
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Theoretical investigation of the ORR on boron–silicon nanotubes (B–SiNTs) as acceptable catalysts in fuel cells
Razieh
Razavi
*a and
Meysam
Najafi
 *b
*b
aDepartment of Chemistry, Faculty of Science, University of Jiroft, Jiroft, Iran. E-mail: R.Razavi@ujiroft.ac.ir
bMedical Biology Research Center, Health Technology Institute, Kermanshah University of Medical Sciences, Kermanshah, Iran. E-mail: iau.mnajafi@yahoo.com
First published on 4th October 2019
Abstract
Here, the potential of boron doped silicon nanotubes (7, 0) as ORR catalysts is examined. Acceptable paths for the ORR on studied catalysts are examined through DFT. The optimum mechanism of the ORR on the surface of B2–SiNT (7, 0) is shown. The ORR on the surface of B2–SiNTs (7, 0) can continue through LH and ER mechanisms. The calculated beginning voltage for the ORR on B2–SiNTs (7, 0) is 0.37 V and it is smaller than the beginning voltage (0.45 V) for platinum-based catalysts. In the acidic solution the beginning voltage for the oxygen reduction process can be evaluated to be 0.97 V, which corresponds to 0.37 V as a minimum overvoltage for the ORR. The B2–SiNTs (7, 0) are suggested as an ORR catalyst in acidic environments.
1. Introduction
Fuel cells as energy machines are important due to their low contamination and great efficiency. The ORR rate in electrodes of cells is slow, therefore the ORR can be evaluated as a significant reason to increase the full cell efficiency.1–4 Platinum-compounds have been used as catalysis in the ORR but platinum-compounds have low ability to endure CO.5–9The potential of various compounds was investigated to find and propose effective catalysts for the ORR. Nanostructures and doped nanostructures with high ability for CO endurance can be used as suitable replacements for platinum-compounds.10–15 The B-nanostructures are acceptable catalysts for the ORR in alkaline conditions and mechanisms of action of B-doped nanostructures in acidic position are not clear.16–23
The nanostructures due to their electrical conductivity and thermal conductivity can be used to product the transistors and non-volatile memory devices.24–28 The electrical conductivity of doped carbon/silicon nanotubes indicated that the adoption of carbon/silicon nanotubes (with various atoms such as B, N, O and some metals) increased their electrical conductivity, significantly. These findings improved the application of carbon/silicon nanotubes in nano-electronic devices and novel catalyst to ORR.29–38 Results demonstrated that the adoption of carbon/silicon nanotubes increased their electrical conductivity and enhanced the ORR efficiency.39–48
Wang et al.49 demonstrated that boron-doped graphene nanoribbons are suitable catalyst to ORR catalyst. Xiao et al.50 proved that the layered silicon–carbon nano sheets represented the high activity in ORR without CO poisoning. Xia and Zhang et al.51,52 investigated the mechanisms of ORR of fuel cells in acidic environment on graphene cathodes. Stevenson et al.53 proved in ORR the O2 in a 2-electron path is reduced to form OOH on carbon nanotubes. Hu and Xiong et al.54,55 confirmed that nitrogen and boron-doped nanostructures as ORR catalysts have low price, great durability and excellent potential. Zhao and Wei et al.56,57 confirmed the doping of carbon nanotubes have vital roles on performance of ORR. Ferrighi et al.58 demonstrated that boron atoms of nano-sheets increase the reactions of oxygen with graphene.
In current study, ORR on B-doped silicon nanotube (7, 0) as acceptable catalysts is examined to find possible mechanisms to ORR on B2–SiNT (7, 0) and to suggest high activity nano-catalysts to ORR.
2. Computational details
In this study the silicon nanotube (length and diameter are 1 and 0.475 nm) is modeled and their open elements are saturated with hydrogen atoms to elude border effects. The geometries of nanotubes and studied molecules (such as OOH, OH, H2O and CO) are optimized by M06-2X/6-311G+ (2d, 2p) in GAMESS package.59–72 The consistent field is investigated by 10−6 Hartree as convergence value. Vibrational frequencies of nanotubes and molecules by M06-2X/6-311G+ (2d, 2p) are calculated.In the density functional, M06 functionals are extremely parameterized proximate exchange functionals theory and they are supported on generalized gradient approximation (meta-GGA). These functionals are used for traditional quantum chemistry, solid-state physics calculations and thermodynamic values of reactions.73–82 M06-2X as the most accurate functional of Minnesota functional is a Global hybrid functional with 54% HF exchange and it is the ascendancy constructor within the 06 functionals for thermochemistry, kinetics and various chemical interactions.73–82 The M06-2X functional as hybrid meta exchange–correlation functionals present 32 empirically improved factors within the exchange–correlation functional.83–87
