
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of upper rim-double-bridged calix[4]arenes bearing seven membered rings and related compounds†
M. Tlustý a,
V. Eignerb,
M. Baborc,
M. Kohouta and
P. Lhoták*a
a,
V. Eignerb,
M. Baborc,
M. Kohouta and
P. Lhoták*a
aDepartment of Organic Chemistry, University of Chemistry and Technology, Prague (UCTP), Technická 5, 166 28 Prague 6, Czech Republic. E-mail: lhotakp@vscht.cz
bInstitute of Physics AS CR v.v.i., Na Slovance 2, 182 21 Prague 8, Czech Republic
cDepartment of Solid State Chemistry, UCTP, Technická 5, 166 28 Prague 6, Czech Republic
First published on 16th July 2019
Abstract
Meta/meta- and meta/para-disubstituted organomercury calix[4]arenes in the cone conformation were transformed into corresponding amino derivatives. Acylation and subsequent intramolecular cyclization using the Bischler–Napieralski reaction provided, in the case of the meta/meta-series, double bridged calixarenes possessing seven membered rings on the upper rim. A similar synthetic strategy applied to meta/para-isomers allowed for the isolation of monobridged compounds bearing an additional trifluoroacetamido group located distally to seven-membered rings. Both series represent inherently chiral systems, which were successfully resolved using preparative chiral HPLC. The pure enantiomers exhibited a recognition ability towards selected chiral guest molecules as documented by the 1H NMR titration experiments. The absolute configuration of the phenyl-substituted enantiomer (meta/meta-) was confirmed by single crystal structure determination (X-ray).
Introduction
Calix[n]arenes,1 a well-known family of macrocyclic oligophenols, possess many properties which make them perfect candidates for the role of building blocks and molecular scaffolds in supramolecular chemistry. Due to their easy multi-gram preparation, simple chemical transformation, well-defined and/or tuneable 3D shapes of the cavity, and very good complexation abilities, these compounds are frequently used in the design of more complex supramolecular systems including various receptors applicable in host–guest chemistry.Over the years, the chemistry of calix[4]arenes in particular has become well-established, and currently many regioselective transformations of the basic skeleton are available. In this context, while the para position of the phenolic moiety is well accessible via electrophilic aromatic substitution (nitration,2 sulfonation,3 halogenation, Friedel–Crafts reactions,4 etc.), the meta positions remained almost unused due to the lack of suitable chemical tools.
Recently we reported direct mercuration of calix[4]arenes leading to meta-substituted organomercury derivatives (Fig. 1).5 This unprecedented regioselectivity enabled transformation of basic skeleton leading to so far inaccessible derivatization patterns. Among them, a direct connection (via a single-bond-bridge) between the meta-positions of two neighbouring aromatic subunits led to a novel type of the upper-rim bridged calix[4]arenes (Fig. 1, structure A).6 Similarly, organomercurials served as a starting point for the introduction of one-atom bridge, represented by carbonyl group (structure B),7 or even two-atoms bridge (structure C).8 All the above mentioned compounds exhibit rigid and highly distorted cavities with interesting complexation properties and surprisingly amended chemical behaviour if compared with the systems without such bridges.
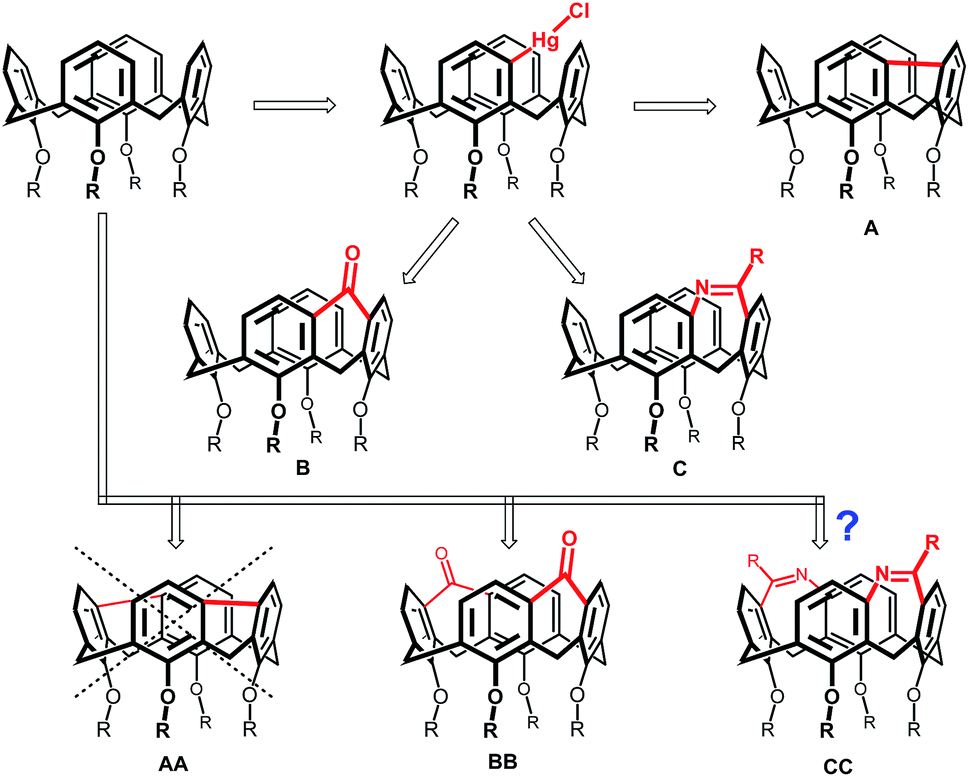 | ||
| Fig. 1 Meta-bridged calix[4]arenes in mono- and di-substituted versions. | ||
Double meta-mercuration9 should enable the construction of bis-bridged calixarenes. Unfortunately, despite our efforts the corresponding single-bond-bridged isomer AA was never isolated. Obviously, two single-bond bridges would impose too high internal strain on the molecule to be stable at common conditions. On the other hand, the isomer BB possessing a well-preorganised cavity, was prepared successfully, and showed the ability to form the solid-state complexes using the cooperative effect of various interactions (hydrogen bonding, CH–π interactions, or halogen bonding).10
Previously, we have reported the synthesis of bridged calixarenes containing a seven membered ring (structure C).8 These compounds with enlarged and rigidified cavities represent inherently chiral systems potentially useful in the design of chiral receptors. In this paper, we report on our continuous synthetic effort to synthesise analogous bis-meta-bridged calix[4]arenes of type CC and some related compounds with previously unknown derivatization patterns in calixarene chemistry.
Results and discussion
As shown in Scheme 1, the starting tetrapropoxy-calix[4]arene 1 immobilized in the cone conformation was reacted (using a recently described procedure)9 with 2.0 equiv. of Hg(TFA)2 providing a mixture of meta/meta- and meta/para-organomercury derivatives 2 and 3 in 34% and 20% yield, respectively. The meta/meta-regioisomer was than transformed into the corresponding nitroso compound 4 by reaction11 with isopentyl nitrite and HCl at 0 °C. A subsequent reduction of the nitroso groups, accomplished by RANEY® nickel and N2H4·H2O in refluxing EtOH, provided smoothly the key diamine 5 in 85% yield.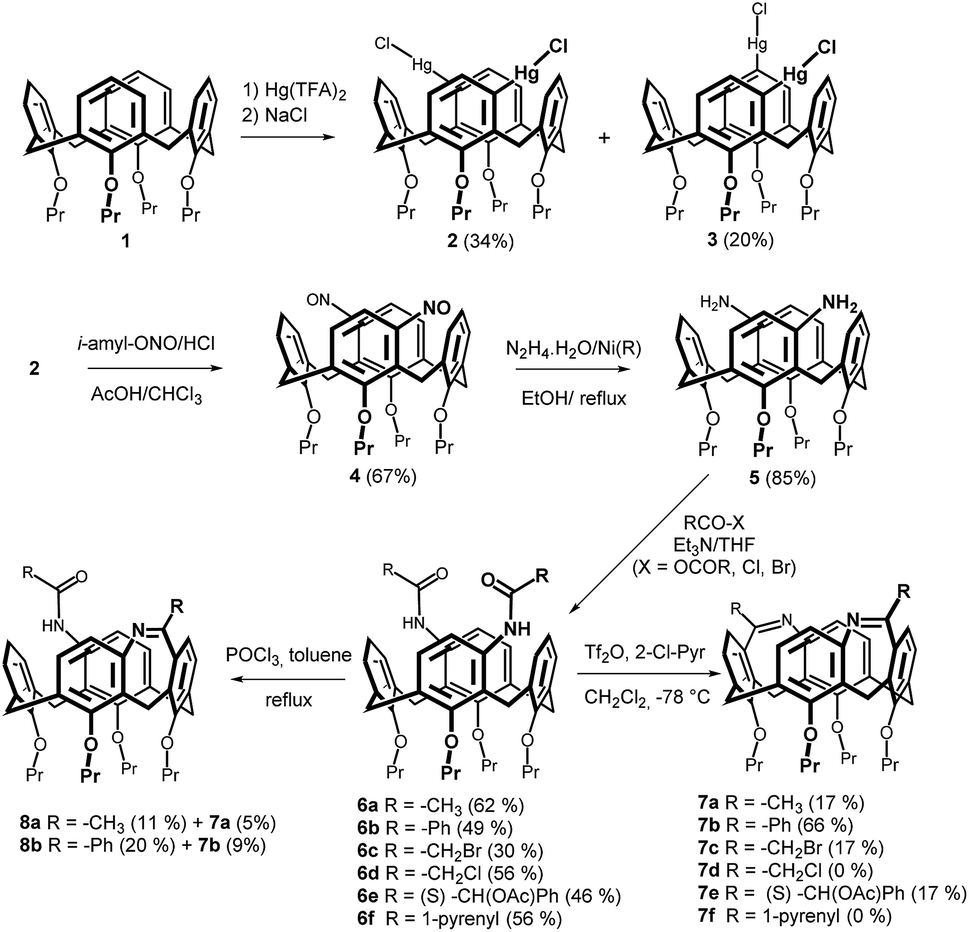 | ||
| Scheme 1 Synthesis of bridged calix[4]arenes (meta/meta-isomers). | ||
The acylation step was carried out using various carboxylic acid derivatives (bromide, chloride, anhydride). Thus, reaction with acetic anhydride in THF in the presence of triethylamine (TEA) provided amide 6a in 62% yield (Scheme 1). A similar reaction using benzoyl chloride or 1-pyrenecarbonyl chloride gave the corresponding amides 6b and 6f in 49% and 56% yields, respectively. To achieve intermediates capable of further derivatization, compounds 6c (30%) and 6d (56%) bearing the halomethyl groups were prepared from bromoacetyl bromide and chloroacetyl chloride. The introduction of stereogenic centrum into the inherently chiral (racemic) amine 5 should lead to a diastereomeric mixture potentially separable by common chromatographic techniques. Accordingly, compound 5 was acylated by (S)-O-acetylmandelic acid using standard DCC coupling conditions (THF, 24 h at rt) to provide amide 6e in 46% yield. Unfortunately, despite our efforts, we were unable to isolate the individual isomers using silica gel chromatography.
The bridging of amidic functions was accomplished using Bischler–Napieralski reaction,8 which is well-known from the synthesis of various heterocyclic systems. The reaction conditions (6a, POCl3 in refluxing toluene) previously applied to monosubstituted derivatives of type C (Fig. 1) led to a rather complex reaction mixture. The preparative TLC on silica gel provided low yields of two compounds 7a (5%) and 8a (11%). As revealed by HRMS ESI+, compound 7a represents the expected bis-bridged derivative (m/z = 671.38433 (predicted) vs. 671.38404 (found) for [M + H]+), while 8a is in agreement with a mono-bridged system (m/z = 689.39490 (predicted) vs. 689.39445 (found) for [M + H]+). A similar result was obtained for the cyclization step of 6b where the products 7b and 8b were isolated in 9% and 20% yields, respectively.
These findings indicated that the reaction conditions were unsuitable for the efficient formation of the expected product. Moreover, the assumed lower stability of products obviously was not compatible with high reaction temperature. To solve the above-mentioned issues we applied much milder reaction conditions reported12 for Bischler–Napieralski reaction, where the cyclization is accomplished using a mixture of triflic anhydride and 2-chloropyridine. Indeed, the reaction of 6b with Tf2O/2-ClPyr in CH2Cl2 at −78 °C afforded the double-bridged product 7b in 66% yield. On the contrary, the corresponding methyl derivative 7a was isolated only in 17% yield, and the same yield was obtained for compounds 7c (R = CH2Br) and 7e (R = (S)-CH2(OAc)Ph), while the reaction of 6d and 6f did not lead to any isolable products.
Although the structures of 7a and 7b were normally assigned using the combination of HRMS ESI+ analysis and the 1H/13C NMR spectroscopy, in the case of compound 7c only MS and 1H NMR spectra were acquired, as compound did not survive the measurement of 13C NMR spectrum. This trend was even more pronounced for derivative 7e, where we obtained successfully only the HRMS analysis, while the attempt to acquire the 1H NMR spectrum was accompanied by very fast decomposition of compound in CDCl3 solution.
