanwhile, the possible intermediates formed in the degradation process were analyzed by HPLC-MS/MS, and the possible degradation pathways were proposed. Finally, the reusability of the Sb-doped SnO2 electrode was also studied.
2. Experimental
2.1. Materials and instruments
The chemicals and reagents were all analytical grade. SnCl4·5H2O, SbCl3, and CIP (>98%) were provided by Aladdin Reagent Co. Ltd. (Shanghai, China). Others were purchased from Sinopharm Group Co. Ltd. (Beijing, China). The titanium plates were obtained from Bojie Co. Ltd. (Baoji, China). Ultrapure water (18.25 MΩ cm) for all the solutions was produced using an Ultrapure Water Purification System from Ulupure Corporation (Sichuan, China). All reagents were used directly without further purification.2.2. Electrode preparation
Titanium plate (20 mm × 20 mm × 1 mm) was polished with sandpaper (1200 mesh) and degreased at 90 °C for 1 hour in 10 wt% NaOH solution. Then, it was etched at 90 °C for 2 hours in 10% boiling oxalic acid solution and finally rinsed with deionized water.25The electrode was prepared by a sol–gel method.32 Citric acid was mixed with ethylene glycol at 70 °C, and then the solution was heated to 90 °C. SnCl4·5H2O and SbCl3 were added to the solution in a molar ratio of citric acid:ethylene glycol:SnCl4·5H2O:SbCl3 of 130:30:9:1. The sol–gel was obtained by holding the solution at 90 °C for 30 min. Then, the sol–gel was applied to coat the prepared titanium plate. Finally, the coated titanium plate was baked in an infrared oven and then sintered for the thermal deposition at 550 °C in a muffle furnace for 10 min. After cooling to room temperature, the above coating-bake-thermal deposition process was repeated several times, and the titanium plate was annealed at 550 °C for 2 hours in the last time. The obtained electrodes were named as SSO-8, SSO-10, SSO-12, SSO-14, SSO-16, and SSO-20, where the numbers represented the times of coating operations.
2.3. Characterization of electrodes
The surface and composition of electrodes were characterized by SEM (Zeiss Sigma HD, Germany) and XRD (Bruker D8 Advance, Germany) with Cu (Kα) radiation at 40 kV/40 mA.The electrochemical performance of as-prepared electrode was evaluated in a three-electrode system at room temperature on CHI604E electrochemical workstation (Shanghai, Chenhua Instrument Co. Ltd., China) with saturated calomel electrode (SCE) as the reference electrode, platinum sheet as the counter electrode. The potentials appearing in the text were referred to SCE. Electrochemical double-layer capacitance was measured using CV in 0.5 M KOH solution, and the potential