The energy and Gibbs free energy (G = E0 + ZPE + ΔH + RT − TS) values of nanotubes are calculated. The E0 and ZPE are electronic energy and zero-point energy and T is 298.15 K.59–72 Adoption energy (Edoped) and Gibbs free energy adoption (Gdoped) of B atoms in SiNT (7, 0) are calculated:
| Edoped = E(B–SiNT) − E(SiNT) − E(B) | (1) |
| Gdoped = G(B–SiNT) − G(SiNT) − G(B) | (2) |
| Edoped = E(B2–SiNT) − E(SiNT) − 2E(B) | (3) |
| Gdoped = G(B2–SiNT) − G(SiNT) − 2G(B) | (4) |
Energy adsorption (ΔEad) and Gibbs free energy adsorption (ΔGad) of molecules (such as OOH, OH, H2O and CO) on surfaces of studied nanotubes (SiNT, B–SiNT, B–B–SiNT and B2–SiNT) are calculated:
| ΔEad = E(molecule–nanotube) − E(nanotube) − E(molecule) | (5) |
| ΔGad = G(molecule–nanotube) − G(nanotube) − G(molecule) | (6) |
Activation energy (ΔEa = ETS − EIS) is difference of energy between transition (ETS) and initial (EIS) studied complexes. In this study, the reaction energy (ΔEa = EFS − EIS) is difference of energy between final (EFS) and initial (EIS) studied complexes.88–95 The ΔG of ORR on B2–SiNT (7, 0) in according to standard hydrogen electrode was evaluated through ΔG = ΔE + ΔZPE − TΔS + ΔGU + ΔGpH (obtained data were reported in Fig. 5).96–104 ΔGpH (ΔGpH = kT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln10 × pH) is modification of proton Gibbs free energy and ΔGU = − neU. The n, e and U are electrons, first charge and electrode potential. The U is requested potential and overvoltage is η = U0 − U.49–58 Conductor like screening method is used to estimate water environment (dielectric constant is 78.54).59–72
ln10 × pH) is modification of proton Gibbs free energy and ΔGU = − neU. The n, e and U are electrons, first charge and electrode potential. The U is requested potential and overvoltage is η = U0 − U.49–58 Conductor like screening method is used to estimate water environment (dielectric constant is 78.54).59–72
3. Results and discussion
3.1. Molecule adsorptions on nanotube
In this section, the B adoption of SiNT (7, 0) were investigated and then interactions of B–SiNT (7, 0) structures with O2, OOH, OH, H2O and CO molecules were investigated. The one Si atom of the SiNT (7, 0) was replaced with one B atom and the B–SiNT (7, 0) was produced (Fig. 1). Also the two Si atoms of the SiNT (7, 0) in two difference positions were replaced with two B atoms and B–B–SiNT (7, 0) and B2–SiNT (7, 0) structures were produced (Fig. 1). Structures of SiNT, B–SiNT, B–B–SiNT, B2–SiNT, O2, OH, H2O, H2O2 and CO are presented in Fig. 1. The adoption energy (Edoped), adoption free Gibbs energy (Gdoped) and bond lengths of B–Si of B–SiNT (7, 0), B–B–SiNT (7, 0) and B2–SiNT (7, 0) were reported in Fig. 1. In the B–SiNT (7, 0) the B atom was connected with three neighboring silicon atoms and the Edoped and Gdoped were −2.18 and −2.10 eV and average of bonds of B–Si in B–SiNT (7, 0) is 1.95 Å. In the B–B–SiNT (7, 0) the B atoms are connected with four neighboring silicon atoms and the Edoped and Gdoped are −2.23 and −2.14 eV and average of bonds of B–Si in B–B–SiNT (7, 0) is 1.93 Å. In the B2–SiNT (7, 0) the B atoms are connected with six neighboring silicon atoms and the Edoped and Gdoped values are −2.28 and −2.17 eV and average of bonds of B–Si in B2–SiNT (7, 0) are 1.92 Å.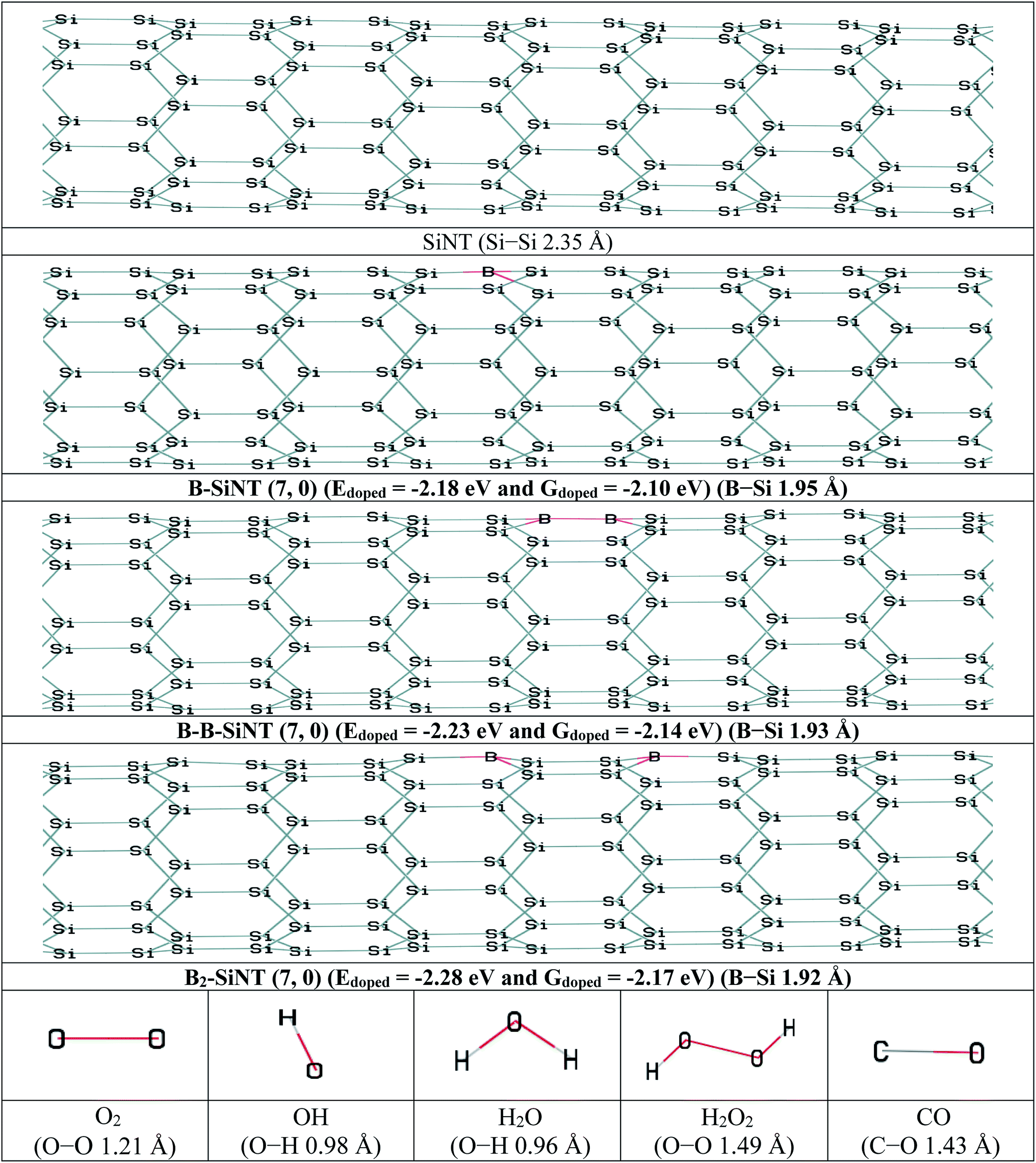 | ||
| Fig. 1 The initial structures of SiNT, B–SiNT, B–B–SiNT and B2–SiNT and O2, OH, H2O, H2O2 and CO molecules. | ||
The q and EHLG of SiNT (7, 0), B–SiNT (7, 0), B–B–SiNT (7, 0) and B2–SiNT (7, 0) are stated in Table 1. EHLG of SiNT (7, 0), B–SiNT (7, 0), B–B–SiNT (7, 0) and B2–SiNT (7, 0) are 1.84, 1.75, 1.68 and 1.64 eV. The q of B–SiNT (7, 0), B–B–SiNT (7, 0) and B2–SiNT (7, 0) are 0.58, 0.69 and 0.73|e|. The |Edoped|, |Gdoped| and q values of B2–SiNT (7, 0) are higher than B–SiNT (7, 0) and B–B–SiNT (7, 0). Results showed that the EHLG value of B2–SiNT (7, 0) is lower than corresponding values on surfaces of B–SiNT (7, 0) and B–B–SiNT (7, 0). Therefore, it can be concluded that the B2–SiNT (7, 0) is the most stable than B–SiNT (7, 0) and B–B–SiNT (7, 0) from thermodynamic view point. The B atoms in B–SiNT (7, 0), B–B–SiNT (7, 0) and B2–SiNT (7, 0) structures are advantageous to adsorption of O2 molecule and these B atoms are active positions of B–SiNT (7, 0), B–B–SiNT (7, 0) and B2–SiNT (7, 0) as catalyst for ORR. Therefore, B atoms are essential location to adsorption of O2 molecule and B atoms can be considered as initiator of first ORR step.