The 1H NMR spectrum (CDCl3) of 7a showed four doublets at 4.59, 4.53, 3.25 and 2.72 ppm in the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:2 ratio possessing typical geminal coupling constants (11.7–12.3 Hz) corresponding to the Ar–CH2–Ar bridges. Moreover, a singlet of the methyl groups from the bridging moieties (2.49 ppm), together with the four doublets with a characteristic ortho-splitting from the aromatic hydrogens, are in a perfect agreement with the splitting pattern expected for the C2 symmetry of the product.
:2:2:2 ratio possessing typical geminal coupling constants (11.7–12.3 Hz) corresponding to the Ar–CH2–Ar bridges. Moreover, a singlet of the methyl groups from the bridging moieties (2.49 ppm), together with the four doublets with a characteristic ortho-splitting from the aromatic hydrogens, are in a perfect agreement with the splitting pattern expected for the C2 symmetry of the product.
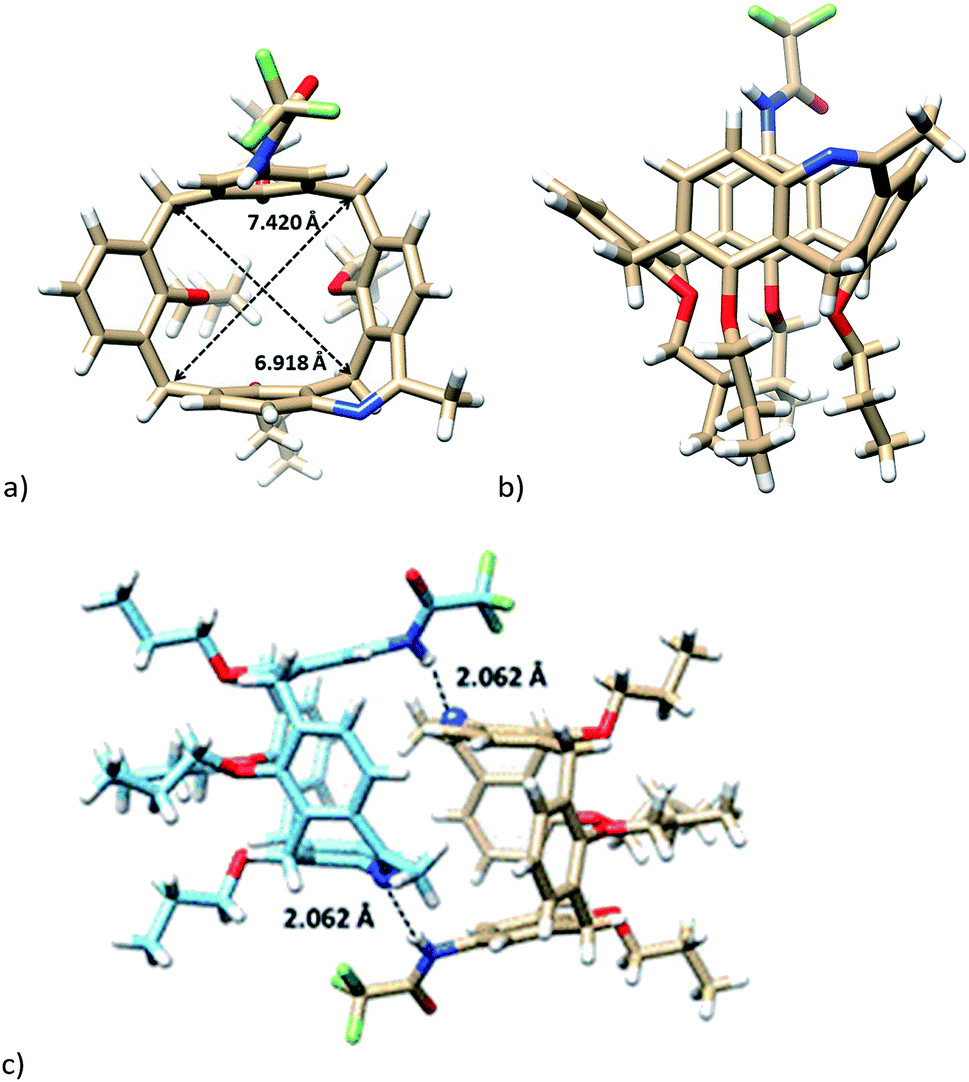
The single crystal X-ray analysis of the first eluting enantiomer from the resolution of 7b (enantiomer assigned as 7b_1, see later for the resolution of the racemate) crystallized in a hexagonal system in the P61 space group. As follows from Fig. 2, the absolute configuration of enantiomer 7b_1 can be assigned as P.13 The presence of the two bridges resulted in a slightly distorted square shape of the cavity, as can be documented by the length of both diagonals (6.944 and 7.352 Å, Fig. 2a). If we define the main plane of the molecule by the four carbon atoms of the CH2 bridges, the corresponding interplanar angles Φ with aromatic subunits were 69.46°, 67.33°, 72.95°, and 71.84°, starting clockwise from aromatic unit bearing amidic moiety (Fig. 2a). This reflects the almost ideal C4v symmetry of the phenolic skeleton creating a rigid cavity suitable for the inclusion. Indeed, the crystal packing consists of the infinite inclusion motif (Fig. 2c) where the methyl group of one propyl moiety is immersed into the cavity of neighbouring calixarene. As shown in Fig. 2d, the methyl group exhibits at least seven close contacts between the C–H bonds and aromatic C atoms of the cavity (distances from 2.714 to 2.889 Å). As a result, the crystal packing of enantiomer (P) 7b is formed by infinite right-handed helices possessing P chirality. The pitch of this helix (the vertical distance between the two consecutive turns) is 36.443 Å, where one turn consists of six calixarene molecules (Fig. 2e).
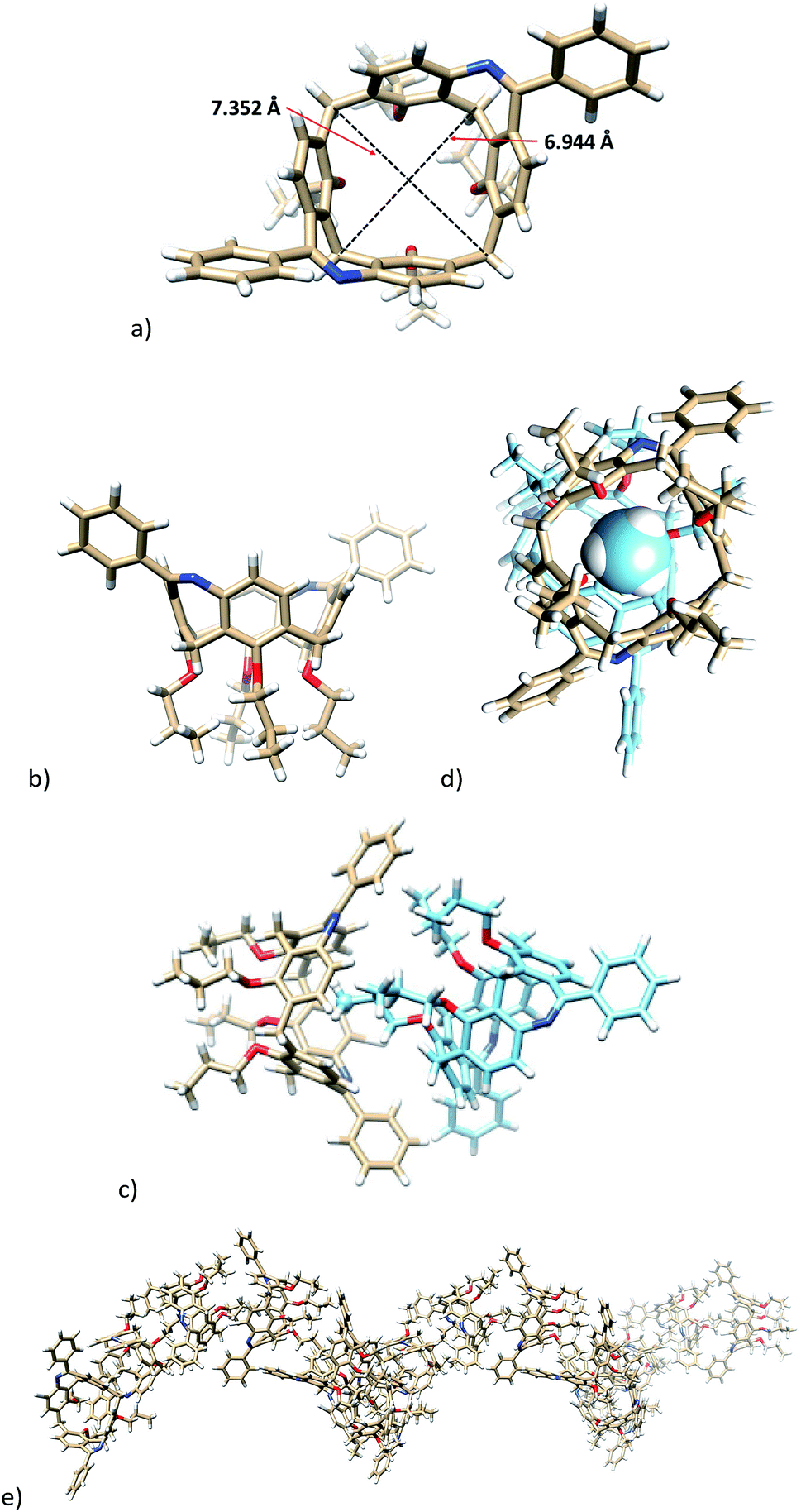 | ||
| Fig. 2 X-ray structure of (P-) enantiomer 7b_1: (a) top view, (b) side view, (c) inclusion motif with the methyl group immersed into the next cavity. (d) Top view of the same motif (methyl group shown as spacefill). (e) The helical arrangement of the molecules. | ||
The exact structure of isomer 8b was also confirmed by the single crystal X-ray analysis. Compound crystallized (EtOH/CH2Cl2) in the monoclinic system with P21/c space group (Fig. 3) and formed a solvate with one molecule of EtOH. The presence of the two-atom-bridge does not impose to the molecule so huge distorsion as observed for a single-bond-bridge derivative of type A.6 It can be demonstrated by almost the same lengths (7.077 Å vs. 7.225 Å) of both diagonals (see Fig. 3a). Consequently, the pinched cone conformation, a typical motif of calix[4]arenes in the cone conformation, is substituted here by much more squared shape with the corresponding interplanar angles Φ (see above) 43.67°, 79.23°, 65.82°, and 74.47°, starting clockwise from aromatic unit bearing amidic moiety (Fig. 3a).
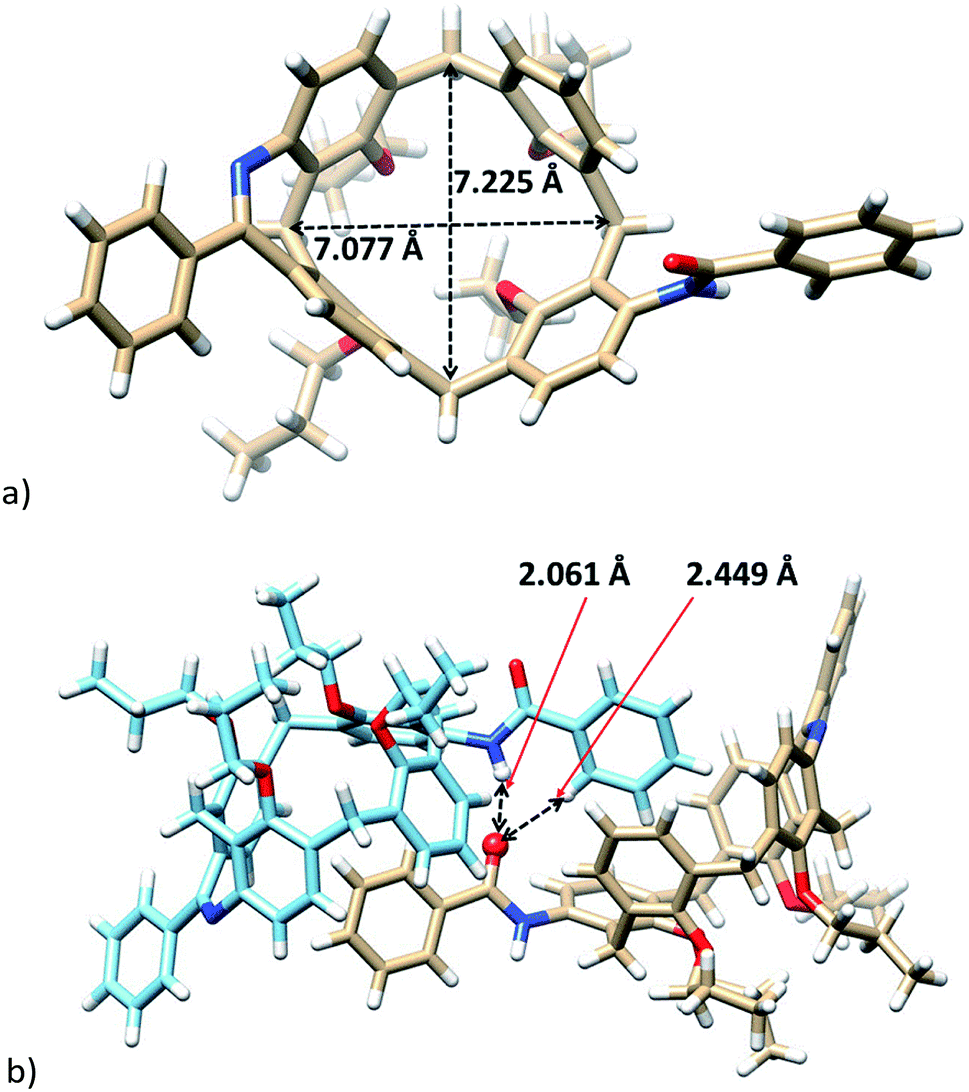 | ||
| Fig. 3 X-ray structure of 8b (molecules of solvent (EtOH) were removed for better clarity): (a) top view; (b) hydrogen bonded dimer. | ||
The crystal packing of 8b shows a dimeric motif held together by hydrogen bonds from the carbonyl group to the amidic proton of the second molecule (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H–N, 2.061 Å). The same carbonyl group is bonded simultaneously to the ortho hydrogen atom (2.449 Å) thus orienting the phenyl moiety into the cavity of calixarene (Fig. 3b). Interestingly, the individual dimeric assemblies within the crystal lattice are formed by the same enantiomers of 8b.