scanning range was from 0.5 V to 0.6 V. The CV was also used to test the electrochemical active area of the electrode in 0.1 M KCl and 1 mM K3Fe(CN)6 solution by sweeping from −0.1 V to 0.5 V. The LSV curves were tested in 0.5 M H2SO4 solution with a sweep rate of 50 mV s−1. The EIS was carried out in the AC frequency range of 100000–0.1 Hz with the amplitude of 5 mV under the applied potential of 2 V and the results were fitted using Zsimpwin software.
2.4. Detection of hydroxyl radical
The terephthalic acid fluorescence method was used to detect the hydroxyl radical.33 The tests were performed in a solution (100 mL) containing 0.5 mM terephthalic acid, 2.0 mM NaOH, and 0.25 M Na2SO4. The as-prepared electrodes and a stainless steel plate were used as anode and cathode, respectively. The applied current density was 15 mA cm−2. During the tests, 2 mL solutions were taken every 5 min for fluorescence spectroscopy. The produced 2-hydroxy terephthalic acid was measured by a fluorescence spectrometer (Hitachi F-7000, Japan) with the excitation wavelength of 315 nm and emission wavelength of 425 nm.2.5. Electrochemical degradation experiment
The CIP solution (50 mL) was degraded in a quartz beaker with Na2SO4 as the supporting electrolyte under a galvanostatic condition. The as-prepared SSO electrodes and a stainless steel plate were used as anode and cathode, respectively, with a submerged geometric surface area of 4 cm2. The distance between the two electrodes was 3 cm. Effects of coating times (8–20), current density (10–25 mA cm−2), electrolyte concentration (10–40 g L−1), initial concentration (10–80 mg L−1), and pH value (3–9) on degradation were investigated. During experiments, samples were taken every 15 min, and UV–vis Spectrophotometer (TU-1901, Beijing, China) was mainly used to monitor the removal rate of CIP in terms of variances in the absorbance, and the main absorption peak (λmax) located at 275 nm.34,35 The removal rate was calculated as follows: removal rate of CIP = (C0 − Ct)/C0 × 100, where C0 and Ct were the concentration of CIP at the initial and sampling time, respectively.Furthermore, the three-dimensional excitation-emission matrix fluorescence spectra (3D EEM) and HPLC-MS/MS were also used to assist the monitoring of the degradation. The 3D EMM were recorded using an Aqualog UV 800 (HORIBA Instruments Incorporated, USA). The final concentration of CIP and possible intermediates during the degradation progress were monitored by an Agilent 6460 triple quadrupole mass spectrometer equipped with an electrospray ionization source, combined with an Agilent 1260 series Liquid Chromatography system (HPLC-MS/MS) (Agilent, Palo Alto, CA, USA). The details were provided in the ESI (Text S1).†
3. Results and discussion
3.1. Characterization of the electrode structure
 | ||
| Fig. 1 SEM images of the prepared electrodes: (a) SSO-8, (b) SSO-10, (c) SSO-12, (d) SSO-14, (e) SSO-16, (f) SSO-20. | ||
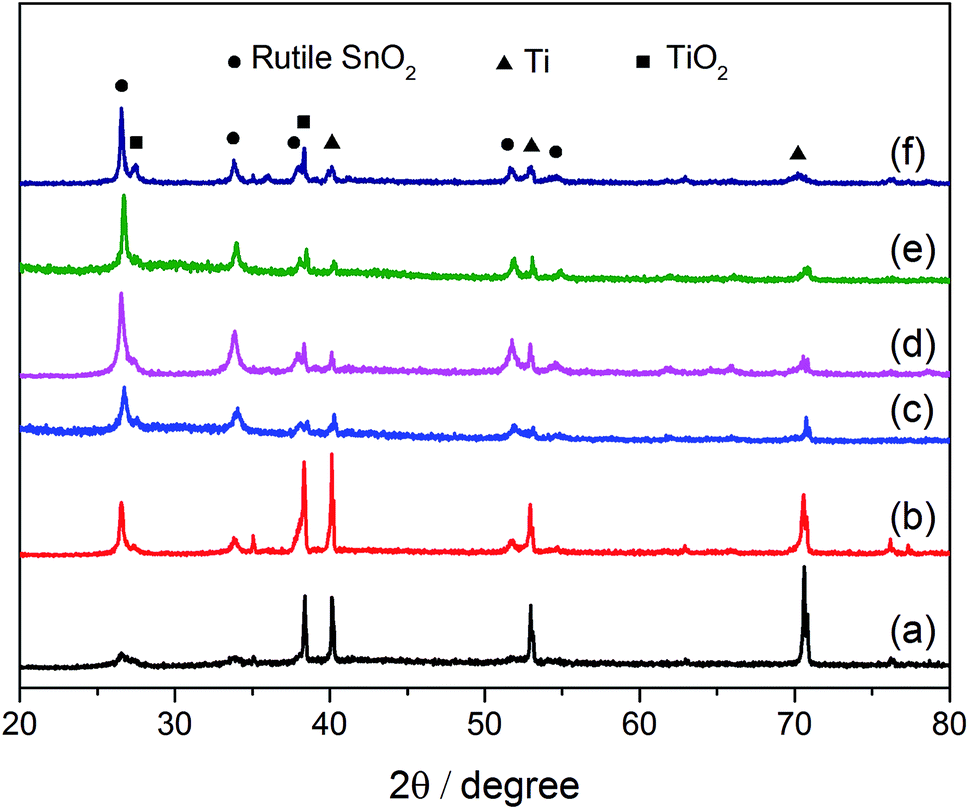 | ||
| Fig. 2 XRD patterns of the prepared electrodes: (a) SSO-8, (b) SSO-10, (c) SSO-12, (d) SSO-14, (e) SSO-16, (f) SSO-20. | ||
The crystal sizes estimated by the Scherer formula and the lattice parameters calculated using Bragg's equation were listed in Table S1.† It could be seen that the crystal size of the SSO-12 (12.2 nm) was the smallest, while those of SSO-14 and SSO-16 were 14.8 and 28.6 nm, respectively. Compared with the lattice parameters of SnO2 (JCPDF 72-1147), these calculated values of the prepared electrodes all decreased. This may be due to that the radius of doped Sb5+ ion was smaller than that of Sn4+ ion.22
3.2. Electrochemical characterization
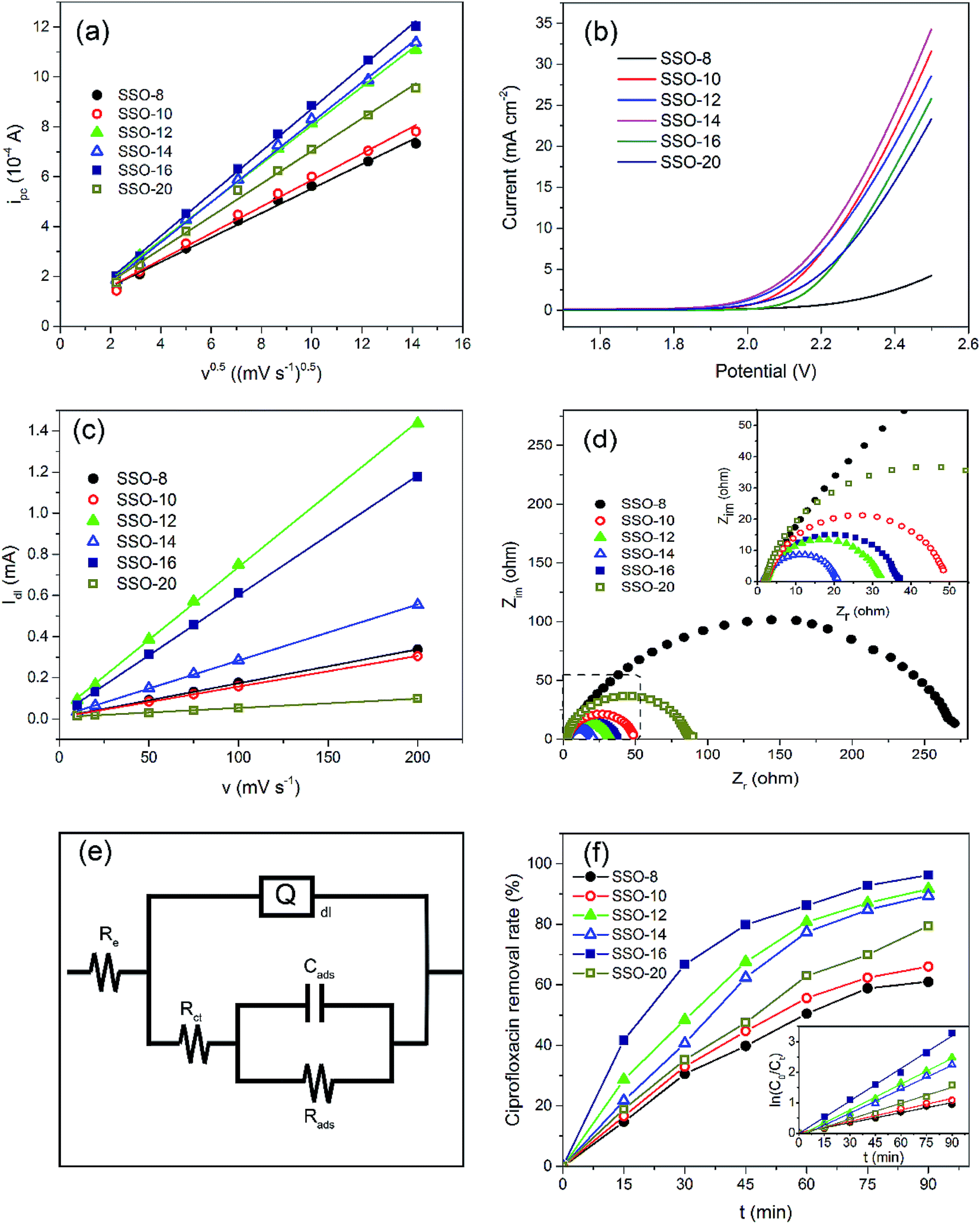 | ||
| Fig. 3 Electrochemical performance of the prepared electrodes: (a) CV peak current ipc in 1 mM K3Fe(CN)6 solution, (b) LSV curves in 0.5 M H2SO4 solution at a scan rate of 50 mV s−1, (c) Idl of CVs in 0.5 M KOH solution, (d) Nyquist plots of the different electrodes and (e) equivalent circuit model, (f) CIP removal (current density = 15 mA cm−2, Na2SO4 concentration = 20 g L−1, initial CIP concentration = 30 mg L−1, and initial pH = 5). Inset shows the kinetic analysis for CIP removal assuming a pseudo-first-order kinetics. | ||
Obviously, the SSO-16 had the largest ipc, indicating that the Fe(CN)63− on the electrode surface could be converted to Fe(CN)64− more quickly. Electrochemical active surface areas were calculated using Randles–Sevcik equation:
| ipc = 268600n1.5CD0.5Av0.5 | (1) |
3.3. Electrochemical degradation of CIP
CIP could produce fluorescence due to the presence of conjugated heterocycles in its molecule.45 In this study, 3D EEM was used to analyze the process of electrocatalytic degradation of CIP with the SSO-16 electrode. As shown in Fig. S3,† there were two fluorescent peaks at Ex/Em = 250–300/400–500 nm and Ex/Em = 300–350/400–500 nm in the initial CIP solution.46 The fluorescence intensity of the two peaks gradually decreased, possibly due to the destruction of the conjugated heterocyclic structure of CIP or its intermediate product.35 When the degradation time reached 90 minutes, no peak was observed, which proved that CIP could be effectively degraded by the SSO-16 electrode. Therefore, the SSO-16 electrode was selected.
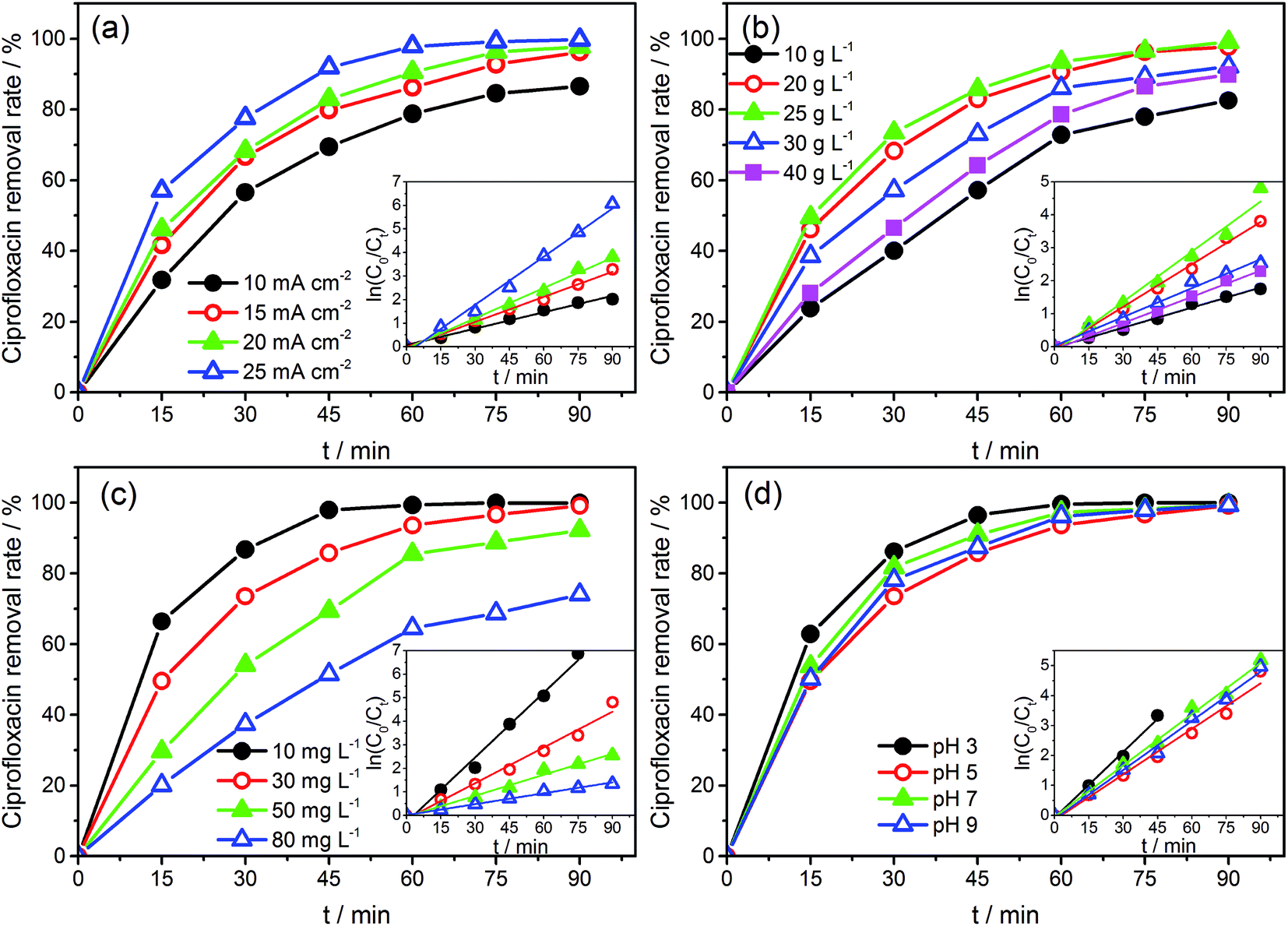 | ||
| Fig. 4 The removal rates of CIP with (a) different current densities (Na2SO4 concentration = 25 g L−1, initial CIP concentration = 30 mg L−1, and initial pH = 5), (b) different electrolyte concentrations (current density = 20 mA cm−2, initial CIP concentration = 30 mg L−1, and initial pH = 5), (c) different initial concentrations (current density = 20 mA cm−2, Na2SO4 concentration = 25 g L−1, and initial pH = 5), and (d) different pH value (current density = 20 mA cm−2, Na2SO4 concentration = 25 g L−1, and initial CIP concentration = 30 mg L−1). Inset shows kinetic analysis for CIP removal assuming a pseudo-first-order kinetics. | ||
The CVs of SSO-16 in 0.1 M Na2SO4 solution with the absence and presence of 30 mg L−1 CIP were conducted and the results were shown in Fig. S4.† When 30 mg L−1 CIP was added into the supporting electrolyte, there was no additional peak, suggesting that no direct electron exchange existed between CIP and the electrode, and the indirect oxidation should account for the majority of the reaction.48 The results of fluorescence detection (Fig. S5†) demonstrated the formation of hydroxyl radicals during electrolysis. Thus, it could be deduced reasonably that the removal of CIP was mainly due to the oxidation of hydroxyl radicals. However, it was reported that the higher current density meant the higher energy consumption, more annoying oxygen evolution, and shorter lifetime of the electrode.49 Correspondingly, 20 mA cm−2 was selected for the subsequent degradation experiment.
Under the optimized operation conditions (i.e., current density of 20 mA cm−2, Na2SO4 concentration of 25 g L−1, and initial pH of 3.0), the removal rate of 30 mg L−1 CIP solution (100 mL) could reach to almost 100% with the SSO-16 electrode within 60 min, which was confirmed by the representative chromatograms of the CIP solution obtained by HPLC-MS/MS (Fig. 5). The TOC removal rate was displayed in Fig. S6,† and it could reach to 93% within 90 min, implying that the mineralization of the target would take longer. It may be related to the removal of the intermediates.
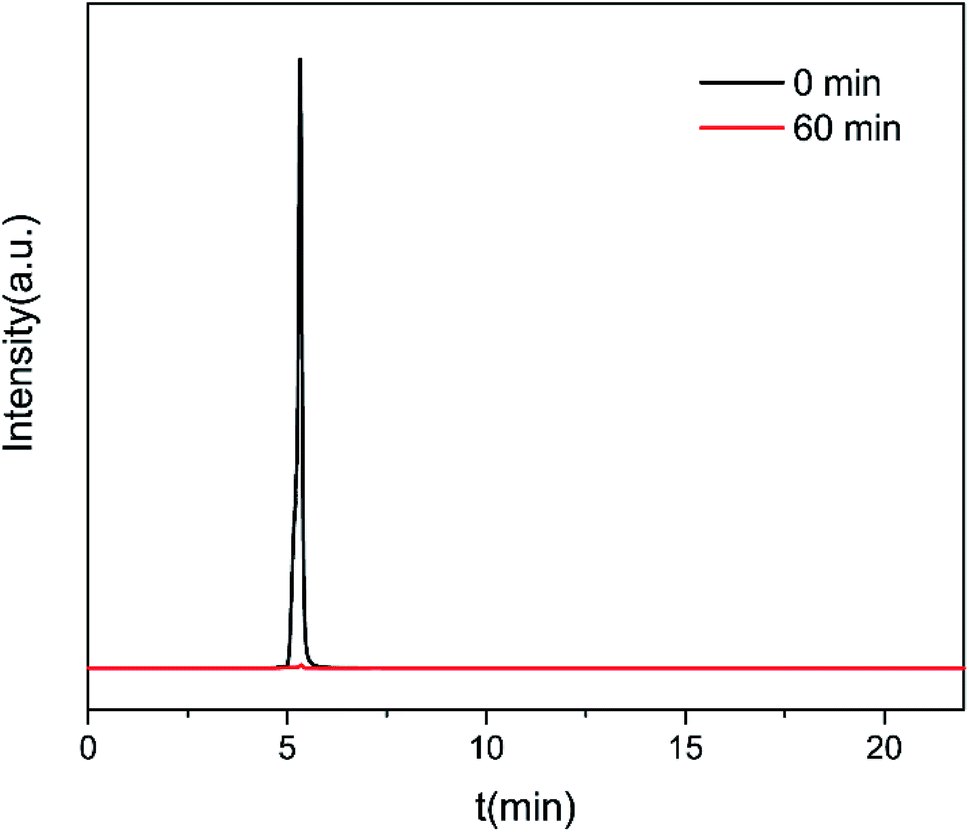 | ||
| Fig. 5 Typical chromatograms of CIP solution at 0 min and 60 min under the optimized electrocatalytic degradation conditions (current density = 20 mA cm−2, Na2SO4 concentration = 25 g L−1, initial CIP concentration = 30 mg L−1, and initial pH = 5). | ||
3.4. Analysis of the degradation pathway of CIP
In order to explore the degradation mechanism of CIP in this system, HPLC-MS/MS was used to identify the main intermediates in the degradation process. The peak areas of those intermediates changed with time (shown in Fig. S7†). In addition, based on the molecular structure of CIP, mass spectrometry information of intermediates and characteristic fragment ions, the structure of intermediates was reasonably inferred (listed in Table S4†). And according to previous studies,11,35,55 the possible degradation pathways of CIP were proposed, as shown in Scheme 1, which mainly included the removal of fluorine and the destruction of piperazine ring of quinolones. The three degradation pathways were staggered, and gradually achieved the goal of degrading CIP into small molecules.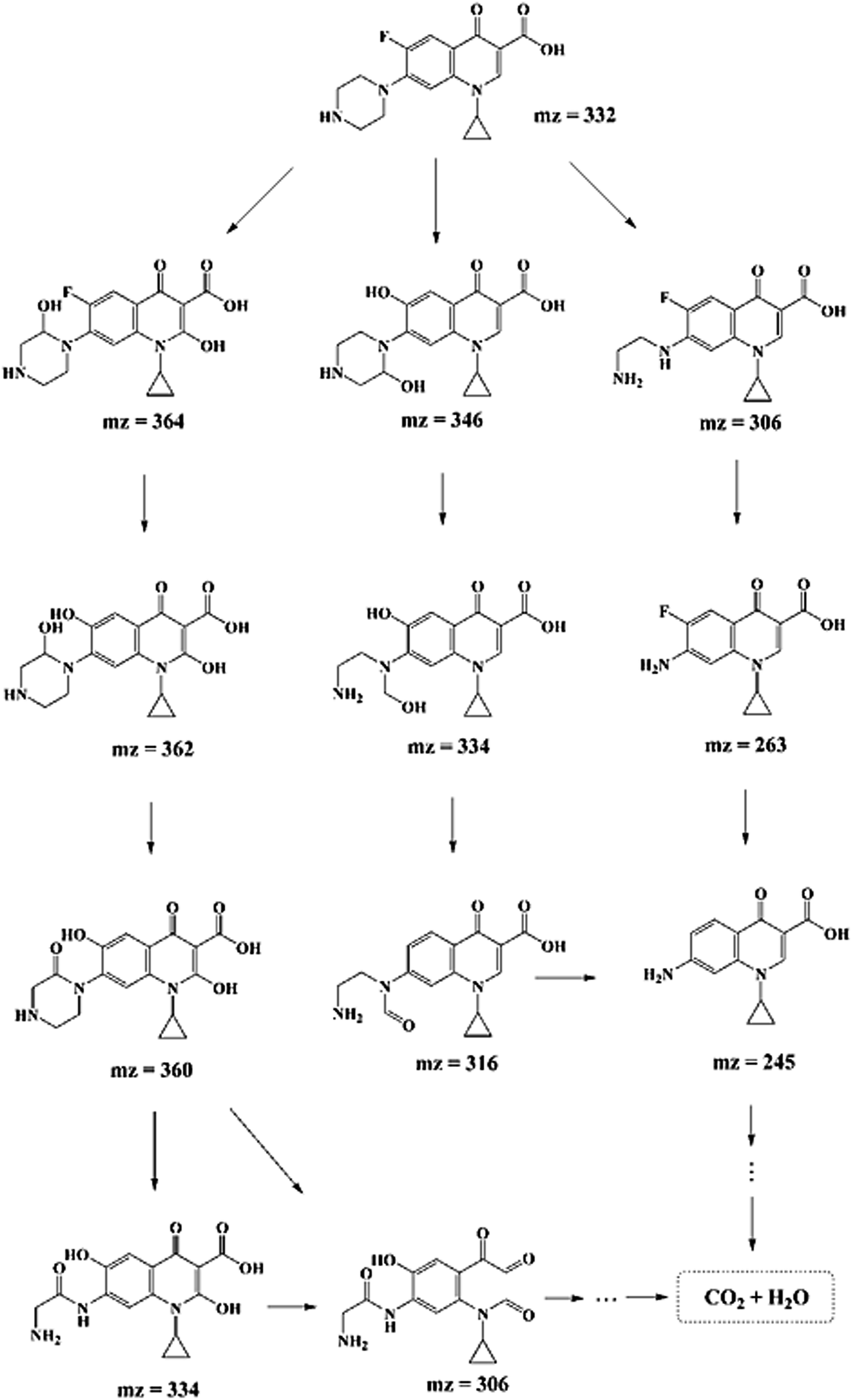 | ||
| Scheme 1 Possible degradation pathways for CIP degradation with SSO-16 electrode. | ||
3.5. Reusability
In order to evaluate the reusability of the electrodes, eight cycles of experiments were performed using the SSO-16 electrode under the optimized electrocatalytic conditions. As shown in Fig. 6, the removal rate of CIP could remain almost 100% with the electrode used 7 times. The removal rate had a slight decrease (94.27%) in the eighth reuse of the electrode. As seen in Fig. S8 and S9,† the particles on the surface of the SSO-16 electrode after the eighth reuse were broken slightly, while its XRD pattern had no obvious change. It indicated that the electrode was stable and reusable.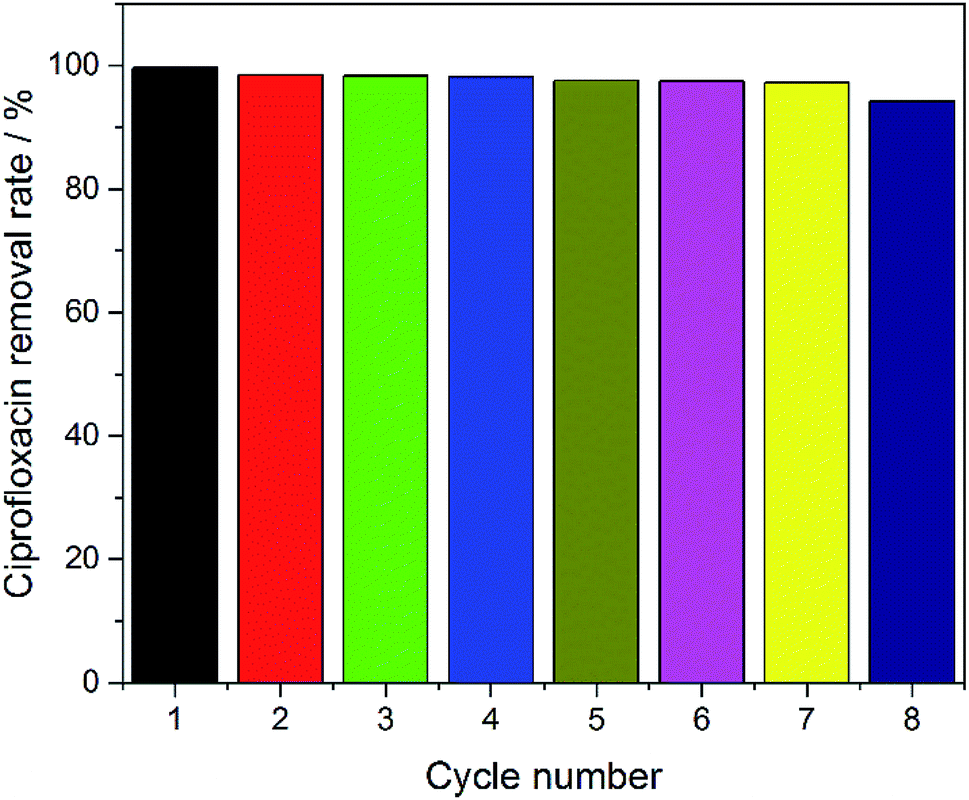 | ||
| Fig. 6 Removal rates of CIP in cycle experiments with SSO-16 electrode. | ||
4. Conclusion
SSO electrodes were successfully prepared by the sol–gel method. The coating times were found to have a remarkable influence on the electrocatalytic activity of the electrodes, and 12 to 16 times were the preferred coating times. The SSO-16 electrode with the highest OEP of 2.2 V exhibited excellent electrocatalytic degradation ability on CIP and reusability. Fluorescence spectroscopy and CV demonstrated that there were hydroxyl radicals produced during the degradation process and that CIP degradation was mainly from the attack of the hydroxyl radicals. Degradation experiments showed that the SSO-16 could remove CIP completely in 60 min with an initial concentration of 30 mg L−1 under the conditions: current density of 20 mA cm−2, Na2SO4 concentration of 25 g L−1 and initial pH value of 3. Based on the results of HPLC-MS/MS, the possible CIP degradation pathway was proposed.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Special Fund for Agro-scientific Research in the Public Interest of China (No. 201503108) and Science & Technology Project of Hunan Province (No. 2017WK2091).References
- Q. Bu, B. Wang, J. Huang, K. Liu, S. Deng, Y. Wang and G. Yu, Chemosphere, 2016, 144, 1384–1390 CrossRef CAS.
- M. Zhang, Y. Shi, Y. Lu, A. C. Johnson, S. Sarvajayakesavalu, Z. Liu, C. Su, Y. Zhang, M. D. Juergens and X. Jin, Sci. Total Environ., 2017, 590, 633–642 CrossRef PubMed.
- E. M. Golet, A. C. Alder and W. Giger, Environ. Sci. Technol., 2002, 36, 3645–3651 CrossRef CAS PubMed.
- E. M. Golet, A. Strehler, A. C. Alder and W. Giger, Anal. Chem., 2002, 74, 5455–5462 CrossRef CAS PubMed.
- H. Lin, H. Li, L. Chen, L. Li, L. Yin, H. Lee and Z. Yang, Ecotoxicol. Environ. Saf., 2018, 147, 530–536 CrossRef CAS PubMed.
- V. N. Binh, N. Dang, N. T. K. Anh, L. X. Ky and P. K. Thai, Chemosphere, 2018, 197, 438–450 CrossRef CAS PubMed.
- C. X. Yan, Y. Yang, J. L. Zhou, M. Liu, M. H. Nie, H. Shi and L. J. Gu, Environ. Pollut., 2013, 175, 22–29 CrossRef CAS PubMed.
- R. Mirzaei, M. Yunesian, S. Nasseri, M. Gholami, E. Jalilzadeh, S. Shoeibi and A. Mesdaghinia, Sci. Total Environ., 2018, 619–620, 446–459 CrossRef CAS PubMed.
- M. B. Ahmed, J. L. Zhou, H. H. Ngo and W. S. Guo, Sci. Total Environ., 2015, 532, 112–126 CrossRef CAS PubMed.
- J. R. Chen, F. X. Qiu, W. Z. Xu, S. S. Cao and H. J. Zhu, Appl. Catal., A, 2015, 495, 131–140 CrossRef CAS.
- X. Y. Zheng, S. P. Xu, Y. Wang, X. Sun, Y. Gao and B. Y. Gao, J. Colloid Interface Sci., 2018, 527, 202–213 CrossRef CAS PubMed.
- B. Tang, J. N. Du, Q. M. Feng, J. Q. Zhang, D. Wu, X. K. Jiang, Y. Dai and J. L. Zou, J. Colloid Interface Sci., 2018, 517, 28–39 CrossRef CAS PubMed.
- J. Chen, Y. Xia and Q. Dai, Electrochim. Acta, 2015, 165, 277–287 CrossRef CAS.
- M. Zhu, C. Zhai, M. Sun, Y. Hu, B. Yan and Y. Du, Appl. Catal., B, 2017, 203, 108–115 CrossRef CAS.
- J. Hu, C. Zhai, H. Gao, L. Zeng, Y. Du and M. Zhu, Sustainable Energy and Fuels, 2019, 3, 439–449 RSC.
- C. Zhai, M. Sun, L. Zeng, M. Xue, J. Pan, Y. Du and M. Zhu, Appl. Catal., B, 2019, 243, 283–293 CrossRef CAS.
- T. Velegraki, G. Balayiannis, E. Diamadopoulos, A. Katsaounis and D. Mantzavinos, Chem. Eng. J., 2010, 160, 538–548 CrossRef CAS.
- I. Sires, E. Brillas, G. Cerisola and M. Panizza, J. Electroanal. Chem., 2008, 613, 151–159 CrossRef CAS.
- J. Lv, Y. Feng, J. Liu, Y. Qu and F. Cui, Appl. Surf. Sci., 2013, 283, 900–905 CrossRef CAS.
- Y. Yao, L. Jiao, N. Yu, F. Guo and X. Chen, J. Solid State Electrochem., 2016, 20, 353–359 CrossRef CAS.
- J. Mora-Gomez, E. Ortega, S. Mestre, V. Perez-Herranz and M. Garcia-Gabaldon, Sep. Purif. Technol., 2019, 208, 68–75 CrossRef CAS.
- L. Yu, Y. Chen, W. Han, X. Sun, J. Li and L. Wang, RSC Adv., 2016, 6, 19848–19856 RSC.
- M. Seo, Y. Akutsu and H. Kagemoto, Ceram. Int., 2007, 33, 625–629 CrossRef CAS.
- M. E. Makgae, M. J. Klink and A. M. Crouch, Appl. Catal., B, 2008, 84, 659–666 CrossRef CAS.
- H. M. Bai, P. He, J. C. Chen, K. L. Liu, H. Lei, X. J. Zhang, F. Q. Dong and H. Li, Water Sci. Technol., 2017, 75, 220–227 CrossRef CAS PubMed.
- H. Y. Ding, Y. J. Feng and J. F. Liu, Mater. Lett., 2007, 61, 4920–4923 CrossRef CAS.
- A. N. S. Rao, V. T. Venkatarangaiah, G. B. Nagarajappa, S. H. Nataraj and P. M. Krishnegowda, J. Environ. Chem. Eng., 2017, 5, 4969–4979 CrossRef.
- P. D. Yao, Q. N. Jin, X. M. Chen, Y. Q. Xing and D. H. Wang, Electrochem. Solid-State Lett., 2008, 11, J37–J39 CrossRef CAS.
- A. S. Huang, G. H. Zhao and H. X. Li, Chin. Chem. Lett., 2007, 18, 997–1000 CrossRef CAS.
- X. Lei, L. Li, Y. Chen and Y. Hu, Environ. Sci. Pollut. Res., 2018, 25, 11683–11693 CrossRef CAS PubMed.
- C. M. Fan, B. Hua, Y. Wang, Z. H. Liang, X. G. Hao, S. B. Liu and Y. P. Sun, Desalination, 2009, 249, 736–741 CrossRef CAS.
- Z. R. Sun, H. Zhang, X. F. Wei, R. Du and X. Hu, J. Electrochem. Soc., 2015, 162, H590–H596 CrossRef CAS.
- H. Ding, Y. Feng, J. Lue and J. Liu, Chin. J. Anal. Chem., 2007, 35, 1395–1399 CAS.
- A. M. El-Kosasy, O. Abdel-Aziz, N. Magdy and N. M. El Zahar, Spectrochim. Acta, Part A, 2016, 157, 26–33 CrossRef CAS PubMed.
- X. J. Wen, C. G. Niu, L. Zhang, C. Liang, H. Guo and G.-M. Zeng, J. Catal., 2018, 358, 141–154 CrossRef CAS.
- Y. Duan, Y. Chen, Q. Wen and T. Duan, J. Electroanal. Chem., 2016, 768, 81–88 CrossRef CAS.
- T. G. Duan, Y. Chen, Q. Wen and Y. Duan, RSC Adv., 2015, 5, 19601–19612 RSC.
- L. Li, Z. Huang, X. Fan, Z. Zhang, R. Dou, S. Wen, Y. Chen, Y. Chen and Y. Hu, Electrochim. Acta, 2017, 231, 354–362 CrossRef CAS.
- P. Z. Duan, X. Hu, Z. Y. Ji, X. M. Yang and Z. R. Sun, Chemosphere, 2018, 212, 594–603 CrossRef CAS.
- M. J. Gira, K. P. Tkacz and J. R. Hampton, Nano convergence, 2016, 3, 6 CrossRef.
- R. Mei, Q. Wei, C. Zhu, W. Ye, B. Zhou, L. Ma, Z. Yu and K. Zhou, Appl. Catal., B, 2019, 245, 420–427 CrossRef CAS.
- S. Ferro, D. Rosestolato, C. A. Martínez-Huitle and A. De Battisti, Electrochim. Acta, 2014, 146, 257–261 CrossRef CAS.
- A. Oury, A. Kirchev and Y. Bultel, Electrochim. Acta, 2012, 63, 28–36 CrossRef CAS.
- Y. Wang, C. Shen, M. Zhang, B.-T. Zhang and Y.-G. Yu, Chem. Eng. J., 2016, 296, 79–89 CrossRef CAS.
- S. N. Yin, T. Yao, T.-H. Wu, Y. Zhang and P. Wang, Talanta, 2017, 174, 14–20 CrossRef CAS PubMed.
- X. J. Wen, C.-G. Niu, L. Zhang, C. Liang and G.-M. Zeng, Appl. Catal., B, 2018, 221, 701–714 CrossRef CAS.
- L. Feng, E. D. van Hullebusch, M. A. Rodrigo, G. Esposito and M. A. Oturan, Chem. Eng. J., 2013, 228, 944–964 CrossRef CAS.
- H. An, H. Cui, W. Zhang, J. Zhai, Y. Qian, X. Xie and Q. Li, Chem. Eng. J., 2012, 209, 86–93 CrossRef CAS.
- D. Shao, J. Liang, X. Cui, H. Xu and W. Yan, Chem. Eng. J., 2014, 244, 288–295 CrossRef CAS.
- H. Alemu and L. Hlalele, Bull. Chem. Soc. Ethiop., 2007, 21, 1–12 CrossRef CAS.
- J. Wang, L. Tang, G. Zeng, Y. Deng, Y. Liu, L. Wang, Y. Zhou, Z. Guo, J. Wang and C. Zhang, Appl. Catal., B, 2017, 209, 285–294 CrossRef CAS.
- C. A. Martínez-Huitle and E. Brillas, Appl. Catal., B, 2009, 87, 105–145 CrossRef.
- L. Ge, J. Chen, X. Wei, S. Zhang, X. Qiao, X. Cai and Q. Xie, Environ. Sci. Technol., 2010, 44, 2400–2405 CrossRef CAS PubMed.
- X. Yu, G. L. Zipp and G. W. R. Davidson Iii, Pharm. Res., 1994, 11, 522–527 CrossRef CAS PubMed.
- T. An, H. Yang, G. Li, W. Song, W. J. Cooper and X. Nie, Appl. Catal., B, 2010, 94, 288–294 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04860j |
| This journal is © The Royal Society of Chemistry 2019 |