| Complex | q | E HLG | Complex | q | E HLG | Complex | q | E HLG | Complex | q | E HLG |
|---|---|---|---|---|---|---|---|---|---|---|---|
| SiNT | —- | 1.84 | B–SiNT | 0.58 | 1.75 | B–B–SiNT | 0.69 | 1.68 | B2–SiNT | 0.73 | 1.64 |
| 2a | 0.36 | 1.69 | 2b | 0.47 | 1.61 | 2c | 0.59 | 1.52 | 2d | 0.64 | 1.48 |
| 2e | 0.29 | 1.75 | 2f | 0.39 | 1.68 | 2g | 0.51 | 1.59 | 2h | 0.58 | 1.55 |
| 2i | 0.32 | 1.72 | 2j | 0.43 | 1.64 | 2k | 0.54 | 1.56 | 2l | 0.61 | 1.52 |
| 2m | 0.49 | 1.35 | 2n | 0.62 | 1.23 | 2o | 0.73 | 1.17 | 2p | 0.82 | 1.14 |
| 3a | 0.41 | 1.54 | 3b | 0.51 | 1.45 | 3c | 0.65 | 1.38 | 3d | 0.73 | 1.25 |
| 3e | 0.59 | 1.14 | 3f | 0.81 | 1.05 | 3g | 0.84 | 0.99 | 3h | 0.91 | 0.95 |
| 3m | 0.11 | 1.80 | 3n | 0.14 | 1.70 | 3o | 0.19 | 1.62 | 3p | 0.21 | 1.60 |
| 3q | 0.08 | 1.82 | 3r | 0.12 | 1.72 | 3s | 0.15 | 1.64 | 3t | 0.18 | 1.63 |
Therefore, O2 adsorption on SiNT (7, 0), B–SiNT (7, 0), B–B–SiNT (7, 0) and B2–SiNT (7, 0) were investigated. The possible positions of SiNT (7, 0), B–SiNT (7, 0), B–B–SiNT (7, 0) and B2–SiNT (7, 0) to O2 adsorption including top position B atom and bridge positions of B–Si, B–B and Si–Si bonds were examined in Fig. 2. The B–O, O–O and Si–O in SiNT (7, 0), B–SiNT (7, 0), B–B–SiNT (7, 0) and B2–SiNT (7, 0) with O2 are presented in Fig. 2 (2a–2l structures). The ΔEad and ΔGad of O2 on SiNT (7, 0), B–SiNT (7, 0), B–B–SiNT (7, 0) and B2–SiNT (7, 0) are displayed in Table 2. |ΔGad| of O2 on B–SiNT, B–B–SiNT and B2–SiNT are greater than SiNT (7, 0). The |ΔEad| and |ΔGad| of O2 on B2–SiNT (7, 0) are greater than B–SiNT (7, 0), B–B–SiNT (7, 0). The bridge position of B–B in B2–SiNT (7, 0) is stable than top position B in B2–SiNT (7, 0) to O2 adsorption.
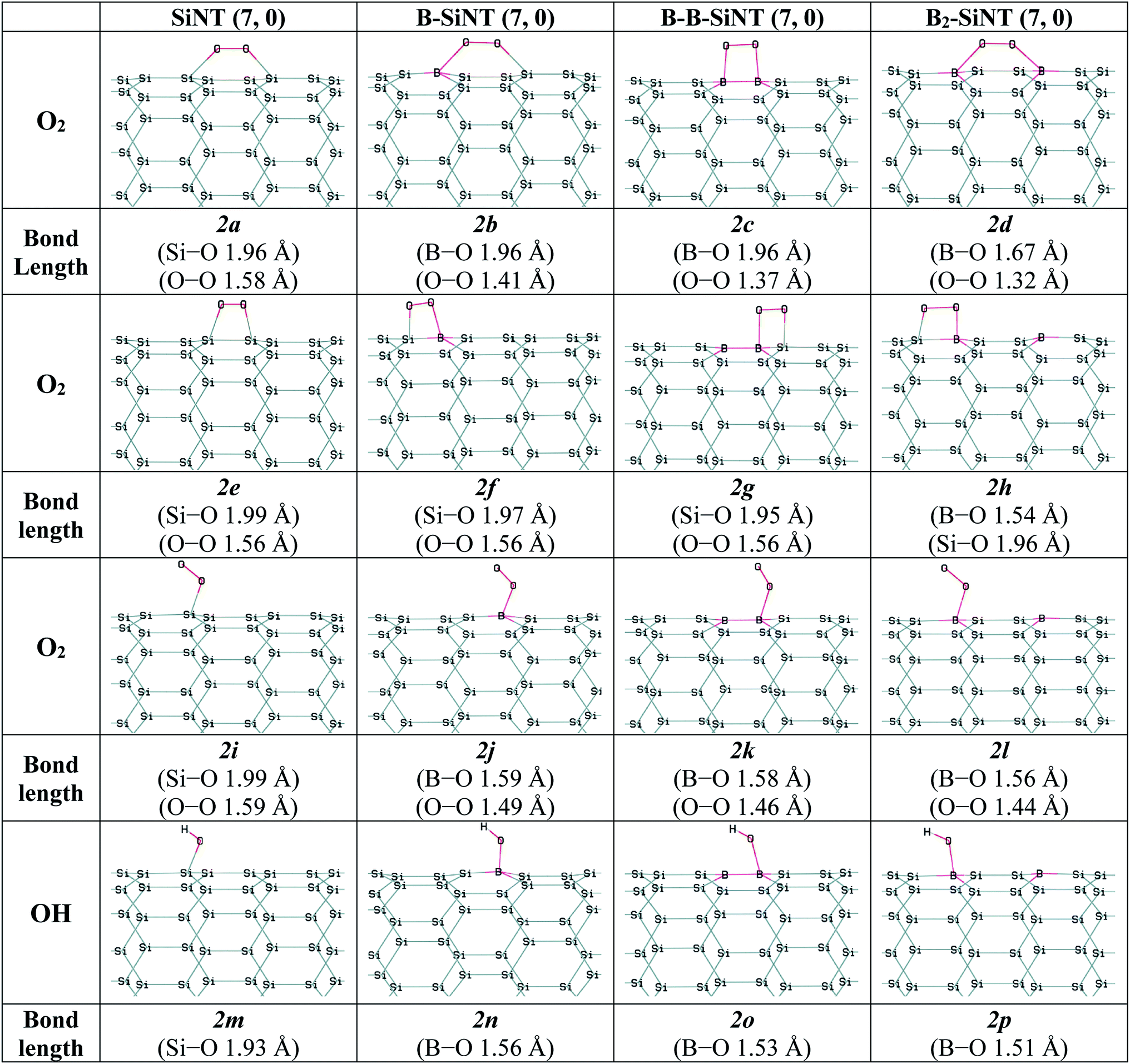 | ||
| Fig. 2 Complexes of the SiNT (7, 0), B–SiNT (7, 0), B–B–SiNT (7, 0) and B2–SiNT (7, 0) with O2 and OH molecules. | ||
| Complex | ΔEad | ΔGad | Complex | ΔEad | ΔGad | Complex | ΔEad | ΔGad | Complex | ΔEad | ΔGad |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2a | −0.41 | −0.36 | 2b | −0.61 | −0.54 | 2c | −0.84 | −0.79 | 2d | −0.88 | −0.81 |
| 2e | −0.39 | −0.33 | 2f | −0.42 | −0.38 | 2g | −0.55 | −0.51 | 2h | −0.61 | −0.55 |
| 2i | −0.27 | −0.24 | 2j | −0.64 | −0.61 | 2k | −0.69 | −0.64 | 2l | −0.73 | −0.68 |
| 2m | −2.07 | −1.97 | 2n | −2.23 | −2.15 | 2o | −2.31 | −2.21 | 2p | −2.39 | −2.28 |
| 3a | −0.87 | −0.81 | 3b | −1.06 | −0.99 | 3c | −1.11 | −1.03 | 3d | −1.17 | −1.12 |
| 3e | −3.11 | −2.97 | 3f | −3.59 | −3.45 | 3g | −3.67 | −3.56 | 3h | −3.79 | −3.67 |
| 3i | −2.24 | −2.13 | 3j | −2.25 | −2.15 | 3k | −2.27 | −2.17 | 3l | −2.31 | −2.21 |
| 3m | −0.15 | −0.10 | 3n | −0.18 | −0.13 | 3o | −0.20 | −0.15 | 3p | −0.24 | −0.19 |
| 3q | −0.09 | −0.05 | 3r | −0.10 | −0.07 | 3s | −0.11 | −0.08 | 3t | −0.14 | −0.12 |
Wang, Xiao and Xia et al.49–51 calculated the O2 adsorption on surfaces of boron-doped graphene nanoribbon, silicon–carbon nano sheets and B and N doped-graphene by theoretical methods, respectively (results reported in Table 4). The ΔEad of O2 on B-doped graphene, silicon–carbon nano-sheets and N-doped graphene are −0.62, −0.53 and −0.60 eV. Therefore ΔEad value (−0.61 eV) of O2 on B2–SiNT (7, 0) in present study is similar to corresponding values of O2 on various nanostructures were calculated in previous theoretical works.49–51
| Path | Studied reaction steps | ΔEa (eV) | ΔEr (eV) |
|---|---|---|---|
| 1 | O2 + B2–SiNT (7, 0) → B2–SiNT (7, 0)–*O2 | — | −0.68 |
| 1 | B2–SiNT (7, 0)–*O2 + H+ + e− → B2–SiNT (7, 0)–*OOH | 0.00 | −1.07 |
| 1 | B2–SiNT (7, 0)–*OOH + H+ + e− → B2–SiNT (7, 0)–*O + H2O | 0.18 | −2.75 |
| 1 | B2–SiNT (7, 0)–*O + H+ + e− → B2–SiNT (7, 0)–*OH | 0.37 | −1.57 |