O⋯H–N, 2.061 Å). The same carbonyl group is bonded simultaneously to the ortho hydrogen atom (2.449 Å) thus orienting the phenyl moiety into the cavity of calixarene (Fig. 3b). Interestingly, the individual dimeric assemblies within the crystal lattice are formed by the same enantiomers of 8b.
The synthesis in meta/para-series (Scheme 2) started with the corresponding nitroso derivative 9 (obtained from chloromercurio derivative 3) which was reduced to yield amine 10 in 94% yield. To achieve the intramolecular Bischler–Napieralski reaction (bridging) in the meta position, the para amino group should be deactivated towards this reaction, otherwise intermolecular reaction cannot be avoided. From our previous study we knew that TFA amide was inert towards the appropriate reaction conditions. Based on this knowledge, the amine 10 was acylated with trifluoroacetic acid anhydride (TFAA)/TEA to provide diamide 11 (94%). A careful hydrolysis of this diamide allowed the isolation of monoamides 12a (para) and 12b (meta) in 22 and 17% yield, respectively. In this context, it is important to carry out this reaction using lower overall conversion (40%), since at higher conversion fully deprotected amine 10 was formed again. Moreover, the para-deprotected isomer 12b can be smoothly recycled to the starting diamide 11 (98% yield) just repeating the acylation step (TFAA/TEA).
 | ||
| Scheme 2 Synthesis of bridged calix[4]arenes (meta/para-isomers). | ||
The protected meta-amine derivative 12a was then acylated using similar reaction conditions described above for the meta/meta- compounds. The corresponding amides 13a–13f were isolated in good to excellent yields (57–97%) depending on the substitution. Finally, the intramolecular bridging was accomplished via the reaction with Tf2O/2-ClPyr at −78 °C in CH2Cl2. As can be seen in Scheme 2, the bridged products 14a–14e were obtained in good yields (51–81%) irrespective of the substitution, the only exception being amide 13f which did not give any reaction. As expected, the trifluoroacetamido group in the para position remained untouched in all the cases.
The structures of the bridged products were confirmed by a combination of NMR and MS techniques. Thus, the HRMS ESI+ analysis of 14b showed signals at m/z = 805.38297 and 827.36360, which were in good agreement with the [M + H]+ (805.38228) and [M + Na]+ (827.36360) cations predicted for the bridged product. The 1H NMR spectrum of 14b (CDCl3) revealed the presence of four doublets at 3.32, 3.25, 3.19 and 2.89 ppm (equatorial) and another four doublets at 4.69, 4.61, 4.48 and 4.43 ppm (axial), representing the CH2 bridges of the calixarene skeleton (J ≅ 12.1–12.5 Hz). This splitting pattern is consistent with the absence of any symmetry elements in the product (Cs symmetry).
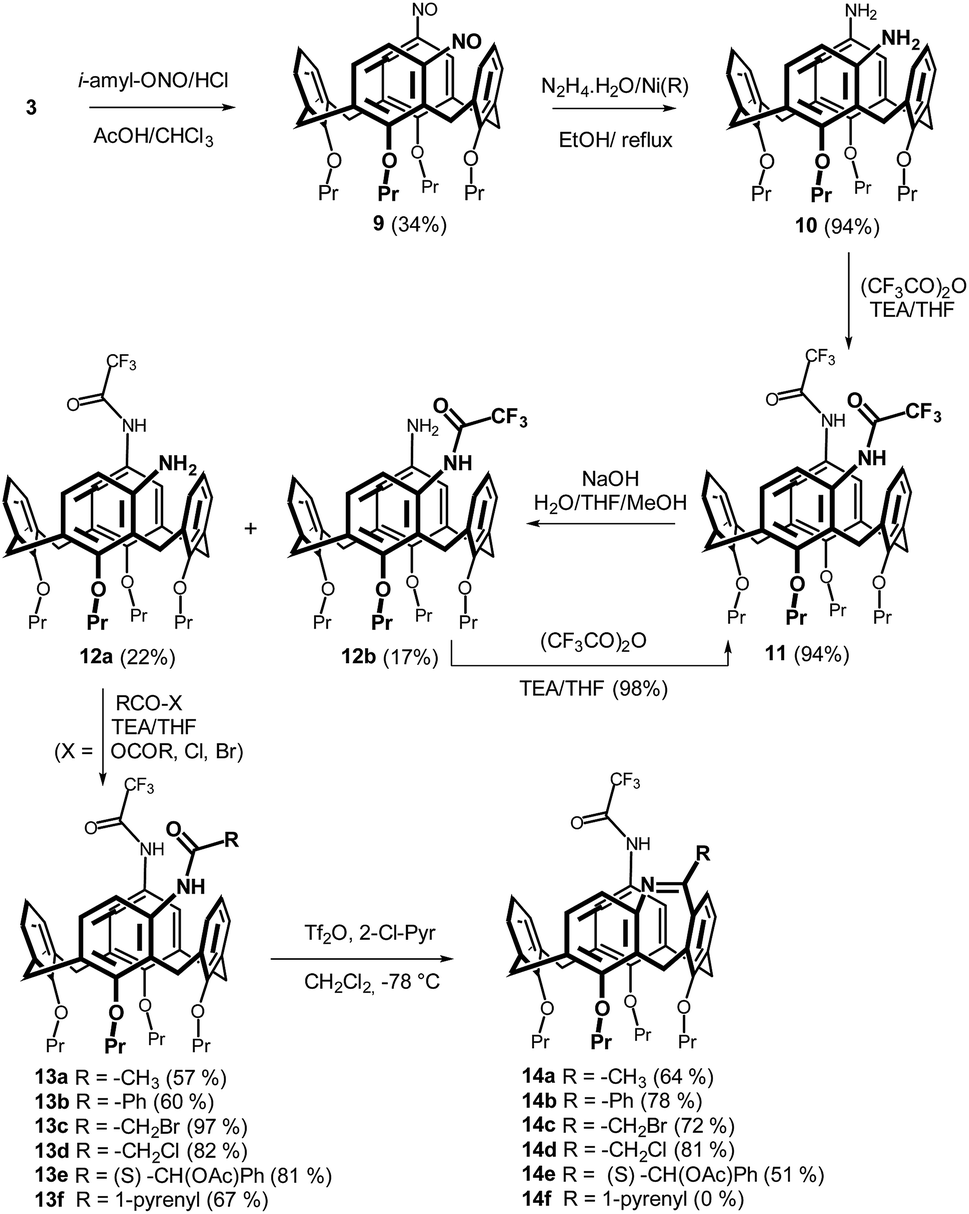
Moreover, the unambiguous structural evidence was obtained by a single crystal X-ray crystallography. Calixarene 14a crystallized (EtOH/CH2Cl2) in the monoclinic system with P 21/c space group. As shown in Fig. 4, the shape of the cavity is slightly distorted by the presence of the additional bridge. The length of the short diagonal (the C⋯C distance of two opposite methylene bridges) was 6.918 Å, while the longer diagonal was 7.420 Å. The corresponding interplanar angles Φ with aromatic subunits (see above) were 82.97°, 62.03°, 83.26°, and 37.46°, starting clockwise from aromatic unit bearing amidic function (Fig. 4a and b). An interesting packing motif is represented (Fig. 4b) by dimeric structure held by CN⋯H–N hydrogen bond (2.062 Å).
 | ||
| Fig. 4 X-ray structure of 14a: (a) top view, (b) side view, (c) dimeric structure with hydrogen bonding interactions. | ||
Chiral separation of 7b was performed using an automated preparative system Autopurification (Waters, USA) consisting of a binary pump module, PDA detector, column manager and fraction collector with separated fluidic ways for preparative and analytical mode. The suitable conditions allowing for efficient enantioseparation were first proven on the analytical scale using chiral polysaccharide column ChiralArt Amylose-SA (250 × 4.6 mm ID, 5 μm). In preparative mode, a polysaccharide column Chiralpak IA (250 × 20 mm ID, 5 μm) was employed using the optimum mobile phase heptane/propan-2-ol (9/1, v/v) with diethylamine 0.1% as a basic additive (Fig. 5a). The enantiomeric character of the separated fractions 7a_1 and 7a_2 was verified with ECD spectroscopy, which showed the typical mirror images (Fig. 5b). The optical purity of both enantiomers was found to be >99.5% ee (see ESI†).
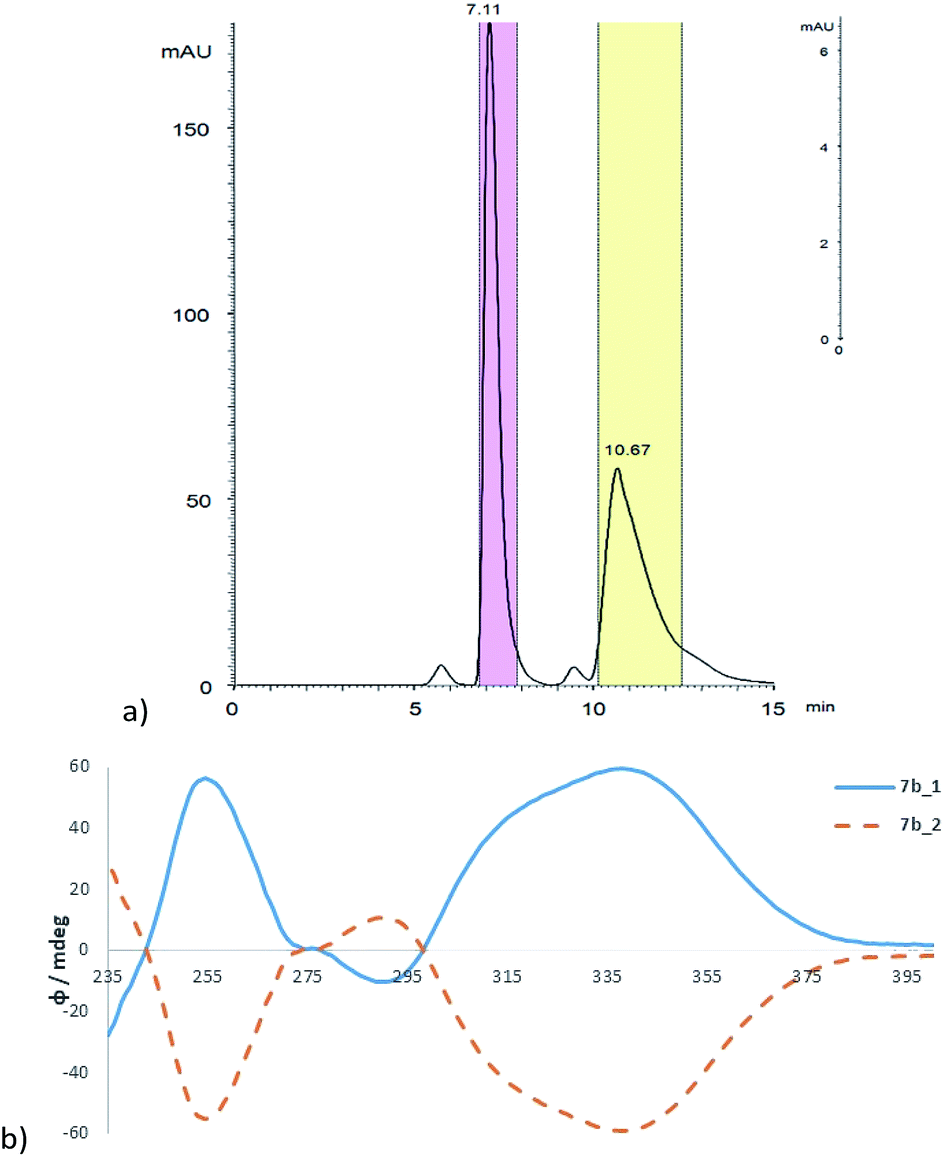 | ||
| Fig. 5 (a) Preparative enantioseparation of 7b on Chiralpak IA (250 × 20 mm i.d., 5 μm) in heptane/propan-2-ol (9/1, v/v) with diethylamine 0.1% as a basic additive at 15 °C, flow rate 15 mL min−1, sample concentration 4.28 mg mL−1, injection volume 0.6 mL, detection wavelength 350 nm. (b) ECD spectra of both separated enantiomers of 7b in MeOH, the first eluting enantiomer 7b_1 full (blue) line, the second eluting enantiomer 7b_2 dashed (orange) line. | ||
Compound 14b was also successfully separated on a preparative column using almost the same conditions as mentioned above (hexane/propan-2-ol (9/1, v/v) with diethylamine 0.05%) to yield two enantiomers 14b_1 and 14b_2.