| 1 | B2–SiNT (7, 0)–*OH + H+ + e− → B2–SiNT (7, 0)* + H2O | 0.07 | −1.34 |
| 2 | O2 + B2–SiNT (7, 0) → B2–SiNT (7, 0)–*O2 | — | −0.68 |
| 2 | B2–SiNT (7, 0)–*O2 + H+ + e− → B2–SiNT (7, 0)–*OOH | 0.00 | −1.07 |
| 2 | B2–SiNT (7, 0)–*OOH + H+ + e− → *OH–B2–SiNT (7, 0)–*OH | 0.24 | −2.97 |
| 2 | *OH–B2–SiNT (7, 0)–*OH + H+ + e− → B2–SiNT (7, 0)–*OH + H2O | 0.35 | −1.20 |
| 2 | B2–SiNT (7, 0)–*OH + H+ + e− → B2–SiNT (7, 0)* + H2O | 0.07 | −1.34 |
The charge transfer (q) and HOMO–LUMO band gap (EHLG) of the complexes of SiNT (7, 0), B–SiNT (7, 0), B–B–SiNT (7, 0) and B2–SiNT (7, 0) with O2 molecule are displayed in Table 1. The EHLG values of O2 adsorption on B–SiNT (7, 0), B–B–SiNT (7, 0) and B2–SiNT (7, 0) are lower than SiNT (7, 0). The EHLG values of O2 adsorption on B2–SiNT (7, 0) are lower than B–SiNT (7, 0), B–B–SiNT (7, 0). The bridge position of B–B in B2–SiNT (7, 0) has higher q and lower EHLG than top position B in B2–SiNT (7, 0) to O2 adsorption. Complex of B2–SiNT (7, 0) with O2 molecule (2d structure) is the most stable than other complexes of B–SiNT (7, 0) and B–B–SiNT (7, 0) with O2 molecule from thermodynamic view point. It can be concluded that O2 adsorbed on B2–SiNT (7, 0) in figure 2d significantly and there are suitable interactions between the O2 molecule and B2–SiNT (7, 0) and the adsorption of O2 molecules on studied surfaces are chemical adsorption processes.
In this study the interactions of important intermediates such as O, H, OOH, OH, H2O and CO molecules with SiNT (7, 0), B–SiNT (7, 0), B–B–SiNT (7, 0) and B2–SiNT (7, 0) surfaces in process of ORR were investigated (Fig. 2 and 3). The bonds of Si–O of SiNT (7, 0), B–SiNT (7, 0), B–B–SiNT (7, 0) and B2–SiNT (7, 0) with molecules are stated. EHLG, q, ΔEad, ΔGad of molecules on SiNT (7, 0), B–SiNT (7, 0), B–B–SiNT (7, 0) and B2–SiNT (7, 0) are reported in Tables 1 and 2. OOH and OH intermediates can be adsorb on B site of B–SiNT (7, 0), B–B–SiNT (7, 0) and B2–SiNT (7, 0). The O intermediate has tendency to adsorb on B–Si and B–B bridge positions of B–SiNT (7, 0), B–B–SiNT (7, 0) and B2–SiNT (7, 0). It can be concluded that the complexes of B2–SiNT (7, 0) with O, H, OOH, OH and H2O molecules are stable than SiNT (7, 0), B–SiNT (7, 0) and B–B–SiNT (7, 0).
 | ||
| Fig. 3 Complexes of the SiNT (7, 0), B–SiNT (7, 0), B–B–SiNT (7, 0) and B2–SiNT (7, 0) with O and H atoms, OOH, H2O and CO molecules. | ||
Wang, Xiao and Xia et al.49–51 calculated the O, OH and OOH adsorption on surfaces of boron-doped graphene nanoribbon, silicon–carbon nano sheets and B and N doped-graphene by theoretical methods, respectively (results reported in Table 4). ΔEad of O on B-doped graphene, silicon–carbon nano-sheets and N-doped graphene were −3.74, −4.11 and −3.55 eV. ΔEad of OH on boron-doped graphene, silicon–carbon nano-sheets and N-doped graphene were −2.38, −2.87 and −2.41 eV. ΔEad of OOH on B-doped graphene, silicon–carbon nano-sheets and N-doped graphene are −1.12, −1.18 and −1.06 eV. ΔEad values of O, OH and OOH (−1.17, −2.39 and −1.17 eV) on B2–SiNT (7, 0) in present study are similar to corresponding values of O, OH and OOH on various nanostructures were calculated in previous theoretical works.49–51
The H2O molecule favored to adsorb on above ring position of SiNT (7, 0), B–SiNT (7, 0), B–B–SiNT (7, 0) and B2–SiNT (7, 0) and the average of ΔEad and ΔGad values are −0.19 and −0.14 eV. The average of q and EHLG values for adsorption of H2O molecule on SiNT (7, 0), B–SiNT (7, 0), B–B–SiNT (7, 0) and B2–SiNT (7, 0) surface is 0.16|e| and 1.68 eV. H2O molecule can be adsorbed on SiNT (7, 0), B–SiNT (7, 0), B–B–SiNT (7, 0) and B2–SiNT (7, 0) surfaces as physical adsorption processes.
In process of ORR the CO can occupy the positions of catalysts and the performance of ORR is reduced and efficiency of catalyst decreases sharply. Previous works showed that reactions between CO molecule and surface of platinum nano-catalyst was powerful (ΔEad is −1.90 eV) and CO poisoning was happen.20,66 The average of ΔEad and ΔGad of CO on SiNT, B–SiNT, B–B–SiNT and B2–SiNT surfaces are −0.11 and −0.08 eV. The average of q and EHLG values for adsorption of CO on SiNT (7, 0), B–SiNT (7, 0), B–B–SiNT (7, 0) and B2–SiNT (7, 0) surface is 0.13|e| and 1.70 eV. The CO molecule can be adsorbed on SiNT (7, 0), B–SiNT (7, 0), B–B–SiNT (7, 0) and B2–SiNT (7, 0) surfaces as physical adsorption processes. It can be concluded that B2–SiNT (7, 0) as acceptable catalyst can be endurance to CO poisoning and it can solve the major problem of platinum nano-catalysts.
Wang, Xiao and Xia et al.49–51 calculated the H2O and CO adsorption on surfaces of boron-doped graphene nanoribbon, silicon–carbon nano sheets and B and N doped-graphene. The ΔEad of H2O on surfaces of B-doped graphene, silicon–carbon nano-sheets and N-doped graphene were −0.24, −0.18 and −0.08 eV. The ΔEad of CO on surfaces of B-doped graphene, silicon–carbon nano-sheets and N-doped graphene were −0.17, −0.07 and −0.12 eV. The ΔEad of H2O and CO (−0.24 and −0.14 eV) on B2–SiNT (7, 0) in present study are similar to corresponding values of H2O and CO on various nanostructures were calculated in previous theoretical works.49–51
3.2. B2–SiNT (7, 0) as catalyst to ORR
Nano-catalysts processed the chemical reactions through the ER and LH paths. The paths for ORR via B2–SiNT (7, 0) as acceptable catalyst through the LH and ER mechanisms were investigated. As start, O2 adsorption is investigated via O2 dissociation or hydrogenation of O2 to create B2–SiNT (7, 0)–*OOH. Firstly, the O2 dissociation process can be defined as B2–SiNT (7, 0)–*O2 → *O–B2–SiNT (7, 0)–*O. The dissociated O atoms were elected to link on B–Si position and activation barrier energy is 0.96 eV (figures 2a (IS), 2b (TS) and 2c (FS)). Secondary, adsorbed O2 can interact via H atom to create B2–SiNT (7, 0)–*OOH as follow: B2–SiNT (7, 0)–*O2 + H+ + e− → B2–SiNT (7, 0)–*OOH, this process has no any activation barrier energy.The OOH adsorption on of B2–SiNT (7, 0) has higher ΔEad than O2ca. 0.29 eV and also O2 dissociation on surface of B2–SiNT (7, 0) has high activation barrier energy. H atom is added into Si in B2–SiNT (7, 0)–*OOH and H atom reacted via B2–SiNT (7, 0)–*OOH. Then the B2–SiNT (7, 0)–*OOH dissociated to *O–B2–SiNT (7, 0)–*OH (figures 2d (IS), 2e (TS) and 2f (FS)), due to great activation barrier energy (1.31 eV) this process is impossible. The creation of B2–SiNT (7, 0)–*OOH in ORR on B2–SiNT (7, 0) is suitable than dissociation of O2 molecule.
The ORR is done through the B2–SiNT–*OOH intermediate as follows:
| B2–SiNT–*OOH → B2–SiNT–*O + H2O | (7) |
| B2–SiNT–*OOH → *OH–B2–SiNT–*OH | (8) |
| B2–SiNT–*OOH → B2–SiNT + H2O2 →*OH–B2–SiNT–*OH | (9) |
In path 1, B2–SiNT (7, 0)–*OOH intermediate was decreased to H2O molecule and B2–SiNT (7, 0)–*O (ΔEa = 0.18 eV). In this process, O–O is fragmented and the first H2O molecule is created (figures 2g (IS), 2h (TS) and 2i (FS)). Then two hydrogenation stages were done and B2–SiNT (7, 0)–*OH (figures 2j (IS), 2k (TS) and 2l (FS)) and the second H2O molecule was created (figures 2m (IS), 2n (TS) and 2o (FS)). The activation barrier energies of these two hydrogenation processes are 0.37 and 0.07 eV, respectively.
In path 2, H atom is linked to O and *OH–B2–SiNT (7, 0)–*OH is created and activation barrier energy is 0.24 eV (figures 2p (IS), 2q (TS) and 2r (FS)). Then, *OH–B2–SiNT (7, 0)–*OH linked to H atom and the first H2O molecule is created and activation barrier energy of this stage is 0.35 eV (figures 2s (IS), 2t (TS) and 2u (FS)). In the end stage of path 2, the B2–SiNT (7, 0)–*OH is hydrogenated and in this step the second H2O molecule is separated.
In the path 3, the B2–SiNT (7, 0)–*OOH is hydrogenated and the H2O2 molecule and B2–SiNT (7, 0) catalyst are created and activation barrier is 0.31 eV (figures 2v (IS), 2w (TS1), 2x (MS), 2y (TS2) and 2z (FS)). The H2O2 molecule creation is a mediated state (MS) on surface of B2–SiNT (7, 0) and it cannot effect on potential of the B2–SiNT (7, 0), significantly. In next stage of path 3, separated H2O2 dissociated into *OH–B2–SiNT (7, 0)–*OH structure and therefore H2O2 dissociation has activation barrier energy about 0.82 eV. The ORR via path 3 continued via two hydrogenation stage as presented in path 1 in Fig. 4 (figures 4s (IS), 4t (TS) and 4u (FS)) and path 2 in Fig. 4 (figures 4m (IS), 4n (TS) and 4o (FS)).