The 1H NMR titration experiments (CDCl3) carried out with racemic 7a indicated that the cavity of double-bridged calixarene can interact with MeCN, although the corresponding complexation constant was very low K = 2.8 ± 0.2 M−1 (see ESI†). Based on this result, we attempted the enantioselective resolution of a chiral guest molecule bearing more acidic CH3 group that could be complexed by the combination of CH–π and/or cation−π interactions.
For this purpose, a natural (S)-nicotine was methylated on pyridine nitrogen atom and the resulting (S)-N-methylnicotinium iodide8 (NMNI) was used as a guest molecule. The 1H NMR titration experiments with resolved enantiomers 7b_1 and 7b_2 as the host molecules and N-methylpyridinium iodide (NMPI) as the guest in 1,1,2,2-C2D2Cl4 revealed that the complexation occurred under fast-exchange conditions. The titration curves (see ESI†) were constructed from the CIS (the Complexation Induced Shift) values of the host aromatic signals and they were analysed using the online available software Bindfit.14
Surprisingly, the titration curves with NMNI gave the best fits using 2:1 (host:guest) stoichiometry15 with very similar overall binding constants: K11 = 330 ± 10 and K21 = 5900 ± 270 for 7b_1; K11 = 450 ± 15 and K21 = 5760 ± 270 for 7b_2. From this point of view, much bigger differences were found during the titration of separated enantiomers of 14b with NMNI: K11 = 720 ± 25 and K21 = 4900 ± 260 for 14b_1, and K11 = 440 ± 20 and K21 = 1150 ± 90 for 14b_2, indicating potential applicability of these compounds in the role of receptors (see ESI†).
A suitable shape of the cavity of 14b (racemic) for the complexation of guest molecules bearing acidic methyl groups was demonstrated on the complexation of acetonitrile as a neutral guest molecule (K = 72 ± 5 M−1) in CDCl3. Moreover, a synchronous effect of CH–π, π–π and/or cation–π interactions was demonstrated using the 1H NMR titrations (1,1,2,2-C2D2Cl4) of 14b with N-methylpyridinium (NMP), N-methylquinolinium (NMQ) and N-methylisoquinolinium (NMIQ) iodides. The analysis of the binding isotherms showed the 1:1 stoichiometry in all cases, with the highest complexation constants for NMP (K = 615 ± 15 M−1) indicating the best fit between the shape of the cavity and the shape of the guest molecule (Fig. 6). The NMQ and NMIQ derivatives showed much worse complexation abilities (K = 395 ± 4 M−1 and K = 211 ± 2 M−1, respectively).
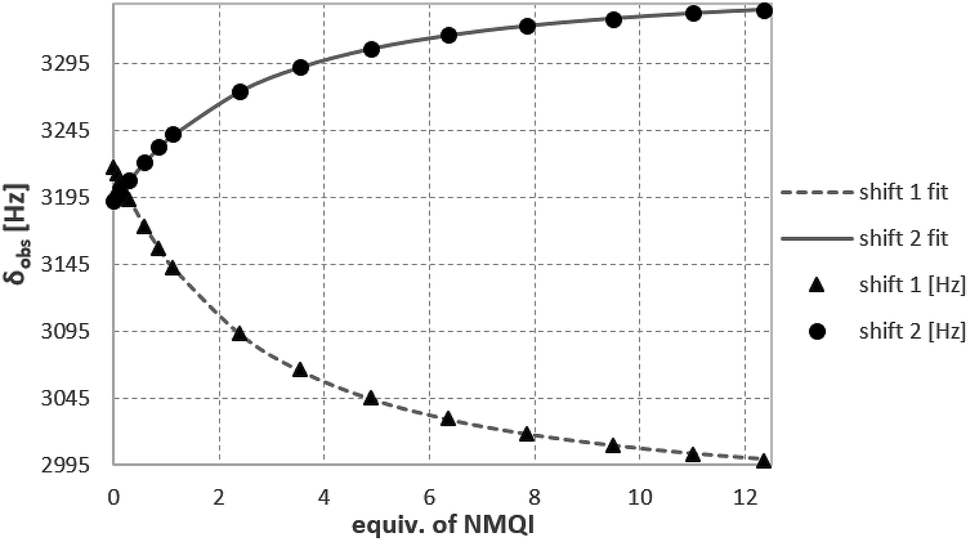 | ||
| Fig. 6 1H NMR titration curve of 14b with N-methylquinolinium iodide (NMQ) (C2D2Cl4, 298 K, 400 MHz). | ||
Conclusion
Meta/meta- and meta/para-disubstituted organomercurials, easily obtainable by direct mercuration of starting calix[4]arene, immobilised in the cone conformation, were transformed into corresponding amino derivatives. Acylation and subsequent intramolecular cyclization using the conditions of Bischler–Napieralski reaction provided in the case of meta/meta-series double bridged calixarenes possessing seven membered rings on the upper rim. A similar synthetic strategy applied to meta/para-isomers allowed for the isolation of monobridged compounds bearing an additional trifluoroacetamido group located distally to seven-membered ring. Both series represent inherently chiral systems, which were successfully resolved using preparative chiral HPLC. The rigidified cavities of bridged calixarenes can interact with guest molecules bearing acidic methyl groups, as documented by the shape selective complexation of N-methylpyridinium, N-methylquinolinium and N-methylisoquinolinium iodides using racemic 14b. Moreover, the 1H NMR titration experiments indicated the recognition ability of pure enantiomers of 14b towards selected chiral guest molecules. The absolute configuration of phenyl-substituted enantiomer (meta/meta-) was confirmed by single crystal structure determination (X-ray).Experimental
General experimental procedures
All chemicals were purchased from commercial sources, and used without further purification. Chloroform, tetrahydrofuran and dichloromethane used for the reactions were dried with CaH2 or MgSO4 and stored over molecular sieves. Melting points were measured on Heiztisch Mikroskop-Polytherm A (Wagner & Munz, Germany) and were not corrected. The IR spectra were measured on FT-IR spectrometer Nicolet 740 in KBr transmission mode. NMR spectra were recorded on spectrometer Agilent 400-MR DDR2 (1H: 400 MHz, 13C: 100 MHz). Chemical shifts (δ) are expressed in parts per million and are referenced to the residual peak of solvent or TMS as an internal standard, coupling constants (J) are in Hertz. The mass analyses were performed using ESI technique on a FT-MS (LTQ Orbitrap Velos) spectrometer. Purity of the substances and the courses of the reactions were monitored by TLC using aluminum sheets with Silica gel 60 F254 (Merck) and analysed at 254 or 365 nm. Preparative TLC chromatography was carried out on a Chromatotron (Harrison Research) with plates covered by Silica gel 60 GF254 (Merck).General procedure for preparation of m,m-diamides
m,m-Diamino calixarene 5 was dissolved in 10 mL of THF at room temperature and Et3N and corresponding carboxylic acid derivative were added. The solution was stirred for 24 h at room temperature, and diluted with dichloromethane (10 mL) was added. The crude reaction mixture was washed with water (3 × 20 mL), and dried over MgSO4. The solvent was removed under reduced pressure to yield crude product which was further purified by preparative TLC on silica gel.:ethyl acetate 2:15, v/v) to give title compound 6a as a colourless amorphous solid (0.080 g, 46%), mp 288–290 °C. 1H-NMR (CDCl3, 400 MHz, 298 K) δ 7.20–6.75 (m, 7H, Ar–H, Ar–NH–CO), 6.73–6.59 (m, 2H, Ar–H), 6.53–6.42 (m, 1H, Ar–H), 6.36–6.08 (m, 2H, Ar–H), 4.49 (d, 2H, J = 14.1 Hz, Ar–CH2−Ar), 4.42 (d, 2H, J = 13.3 Hz, Ar–CH2–Ar), 4.16–3.99 (m, 2H, O–CH2), 3.97–3.80 (m, 2H, O–CH2), 3.77–3.61 (m, 4H, O–CH2), 3.23 (d, 2H, J = 14.5 Hz, Ar–CH2−Ar), 3.16 (d, 2H, J = 13.7 Hz, Ar−CH2–Ar), 1.99 (s, 3H, CO–CH3), 1.97–1.76 (m, 8H, O–CH2–CH2), 1.12–1.00 (m, 6H, O–CH2–CH2–CH3), 0.94–0.81 (m, 6H, O–CH2–CH2–CH3) ppm.13C-NMR (CDCl3, 100 MHz, 298 K) δ 168.0, 157.9, 156.6, 134.8 (2×), 134.3, 134.2, 133.6, 129.0, 128.7, 127.3, 121.8, 120.1, 76.9, 76.7, 31.1, 29.7, 23.9, 23.4, 22.7, 10.6, 9.9 ppm.IR (KBr) ν 2961.0, 2934.4, 2874.8, 1661.9, 1521.9, 1455.4, 1206.3 cm−1. HRMS (ESI+) calcd for C44H54N2O6 729.38741 [M + Na]+, 745.36135 [M + K]+, found m/z 729.38820 [M + Na]+ (100%), 745.36046 [M + K]+ (5%).
:ethyl acetate 5:1, v/v) to give title compound 6b as a colourless amorphous solid (0.056 g, 41%) mp 314–317 °C. 1H-NMR (CDCl3, 400 MHz, 298 K) δ 7.72–7.62 (m, 4H, Ar–H), 7.57–7.52 (m, 2H, Ar–H), 7.49–7.44 (m, 2H, Ar–H), 7.21 (br s, 2H, Ar–NH–CO), 7.10–6.94 (m, 2H, Ar–H), 6.90–6.67 (m, 6H, Ar–H), 6.46–6.32 (m, 2H, Ar–H), 4.57–4.45 (m, 4H, Ar–CH2–Ar), 4.16–4.05 (m, 2H, O–CH2), 3.92–3.70 (m, 6H, O–CH2), 3.29 (d, 2H, J = 14.5 Hz, Ar–CH2–Ar), 3.23 (d, 2H, J = 13.7 Hz, Ar–CH2–Ar), 1.98–1.78 (m, 8H, O–CH2–CH2), 1.06 (t, 6H, J = 7.4 Hz, O–CH2–CH2–CH3), 0.90 (t, 6H, J = 7.4 Hz, O–CH2–CH2–CH3) ppm. 13C-NMR (CDCl3, 100 MHz, 298 K) δ 165.7, 156.8, 145.5, 134.8, 134.2, 134.1, 133.6, 131.6, 129.0 (2×), 128.6 (2×), 127.4, 122.2, 120.2, 77.0, 76.8, 31.3, 31.2, 23.4, 22.7, 10.6, 9.9 ppm. IR (KBr) ν 3300.8, 2959.7, 2927.8, 2873.5, 1649.8, 1580.7, 1516.3, 1487.6, 1268.3 cm−1. HRMS (ESI+) calcd for C54H58N2O6 853.41871 [M + Na]+, 869.39265 [M + K]+, found m/z 853.41870 [M + Na]+ (100%), 869.39185 [M + K]+ (13%).:ethyl acetate 1:1, v/v) to give title compound 6c as a red amorphous solid (0.042 g, 30%), mp 273–276 °C. 1H-NMR (CDCl3, 400 MHz, 298 K) δ 7.46 (br s, 2H, Ar–NH–CO), 6.96–6.29 (m, 10H, Ar–H), 4.50–4.39 (m, 4H, Ar–CH2–Ar), 4.02–3.71 (m, 10H, O–CH2, CO–CH2–Br), 3.28 (d, 2H, J = 14.1 Hz, Ar–CH2–Ar), 3.17 (d, 2H, J = 13.7 Hz, Ar–CH2–Ar), 1.98–1.78 (m, 8H, O–CH2–CH2), 1.01 (t, 6H, J = 7.4 Hz, O–CH2–CH2–CH3), 0.94 (t, 6H, J = 7.4 Hz, O–CH2–CH2–CH3) ppm. 13C-NMR (CDCl3, 100 MHz, 298 K) δ 163.7, 157.2, 157.1, 133.6, 133.5, 132.8, 132.7, 128.7, 128.3, 127.8, 122.3, 119.6, 76.9, 76.8, 31.0, 29.7, 29.4, 23.3, 22.8, 10.4, 10.1 ppm. IR (KBr) ν 3261.1, 2960.1, 2925.7, 2873.9, 1663.0, 1527.0, 1454.9, 1207.3 cm−1. HRMS (ESI+) calcd for C44H52Br2N2O6 887.20639 [M + Na]+, 903.18032 [M + K]+, found m/z 887.20630 [M + Na]+ (100%), 903.17920 [M + K]+ (17%).:ethyl acetate 1:1, v/v) to give title compound 6d as a colourless solid (0.071 g, 56%), mp 240–243 °C.1H-NMR (CDCl3, 400 MHz, 298 K) δ 7.82–7.61 (m, 2H, Ar–H), 6.95–6.82 (m, 2H, Ar–H), 6.77–6.46 (m, 8H, Ar–H, Ar–NH–CO), 4.51–4.39 (m, 4H, Ar–CH2–Ar), 4.15–4.10 (m, 2H, CO–CH2–Cl), 4.02–3.67 (m, 8H, O–CH2), 3.29 (d, 2H, J = 14.5 Hz, Ar–CH2–Ar), 3.18 (d, 2H, J = 13.7 Hz, Ar–CH2–Ar), 1.99–1.79 (m, 8H, O–CH2–CH2), 1.05–0.89 (m, 12H, O–CH2–CH2–CH3) ppm. 13C-NMR (CDCl3, 100 MHz, 298 K) δ 164.2, 157.3, 156.7, 143.4, 133.9, 133.3, 132.5, 129.3, 128.7, 128.3, 127.9, 122.3, 119.7, 76.9, 76.8, 42.9, 31.0, 30.8, 23.3, 22.9, 10.4, 10.2 ppm. IR (KBr) ν 3282.0, 2960.9, 2932.5, 2874.6, 1671.1, 1520.0, 1455.7, 1207.3, 1085.1 cm−1.