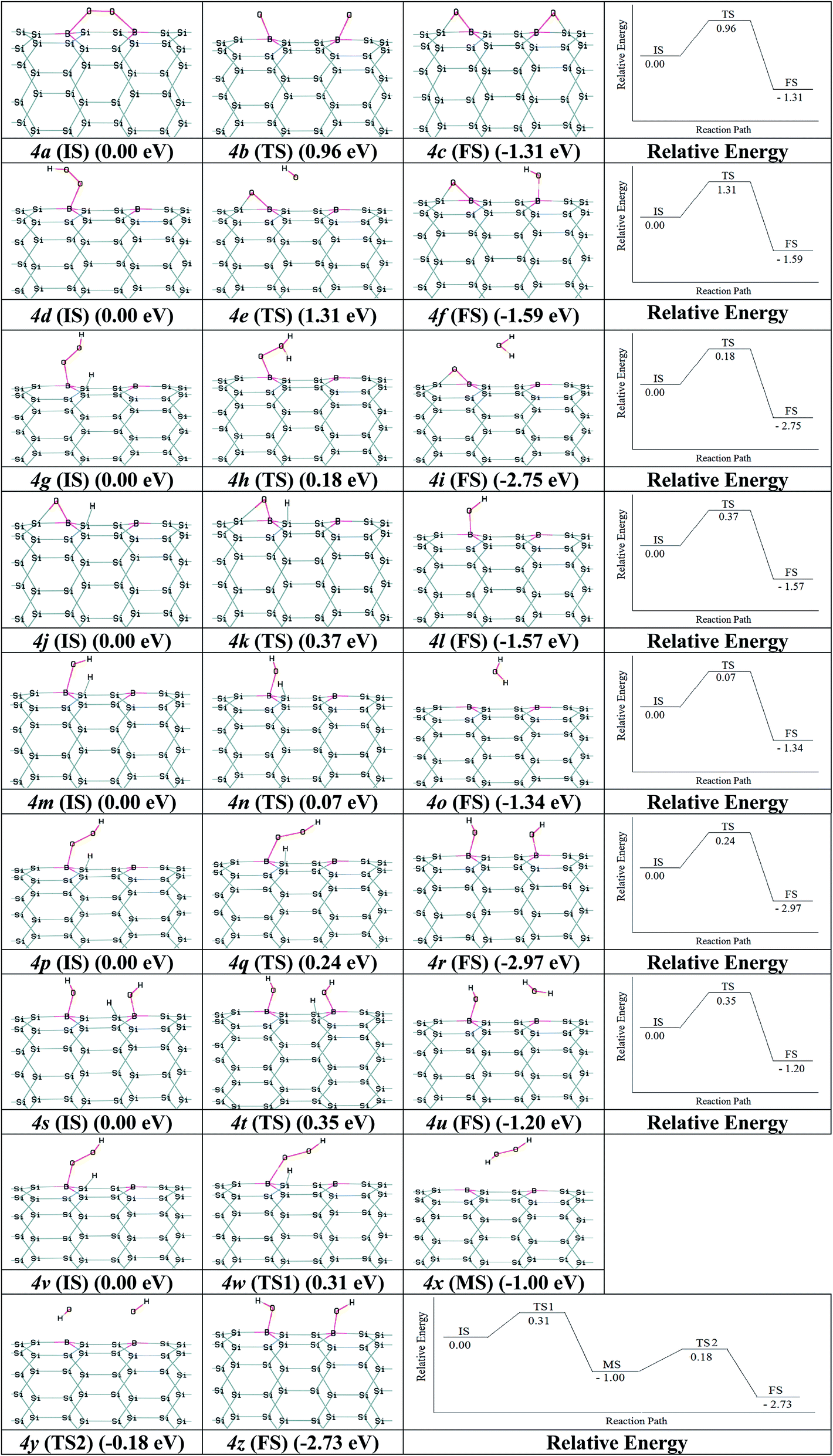 | ||
| Fig. 4 The intermediates of ORR and relative energies: (1) B2–SiNT (7, 0)–*O2 → *O–B2–SiNT (7, 0)–*O; (2) B2–SiNT (7, 0)–*OOH → *O–B2–SiNT (7, 0)–*OH; (3) B2–SiNT (7, 0)–*OOH → B2–SiNT (7, 0)–*O + H2O; (4) B2–SiNT (7, 0)–*O → B2–SiNT (7, 0)–*OH; (5) B2–SiNT (7, 0)–*OH → B2–SiNT (7, 0)–* + H2O; (6) B2–SiNT (7, 0)–*OOH → *OH–B2–SiNT (7, 0)–*OH; (7) *OH–B2–SiNT (7, 0)–*OH → *OH–B2–SiNT (7, 0) + H2O; (8) B2–SiNT (7, 0)–*OOH → B2–SiNT (7, 0) + H2O2 →*OH–B2–SiNT (7, 0)–*OH. | ||
The parameters of two acceptable paths about reduction of B2–SiNT (7, 0)–*OOH structure are stated in Table 3. In path 1, rate-determining stage (ΔEa = 0.37 eV) on B2–SiNT (7, 0) surface is creation of B2–SiNT (7, 0)–*OH. In path 2, creation of B2–SiNT (7, 0)–*OH structure and H2O molecule is rate-determining stage (ΔEa = 0.35 eV). In path 2, creation of *OH–B2–SiNT (7, 0)–*OH has higher ΔEa than formation of B2–SiNT (7, 0)–*O + H2O in path 1 ca. 0.06 eV and so path 1 can be considered as optimal pathway to ORR.
The over-potential of ORR on Pt-based compounds and graphene are 0.44 and 0.45 V.49–59 The experimental researchers investigated the onset-potential for the ORR performed on several catalysts105–110 and results are stated in Table 5. The G of ORR steps are stated in Fig. 5. The level of the final produce (B2–SiNT (7, 0)–* + 2H2O) is considered as reference step and ORR steps in U = 0 V is downhill. Reaction steps become downward that U is decreased to 0.97 V and beginning voltage for ORR is 0.97 V. The B2–SiNT (7, 0) is suggested as suitable ORR catalyst.
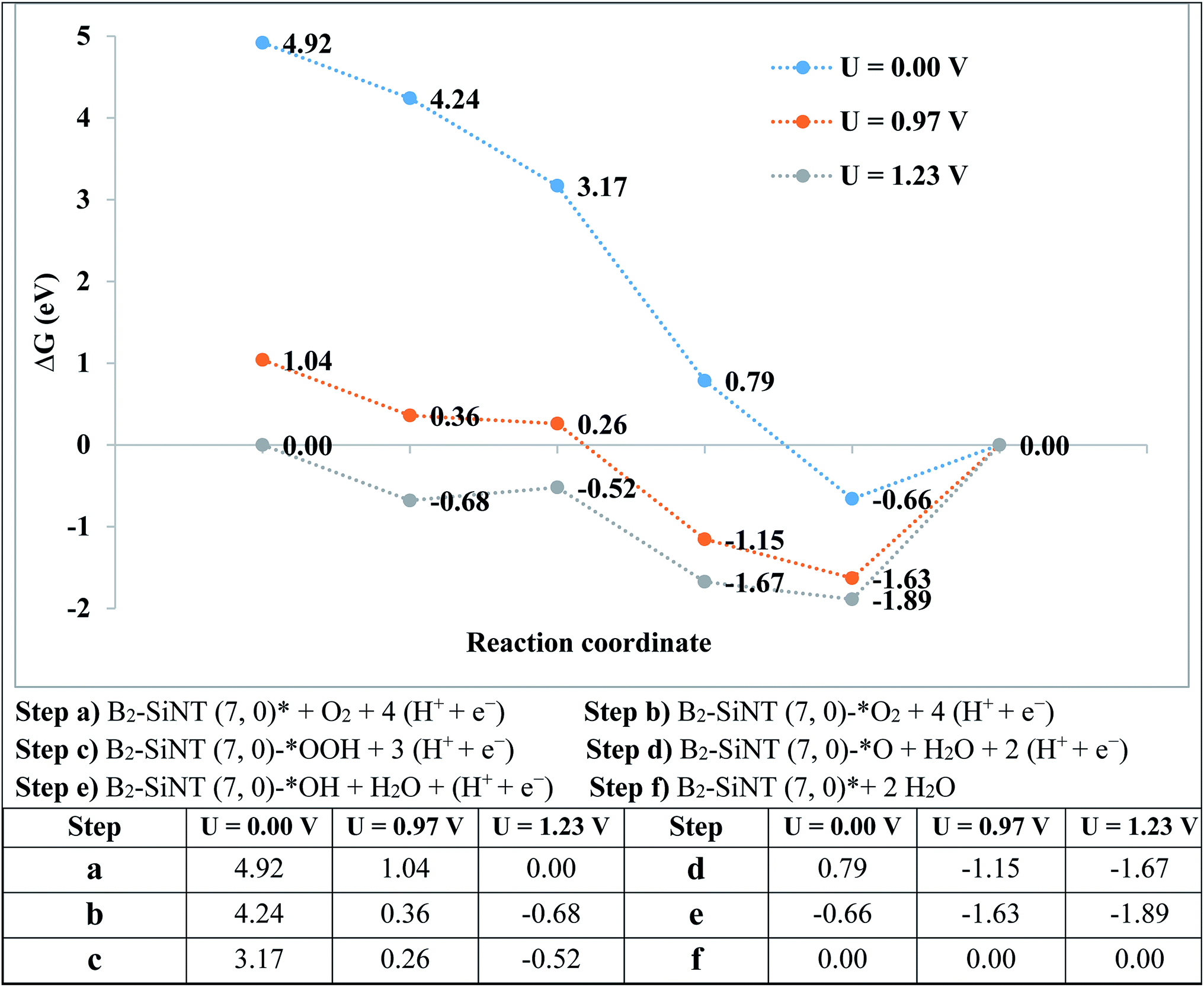 | ||
| Fig. 5 The G values for ORR on B2–SiNT (7, 0). | ||
4. Conclusions
Performances of boron–silicon nanotube (7, 0) as novel catalyst to ORR are investigated. The ORR on surface of B2–SiNT can be continued through LH and ER mechanisms. The rate-determining stage (ΔEa = 0.35 eV) for ORR on B2–SiNT (7, 0) surface is creation of B2–SiNT (7, 0)–*OH structure. The calculated beginning voltage to ORR on surface of the B2–SiNT (7, 0) is 0.37 V. In the acidic solution the beginning voltage to oxygen reduction process can be evaluated to 0.97 V. Results indicated that the B2–SiNT (7, 0) is suggested as catalyst to ORR with suitable efficiency.Conflicts of interest
There are no conflicts to declare.References
- H. A. Gasteiger and N. M. Marković, Science, 2009, 324, 48 CrossRef CAS.
- J. B. Lee and Y. K. Park, J. Power Sources, 2006, 158, 1251 CrossRef CAS.
- N. M. Marković, T. Schmidt, J. Stamenkovic and V. Ross, Fuel Cells, 2001, 1, 105 CrossRef.
- Z. W. Liu, F. Peng, H. J. Wang and H. Yu, Catal. Commun., 2012, 29, 11 CrossRef CAS.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729 CrossRef CAS.
- P. H. Matter and L. Zhang, J. Catal., 2006, 239, 83 CrossRef CAS.
- M. Winter, Chem. Rev., 2004, 104, 4245 CrossRef CAS.
- V. I. Zaikovskii, J. Phys. Chem. B, 2006, 110, 6881 CrossRef CAS.
- W. Song and E. J. M. Hensen, ACS Catal., 2014, 4, 1885 CrossRef CAS.
- J. Liu, ACS Catal., 2016, 7, 34 CrossRef.
- Y. Nie, L. Li and Z. D. Wei, Chem. Soc. Rev., 2015, 44, 2168 RSC.
- M. E. Scofield, H. Q. Liu and S. S. Wong, Chem. Soc. Rev., 2015, 44, 5836 RSC.
- M. Zhou, H. L. Wang and S. Guo, Chem. Soc. Rev., 2016, 45, 1273 RSC.
- S. B. Yang, L. J. Zhi, K. Tang, X. L. Feng and J. Maier, Adv. Funct. Mater., 2012, 22, 3634 CrossRef CAS.
- R. Li, Z. D. Wei, X. L. Gou and W. Xu, RSC Adv., 2013, 3, 9978 RSC.
- K. Gong, P. Du, F. Xia and Z. H. Durstock, Science, 2009, 323, 760 CrossRef CAS.
- M. Li, L. Zhang, Q. Xu, J. Niu and Z. J. Xia, J. Catal., 2014, 314, 66 CrossRef CAS.
- A. Wang, B. Lin and H. Zhang, Catal. Sci. Technol., 2017, 7, 2362 RSC.
- I. A. Pasti, N. M. Gavrilov and A. S. Dobrota, Electrocatalysis, 2015, 6, 498 CrossRef CAS.
- X. J. Bo and L. P. Guo, Phys. Chem. Chem. Phys., 2013, 15, 2459 RSC.
- B. Delley, J. Chem. Phys., 2000, 113, 7756 CrossRef CAS.
- J. P. Perdew and K. Burke, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- A. A. Verma, S. A. Bates, T. Anggara and C. Paolucci, J. Catal., 2014, 312, 179 CrossRef CAS.
- S. Ata, H. Yoon and C. Subramaniam, Polymer, 2014, 55, 5276 CrossRef CAS.
- D. Janas and A. P. Herman, Carbon, 2014, 73, 225 CrossRef CAS.
- C. C. Lawlor and C. M. Schauerman, J. Mater. Chem. C, 2015, 3, 10256 RSC.
- J. Alvarenga, P. R. Jarosz and C. M. Schauerman, Appl. Phys. Lett., 2010, 97, 19 CrossRef.
- I. Rubinstein and E. Gileadi, J. Electroanal. Chem., 2017, 108, 191 CrossRef.
- D. Banham and S. Ye, J. Power Sources, 2015, 285, 334 CrossRef CAS.
- G. Wu, K. I. More and P. Xu, Chem. Commun., 2013, 49, 329 Search PubMed.
- S. Gong and Z. H. Zhu, J. Appl. Phys., 2013, 114, 74303 CrossRef.
- K. Kordek and L. Jiang, Adv. Energy Mater., 2018, 2018, 1802936 Search PubMed.
- J. Liang, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 11496 CrossRef CAS.
- B. C. Han, Phys. Rev. B: Condens. Matter Mater. Phys, 2008, 77, 075410 CrossRef.
- K. Shinozaki, J. W. Zack, S. Pylypenko, B. S. Pivovar and S. S. Kocha, Kinet. Catal., 2015, 54, 255 Search PubMed.
- A. N. Valisi, Electrocatalysis, 2013, 3, 108 CrossRef.
- N. V. Smirnova, J. Electrochem. Soc., 2013, 162, 1 Search PubMed.
- H. A. Gasteiger, Appl. Catal., B, 2005, 56, 9 CrossRef CAS.
- K. Qu, Y. Zheng, S. Dai and S. Z. Qiao, Nanoscale, 2015, 7, 12598 RSC.
- V. Datsyuk, M. Kalyva and K. Papagelis, Carbon, 2008, 46, 833 CrossRef CAS.
- J. C. Li and Z. Q. Yang, NPG Asia Mater., 2018, 10, 461 CrossRef.
- C. Zhu, S. Fu, J. Song, Q. Shi and D. Su, Small, 2017, 13, 1603407 CrossRef.
- C. Zhu, S. Fu, Q. Shi and D. Du, Angew. Chem., Int. Ed., 2017, 56, 13944 CrossRef CAS.
- B. Bayatsarmadi, Y. Zheng and A. Vasileff, Small, 2017, 13, 1702002 CrossRef PubMed.
- J. C. Li, S. Y. Zhao and P. X. Hou, Nanoscale, 2015, 7, 19201 RSC.
- N. S. Parimi, Y. Umasankar and P. Atanassov, ACS Catal., 2012, 2, 38 CrossRef CAS.
- F. Jaouen, Electrochim. Acta, 2007, 52, 5975 CrossRef CAS.