HRMS (ESI+) calcd for C44H52Cl2N2O6 797.30946 [M + Na]+, 813.28340 [M + K]+, found m/z 797.30895 [M + Na]+ (100%), 813.28276 [M + K]+ (25%).
:ethylacetate 2:1, v/v) to give title compound 6e as a colourless amorphous solid (0.092 g, 46%), mp > 330 °C. 1H-NMR (CDCl3, 400 MHz, 298 K) δ 7.58–7.26 (m, 23H, Ar–H), 7.14 (m, 16H, Ar–H, Ar–NH–CO), 6.17–6.03 (m, 2H, Ar–H), 5.93–5.70 (m, 2H, Ar–H), 4.51–4.32 (m, 8H, Ar–CH2–Ar), 4.10–3.91 (m, 4H, O–CH2), 3.88–3.59 (m, 14H, O–CH2, CO–CH(OAc)–Ar), 3.27–3.08 (m, 8H, Ar–CH2–Ar), 2.22 (s, 3H, O–CO–CH3), 2.19 (s, 3H, O–CO–CH3), 2.00–1.72 (m, 16H, O–CH2–CH2), 1.08–0.96 (m, 12H, O–CH2–CH2–CH3), 0.93 (t, 6H, J = 7.4 Hz, O–CH2–CH2–CH3), 0.85 (t, 6H, J = 7.4 Hz, O–CH2–CH2–CH3) ppm. 13C-NMR (CDCl3, 100 MHz, 298 K) δ 169.9, 169.1, 166.4, 166.3, 158.2, 158.1, 157.1, 156.5, 135.4, 134.7, 134.2, 133.7, 133.2, 133.0, 132.5 (2×), 132.1, 132.0, 129.5, 129.4, 129.2, 129.0, 128.9, 128.8, 128.6, 128.1, 127.7, 127.6, 127.4, 127.1, 122.2, 122.0, 119.8, 119.4, 76.9 (2×), 76.8, 76.7, 75.8, 75.7, 31.1, 31.0, 29.7, 28.5, 23.3, 23.2, 22.8, 22.5, 21.2, 20.9, 10.6, 10.4, 10.1, 9.8 ppm. IR (KBr) ν 3369.4, 2961.3, 2933.0, 2875.0, 1746.3, 1686.3, 1517.1, 1455.2, 1233.0 cm−1. HRMS (ESI+) calcd for C60H66N2O10 997.46097 [M + Na]+, 1013.43490 [M + K]+, found m/z 997.46149 [M + Na]+ (100%), 1013.43352 [M + K]+ (10%).:ethyl acetate 5:1, v/v) to give title compound 6f as a yellow amorphous solid (0.142 g, 56%), mp 187–190 °C. Compound 6f was also prepared using DCC as coupling reagent. 1-pyrenecarboxylic acid (0.120 g, 0.49 mmol) was dissolved in 5 mL of THF at room temperature. DCC (0.100 g, 0.64 mmol) was added and the solution was stirred for 10 minutes. Calixarene 5 (0.100 g, 0.16 mmol) was added afterwards and the solution was stirred for 24 h at room temperature. 10 mL of dichloromethane were added. The crude reaction mixture was washed with water (3 × 20 mL) and dried over MgSO4. The solvent was removed under reduced pressure to yield crude product which was further purified by preparative TLC (cyclohexane:ethyl acetate 5:1, v/v) to give title compound 6f (0.023 g, 11%). 1H-NMR (CDCl3, 400 MHz, 298 K) δ 8.61 (d, 2H, J = 9.4 Hz, Ar–H), 8.32–8.00 (m, 15H, Ar–H), 7.47–7.41 (m, 3H, Ar–H), 6.81 (br s, 2H, Ar–NH–CO), 6.66–6.19 (m, 6H, Ar–H), 6.14–6.02 (m, 2H, Ar–H), 4.59 (d, 2H, J = 14.1 Hz, Ar–CH2–Ar), 4.48 (d, 2H, J = 13.7 Hz, Ar–CH2–Ar), 4.21–4.00 (m, 2H, O–CH2), 3.93–3.70 (m, 6H, O–CH2), 3.33 (d, 2H, J = 14.1 Hz, Ar–CH2–Ar), 3.22 (d, 2H, J = 13.3 Hz, Ar–CH2–Ar), 2.01–1.75 (m, 8H, O–CH2–CH2), 1.07 (t, 6H, J = 7.0 Hz, O–CH2–CH2–CH3), 0.86 (t, 6H, J = 7.0 Hz, O–CH2–CH2–CH3) ppm. 13C-NMR (CDCl3, 100 MHz, 298 K) δ 167.9, 156.9, 156.8, 134.1, 133.8 (2×), 132.7, 131.2, 131.1, 130.8, 130.7 (2×), 139.0, 128.8 (2×), 128.7, 128.4, 127.7, 127.2, 126.3, 125.8 (2×), 124.9, 124.8, 124.4 (2×), 124.3, 122.2, 119.4, 76.8, 76.7, 33.9, 31.2, 23.4, 22.7, 10.6, 9.9 ppm. IR (KBr) ν 3372.2, 2959.6, 2928.5, 2874.0, 1650.1, 1588.4, 1511.8, 1455.7, 1087.8 cm−1. HRMS (ESI+) calcd for C74H66N2O6 1101.48131 [M + Na]+, 1117.45525 [M + K]+, found m/z 1101.48226 [M + Na]+ (100%), 1117.45522 [M + K]+ (35%).General procedure for preparation of bis-bridged calixarenes
Calixarene 6a–f was dissolved under argon atmosphere in 5 mL of dry CH2Cl2. The solution was cooled down to −78 °C, and 2-chloropyridine and triflic anhydride were added. The mixture was stirred for 5 min, then the solution was warmed to 0 °C and stirred for another 5 min. After that, the mixture was warmed to room temperature and stirred for one more hour. The reaction was quenched by a solution of NaHCO3. The organic layer was separated, washed with water (10 mL) and dried over MgSO4. The solvent was removed under reduced pressure to yield crude product, which was further purified by preparative TLC on silica gel.Alternative procedure was also examined. Corresponding calixarene was dissolved in 5 mL of toluene. POCl3 was added afterwards, and the reaction mixture was heated to reflux and stirred for 24 hours. The solution was washed with NaHCO3 (10 mL) and then with water (2 × 10 mL). The separated organic layer was dried over MgSO4. The solvent was removed under reduced pressure to yield crude product, which was further purified by preparative TLC on silica gel.
:ethyl acetate 4:5, v/v) to give title compound 7a as a yellow amorphous solid (0.015 g, 17%), mp 130–133 °C.Alternative procedure was also examined by reacting calixarene 6a (0.066 g, 0.09 mmol) and POCl3 (0.085 mL, 0.91 mmol). The crude reaction mixture was purified by preparative TLC on silica gel (cyclohexane:ethyl acetate 1:1, v/v) to give title compound as a yellow amorphous solid (0.003 g, 5%).
1H-NMR (CDCl3, 400 MHz, 298 K) δ 6.97 (d, 2H, J = 7.8 Hz, Ar–H), 6.87 (d, 2H, J = 8.2 Hz, Ar–H), 6.79 (d, 2H, J = 7.8 Hz, Ar–H), 6.58 (d, 2H, J = 8.2 Hz, Ar–H), 4.59 (d, 2H, J = 12.1 Hz, Ar–CH2–Ar), 4.53 (d, 2H, J = 11.7 Hz, Ar–CH2–Ar), 3.94–3.72 (m, 8H, O–CH2), 3.25 (d, 2H, J = 11.7 Hz, Ar–CH2–Ar), 2.72 (d, 2H, J = 11.7 Hz, Ar–CH2–Ar), 2.49 (s, 6H, NC(Ar)–CH3), 2.07–1.85 (m, 8H, O–CH2–CH2), 1.14 (t, 6H, J = 7.4 Hz, O–CH2–CH2–CH3), 1.09 (t, 6H, J = 7.4 Hz, O–CH2–CH2–CH3) ppm. 13C-NMR (CDCl3, 100 MHz, 298 K) δ 167.3, 153.0, 152.6, 146.2, 138.3, 136.1, 133.7, 131.9, 127.3, 127.2, 126.7, 121.1, 118.9, 77.4, 77.5, 30.7, 29.7, 27.5, 23.4, 23.3, 10.5 (2×) ppm. IR (KBr) ν 3363.1, 2959.3, 2928.3, 2874.8, 1418.8, 1066.7 cm−1. HRMS (ESI+) calcd for C44H50N2O4 671.38433 [M + H]+, 693.36628 [M + Na]+, found m/z 671.38404 [M + H]+ (100%), 693.36582 [M + Na]+ (60%).
:ethyl acetate 6:1, v/v) to give title compound 7b as a yellow amorphous solid (0.043 g, 66%), mp 175–178 °C.Alternative procedure was also examined by reacting calixarene 6b (0.056 g, 0.07 mmol) and POCl3 (0.062 mL, 0.66 mmol). The crude reaction mixture was purified by preparative TLC on silica gel (cyclohexane:ethyl acetate 7:1, v/v) to give title compound as a yellow amorphous solid (0.005 g, 9%).
1H-NMR (CDCl3, 400 MHz, 298 K) δ 7.84–7.78 (m, 4H, Ar–H), 7.49–7.35 (m, 6H, Ar–H), 6.97 (d, 2H, J = 7.8 Hz, Ar–H), 6.94 (d, 2H, J = 8.2 Hz, Ar–H), 6.73 (d, 2H, J = 8.2 Hz, Ar–H), 6.59 (d, 2H, J = 8.2 Hz, Ar–H), 4.72 (d, 2H, J = 12.1 Hz, Ar–CH2–Ar), 4.62 (d, 2H, J = 11.7 Hz, Ar–CH2–Ar), 4.07–3.99 (m, 2H, O–CH2), 3.93–3.79 (m, 6H, O–CH2), 3.32 (d, 2H, J = 12.1 Hz, Ar–CH2–Ar), 2.86 (d, 2H, J = 12.1 Hz, Ar–CH2–Ar), 2.17–1.88 (m, 8H, O–CH2–CH2), 1.22 (t, 6H, J = 7.4 Hz, O–CH2–CH2–CH3), 1.11 (t, 6H, J = 7.4 Hz, O–CH2–CH2–CH3) ppm. 13C-NMR (CDCl3, 100 MHz, 298 K) δ 166.3, 153.0, 152.8, 146.4, 140.4, 138.5, 137.6, 132.2, 131.6, 129.9, 129.5, 128.0, 127.4, 126.8, 126.7, 123.9, 119.4, 77.5, 77.4, 30.7, 23.9, 23.6, 23.3, 10.7, 10.6 ppm. IR (KBr) ν 2959.6, 1933.0, 2875.1, 1572.0, 1466.4, 1417.5, 1217.1, 1062.6 cm−1. HRMS (ESI+) calcd for C54H54N2O4 795.41563 [M + H]+, 817.39758 [M + Na]+, found m/z 795.41581 [M + H]+ (100%), 817.39702 [M + Na]+ (35%).
:ethylacetate 3:1, v/v) to give title compound 7c as a red amorphous solid (0.009 g, 17%). 1H-NMR (CDCl3, 400 MHz, 298 K) δ 7.00 (d, 2H, J = 8.2 Hz, Ar–H), 6.90 (d, 2H, J = 8.2 Hz, Ar–H), 6.84 (d, 2H, J = 8.2 Hz, Ar–H), 6.58 (d, 2H, J = 8.2 Hz, Ar–H), 4.62 (d, 2H, J = 12.1 Hz, Ar–CH2–Ar), 4.61 (d, 2H, J = 9.8 Hz, NC(Ar)–CH2–Br), 4.55 (d, 2H, J = 11.7 Hz, Ar–CH2–Ar), 4.28 (d, 2H, J = 9.8 Hz, NC(Ar)–CH2–Br), 3.98–3.91 (m, 2H, O–CH2), 3.88–3.75 (m, 6H, O–CH2), 3.28 (d, 2H, J = 11.7 Hz, Ar–CH2–Ar), 2.80 (d, 2H, J = 12.1 Hz, Ar–CH2–Ar), 2.06–1.86 (m, 8H, O–CH2–CH2), 1.16 (t, 6H, J = 7.4 Hz, O–CH2–CH2–CH3), 1.08 (t, 6H, J = 7.4 Hz, O–CH2–CH2–CH3) ppm. HRMS (ESI+) calcd for C44H48Br2N2O4 829.20331 [M + H]+, 851.18526 [M + Na]+, found m/z 829.20333 [M + H]+ (30%), 851.18539 [M + Na]+ (100%).:ethyl acetate 3:1, v/v) to give the title compound 7e as a yellow amorphous solid (0.016 g, 17%). HRMS (ESI+) calcd for C60H62N2O8 939.45789 [M + H]+, 961.43984 [M + Na]+, found m/z 939.45758 [M + H]+ (75%), 961.43933 [M + Na]+ (100%).C(Ar)–CH3), 2.29–1.82 (m, 8H, O–CH2–CH2), 2.12 (s, 3H, Ar–CO–CH3), 1.17–0.92 (m, 12H, O–CH2–CH2–CH3) ppm. IR (KBr) ν 2919.3, 2850.7, 1622.4, 1407.7, 1118.7, 1046.1 cm−1. HRMS (ESI+) calcd for C44H52N2O5 689.39490 [M + H]+, 711.37684 [M + Na]+, found m/z 689.39445 [M + H]+ (95%), 711.37628 [M + Na]+ (100%).:ethyl acetate 5:1, v/v) to give title compound 11 as a yellow amorphous solid (0.829 g, 82%), mp 236–238 °C. 1H-NMR (CDCl3, 400 MHz, 298 K) δ 8.47 (br s, 1H, Ar–NH–CO), 7.38 (br s, 1H, Ar–NH–CO), 7.19 (dd, 1H, J = 7.4, 1.6 Hz, Ar–H), 7.14 (dd, 1H, J = 7.4, 1.6 Hz, Ar–H), 7.11–7.06 (m, 2H, Ar–H), 7.00 (t, 1H, J = 7.4 Hz, Ar–H), 6.90 (t, 1H, J = 7.4 Hz, Ar–H), 6.36 (d, 1H, J = 6.36 Hz, Ar–H), 6.23 (d, 1H, J = 2.7 Hz, Ar–H), 6.13 (d, 1H, J = 8.6 Hz, Ar–H), 5.83 (d, 1H, J = 2.7 Hz, Ar–H), 4.60 (d, 1H, J = 14.5 Hz, Ar–CH2–Ar), 4.50–4.41 (m, 3H, Ar–CH2–Ar), 4.12–4.03 (m, 2H, O–CH2), 3.99–3.88 (m, 2H, O–CH2), 3.71–3.57 (m, 4H, O–CH2), 3.26–3.10 (m, 4H, Ar–CH2–Ar), 1.94–1.80 (m, 8H, O–CH2–CH2), 1.14–1.08 (m, 6H, O–CH2–CH2–CH3), 0.87 (t, 3H, J = 7.4 Hz, O–CH2–CH2–CH3), 0.81 (t, 3H, J = 7.4 Hz, O–CH2–CH2–CH3) ppm. 13C-NMR (CDCl3, 100 MHz, 298 K) δ 158.1, 157.8, 156.5, 153.7, 137.3, 137.2, 136.0, 134.5 (2×), 133.6, 133.2, 130.3, 130.0, 129.6, 129.4, 129.3, 129.2, 128.5, 127.8, 122.3, 121.6, 121.5, 120.6, 119.4, 117.5, 117.4, 114.6, 114.5, 77.2, 76.9, 76.8, 76.1, 31.4, 31.3, 30.6, 28.9, 23.5 (2×), 23.1, 22.4, 10.9, 10.8, 9.8, 9.6 ppm. IR (KBr) ν 3289.8, 2963.1, 1932.8, 2876.3, 1706.5, 1462.9, 1200.0, 1159.4 cm−1. HRMS (ESI+) calcd for C44H48F6N2O6 837.33088 [M + Na]+, 853.30481 [M + K]+, found m/z 837.33243 [M + Na]+ (100%), 853.30555 [M + K]+ (60%).:THF (1:1, v/v) at room temperature. NaOH (0.110 g, 2.75 mmol) and water (0.500 mL, 27.78 mmol) were added. The solution was stirred for 50 h at room temperature. 20 mL of dichloromethane were added. The crude reaction mixture was washed with water (3 × 20 mL) and dried over magnesium sulphate. The solvent was removed under reduced pressure to yield crude product which was further purified by column chromatography on silica gel (cyclohexane:ethyl acetate 5:1, v/v) to give title compound 12a as a yellow amorphous solid (0.426 g, 22%), mp 215–218 °C. 1H-NMR (CDCl3, 400 MHz, 298 K) δ 7.45 (br s, 1H, Ar–NH–CO), 7.02 (d, 1H, J = 7.4 Hz, Ar–H), 6.94–6.84 (m, 8H, Ar–H), 6.82–6.75 (m, 2H, Ar–H), 6.56 (d, 1H, J = 2.0 Hz, Ar–H), 6.26 (d, 1H, J = 2.0 Hz, Ar–H), 6.21 (d, 1H, J = 8.2 Hz, Ar–H), 5.80 (d, 1H, J = 8.2 Hz, Ar–H), 4.52–4.44 (m, 3H, Ar–CH2–Ar), 4.34 (d, 1H, J = 13.3 Hz, Ar–CH2–Ar), 4.09–3.99 (m, 1H, O–CH2), 3.96–3.82 (m, 3H, O–CH2), 3.80–3.62 (m, 4H, O–CH2), 3.22–3.06 (m, 4H, Ar–CH2–Ar), 1.95–1.81 (m, 8H, O–CH2–CH2), 1.10–1.01 (m, 6H, O–CH2–CH2–CH3), 0.97–0.86 (m, 6H, O–CH2–CH2–CH3) ppm. 13C-NMR (CDCl3, 100 MHz, 298 K) δ 157.7, 157.3, 157.1, 154.5, 143.2, 136.8, 136.5, 136.1, 135.6, 135.5, 134.4, 129.0, 128.9, 128.8 (2×), 128.2, 128.1, 124.5, 122.0 (3×), 119.9, 117.3, 114.4, 110.7, 76.8 (3×), 31.5, 31.0, 30.5, 27.8, 23.4, 23.3, 23.2, 22.6, 10.6 (2×), 10.1, 9.9 ppm. IR (KBr) ν 3312.4, 2962.3, 2933.3, 2875.7, 1712.6, 1463.4, 1216.1, 1184.0 cm−1. HRMS (ESI+) calcd for C42H49F3N2O5 719.36663 [M + H]+, 741.34858 [M + Na]+, 757.32252 [M + K]+, found m/z 719.36658 [M + H]+ (100%), 741.34772 [M + Na]+ (15%), 757.32153 [M + K]+ (7%).General procedure for preparation of p-trifluoroamido-m-amides
p-Trifluoroamido-m-amino calixarene 12a was dissolved in 10 mL of THF at room temperature. Et3N and corresponding carboxylic acid derivative were added. The solution was stirred for 24 h at room temperature. 10 mL of dichloromethane were added. The crude reaction mixture was washed with water (3 × 20 mL) and dried over magnesium sulphate. The solvent was removed under reduced pressure to yield crude product which was further purified by preparative TLC on silica gel.:ethyl acetate 5:1, v/v) to give title compound 13b as a colourless amorphous solid (0.042 g, 60%), mp 280–283 °C. 1H-NMR (CDCl3, 400 MHz, 298 K) δ 9.65 (br s, 1H, Ar–NH–CO), 7.76 (d, 2H, J = 7.0 Hz, Ar–H), 7.61–7.55 (m, 1H, Ar–H), 7.55–7.48 (m, 2H, Ar–H), 7.25 (s, 1H, Ar–NH–CO), 7.20–7.13 (m, 2H, Ar–H), 6.99 (t, 1H, J = 7.4 Hz, Ar–H), 6.94 (dd, 1H, J = 7.4, 1.2 Hz, Ar–H), 6.72 (dd, 1H, J = 7.4, 1.2 Hz, Ar–H), 6.60 (d, 1H, J = 8.22 Hz, Ar–H), 6.31 (t, 1H, J = 7.4 Hz, Ar–H), 6.27 (d, 1H, J = 2.4 Hz, Ar–H), 6.14 (d, 1H, J = 8.2 Hz, Ar–H), 5.78 (d, 1H, J = 2.4 Hz, Ar–H), 4.61 (d, 1H, J = 14.5 Hz, Ar–CH2–Ar), 4.52–4.40 (m, 3H, Ar–CH2–Ar), 4.14–4.03 (m, 2H, O–CH2), 4.01–3.87 (m, 2H, O–CH2), 3.74–3.57 (m, 4H, O–CH2), 3.27–3.19 (m, 2H, Ar–CH2–Ar), 3.17–3.08 (m, 2H, Ar–CH2–Ar), 1.97–1.79 (m, 8H, O–CH2–CH2), 1.15–1.08 (m, 6H, O–CH2–CH2–CH3), 0.88 (t, 3H, J = 7.4 Hz, O–CH2–CH2–CH3), 0.81 (t, 3H, J = 7.4 Hz, O–CH2–CH2–CH3) ppm. 13C-NMR (CDCl3, 100 MHz, 298 K) δ 164.6, 158.2, 157.8, 156.2, 155.1, 154.8, 153.6, 137.3, 137.2, 136.2, 134.7, 134.4, 134.3, 133.7, 133.1, 131.6, 131.6, 130.6, 130.1, 129.4 (2×), 129.2, 128.5, 127.7, 127.2, 126.5, 122.3, 122.2, 121.1, 120.9, 119.2, 77.1, 76.8, 76.7, 76.1, 31.3 (2×), 30.6, 29.4, 23.5 (2×), 23.0, 22.4, 10.9, 10.8, 9.8, 9.6 ppm. IR (KBr) ν 3250.8, 3066.0, 2962.5, 2933.5, 2875.7, 1706.6, 1463.6, 1214.7, 1004.4 cm−1. HRMS (ESI+) calcd for C49H53F3N2O6 845.37479 [M + Na]+, 861.34873 [M + K]+, found m/z 845.37536 [M + Na]+ (100%), 861.34718 [M + K]+ (8%).HRMS (ESI+) calcd for C44H50ClF3N2O6 817.3202 [M + Na]+, 833.2941 [M + K]+, found m/z 817.3207 [M + Na]+ (100%), 833.2940 [M + K]+ (90%).
:ethyl acetate 3:1, v/v) to give title compound 13e as a colourless amorphous solid (0.101 g, 81%), mp 124–127 °C. 1H-NMR (CDCl3, 400 MHz, 298 K) δ 9.34 (br s, 1H, Ar–NH–CO), 8.61 (br s, 1H, Ar–NH–CO), 7.53–7.47 (m, 3H, Ar–H), 7.44–7.32 (m, 7H, Ar–H), 7.19–6.83 (m, 13H, Ar–H), 6.67 (d, 1H, J = 8.6 Hz, Ar–H), 6.35–6.30 (m, 3H, Ar–H), 6.08 (s, 1H, Ph–CH(OAc)–CO), 6.06 (t, 2H, J = 7.8 Hz, Ar–H), 5.96 (d, 1H, J = 2.4 Hz, Ar–H), 5.85 (s, 1H, Ph–CH(OAc)–CO), 5.62 (d, 1H, J = 2.4 Hz, Ar–H), 4.63–4.53 (m, 2H, Ar–CH2–Ar), 4.50–4.39 (m, 6H, Ar–CH2–Ar), 4.13–4.00 (m, 4H, O–CH2), 3.98–3.87 (m, 4H, O–CH2), 3.73–3.51 (m, 8H, O–CH2), 3.27–3.05 (m, 8H, Ar–CH2–Ar), 2.34 (s, 3H, CO–CH3), 2.18 (s, 3H, CO–CH3), 1.95–1.77 (m, 16H, O–CH2–CH2), 1.14–1.03 (m, 12H, O–CH2–CH2–CH3), 0.91–0.77 (m, 12H, O–CH2–CH2–CH3) ppm. 13C-NMR (CDCl3, 100 MHz, 298 K) δ 170.2, 168.6, 165.6, 165.5, 158.2, 158.1 (3×), 156.3, 156.0, 154.8, 154.6, 154.4, 153.7, 153.4, 137.8, 137.5, 137.4, 136.9, 136.5, 135.9 (2×), 135.7, 135.0, 134.5, 134.4, 134.3, 134.2, 134.0, 133.3, 132.5, 132.1, 131.6, 131.1, 129.8, 129.5 (3×), 129.4, 129.3, 129.2 (3×), 128.8, 128.5, 128.1 (2×), 127.7, 127.4, 127.1, 125.5, 122.3 (2×), 122.0, 121.6, 121.5, 120.9, 120.8, 119.9 (2×), 117.6 (2×), 114.6, 114.5, 77.1, 77.0, 76.9, 76.8 (2×), 76.6, 76.1 (2×), 75.4, 75.2, 31.4, 31.3 (2×), 31.2, 30.6, 30.5, 29.6, 29.2, 23.5 (2×), 23.4 (2×), 23.0 (2×), 22.4, 22.2, 21.3, 20.9, 10.9, 10.8 (3×), 9.8 (2×), 9.6, 9.5 ppm. IR (KBr) ν 3260.5, 2962.8, 2934.0, 2875.8, 1711.3, 1675.1, 1517.6, 1464.1, 1216.5 cm−1. HRMS (ESI+) calcd for C52H57F3N2O8 917.39592 [M + Na]+, 933.36986 [M + K]+, found m/z 917.39736 [M + Na]+ (100%), 933.37037 [M + K]+ (55%).:ethyl acetate 4:1, v/v) to give title compound 13f as a yellow amorphous solid (0.088 g, 67%), mp 160–163 °C. 1H-NMR (CDCl3, 400 MHz, 298 K) δ 9.89 (br s, 1H, Ar–NH–CO), 8.54 (d, 1H, J = 9.4 Hz, Ar–H), 8.31–8.07 (m, 8H, Ar–H), 7.35 (br s, 1H, Ar–NH–CO), 7.25–7.19 (m, 2H, Ar–H), 7.08–7.02 (m, 2H, Ar–H), 6.98–6.94 (m, 1H, Ar–H), 6.42 (d, 1H, J = 7.4 Hz, Ar–H), 6.39 (d, 1H, J = 2.4 Hz, Ar–H), 6.28 (d, 1H, J = 8.2 Hz), 5.98 (d, 1H, J = 2.0 Hz, Ar–H), 5.93 (t, 1H, J = 7.4 Hz, Ar–H), 4.64 (d, 1H, J = 14.5 Hz, Ar–CH2–Ar), 4.57–4.47 (m, 3H, Ar–CH2–Ar), 4.16–3.89 (m, 4H, O–CH2), 3.79–3.61 (m, 4H, O–CH2), 3.31 (d, 1H, J = 13.7 Hz, Ar–CH2–Ar), 3.24–3.18 (m, 2H, Ar–CH2–Ar), 3.16 (d, 1H, J = 14.9 Hz, Ar–CH2–Ar), 2.00–1.79 (m, 8H, O–CH2–CH2), 1.18–1.09 (m, 6H, O–CH2–CH2–CH3), 0.91 (t, 3H, J = 7.4 Hz, O–CH2–CH2–CH3), 0.81 (t, 3H, J = 7.4 Hz, O–CH2–CH2–CH3) ppm. 13C-NMR (CDCl3, 100 MHz, 298 K) δ 167.0, 158.2, 157.9, 156.3, 155.6, 155.2, 153.8, 137.3 (2×), 136.3, 136.6, 134.5, 133.8, 133.5, 133.1, 131.2, 131.1, 130.7, 130.2, 129.5 (2×), 129.3, 129.2, 129.0, 128.6, 128.0, 127.2, 126.4, 126.0, 125.9, 125.5, 125.4, 124.9 (2×), 124.4, 124.3, 123.0, 122.4, 121.7, 121.0, 117.9, 117.7, 114.8, 77.1, 76.9, 76.7, 76.2, 31.4, 31.3, 30.7, 29.3, 23.5 (2×), 23.1, 22.3, 10.9, 10.8, 9.9, 9.6 ppm. IR (KBr) ν 3248.1, 3050.6, 2961.6, 2932.3, 2875.2, 1713.3, 1654.2, 1464.0, 1214.2 cm−1. HRMS (ESI+) calcd for C59H57F3N2O6 969.40609 [M + Na]+, 985.38003 [M + K]+, found m/z 969.40613 [M + Na]+ (100%), 985.37960 [M + K]+ (80%).General procedure for preparation of bridged calixarenes
Corresponding calixarene was dissolved in the argon atmosphere in 5 mL of dry CH2Cl2. The solution was cooled down to −78 °C. 2-chloropyridine and trifluoromethanesulfonic anhydride were added afterwards. After 5 minutes of stirring the solution was warmed to 0 °C and stirred for another 5 min. The mixture was then warmed to room temperature and stirred for one more hour. The reaction was quenched by a solution of NaHCO3. The organic layer was separated, washed with water (1×10 mL) and dried over MgSO4. The solvent was removed under reduced pressure to yield crude product which was further purified by preparative TLC on silica gel.:ethyl acetate 3:1, v/v) to give title compound 14a as a yellow amorphous solid (0.027 g, 64%), mp 122–125 °C. 1H-NMR (CDCl3, 400 MHz, 298 K) δ 8.22 (br s, 1H, Ar–NH–CO), 7.16 (d, 1H, J = 2.4 Hz, Ar–H), 7.07–6.99 (m, 2H, Ar–H), 6.96 (dd, 1H, J = 7.4, 1.2 Hz, Ar–H), 6.86 (d, 1H, J = 7.8 Hz, Ar–H), 6.80 (d, 1H, J = 2.4 Hz, Ar–H), 6.72 (t, 1H, J = 7.4 Hz, Ar–H), 6.61 (d, 1H, J = 8.2 Hz, Ar–H), 6.20 (d, 1H, J = 8.2 Hz, Ar–H), 4.61–4.54 (m, 2H, Ar–CH2–Ar), 4.45–4.37 (m, 2H, Ar–CH2–Ar), 4.01–3.68 (m, 8H, O–CH2), 3.31 (d, 1H, J = 12.1 Hz, Ar–CH2–Ar), 3.23–3.15 (m, 2H, Ar–CH2–Ar), 2.75 (d, 1H, J = 11.7 Hz, Ar–CH2–Ar), 2.51 (s, 3H, NC(Ar)–CH3), 2.29 (sex, 2H, J = 7.8 Hz, O–CH2–CH2), 2.05–1.83 (m, 6H, O–CH2–CH2), 1.14 (t, 3H, J = 7.4 Hz, O–CH2–CH2–CH3), 1.09–0.96 (m, 9H, O–CH2–CH2–CH3) ppm.13C-NMR (CDCl3, 100 MHz, 298 K) δ 166.9, 156.1, 153.6, 153.2 (2×), 143.2, 140.3, 136.9, 135.8, 135.4, 135.3, 135.1, 135.0, 133.8, 130.6, 129.3, 128.8, 127.4, 127.1, 126.8, 123.0, 121.8, 121.2, 120.4, 118.1, 117.3, 115.5, 77.9, 77.8, 77.6, 76.3, 31.0, 30.5, 26.8, 23.5, 23.3 (2×), 23.2, 23.0, 22.7, 10.7, 10.3 (2×), 9.9 ppm. IR (KBr) ν 2960.6, 2930.2, 2874.8, 1721.0, 1610.7, 1463.4, 1215.5 cm−1. HRMS (ESI+) calcd for C44H49F3N2O5 743.36663 [M + H]+, 765.34858 [M + Na]+, found m/z 743.36721 [M + H]+ (100%), 765.34773 [M + Na]+ (27%).:ethyl acetate 3:1, v/v) to give title compound 14b as a yellow amorphous solid (0.032 g, 78%), mp 127–130 °C. 1H-NMR (CDCl3, 400 MHz, 298 K) δ 7.89–7.84 (m, 2H, Ar–H), 7.54 (bs s, 1H, Ar–NH–CO), 7.45–7.35 (m, 3H, Ar–H), 7.17 (d, 1H, J = 2.4 Hz, Ar–H), 7.05 (dd, 1H, J = 7.8, 1.6 Hz, Ar–H), 6.99 (d, 2H, J = 7.8 Hz. Ar–H), 6.85 (d, 1H, J = 2.7 Hz, Ar–H), 6.80–6.74 (m, 2H, Ar–H), 6.67–6.63 (m, 2H, Ar–H), 4.69 (d, 1H, J = 12.1 Hz, Ar–CH2–Ar), 4.61 (d, 1H, J = 12.1 Hz, Ar–CH2–Ar), 4.48 (d, 1H, J = 12.5 Hz, Ar–CH2–Ar), 4.43 (d, 1H, J = 12.5 Hz, Ar–CH2–Ar), 4.05–3.94 (m, 2H, O–CH2), 3.92–3.81 (m, 2H, O–CH2), 3.81–3.69 (m, 4H, O–CH2), 3.32 (d, 1H, J = 12.1 Hz, Ar–CH2–Ar), 3.25 (d, 1H, J = 12.5 Hz, Ar–CH2–Ar), 3.19 (d, 1H, J = 12.5 Hz, Ar–CH2–Ar), 2.89 (d, 1H, J = 12.1 Hz, Ar–CH2–Ar), 2.32 (sex, 2H, J = 7.4 Hz, O–CH2–CH2), 2.10–1.99 (m, 4H, O–CH2–CH2), 1.94–1.84 (m, 2H, O–CH2–CH2), 1.18 (t, 3H, J = 7.4 Hz, O–CH2–CH2–CH3), 1.10–0.99 (m, 9H, O–CH2–CH2–CH3) ppm. 13C-NMR (CDCl3, 100 MHz, 298 K) δ 165.9, 156.2, 154.5, 153.6, 153.4, 153.3, 145.9, 139.8, 136.9, 135.9, 135.4, 135.2, 135.0, 131.8, 130.8, 130.1, 129.4, 129.2, 128.8 (2×), 128.0, 127.1, 127.0, 126.9, 123.3, 123.0, 121.3, 120.8, 118.6, 117.2, 114.3, 78.0, 77.8, 77.6, 76.4, 31.1, 30.6, 30.5, 23.8, 23.6, 23.3, 23.0, 22.8, 10.7, 10.4, 10.3, 9.9 ppm. IR (KBr) ν 3331.0, 2924.0, 2872.4, 1459.9, 1383.6, 1212.5, 1195.5, 1056.2 cm−1. HRMS (ESI+) calcd for C49H51F3N2O5 805.38228 [M + H]+, 827.36423 [M + Na]+, found m/z 805.38297 [M + H]+ (100%), 827.36360 [M + Na]+ (35%).:ethyl acetate 4:1, v/v) to give title compound 14c as a yellow amorphous solid (0.050 g, 72%), mp 145–148 °C. 1H-NMR (CDCl3, 400 MHz, 298 K) δ 7.70 (br s, 1H, Ar–NH–CO), 7.19 (d, 1H, J = 2.4 Hz, Ar–H), 7.07 (d, 1H, J = 7.8 Hz, Ar–H), 7.02 (dd, 1H, J = 7.4, 1.6 Hz, Ar–H), 6.98 (dd, 1H, J = 7.4, 1.6 Hz, Ar–H), 6.88 (d, 1H, J = 8.2 Hz, Ar–H), 6.82 (d, 1H, J = 2.7 Hz, Ar–H), 6.78–6.72 (m, 2H, Ar–H), 6.42 (d, 1H, J = 7.8 Hz, Ar–H), 4.66–4.60 (m, 2H, Ar–CH2–Ar, CO–CH2–Br), 4.58 (d, 1H, J = 12.1 Hz, Ar–CH2–Ar), 4.46 (d, 1H, J = 12.1 Hz, Ar–CH2–Ar), 4.41 (d, 1H, J = 12.5 Hz, Ar–CH2–Ar), 4.24 (d, 1H, J = 10.2 Hz, CO–CH2–Br), 4.03–3.90 (m, 2H, O–CH2), 3.87–3.68 (m, 6H, O–CH2), 3.33 (d, 1H, J = 12.1 Hz, Ar–CH2–Ar), 3.23 (d, 1H, J = 12.5 Hz, Ar–CH2–Ar), 3.18 (d, 1H, J = 12.5 Hz, Ar–CH2–Ar), 2.86 (d, 1H, J = 12.1 Hz, Ar–CH2–Ar), 2.33–2.23 (m, 2H, O–CH2–CH2), 2.07–1.82 (m, 6H, O–CH2–CH2), 1.14 (t, 3H, J = 7.4 Hz, O–CH2–CH2–CH3), 1.10–0.97 (m, 9H, O–CH2–CH2–CH3) ppm. 13C-NMR (CDCl3, 100 MHz, 298 K) δ 164.6, 156.2, 153.6 (2×), 153.3, 144.0, 137.9, 136.6, 135.7, 135.3, 135.2, 134.9, 131.9, 131.0, 129.3, 128.9, 128.8, 127.6, 127.2, 127.1, 123.1, 121.8, 121.4, 119.5, 118.2, 117.2, 114.3, 78.0, 77.9, 77.6, 76.3, 35.2, 31.1, 30.6, 30.5, 23.6, 23.3, 23.2, 23.1, 22.7, 10.7, 10.6, 10.3, 9.9 ppm. IR (KBr) ν 3300.8, 2962.1, 2934.4, 2875.7, 1721.8, 1464.2, 1422.7, 1217.4, 1158.1 cm−1. HRMS (ESI+) calcd for C44H48BrF3N2O5 821.2771 [M + H]+, 843.2591 [M + Na]+, 861.2315 [M + K]+, found m/z 821.2768 [M + H]+ (100%), 843.2585 [M + Na]+ (60%), 861.2330 [M + K]+ (30%).:ethyl acetate 4:1, v/v) to give title compound 14d as a yellow amorphous solid (0.052 g, 81%), mp 153–156 °C. 1H-NMR (CDCl3, 400 MHz, 298 K) δ 7.16 (d, 1H, J = 2.7 Hz, Ar–H), 7.08 (d, 1H, J = 8.2 Hz, Ar–H), 7.03 (dd, 1H, J = 7.4, 1.6 Hz, Ar–H), 6.98 (dd, 1H, J = 7.4, 1.6 Hz, Ar–H), 6.87 (d, 1H, H = 7.8 Hz, Ar–H), 6.82 (d, 1H, J = 2.7 Hz, Ar–H), 6.74 (t, 1H, J = 7.4 Hz, Ar–H), 6.68 (d, 1H, J = 8.2 Hz, Ar–H), 6.28 (d, 1H, J = 7.8 Hz, Ar–H), 4.71 (d, 1H, J = 11.7 Hz, CO–CH2–Br), 4.64 (d, 1H, J = 12.1 Hz, Ar–CH2–Ar), 4.59 (d, 1H, J = 12.1 Hz, Ar–CH2–Ar), 4.46 (d, 1H, J = 12.5 Hz, Ar–CH2–Ar), 4.41 (d, 1H, J = 12.5 Hz, Ar–CH2–Ar), 4.33 (d, 1H, J = 11.7 Hz, CO–CH2–Br), 4.03–3.92 (m, 2H, O–CH2), 3.87–3.68 (m, 6H, O–CH2), 3.33 (d, 1H, J = 12.1 Hz, Ar–CH2–Ar), 3.23 (d, 1H, J = 12.5 Hz, Ar–CH2–Ar), 3.18 (d, 1H, J = 12.5 Hz, Ar–CH2–Ar), 2.81 (d, 1H, J = 12.1 Hz, Ar–CH2–Ar), 2.35–2.24 (m, 2H, O–CH2–CH2), 2.07–1.96 (m, 4H, O–CH2–CH2), 1.95–1.82 (m, 2H, O–CH2–CH2), 1.15 (t, 3H, O–CH2–CH2–CH3), 1.10–0.98 (m, 9H, O–CH2–CH2–CH3) ppm. 13C-NMR (CDCl3, 100 MHz, 298 K) δ 164.3, 156.1, 153.6, 153.5, 153.3, 144.1, 137.9, 136.6, 135.7, 135.3, 135.1, 134.9, 131.8, 130.8, 129.3, 128.9, 128.8, 127.6, 127.2, 127.1, 123.1, 121.7, 121.2, 119.8, 118.2, 117.2, 114.3, 78.0, 77.9, 77.6, 76.4, 47.7, 31.1, 30.6, 30.5, 23.6, 23.3 (2×), 23.0, 22.7, 10.7, 10.6, 10.3, 9.9 ppm. IR (KBr) ν 3300.8, 2962.1, 2934.4, 2875.7, 1721.8, 1464.2, 1217.4, 1158.1, 1005.5 cm−1. HRMS (ESI+) calcd for C44H48ClF3N2O5 777.32766 [M + H]+, 799.30961 [M + Na]+, 815.28354 [M + K]+, found m/z 777.32712 [M + H]+ (20%), 799.30930 [M + Na]+ (100%), 815.28301 [M + K]+ (55%).:ethyl acetate 3:2, v/v) to give title compound 14e as a yellow amorphous solid (0.018 g, 16%), mp 130–133 °C. 1H-NMR (CDCl3, 400 MHz, 298 K) δ 8.15–8.11 (m, 2H, Ar–H), 7.58 (tt, 1H, J = 7.4, 1.2 Hz, Ar–H), 7.55 (br s, 1H, Ar–NH–CO), 7.49–7.44 (m, 2H, Ar–H), 7.11 (d, 1H, J = 2.7 Hz, Ar–H), 7.03 (dd, 1H, J = 7.8, 1.6 Hz, Ar–H), 7.00 (d, 2H, J = 7.8 Hz, Ar–H), 6.93 (d, 1H, J = 2.7 Hz, Ar–H), 6.86 (d, 1H, J = 8.2 Hz, Ar–H), 6.78 (d, 2H, J = 7.8 Hz, Ar–H), 6.73 (d, 1H, J = 8.2 Hz, Ar–H), 4.76 (d, 1H, J = 12.1 Hz, Ar–CH2–Ar), 4.59 (d, 1H, J = 12.1 Hz, Ar–CH2–Ar), 4.51 (d, 1H, J = 12.1 Hz, Ar–CH2–Ar), 4.43 (d, 1H, J = 12.9 Hz, Ar–CH2–Ar), 4.08–3.90 (m, 3H, O–CH2), 3.89–3.70 (m, 6H, O–CH2, AcO–CH(Ph)–CN), 3.31 (d, 1H, J = 12.1 Hz, Ar–CH2–Ar), 3.28 (d, 1H, J = 12.5 Hz, Ar–CH2–Ar), 3.20 (d, 1H, J = 12.9 Hz, Ar–CH2–Ar), 2.95 (d, 1H, J = 12.1 Hz, Ar–CH2–Ar), 2.32–2.25 (m, 2H, O–CH2–CH2), 2.17 (s, 3H, O–CO–CH3), 2.06–1.86 (m, 6H, O–CH2–CH2), 1.15 (t, 3H, J = 7.4 Hz, O–CH2–CH2–CH3), 1.08 (t, 3H, J = 7.4 Hz, O–CH2–CH2–CH3), 1.03 (t, 3H, J = 7.4 Hz, O–CH2–CH2–CH3), 1.02 (t, 3H, J = 7.4 Hz, O–CH2–CH2–CH3) ppm. 13C-NMR (CDCl3, 100 MHz, 298 K) δ 194.3, 163.9, 156.1, 153.6, 153.5, 144.5, 138.0, 136.7, 136.1, 135.4 (2×), 135.1, 134.9, 133.4, 133.3, 130.8, 130.7, 129.4, 129.0, 128.7, 128.4, 127.4, 127.3, 127.2, 123.1, 121.7, 121.3, 120.7, 119.4, 113.9, 78.0 (2×), 77.6, 76.7, 76.4, 31.2 (2×), 30.8, 30.5, 23.5 (2×), 23.3, 23.0, 22.8, 10.6 (2×), 10.3, 9.9 ppm. IR (KBr) ν 2961.3, 2929.7, 2875.6, 1724.0, 1465.2, 1220.0 cm−1. HRMS (ESI+) calcd for C52H55F3N2O7 877.40341 [M + H]+, 899.38536 [M + Na]+, 915.35929 [M + K]+, found m/z 877.40344 [M + H]+ (20%), 899.38502 [M + Na]+ (100%), 915.35644 [M + K]+ (25%).X-ray crystallography
623 independent reflections (θmax = 68.5°), 1456 parameters and 491 restrains. The disordered propoxy groups and solvent were refined with restrained geometry and thermal parameters. The sum occupancy of disordered positions was restrained to 1 for each group. The hydrogen atoms attached to carbon atoms were placed in calculated positions. The hydrogen atoms attached to oxygen and nitrogen atoms were found in difference electron density maps. In both cases were the hydrogen atoms refined with riding constrains after initial refinement of geometry. The MCE program20 was used for visualization of residual electron density maps. The structure was deposited into Cambridge Structural Database under number CCDC 1918371.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Czech Science Foundation (Grant 18-08680J). Financial support from specific university research (21-SVV/2019) is also acknowledged.Notes and references
- For selected books on calixarenes and their applications, see: (a) P. Neri, J. L. Sessler and M. X. Wang, Calixarenes and Beyond, Springer, Cham, Switzerland, 2016 CrossRef; (b) C. D. Gutsche, Calixarenes: An Introduction, Royal Society of Chemistry, Cambridge, U.K., 2nd edn, 2008 Search PubMed; (c) J. Vicens, J. Harrowfield and L. Backlouti, Calixarenes in the Nanoworld, Springer, Dordrecht, The Netherlands, 2007 CrossRef; (d) Z. Asfari, V. Böhmer, J. Harrowfield and J. Vicens, Calixarenes 2001, Kluwer, Dordrecht, The Netherlands, 2001 Search PubMed; (e) L. Mandolini and R. Ungaro, Calixarenes in Action, Imperial College Press, London, 2000 CrossRef.
- E. Kelderman, W. Verboom, J. F. J. Engbersen, D. N. Reinhoudt, G. J. T. Heesink, N. F. van Hulst, L. Derhaeg and A. Persoons, Angew. Chem., Int. Ed. Engl., 1992, 31, 1075–1077 CrossRef.
- S. Shinkai, K. Araki, T. Tsubaki, T. Arimura and O. Manabe, J. Chem. Soc., Perkin Trans. 1, 1987, 2297–2299 RSC.
- C. D. Gutsche and P. F. Pagoria, J. Org. Chem., 1985, 50, 5795–5802 CrossRef CAS.
- P. Slavik, M. Dudic, K. Flidrova, J. Sykora, I. Cisarova, S. Böhm and P. Lhotak, Org. Lett., 2012, 14, 3628–3631 CrossRef CAS.
- K. Flídrová, P. Slavík, V. Eigner, H. Dvořáková and P. Lhoták, Chem. Commun., 2013, 49, 6749–6751 RSC.
- P. Slavík, V. Eigner and P. Lhoták, Org. Lett., 2015, 17, 2788–2791 CrossRef.
- M. Tlustý, P. Slavík, M. Kohout, V. Eigner and P. Lhoták, Org. Lett., 2017, 19, 2933–2936 CrossRef.
- K. Flídrová, S. Böhm, H. Dvořáková, V. Eigner and P. Lhoták, Org. Lett., 2014, 16, 138–141 CrossRef.
- P. Slavík, V. Eigner and P. Lhoták, CrystEngComm, 2016, 18, 4964–4970 RSC.
- L. Vrzal, K. Flídrová, T. Tobrman, H. Dvořáková and P. Lhoták, Chem. Commun., 2014, 50, 7590–7592 RSC.
- M. Movassaghi and M. D. Hill, Org. Lett., 2008, 10, 3485–3488 CrossRef CAS.
- For selected reviews on the inherent chirality of calixarenes, see: (a) G. E. Arnott, Chem.–Eur. J., 2018, 24, 1744–1754 CrossRef CAS; (b) P. Neri, J. L. Sessler and M. X. Wang, Calixarenes and Beyond, Springer, Cham, Switzerland, 2016 CrossRef; (c) S. Y. Li, Y. W. Xu, J. M. Liu and C. Y. Su, Int. J. Mol. Sci., 2011, 12, 429–455 CrossRef CAS PubMed; (d) A. Szumna, Chem. Soc. Rev., 2010, 39, 4274–4285 RSC.
- The binding constants were calculated using the Bindfit application freely available at: http://supramolecular.org.
- For measuring of association constants and determining the stoichiometry of the complexes from titration experiments, see: (a) D. B. Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC; (b) F. Ulatowski, K. Dabrowa, T. Balakier and J. Jurczak, J. Org. Chem., 2016, 81, 1746–1756 CrossRef CAS; (c) P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435–436 Search PubMed.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS.
- M. Lusi, CrystEngComm, 2018, 20, 7042–7052 RSC.
- J. Rohlicek and M. Husak, J. Appl. Crystallogr., 2007, 40, 600–601 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full characterization of new compounds and the results of the complexation studies. CCDC 1918213, 1918371, and 1918372. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra05075b |
| This journal is © The Royal Society of Chemistry 2019 |