- Y. Chu, L. Gu, X. Ju and H. Du, Catalysts, 2018, 8, 245 CrossRef.
- L. Wang and H. Dong, J. Phys. Chem. C, 2016, 1203, 117427 Search PubMed.
- P. Zhang and B. Xiao, Sci. Rep., 2014, 4, 3821 CrossRef CAS PubMed.
- L. Zhang and Z. Xia, J. Phys. Chem. C, 2011, 115, 11170 CrossRef CAS.
- P. Zhang and J. S. Lian, Phys. Chem. Chem. Phys., 2012, 14, 11715 RSC.
- J. D. Wiggins-Camacho and K. J. Stevenson, J. Phys. Chem. C, 2011, 115, 20002 CrossRef CAS.
- D. Xiong and X. Li, Catalysts, 2018, 8, 301 CrossRef.
- X. Hu and C. Liu, New J. Chem., 2011, 35, 2601 RSC.
- Q. Wei and X. Tong, Catalysts, 2015, 5, 1574 CrossRef CAS.
- Y. Zhao and L. Yang, J. Am. Chem. Soc., 2013, 135, 1201 CrossRef CAS.
- L. Ferrighi and M. Datteo, J. Phys. Chem. C, 2014, 118, 223 CrossRef CAS.
- A. Bruix, K. M. Neyman and F. Illas, J. Phys. Chem. C, 2010, 114, 14202 CrossRef CAS.
- J. Cai, Nature, 2010, 466, 470 CrossRef CAS.
- T. Wassmann, A. P. Seitsonen and A. M. Saitta, Phys. Rev. Lett., 2008, 101, 096402 CrossRef.
- Y. H. Lu, R. Q. Wu, L. Shen, M. Yang and Z. D. Sha, Appl. Phys. Lett., 2009, 94, 122111 CrossRef.
- R. S. Mulliken, J. Chem. Phys., 1955, 23, 18330 Search PubMed.
- N. Govind, Comput. Mater. Sci., 2003, 28, 250 CrossRef CAS.
- C. T. Campbell and J. R. V. Sellers, Faraday Discuss., 2013, 162, 9 RSC.
- J. K. Nørskov and J. Rossmeisl, J. Phys. Chem. B, 2004, 108, 17886 CrossRef.
- S. Royer and D. Duprez, ChemCatChem, 2011, 3, 24 CrossRef CAS.
- M. S. Chen, Y. Cai and Z. Yan, Surf. Sci., 2007, 601, 5326 CrossRef CAS.
- M. A. Yu, Y. Feng, L. Gao and S. Lin, Phys. Chem. Chem. Phys., 2018, 20, 20661 RSC.
- J. A. Keith and T. Jacob, Angew. Chem., Int. Ed., 2010, 49, 9521 CrossRef CAS.
- Y. Sha, T. H. Yu, Y. Liu and B. V. Merinov, J. Phys. Chem. Lett., 2010, 1, 856 CrossRef CAS.
- X. W. Yu and S. Y. Ye, J. Power Sources, 2007, 172, 145 CrossRef CAS.
- A. J. Cohen, P. Mori-Sánchez and W. Yang, Chem. Rev., 2012, 112, 289 CrossRef CAS PubMed.
- E. Skulason, Phys. Chem. Chem. Phys., 2007, 9, 3241 RSC.
- Y. X. Chen, Angew. Chem., Int. Ed., 2012, 51, 8500 CrossRef CAS.
- W. Y. Zhang, Electrochim. Acta, 2016, 200, 131 CrossRef CAS.
- H. Jeon, J. Power Sources, 2010, 195, 5929 CrossRef CAS.
- J. Shui, Sci. Adv., 2015, 1, 1 Search PubMed.
- C. Bianchini, Chem. Rev., 2009, 109, 4183 CrossRef CAS.
- E. DeCarlos and J. G. Ángyán, J. Chem. Phys., 2016, 145, 124105 CrossRef.
- P. Kasper, J. Phys. Chem. A, 2017, 121, 2022 CrossRef.
- G. Michael and I. S. Bushmarinov, Science, 2017, 355, 49 CrossRef.
- M. Nielsen, Surf. Sci., 2015, 631, 2 CrossRef CAS.
- M. H. Shao, J. Power Sources, 2011, 196, 2433 CrossRef CAS.
- D. H. Guo, Science, 2016, 351, 361 CrossRef CAS.
- S. E. Wheeler, A. Moran, S. N. Pieniazek and K. N. Houk, J. Phys. Chem. A, 2009, 113, 10376 CrossRef CAS.
- J. Hohenstein, Chem. Phys., 2006, 15, 128 Search PubMed.
- G. Fazio, L. Ferrighi and C. Di Valentin, J. Catal., 2014, 318, 203 CrossRef CAS.
- V. Tripkovic and E. Skulason, Electrochim. Acta, 2010, 55, 7975 CrossRef CAS.
- M. J. Janik, C. D. Taylor and M. Neurock, J. Electrochem. Soc., 2009, 156, 126 CrossRef.
- J. X. Zhao, C. R. Cabrera, Z. H. Xia and Z. F. Chen, Carbon, 2016, 104, 56 CrossRef CAS.
- L. H. Gan and J. Q. Zhao, Phys. E, 2009, 41, 1249 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553 CrossRef CAS.
- L. Ma, J. M. Zhang, K. W. Xu and V. Ji, Appl. Surf. Sci., 2015, 343, 121 CrossRef CAS.
- B. Luo, Small, 2012, 8, 630 CrossRef CAS PubMed.
- J. B. Zhu, J. Mater. Chem. A, 2016, 4, 7422 RSC.
- G. Kresse and M. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys, 1993, 47, 558 CrossRef CAS PubMed.
- X. K. Kong, Chem. Soc. Rev., 2014, 43, 2841 RSC.
- J. C. Liu, Y. G. Wang and J. Li, J. Am. Chem. Soc., 2017, 139, 6190 CrossRef CAS.
- Z. Qi, J. Power Sources, 2011, 196, 5823 CrossRef CAS.
- M. Branda, N. C. Hernández, J. F. Sanz and F. Illas, J. Phys. Chem. C, 2010, 114, 1934 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901 CrossRef CAS.
- A. J. Binder, T. J. Toops and R. R. Unocic, Angew. Chem., Int. Ed., 2015, 54, 13263 CrossRef CAS.
- E. Y. Ko, E. D. Park, H. C. Lee and D. Lee, Angew. Chem., Int. Ed., 2007, 46, 734 CrossRef CAS.
- S. Zhao, H. Zhang, S. D. House, R. Jin and R. Jin, ChemElectroChem, 2016, 3, 1225 CrossRef CAS.
- G. Ramos-Sánchez and H. Yee-Madeira, Int. J. Hydrogen Energy, 2008, 33, 3596 CrossRef.
- M. Neergat, V. Gunasekar and R. Rahul, J. Electroanal. Chem., 2011, 658, 25 CrossRef CAS.
- Y. Dai, P. Yu, Q. Huang and K. Sun, Fuel Cells, 2016, 16, 165 CrossRef CAS.
- L. Xiong, Y. X. Huang, X. W. Liu and W. W. Li, Electrochim. Acta, 2013, 89, 24 CrossRef CAS.
- W. E. Mustain, K. Kepler and M. Prakash, Electrochem. Commun., 2006, 8, 406 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |