
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural versatility of the quasi-aromatic Möbius type zinc(II)-pseudohalide complexes – experimental and theoretical investigations†
Mariusz P. Mitoraj *a,
Farhad Akbari Afkhamib,
Ghodrat Mahmoudi*c,
Ali Akbar Khandarb,
Atash V. Gurbanovde,
Fedor I. Zubkovf,
Rory Watermang,
Maria G. Babashkinah,
Dariusz W. Szczepanika,
Himanshu S. Jenai and
Damir A. Safin*h
*a,
Farhad Akbari Afkhamib,
Ghodrat Mahmoudi*c,
Ali Akbar Khandarb,
Atash V. Gurbanovde,
Fedor I. Zubkovf,
Rory Watermang,
Maria G. Babashkinah,
Dariusz W. Szczepanika,
Himanshu S. Jenai and
Damir A. Safin*h
aDepartment of Theoretical Chemistry, Faculty of Chemistry, Jagiellonian University, Gronostajowa 2, 30-387 Cracow, Poland. E-mail: mitoraj@chemia.uj.edu.pl
bDepartment of Inorganic Chemistry, Faculty of Chemistry, University of Tabriz, 51666-16471, Tabriz, Iran
cDepartment of Chemistry, Faculty of Science, University of Maragheh, P.O. Box 55181-83111, Maragheh, Iran. E-mail: mahmoudi_ghodrat@yahoo.co.uk
dDepartment of Chemistry, Baku State University, Z. Xalilov Str. 23, AZ1148, Baku, Azerbaijan
eCentro de Química Estrutural, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001, Lisboa, Portugal
fOrganic Chemistry Department, Faculty of Science, Peoples' Friendship University of Russia (RUDN University), 6 Miklukho-Maklaya St., Moscow, 117198, Russian Federation
gDepartment of Chemistry, University of Vermont, 82 University Place, Burlington, VT 05405, USA
hInstitute of Chemistry, University of Tyumen, Perekopskaya Str. 15a, 625003 Tyumen, Russian Federation. E-mail: damir.a.safin@gmail.com; d.a.safin@utmn.ru
iCOMOC, Department of Chemistry, Ghent University, Krijgslaan 281-S3B, Ghent-9000, Belgium
First published on 31st July 2019
Abstract
In this contribution we report for the first time fabrication, isolation, structural and theoretical characterization of the quasi-aromatic Möbius complexes [Zn(NCS)2LI] (1), [Zn2(μ1,1-N3)2(LI)2][ZnCl3(MeOH)]2·6MeOH (2) and [Zn(NCS)LII]2[Zn(NCS)4]·MeOH (3), constructed from 1,2-diphenyl-1,2-bis((phenyl(pyridin-2-yl)methylene)hydrazono)ethane (LI) or benzilbis(acetylpyridin-2-yl)methylidenehydrazone (LII), respectively, and ZnCl2 mixed with NH4NCS or NaN3. Structures 1–3 are dictated by both the bulkiness of the organic ligand and the nature of the inorganic counter ion. As evidenced from single crystal X-ray diffraction data species 1 has a neutral discrete heteroleptic mononuclear structure, whereas, complexes 2 and 3 exhibit a salt-like structure. Each structure contains a ZnII atom chelated by one tetradentate twisted ligand LI creating the unusual Möbius type topology. Theoretical investigations based on the EDDB method allowed us to determine that it constitutes the quasi-aromatic Möbius motif where a metal only induces the π-delocalization solely within the ligand part: 2.44|e| in 3, 3.14|e| in 2 and 3.44|e| in 1. It is found, that the degree of quasi-aromatic π-delocalization in the case of zinc species is significantly weaker (by ∼50%) than the corresponding estimations for cadmium systems – it is associated with the Zn–N bonds being more polar than the related Cd–N connections. The ETS-NOCV showed, that the monomers in 1 are bonded primarily through London dispersion forces, whereas long-range electrostatic stabilization is crucial in 2 and 3. A number of non-covalent interactions are additionally identified in the lattices of 1–3.
Introduction
Helical molecules are highly favoured by nature.1 Such molecules are of great importance, which is also supported by the structure of deoxyribonucleic acid first discovered in 1953.2On the other hand, zinc(II) (ZnII) ions are found in all six main classes of metalloenzymes and are essential for living organisms.3,4 Moreover, the dinuclear ZnII complex fabricated from doubly deprotonated octaethyl formylbiliverdine is the first established helical doublestranded structure, which was reported in 1976.5 Following this discovery, strategies towards helical structure as well as their self-assembly have been the focus of researchers.6,7 Obviously, the most powerful strategy towards metal-based helical structures is the smart predesign of parent ligands. The other strategy, which, however, is less predictable and thus less efficient, is the choice of a metal-containing precursor. The latter is much less investigated.5–7
Some efforts have been focused on the design and preparation of helical metal complexes by applying chelating ligands with suitable donor sites.7–16 The point is that the ligand should produce a helical topology upon binding to the metal ions. In some cases, the coordination features of the cations dictate the wrapping of non-helical chelating ligands around them in such a manner that they can be twisted and eventually form helical complexes.7,9–11,13,15 However, synthesis of organic ligands with a helical topology is more difficult than metallo-organic compounds and there are only a few reported synthetic helical organic molecules.8,12,14,16,17 Researchers mainly focused on the design and construction of metal complexes with synthetic helical chelating ligands.18–20 Recently, we have also directed our attention to Schiff bases comprising two pyridyl-imine functions obtained from benzyldihydrazone.21–27 These ligands were found to be efficient for helical structures upon coordination to metal centers. Particularly our comprehensive efforts were directed to various CdII salts as complexing agents.23–27 Moreover, we were able to demonstrate for the first time that the helical motif in the obtained complexes together with the chelate metalloring correspond to a quasi-aromatic Möbius object.24,27
Herein, we report Zn(NCS)2- and Zn(N3)2-derived structures with 1,2-diphenyl-1,2-bis((phenyl(pyridin-2-yl)methylene)hydrazono)ethane (LI) and benzilbis(acetylpyridin-2-yl)methylidenehydrazone (LII). Using thiocyanate (NCS−) and azide (N3−) counterions is intriguing and of great interest since both anions are known to be ambidentate ligands, which can bind metal centers in different coordination modes.28,29 As a result were able to isolate the unique quasi-aromatic Möbius type zinc complexes [Zn(NCS)2LI] (1), [Zn2(μ1,1-N3)2(LI)2][ZnCl3(MeOH)]2·6MeOH (2) and [Zn(NCS)LII]2[Zn(NCS)4]·MeOH (3), respectively. Notably, our numerous attempts to isolate crystals of the reaction product of Zn(N3)2 and LI failed regardless using a great number of ZnII and N3− sources. DFT experiments were additionally performed to estimate the stability and aromaticity of the obtained species.
Results and discussion
Interaction of ZnCl2 mixed with NH4NCS or NaN3 with LI or LII in MeOH at 60 °C has allowed to isolate complexes 1–3 (Scheme 1 and ESI†). The elemental analysis data supports their compositions. Notably, the same one-pot reaction of LI or LII with Cd(NO3)2 in the presence of NH4NCS produced a dinuclear structure [Cd2(μ1,3-NCS)2(NCS)2(LIII)2]·4MeOH (4), where LIII is formed upon hydrolysis of one of the 2-PyC(Ph) functions of LI,27 and neutral mononuclear complex [Cd(NCS)2(LII)(MeOH)] (5),24 respectively.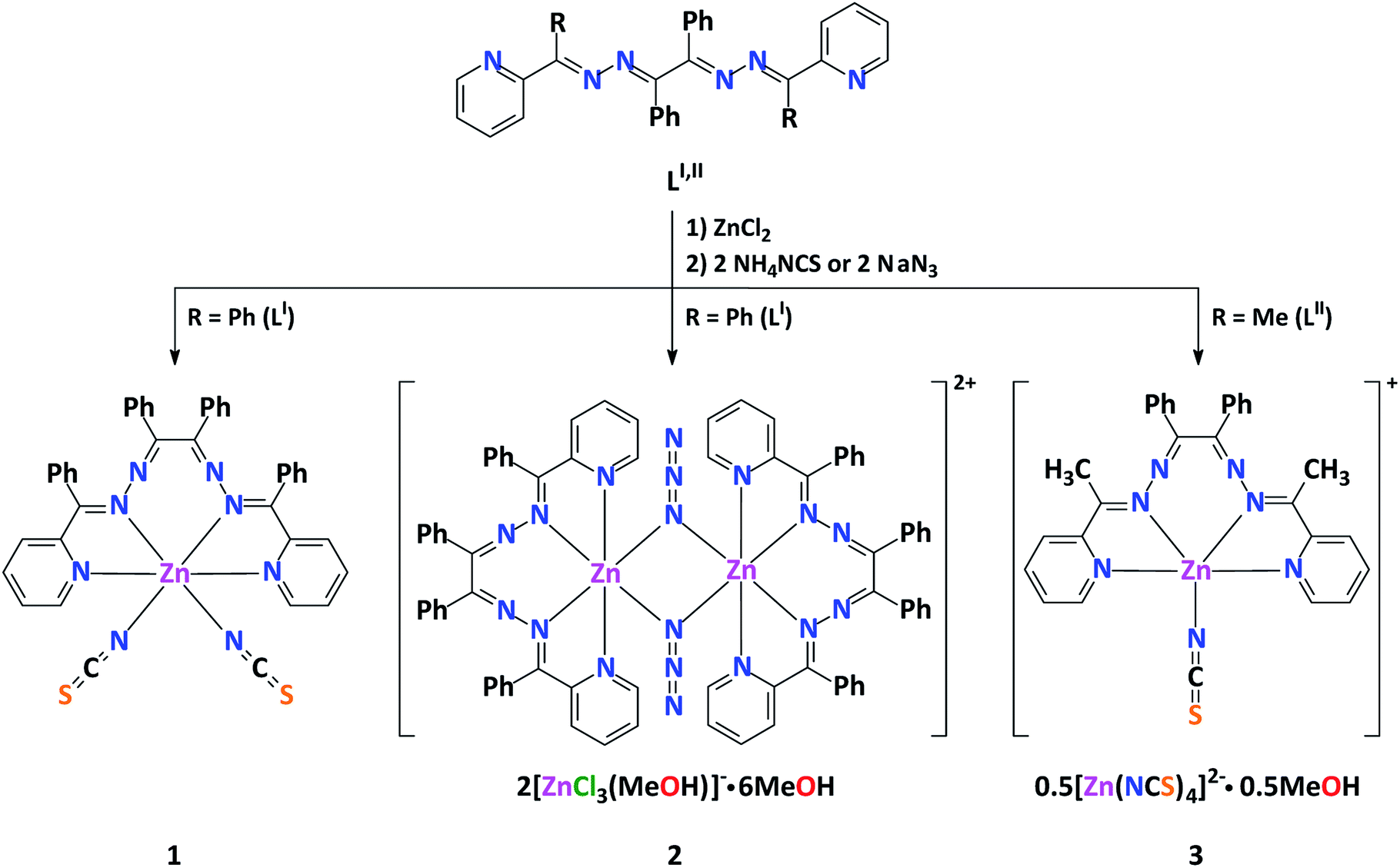 | ||
| Scheme 1 Synthesis of complexes 1–3. | ||
The FTIR spectrum of 1 contains a characteristic intense band at 2073 cm−1 attributed to the CN stretching of NCS−. The same stretching mode in the IR spectrum of 3 is shown as two clearly defined bands at 2066 and 2112 cm−1, corresponding to two different types of the NCS− ligands (Scheme 1). These bands are in the typical region for the N-linked terminal NCS− ions.30 The N3− anions in the FTIR spectrum of 2 are shown as an intense band at 2054 cm−1 arising from the νasym stretching vibration, as well as a band at 1441 cm−1 corresponding to the νasym stretching vibration. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration is at a lower energy by 40 cm−1 for complexes 1 and 2 compared to the free ligand LI, whilst a similar 20 cm−1 difference is observed in this vibration between compound 3 and LII.22 This firmly confirms the participation of the azomethine nitrogen atoms in chelate formation. The FTIR spectra of 2 and 3 further contain a broad band for the methanol at 3353 to 3443 cm−1, respectively.
N stretching vibration is at a lower energy by 40 cm−1 for complexes 1 and 2 compared to the free ligand LI, whilst a similar 20 cm−1 difference is observed in this vibration between compound 3 and LII.22 This firmly confirms the participation of the azomethine nitrogen atoms in chelate formation. The FTIR spectra of 2 and 3 further contain a broad band for the methanol at 3353 to 3443 cm−1, respectively.
Complex 1 crystallizes in the monoclinic space group P21/n, while complexes 2 and 3 each crystallize in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . It is worthy to note, that 1 is isostructural to its MnII and CoII analogues.22,31
. It is worthy to note, that 1 is isostructural to its MnII and CoII analogues.22,31
Complex 1 has a neutral discrete heteroleptic mononuclear structure, where the ZnII metal center is coordinated by one ligand LI via its two pyridyl-imine chelate functions as well as two N-bound NCS− anions giving rise to the ZnN6 chromophore with a distorted trigonal-prismatic coordination environment around the cation (Fig. 1, Table S1 in the ESI†), which has been proven by the SHAPE 2.1 software.32,33
 | ||
| Fig. 1 Crystal structure of 1 (H-hydrogen atoms are omitted for clarity). Color code: C = gold, N = blue, S = orange, Zn = magenta. | ||
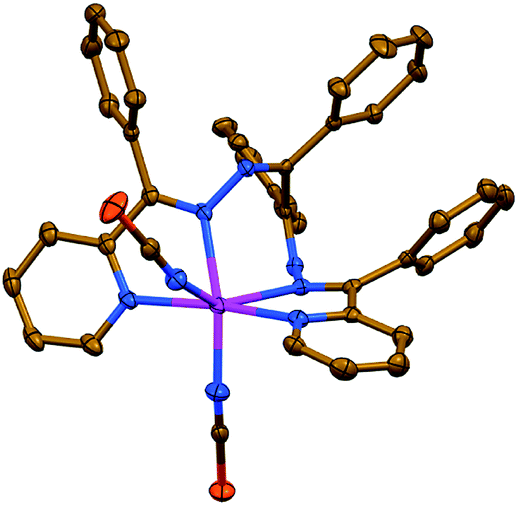
Complexes 2 and 3 each exhibit a salt-like structure (Scheme 1), as opposed to our previously studied CdII based counterparts.23–27 In 2, the cationic part exhibits a doubly charged centrosymmetric dinuclear structure, were two ZnII centers are interlinked via two μ1,1-N3− anions and the coordination domain of each metal is filled by the tetracoordinated ligand LI (Fig. 2). Here a coordination geometry is best described as a distorted octahedron (Table S1 in the ESI†).32,33 The anionic part represents a discrete mononuclear structure of the composition [ZnCl3(MeOH)]− with a tetracoordinate Cl3O environment around the metal atom (Fig. 2). As evidenced from the so-called distortion index τ4 = 0.9513,34 the coordination core of [ZnCl3(MeOH)]− is almost a perfect tetrahedron. This is also supported by the SHAPE 2.1 software (Table S1 in the ESI†).32,35
 | ||
| Fig. 2 (top) Crystal structure of the cationic part [Zn2(μ1,1-N3)2(LI)2]2+ of 2. Hydrogen atoms are omitted for clarity. Color code: C = gold, N = blue, Zn = magenta. (bottom) Crystal structure of the hydrogen bonded synthon of motif R88(20) of the ([ZnCl3(MeOH)]2)2−·6MeOH composition, constructed from the [ZnCl3(MeOH)]− anionic part and lattice MeOH molecules of 2. Color code: H = black, C = gold, Cl = green, O = red, Zn = magenta. | ||
The anionic part of 2 via one of its chlorine atoms and the methanol OH hydrogen atom is engaged in intermolecular hydrogen bonds with the lattice MeOH molecules yielding a synthon of motif R88(20) of the ([ZnCl3(MeOH)]2)2−·6MeOH composition (Fig. 2, Table S2 in the ESI†).
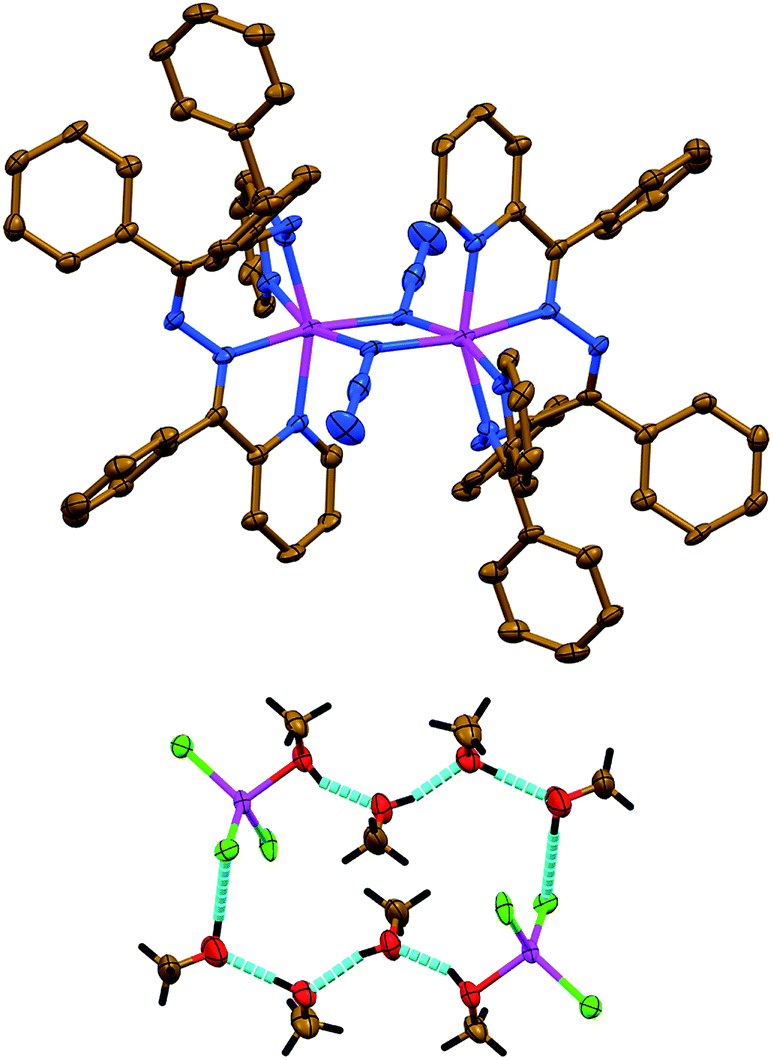
The salt like structure of 3 is built from two [Zn(NCS)LII]+ cations, where the ZnII metal center is, similar to 1 and 2, chelated by two pyridyl-imine fragments of one parent ligand LII, and further bound by one N-linked NCS− anion, exhibiting a pentacoordinated geometry (Fig. 3). The distortion index τ534 is 0.693 and 0.653 for two [Zn(NCS)LII]+ cations. These values are best described as being about 31% and 35%, respectively, along the pathway of distortion from the ideal trigonal bipyramidal structure towards square pyramidal structure. The trigonal bipyramidal coordination environment is also evidenced from the SHAPE 2.1 software (Table S1 in the ESI†).32,36
 | ||
| Fig. 3 Crystal structure of 3 (hydrogen atoms and MeOH molecules are omitted for clarity). Color code: C = gold, N = blue, S = orange, Zn = magenta. | ||
The anionic part of 3 exhibits a doubly charged [Zn(NCS)4]2− species, where four NCS− anions are bound via their N-atoms yielding a tetracoordinated coordination geometry around the metal atom (Fig. 3). The τ4 value of 0.9594 indicates almost a perfect tetrahedron, which has also been supported by the SHAPE 2.1 software (Table S1 in the ESI†).32,35 Notably, the CS fragment of one of the NCS− anions is disordered over three positions with a ratio of 35%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :35%:30% (Fig. 3).
:35%:30% (Fig. 3).
The Zn–N bonds in 1–3, formed by four nitrogen atoms of the corresponding organic ligands, are in the range from 2.073(3) Å to 2.2960(15) Å, maintaining that Zn–NPy < Zn–Nimine (Table 1). It is worthy to note, that the Cd(L)–NCS bonds (1.970(3)–2.0289(18) Å) in 1 and 3 are remarkably shorter than the Zn–NPy and Zn–Nimine bonds, while the Zn(L)–N3 bonds in 2 are similar to those within the corresponding organic ligand. This is obviously explained by the terminal coordination mode of the NCS− anions in contrast to the μ1,1-coordination mode of N3−. The Zn–NCS bonds within the [Zn(NCS)4]2− anion in 3 are 1.953(6)–1.983(4) Å. All the NCS− and N3− ligands are almost linear (Table 1). The Zn⋯Zn separation within the dinuclear molecule of 2 is 3.3729(15) Å.
| Complex 1 | Complex 2 | Complex 3 | |
|---|---|---|---|
| a Torsion angles must be compared by their magnitudes. | |||
| Bond lengths | |||
| Zn–NPy | 2.2165(16), 2.2755(17) | 2.075(5), 2.168(5) | 2.073(3), 2.079(4), 2.100(3) |
| Zn–Nimine | 2.2692(16), 2.2960(15) | 2.131(5), 2.265(5) | 2.107(3), 2.118(3), 2.119(3), 2.123(4) |
| Zn(L)–NCS | 2.0046(18), 2.0289(18) | — | 1.970(3), 1.971(3) |
| Zn(L)–N3 | — | 2.154(5), 2.225(4) | — |
| Zn–NCS | — | — | 1.953(6), 1.955(5), 1.971(5), 1.983(4) |
| Zn(L)⋯Zn(L) | — | 3.3729(15) (intramolecular) | — |
|
|||
| Bond angles | |||
| NPy–Zn–NPy | 170.55(6) | 103.76(17) | 104.50(14), 104.63(15) |
| NPy–Zn–Nimine | 69.96(6), 70.63(6), 116.80(6), 118.40(6) | 75.27(17), 75.95(17), 102.79(17), 156.78(18) | 76.44(12), 76.71(13), 78.20(14), 78.36(14), 119.93(12), 121.35(14), 163.40(13), 164.34(13) |
| Nimine–Zn–Nimine | 76.13(6) | 81.62(17) | 86.81(12), 87.76(12) |
| NPy–Zn–NCS | 86.22(7), 87.06(7), 87.39(6), 89.72(7) | — | 97.89(14), 98.68(13), 122.76(15), 124.22(15) |
| Nimine–Zn–NCS | 88.08(6), 93.14(7), 142.43(6), 144.78(7) | — | 94.12(13), 94.20(13), 114.07(15), 114.42(16) |
| NPy–Zn–N3 | — | 89.25(17), 92.07(17), 98.01(18), 161.65(17) | — |
| Nimine–Zn–N3 | — | 89.89(17), 93.84(17), 105.15(17), 167.47(17) | — |
| NCS–Zn(L)–NCS | 117.54(7) | — | — |
| NCS–Zn–NCS | — | — | 105.0(2), 105.3(2), 110.0(2), 112.00(17), 112.0(2), 112.73(17) |
| N3–Zn–N3 | — | 79.26(17) | — |
| Zn(L)–N–C(S) | 158.37(16), 156.48(18) | — | 165.7(4), 166.8(4) |
| Zn–N–C(S) | — | — | 149.5(11), 150.9(15), 158(3), 163.5(5), 169.6(4), 174.5(4) |
| Zn–N–N(N) | — | 121.8(4), 123.4(4) | — |
| N–C–S | 178.62(19), 178.9(2) | — | 178.6(4), 179.2(4) |
| N–N–N | — | 178.8(7) | — |
| Zn–N–Zn | — | 100.7(2) | — |
|
|||
| Torsion anglesa | |||
| N–C(Ph)–C(Ph)–N | −67.4(3) | −64.7(9) | 66.1(6), 67.6(6) |
| C(Ph)–N–N–C(Ph) | −87.0(2), −114.85(19) | −96.7(7), −143.5(6) | — |
| C(Me)–N–N–C(Ph) | — | — | 138.5(4), 140.0(4), 152.4(4), 152.9(4) |
| Py⋯Py | 64.19(10) | 51.1(3) | 56.6(2), 58.0(2) |
| Zn–N–N–C(Ph) | 81.04(18), 86.13(17) | 49.8(7), 81.7(5) | −47.0(5), −48.3(5), −71.2(3), −72.4(4) |
Organic ligands in the structures of 1–3 each produce a twisted geometry of different extent. As a result of this conformation, the N–C(Ph)–C(Ph)–N fragments adopt a torsion angle of about 65° (Table 1), which is significantly lower than in the corresponding CdII analogues.23–27 This is also reflected in the Zn–N–N–C(Ph), C(Ph)–N–N–C(Ph) in 1 and 2, and C(Me)–N–N–C(Ph) in 3 torsion angles. Particularly, a simultaneous influence of a pentacoordination mode of the metal center together with the presence of less bulkier Me substituents in complex 3 leads to the C(Me)–N–N–C(Ph) torsion angles of close values (∼138–153°), while a hexacoordination mode of ZnII and the presence of the organic ligand, which is highly enriched by four phenyl fragments, induces significantly different C(Ph)–N–N–C(Ph) torsion angles within single species (Table 1). The Zn–N–N–C(Ph) torsion angles are very similar in 1, while the same angles in 2 and 3 are significantly different (Table 1). The torsion angle between two pyridyl rings ranges from about 51° to 64°, increasing from 2 through 3 to 1 (Table 1).
Due to a twisted helical topology of organic ligands in the mononuclear structures of 1 and 3, enantiomers of the coordination species can be expected. Indeed, both structures exhibit molecules with Δ and Λ helicity. The overall structure of 1 and 3 is a racemic mixture. The molecule of 2, although also containing organic ligands with a twisted helical topology, is constructed from two chiral centres, resulting in the formation of the achiral meso-form.
The crystal packing of 1–3 is described by a network of face-to-face π⋯π stacking between the aromatic rings (Table S3 in the ESI†). The structures of 1 and 2 are also dictated by C–H⋯π interactions (Table S4 in the ESI†).
For more detailed analyses of non-covalent interactions in 1–3 the charge and energy decomposition scheme ETS-NOCV37 is applied as available in the ADF program.38 We have applied BLYP-D3/TZP since they provide reliable results for non-covalent interactions.39 The X-ray models are considered.
We have determined, that the neutral monomers of [Zn(NCS)2LI] in 1 are efficiently bonded to each other with the interaction energy ΔEint = −23.78 kcal mol−1 (Fig. 4). The main gluing force (55% of the overall stabilization) is the dispersion term (ΔEdisp = −23.20 kcal mol−1) due to the presence of C–H⋯S and C–H⋯π contacts (Fig. 4). Such close contacts enforce additionally less important electrostatic (28.5%, ΔEelstat = −12.01 kcal mol−1) and charge delocalisation (16.5%, ΔEorb = −6.95 kcal mol−1) constituents (Fig. 4). The prevalence of the ΔEdisp term is consistent with recent findings, which rediscover the importance of London dispersion forces in small and sizeable species.24,27,40–56
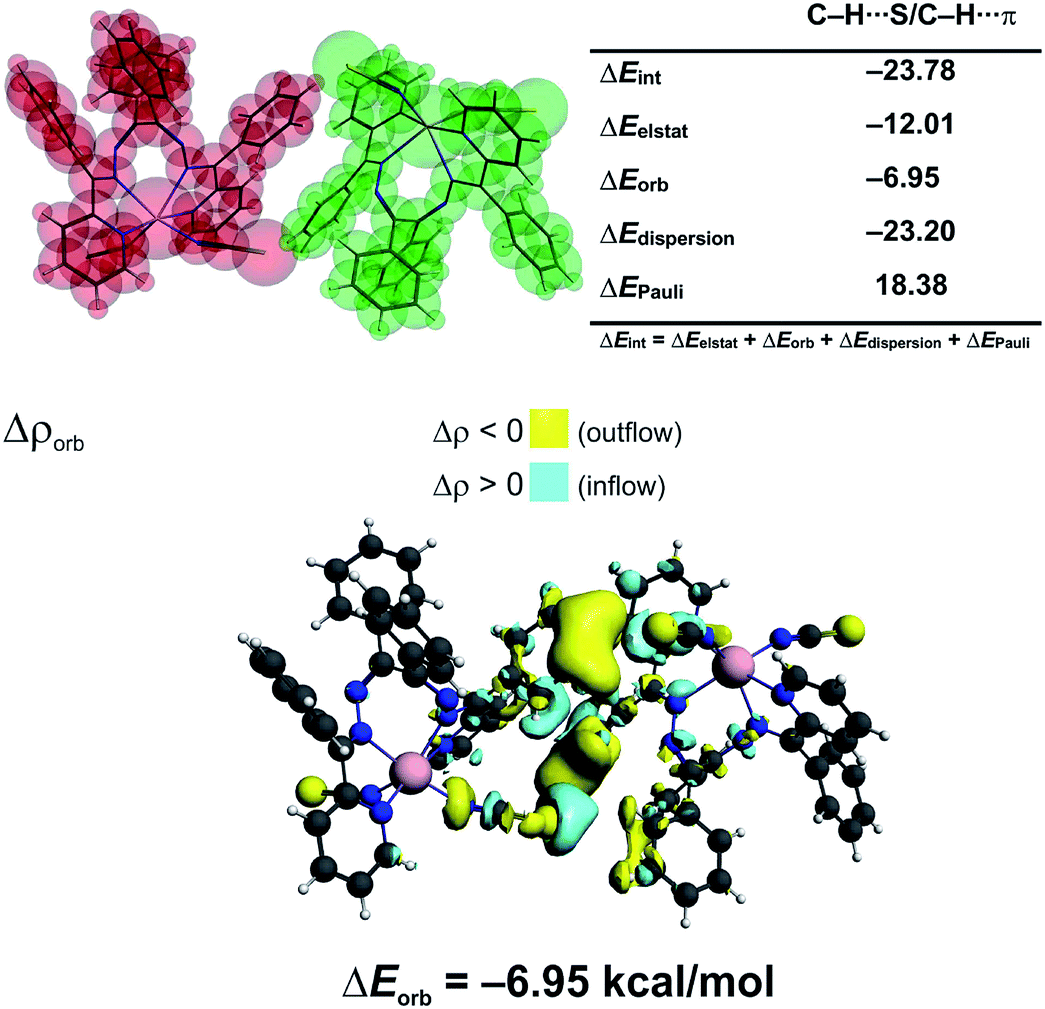 | ||
| Fig. 4 (top) ETS-NOCV outcomes scrutinizing the nature of bonding between the [Zn(NCS)2LI] monomers in 1. (bottom) The overall change in electron density Δρorb with the corresponding energy ΔEorb. | ||
In 3 the [Zn(NCS)4]2− anion sticks very strongly (ΔEint = −193.21 kcal mol−1) to two neighboring stacked [Zn(NCS)LII]+ units primarily through electrostatic forces (75.3% of the overall stabilization) (Fig. 5). Quite notable (14.8%) is the charge delocalization term ΔEorb = −31.10 kcal mol−1 mostly due to C–H⋯π contacts followed by the least important (9.90%) dispersion term ΔEdisp = −20.62 kcal mol−1 (Fig. 5). It is interesting to emphasize, that such ionic bonds are of crucial importance for the overall stability of 3 since the pure π⋯π stacking between the [Zn(NCS)LII]+ units, though containing a significant portion of dispersion stabilization (ΔEdisp = −31.76 kcal mol−1), is found to be repulsive with ΔEint = 7.86 kcal mol−1 caused predominantly by the unfavourable electrostatic constituent ΔEelstat = 29.1 kcal mol−1 (Fig. 6). In 2 the electrostatically dominated stabilizing interactions occur between [ZnCl3(MeOH)]− and [Zn2(μ1,1-N3)2(LI)2]2+ (Fig. S1 in the ESI†). Additionally, [ZnCl3(MeOH)]− forms primarily O–H⋯O as well as a series of ancillary C–H⋯Cl hydrogen bonds with the neighbouring methanol species. Such cooperative interactions, leading to ΔEint = −13.17 kcal mol−1, are found to be determined mostly by the electrostatic factor (50%) followed by the charge delocalization (27%) and dispersion (23%) constituents (Fig. S2 in the ESI†). The synthon [Zn2(μ1,1-N3)2(LI)2]2+ was found to be stable due to electrostatically dominated (ΔEelstat = −63.33 kcal mol−1) dative-covalent Zn–N connections with ΔEint = −31.63 kcal mol−1 constituted additionally from the significant portion of London dispersion forces (ΔEdisp = −37.25 kcal mol−1) (Fig. S3 in the ESI†).
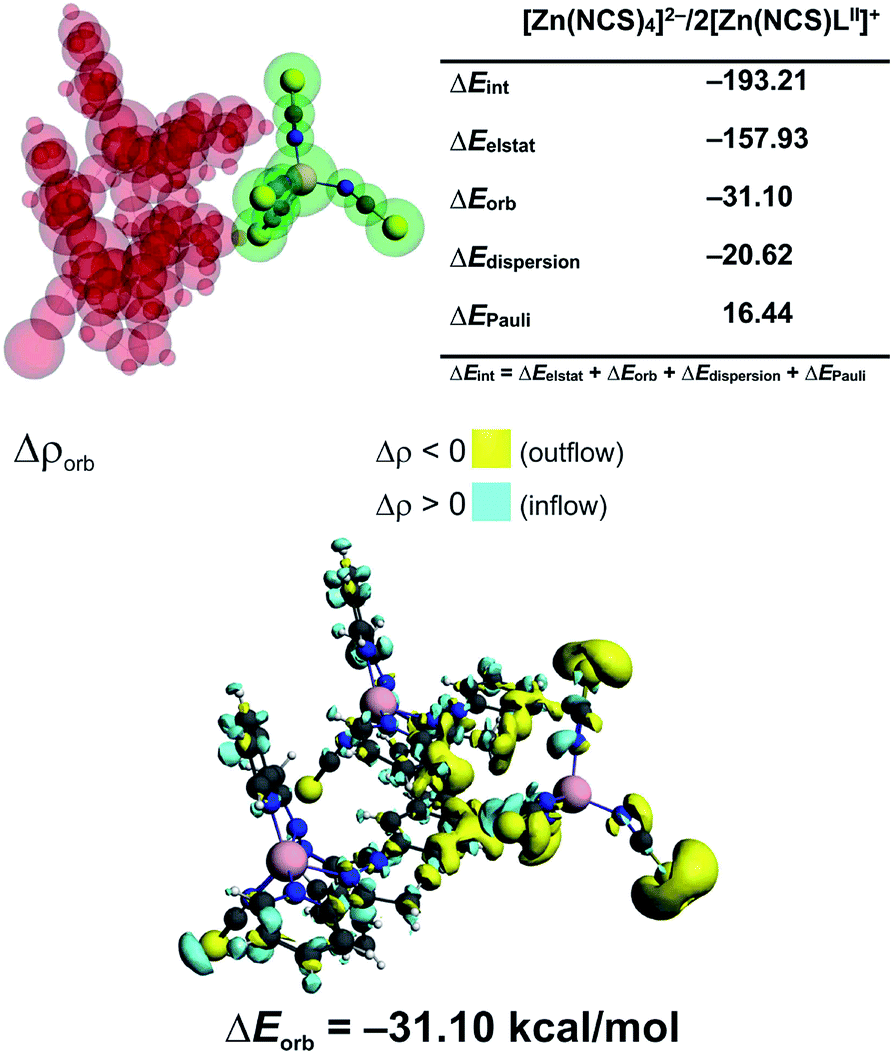 | ||
| Fig. 5 (top) ETS-NOCV outcomes scrutinizing the nature of ionic interaction between [Zn(NCS)4]2− and two [Zn(NCS)LII]+ in 3. (bottom) The overall change in electron density Δρorb with the corresponding energy ΔEorb. | ||
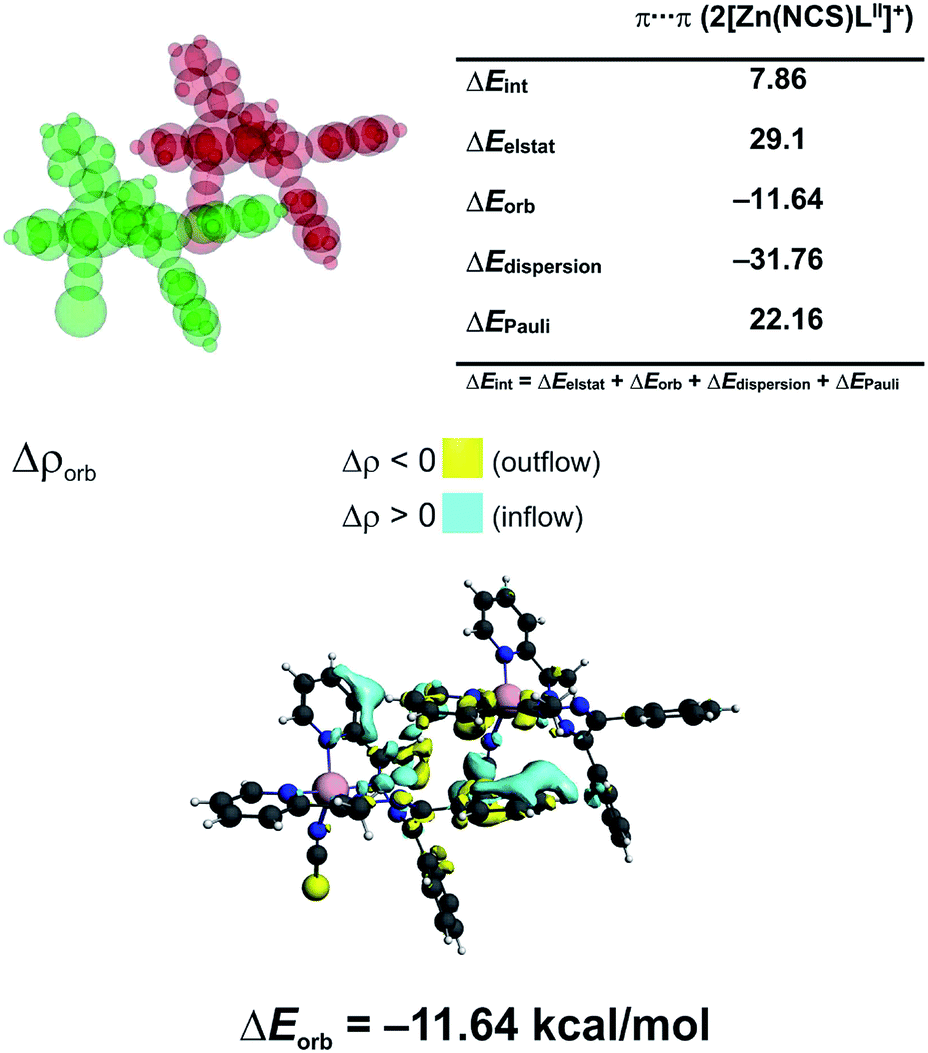 | ||
| Fig. 6 (top) ETS-NOCV outcomes scrutinizing the nature of ionic interaction between two π-stacked [Zn(NCS)LII]+ units in 3. (bottom) The overall change in electron density Δρorb with the corresponding energy ΔEorb. | ||
In order to evaluate aromaticity in 1–3, we have applied the electron density of delocalized bonds (EDDB) method, which has been proposed to visualize and quantify aromaticity and chemical resonance in a wide range of chemical species.57–60
Moreover, it has recently been shown that, in the case of organometallics, the EDDB method provides very useful data on the role of the transition metal d-orbitals in electron delocalization,24,27,50,61 which is inaccessible by means of such popular and commonly used aromaticity descriptors as the nucleus-independent chemical shift (NICS)62 or the anisotropy of the induced current density (ACID).63
The global EDDB isocontours and the corresponding electron populations of 1–3 are collected in Fig. S4 in the ESI.† Here, we focus our attention on the characteristic seven-membered quasi-aromatic motif (7-MR), encompassing the twisted 1,1′-(1,2-ethenediyl)bis-diazene (BDA) fragment and the metal atom (abbreviated as BDA–Zn). The BDA-based complexes with cadmium have recently been demonstrated to exhibit a unique type of transition-metal induced Möbius-like aromaticity in which the metal d-orbitals themselves do not contribute to the π-conjugation occurring at BDA.24,27 Since the quantitative study of aromaticity/electron delocalization in large systems is very difficult in practice, we have decided to consider the simplified BDA–Zn models adopting the exact fragments geometries from crystals of 1–3 (Fig. 7). The calculated total EDDB contours and electron populations have been dissected (according to the orbital symmetry) to get the strict π-contributions to quasi-aromaticity; natural atomic charges on the metal and the two closest nitrogen atoms have been added together with the average dihedral angles and the calculated electric dipole moments (EDM). It was found, that the number of π-electrons delocalized in the quasi-aromatic rings, particularly in 1 and 2 (on average ∼3.3|e|), resembles pretty much the values found for the previously studied BDA–Cd complexes, despite different configurations of the phenyl units and applying other ligands.24,27 The most twisted 7-MR in 1 (containing the most bulky substituents) is at the same time the most stabilized by quasi-aromaticity (3.44|e|, i.e. ∼0.6|e| per a quasi-ring member, which is comparable to the corresponding value for pyrrole60). Interestingly, it is found for the first time, that the systematic increase of the Zn–N bond polarization when going from 1 to 3 reveals a strict correlation between EDM (i.e. indirectly the topology and the metal to BDA charge transfer) and π-electron delocalization: R = −0.986. In other words, the more twisted is the 7-MR, the more quasi-aromatic character is observed (Fig. 7). It demonstrates the two-folded role of bulky substituents: they are not only dispersion donors,40–56 but they also lead to amplification of the 7-MR twist (and enhanced quasi-aromaticity). Previously only the former feature has been recognized.23–27 Interestingly, the optimized BDA–Zn structure (without steric effects from the Ph units) has significantly reduced quasi-aromaticity compared to 1 and 2 (2.43|e|, i.e. ∼0.4|e| per a quasi-ring member, which is comparable to the corresponding value for furan),60 but at the same time, it is almost twice less aromatically-stabilized than its optimized BDA–Cd analogue (4.74|e|, i.e. ∼0.8|e| per a quasi-ring member, which is exactly between the corresponding values for pyrrole and benzene).60 Since both equilibrium structures have comparable average dihedral angles N–N–M–N, it is clearly the larger metal–nitrogen bond polarization (the charges qZn = +1.15, qN = −0.9, EDM = 1.26 D in BDA–Zn compared to qZn = +0.72, qN = −0.7, EDM = 0.68 D in BDA–Cd) that limits the π-electron delocalization in the 7-memberd quasi-aromatic unit (changes in the effectiveness of π-conjugation involving the 2pz orbitals of nitrogen atoms at close proximity of the metals are well marked in the EDDBπ(r) isocontours, Fig. 7). Such inter-relation between the nature of metal–ligand bonding and quasi-aromaticity of the ligand has not been known before.23–27
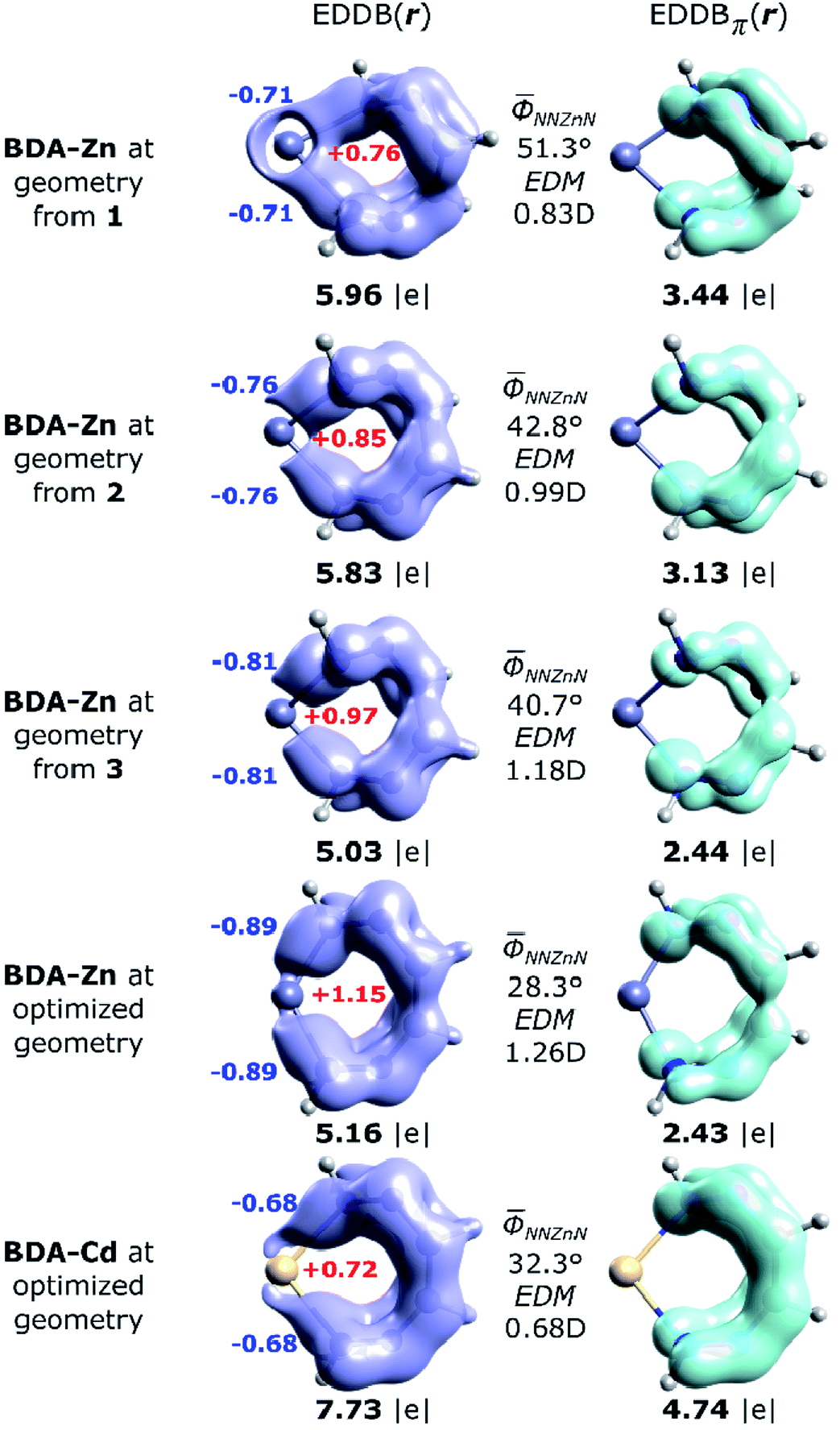 | ||
| Fig. 7 Imagining of the EDDB(r) and EDDBπ(r) functions with the corresponding electron populations (in |e|) for the isolated 7-MR model systems at geometries adopted from the corresponding crystals of 1–3, as well as the fully optimized units: BDA–Zn and BDA–Cd.27 The natural atomic charges (colored bold numbers), average dihedral angles and the calculated electric dipole moments (EDM) have been added for comparison. | ||
Conclusions
In summary, we successfully isolated and characterized the quasi-aromatic Möbius type zinc complexes [Zn(NCS)2LI] (1), [Zn2(μ1,1-N3)2(LI)2][ZnCl3(MeOH)]2·6MeOH (2) and [Zn(NCS)LII]2[Zn(NCS)4]·MeOH (3), fabricated from 1,2-diphenyl-1,2-bis((phenyl(pyridin-2 yl)methylene)hydrazono)ethane (LI) or benzilbis(acetylpyridin-2-yl)methylidenehydrazone (LII), respectively, and ZnCl2 mixed with NH4NCS or NaN3. The creation of 1–3 is dictated by both the bulkiness of the organic ligand and the nature of inorganic counter ion.Complex 1 has a neutral discrete heteroleptic mononuclear structure with the ZnII metal atom being chelated by one tetradentate ligand LI and two N-bound NCS− anions with the formation of a distorted trigonal-prismatic ZnN6 coordination core. The [Zn(NCS)2LI] monomers were found (due to the ETS-NOCV calculations) to be bonded to each other primarily through London dispersion forces exerted by the presence of bulky hydrophobic substituents. Contrary, complexes 2 and 3 exhibit a salt-like structure where the long-range electrostatic forces were found to be of prime importance additionally to more typical non-covalent interactions (O–H⋯O, C–H⋯Cl, C–H⋯S, C–H⋯π, π⋯π). In 2, the cation is a doubly charged centrosymmetric dinuclear structure with two ZnII atoms linked via two μ1,1-N3− anions. Each metal center is further linked by the tetracoordinate ligand LI. The anionic part has a discrete mononuclear composition [ZnCl3(MeOH)]−. Notably, the anionic part of 2 together with the lattice MeOH molecules produces a synthon of motif R88(20) stabilized mostly by O–H⋯O and C–H⋯Cl interactions. Complex 3 is composed from two [Zn(NCS)LII]+ cations with the ZnII atoms each being chelated by two pyridyl-imine fragments of LII and further bound by one N-linked NCS− anion. The anionic part of 3 is a doubly charged [Zn(NCS)4]2− species. Long range electrostatic forces between [Zn(NCS)4]2− and [Zn(NCS)LII]+ are responsible for the stability of 3 since pure π⋯π stacking between the [Zn(NCS)LII]+ units appeared to be repulsive. Finally, we have proven, by means of the EDDB57–61 study, that the seven-membered rings in 1–3 constitute a quasi-aromatic Möbius-type motif, though the absolute magnitude of such π-delocalization is notably weaker than in the corresponding cadmium-based analogs.24,27 Bulkiness of the ligands (L) are found not only to amplify London dispersion stabilization,24,27,40–56 but also influence the magnitude of quasi-π-delocalization (of Möbius-type) through modification of the polarity of Zn–L bonding.
Experimental
Materials
All chemicals and solvents were used from commercial sources without further purification. LI and LII were synthesized according to a literature method.20Physical measurements
FTIR spectra were recorded on a Bruker Tensor 27 FTIR spectrometer. Microanalyses were performed using a ElementarVario EL III analyzer.Synthesis
ZnCl2 (0.068 g, 0.5 mmol), NH4NCS (0.076 g, 1 mmol) or NaN3 (0.065 g, 1 mmol) and LI or LII (0.284 and 0.222 g, respectively; 0.5 mmol) were placed in the main arm of a branched tube. MeOH (15 mL) was carefully added to fill the arms. The tube was sealed and immersed in an oil bath at 60 °C while the branched arm was kept at ambient temperature. X-ray suitable crystals were formed during the next days in the cooler arm and were filtered off.(1) Colorless block-like crystals. Yield: 0.248 g (66%). Anal. calc. for C40H28N8S2Zn (750.22) (%): C 60.04, H 3.76 and N 14.94; found: C 60.29, H 3.83 and N 14.77.
(2) Yellow block-like crystals. Yield: 0.156 g (64%). Anal. calc. for C84H88Cl6N18O8Zn4 (1951.98) (%): C 51.69, H 4.54 and N 12.92; found: C 51.56, H 4.61 and N 12.81.
(3) Yellow prism-like crystals. Yield: 0.174 g (73%). Anal. calc. for C63H52N18OS6Zn3 (1465.74) (%): C 51.62, H 3.58 and N 17.20; found: C 51.76, H 3.48 and N 17.33.
ETS-NOCV charge and energy decomposition method
The Natural Orbitals for Chemical Valence (NOCV) ψi constitute the canonical representation for a differential density matrix ΔP (it is formed by subtracting the appropriate molecular fragments density matrices from a density matrix of a molecule under consideration) in which ΔP adopts a diagonal form. It gives rise to the corresponding eigenvalues vi and the related vectors ψi. NOCVs occur in pairs (ψ−k,ψk) related to |vk| and they decompose overall deformation density Δρ into bonding components with different symmetries (Δρk):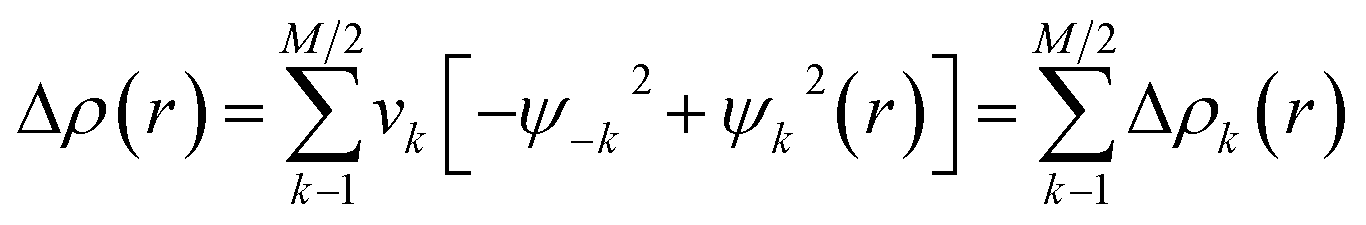
Usually, a few k allow to recover a major shape of Δρ. By combining NOCVs with ETS scheme in ETS-NOCV, one can obtain the related energetics, ΔEorb(k), in addition to qualitative picture emerging from Δρk. ETS originally divides the total bonding energy, between fragments, ΔEtotal, into four distinct components: ΔEtotal = ΔEelstat + ΔEPauli + ΔEorb + ΔEdispersion. The ΔEelstat is an energy of quasi-classical electrostatic interaction between fragments. The next term, ΔEPauli, is responsible for repulsive Pauli interaction between occupied orbitals on the two fragments. The third component, ΔEorb, is stabilizing and shows formation of a chemical bond (including polarizations). In the ETS-NOCV scheme ΔEorb is expressed in terms of the eigenvalues vk and diagonal Fock energy matrix elements FTSi,i (transformed into NOCV representation) as:
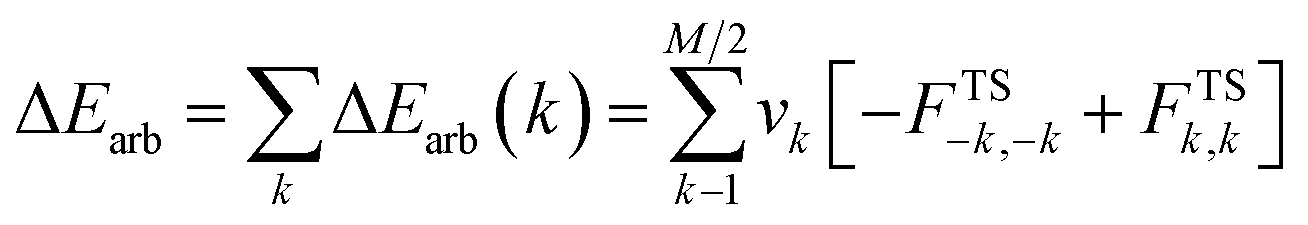
Finally, ΔEdispersion denotes the semiempirical Grimme dispersion correction (D3).
Single-crystal X-ray diffraction
The X-ray data were collected on a Bruker APEX-II CCD single crystal diffractometer using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). The collected frames were integrated with the Saint64 software using a narrow-frame algorithm. Data were corrected for absorption effects using the multi-scan method in SADABS.65 The space groups were assigned using XPREP of the Bruker ShelXTL66 package, solved with ShelXT66 and refined with ShelXL66 and the graphical interface ShelXle.67 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms attached to carbon were positioned geometrically and constrained to ride on their parent atoms. Figures were generated using the program Mercury.68, a = 11.836(4), b = 14.385(4), c = 14.807(4) Å, α = 68.045(3), β = 83.861(3), γ = 78.264(3)°, V = 2288.0(12) Å3, Z = 2, ρ = 1.417 g cm−3, μ(Mo-Kα) = 1.274 mm−1, reflections: 15639 collected, 5424 unique, Rint = 0.053, R1(all) = 0.0648, wR2(all) = 0.1386, S = 1.092., a = 13.3235(9), b = 15.6906(10), c = 18.6880(13) Å, α = 65.795(2), β = 71.872(2), γ = 77.010(2)°, V = 3365.1(4) Å3, Z = 2, ρ = 1.443 g cm−3, μ(Mo-Kα) = 1.302 mm−1, reflections: 45317 collected, 15733 unique, Rint = 0.060, R1(all) = 0.1102, wR2(all) = 0.1712, S = 1.024.Contributions
Mariusz P. Mitoraj has planned and partially performed (ETS-NOCV) the theoretical calculations, written the manuscript text and analyzed the entire data. Farhad Akbari Afkhami has primarily done the experimental part. Ghodrat Mahmaoudi has planned the experimental research. Ali Akbar Khandar has supported the work, whereas Atash V. Gurbanov has synthesized the compounds. Fedor I. Zubkov has also participated in the synthesis of the compounds. Rory Waterman is a crystallographer of compound 1-2, whereas Himanshu Sekhar Jena is a crystallographer of system 3. D W. Szczepanik has done the aromaticity calculations. Damir A. Safin and Maria G. Babashkina have analysed and discussed the results as well as have written the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the University of Maragheh for the financial support of this research. We also thank “RUDN University Program 5-100” for the support. This work was also partially supported by the National Science Centre, Poland (grant no. 2015/17/D/ST4/00558, D. W. S.). M. P. M. acknowledges the financial support of the Polish National Science Center within the Sonata Bis Project 2017/26/E/ST4/00104. X-ray facilities were provided by the U. S. National Science Foundation (CHE-1039436 to RW). H. S. J. thanks FWO [PEGASUS]2 Marie Sklodowska-Curie grant agreement no. 665501 for Incoming postdoctoral fellowship. D. W. S. acknowledges the European Union's Framework Programme for Research and Innovation Horizon 2020 (2014–2020) under the Marie Skłodowska-Curie Grant Agreement No. 797335 “MulArEffect”.References
- V. G. Machado, P. N. W. Baxter and J.-M. Lehn, J. Braz. Chem. Soc., 2001, 12, 431–462 CrossRef CAS.
- J. D. Watson and F. H. C. Crick, Nature, 1953, 171, 737–738 CrossRef CAS PubMed.
- J. Anastassopoulou and T. Theophanides, The Role of Metal Ions in Biological Systems and Medicine in Bioinorganic Chemistry. NATO ASI Series (Series C: Mathematical and Physical Sciences), Springer, Dordrecht, 1995, vol. 459, pp. 209–218 Search PubMed.
- F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochmann, Advanced Inorganic Chemistry, John Wiley and sons, New York, 6th edn, 1999 Search PubMed.
- G. Struckmeier, U. Thewalt and J.-H. Fuhrhop, J. Am. Chem. Soc., 1976, 98, 278–279 CrossRef CAS.
- C. Piguet, G. Bernardinelli and G. Hopfgartner, Chem. Rev., 1997, 97, 2005–2062 CrossRef CAS PubMed.
- M. Albrecht, Chem. Rev., 2001, 101, 3457–3498 CrossRef CAS PubMed.
- M. G. B. Drew, D. Parui, S. De, J. P. Naskar and D. Datta, Eur. J. Inorg. Chem., 2006, 4026–4028 CrossRef CAS.
- T. Riss-Johannessen, L. P. Harding, J. C. Jeffery, R. Moon and R. C. Rice, Dalton Trans., 2007, 1577–1587 RSC.
- Y. Wang, H. Fu, F. Shen, X. Sheng, A. Peng, Z. Gu, H. Ma, J. S. Ma and J. Yao, Inorg. Chem., 2007, 46, 3548–3556 CrossRef CAS PubMed.
- A. M. Stadler, N. Kyritsakas, G. Vanghan and J.-M. Lehn, Chem.–Eur. J., 2007, 13, 59–68 CrossRef CAS PubMed.
- M. G. B. Drew, S. De and D. Datta, Inorg. Chim. Acta, 2009, 362, 2487–2491 CrossRef CAS.
- T. Kaczorowski, I. Justyniak, T. Lipińska, J. Lipkowski and J. Lewiński, J. Am. Chem. Soc., 2009, 131, 5393–5395 CrossRef CAS PubMed.
- S. De, M. G. B. Drew and D. Datta, Inorg. Chim. Acta, 2010, 363, 4123–4126 CrossRef CAS.
- C. S. Tsang, C. C. Yee, S. M. Yiu, W. T. Wong and H. L. Kwong, Polyhedron, 2014, 83, 167–177 CrossRef CAS.
- M. Enamullah, M. A. Quddus, M. R. Hasan, G. Pescitelli, R. Berardozzi, G. Makhloufi, V. Vasylyeva and C. Janiak, Dalton Trans., 2016, 45, 667–680 RSC.
- M. Hasegawa, H. Ohtsu, D. Kodama, T. Kasai, S. Sakurai, A. Ishiia and K. Suzuki, New J. Chem., 2014, 23, 1225–1234 RSC.
- S. Chowdhury, M. G. B. Drew and D. Datta, New J. Chem., 2003, 27, 831–835 RSC.
- Q. Sun, Y. Bai, G. He, C. Duan, Z. Lin and Q. Meng, Chem. Commun., 2006, 2777–2779 RSC.
- M. G. B. Drew, D. Parui, S. De, S. Chowdhury and D. Datta, New J. Chem., 2007, 31, 1763–1768 RSC.
- G. Mahmoudi, V. Stilinović, M. S. Gargari, A. Bauzá, G. Zaragoza, W. Kaminsky, V. Lynch, D. Choquesillo-Lazarte, K. Sivakumar, A. A. Khandar and A. Frontera, CrystEngComm, 2015, 17, 3493–3502 RSC.
- A. Masoumi, M. S. Gargari, G. Mahmoudi, B. Machura, V. Lynch, G. Giester, M. Abedi and P. Hazendonk, Z. Anorg. Allg. Chem., 2015, 641, 1176–1181 CrossRef CAS.
- F. A. Afkhami, G. Mahmoudi, A. V. Gurbanov, F. I. Zubkov, F. Qu, A. Gupta and D. A. Safin, Dalton Trans., 2017, 46, 14888–14896 RSC.
- G. Mahmoudi, F. A. Afkhami, A. Castiñeiras, I. García-Santos, A. Gurbanov, F. I. Zubkov, M. P. Mitoraj, M. Kukułka, F. Sagan, D. W. Szczepanik, I. A. Konyaeva and D. A. Safin, Inorg. Chem., 2018, 57, 4395–4408 CrossRef CAS PubMed.
- F. A. Afkhami, G. Mahmoudi, J. M. White, J. Lipkowski, I. A. Konyaeva and D. A. Safin, Inorg. Chim. Acta, 2019, 484, 481–490 CrossRef.
- F. A. Afkhami, G. Mahmoudi, A. A. Khandar, J. M. White, I. A. Konyaeva and D. A. Safin, J. Mol. Struct., 2019, 1176, 743–750 CrossRef CAS.
- M. P. Mitoraj, G. Mahmoudi, F. Afkhami, A. Castineiras, G. Giester, I. A. Konyaeva, A. A. Khandar, F. Qu, A. Gupta, F. Sagan, D. W. Szczepanik and D. A. Safin, Cryst. Growth Des., 2019, 19, 1649–1659 CrossRef CAS.
- H. Zhang, X. M. Wang, K. C. Zhang and B. K. Teo, Coord. Chem. Rev., 1999, 183, 157–195 CrossRef CAS.
- W. P. Fehlhammer and W. Z. Beck, Z. Anorg. Allg. Chem., 2013, 639, 1053–1082 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part B, Wiley, N. Y., 5th edn, 1997, p. 116 Search PubMed.
- G. Mahmoudi, J. K. Zaręba, A. V. Gurbanov, A. Bauzá, F. I. Zubkov, M. Kubicki, V. Stilinović, V. Kinzhybalo and A. Frontera, Eur. J. Inorg. Chem., 2017, 4763–4772 CrossRef CAS.
- http://www.ee.ub.edu/index.php?option=com_content%26view=article%26id=72%26Itemid=469.
- S. Alvarez, D. Avnir, M. Llunell and M. Pinsky, New J. Chem., 2002, 26, 996–1009 RSC.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- J. Cirera, P. Alemany and S. Alvarez, Chem.–Eur. J., 2004, 10, 190–207 CrossRef CAS PubMed.
- S. Alvarez and M. Llunell, J. Chem. Soc., Dalton Trans., 2000, 3288–3303 RSC.
- M. P. Mitoraj, A. Michalak and T. Ziegler, J. Chem. Theory Comput., 2009, 5, 962–975 CrossRef CAS PubMed.
- E. J. Baerends, J. Autschbach, D. Bashford, A. Bérces, F. M. Bickelhaupt, C. Bo, P. M. Boerrigter, L. Cavallo, D. P. Chong, L. Deng, R. M. Dickson, D. E. Ellis, M. van Faassen, L. Fan, T. H. Fischer, C. Fonseca Guerra, A. Ghysels, A. Giammona, S. J. A. van Gisbergen, A. W. Götz, J. A. Groeneveld, O. V. Gritsenko, M. Grüning, F. E. Harris, P. van den Hoek, C. R. Jacob, H. Jacobsen, L. Jensen, G. van Kessel, F. Kootstra, M. V. Krykunov, E. van Lenthe, D. A. McCormack, A. Michalak, M. Mitoraj, J. Neugebauer, V. P. Nicu, L. Noodleman, V. P. Osinga, S. Patchkovskii, P. H. T. Philipsen, D. Post, C. C. Pye, W. Ravenek, J. I. Rodríguez, P. Ros, P. R. T. Schipper, G. Schreckenbach, M. Seth, J. G. Snijders, M. Solà, M. Swart, D. Swerhone, G. te Velde, P. Vernooijs, L. Versluis, L. Visscher, O. Visser, F. Wang, T. A. Wesolowski, E. M. van Wezenbeek, G. Wiesenekker, S. K. Wolff, T. K. Woo, A. L. Yakovlev and T. Ziegler, ADF2012.01, Theoretical Chemistry, Vrije Universiteit, Amsterdamsoft Search PubMed.
- O. A. Stasyuk, R. Sedlak, C. Fonseca Guerra and P. Hobza, J. Chem. Theory Comput., 2018, 14, 3440–3450 CrossRef CAS PubMed.
- I. Cukrowski, K. K. Govender, M. P. Mitoraj and M. Srebro, J. Phys. Chem. A, 2011, 115, 12746–12757 CrossRef CAS PubMed.
- I. Cukrowski, J. H. de Lange and M. P. Mitoraj, J. Phys. Chem. A, 2014, 118, 623–637 CrossRef CAS PubMed.
- J. P. Wagner and P. R. Schreiner, Angew. Chem., Int. Ed., 2015, 54, 12274–12296 CrossRef CAS PubMed.
- F. Sagan, Ł. Piękoś, M. Andrzejak and M. P. Mitoraj, Chem.–Eur. J., 2015, 21, 15299–15307 CrossRef CAS PubMed.
- D. A. Safin, M. G. Babashkina, K. Robeyns, M. P. Mitoraj, P. Kubisiak and Y. Garcia, Chem.–Eur. J., 2015, 21, 16679–16687 CrossRef CAS PubMed.
- F. Sagan, R. Filas and M. P. Mitoraj, Crystals, 2016, 6, 28 CrossRef.
- I. Cukrowski, F. Sagan and M. P. Mitoraj, J. Comput. Chem., 2016, 37, 2783–2798 CrossRef CAS PubMed.
- D. J. Liptrot and P. P. Power, Nat. Rev. Chem., 2017, 1, 0004 CrossRef.
- G. Bistoni, A. A. Auer and F. Neese, Chem.–Eur. J., 2017, 23, 865–873 CrossRef CAS PubMed.
- Q. Lu, F. Neese and G. Bistoni, Angew. Chem., Int. Ed., 2018, 57, 4760–4764 CrossRef CAS PubMed.
- M. P. Mitoraj, M. G. Babashkina, A. Y. Isaev, Y. M. Chichigina, K. Robeyns, Y. Garcia and D. A. Safin, Cryst. Growth Des., 2018, 18, 5385–5397 CrossRef CAS.
- P. M. Nowak, K. Olesek, M. Woźniakiewicz, M. Mitoraj, F. Sagan and P. Kościelniak, J. Chromatogr. A, 2018, 1580, 142–151 CrossRef CAS PubMed.
- G. Mahmoudi, E. Zangrando, M. P. Mitoraj, A. V. Gurbanov, F. I. Zubkov, M. Moosavifar, I. A. Konyaeva, A. M. Kirillov and D. A. Safin, New J. Chem., 2018, 42, 4959–4971 RSC.
- M. P. Mitoraj, F. Sagan, M. G. Babashkina, A. Y. Isaev, Y. M. Chichigina and D. A. Safin, Eur. J. Org. Chem., 2019, 493–503 CrossRef CAS.
- M. P. Mitoraj, M. G. Babashkina, K. Robeyns, F. Sagan, D. W. Szczepanik, Y. V. Seredina, Y. Garcia and D. A. Safin, Organometallics, 2019, 38, 1973–1981 CrossRef CAS.
- F. Sagan and M. P. Mitoraj, J. Phys. Chem. A, 2019, 123, 4616–4622 CrossRef CAS PubMed.
- F. Sagan and M. P. Mitoraj, Non-covalent Interactions in Selected Transition Metal Complexes in Transition Metals in Coordination Environments, Springer, 2019 Search PubMed.
- D. W. Szczepanik, E. J. Zak, K. Dyduch and J. Mrozek, Chem. Phys. Lett., 2014, 593, 154–159 CrossRef CAS.
- D. W. Szczepanik, M. Andrzejak, K. Dyduch, E. J. Zak, M. Makowski, G. Mazur and J. Mrozek, Phys. Chem. Chem. Phys., 2014, 16, 20514–20523 RSC.
- D. W. Szczepanik, Comput. Theor. Chem., 2016, 1080, 33–37 CrossRef CAS.
- D. W. Szczepanik, M. Andrzejak, J. Dominikowska, B. Pawełek, T. M. Krygowski, H. Szatylowicz and M. Solà, Phys. Chem. Chem. Phys., 2017, 19, 28970–28981 RSC.
- D. W. Szczepanik and M. Solà, ChemistryOpen, 2019, 8, 219–227 CrossRef CAS PubMed.
- R. Gershoni-Porannea and A. Stanger, Chem. Soc. Rev., 2015, 44, 6597–6615 RSC.
- D. Geuenich, K. Hess, F. Köhler and R. Herges, Chem. Rev., 2005, 105, 3758–3772 CrossRef CAS PubMed.
- Bruker Saint Plus, Saint Plus 8.34A, Bruker AXS Inc., Madison, Wisconsin, USA, 2007 Search PubMed.
- Bruker, SADABS, TWINABS, SADABS 2012/1, Bruker AXS Inc., Madison, Wisconsin, USA, 2001 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2008, 64, 112–122 CrossRef CAS.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S4, Tables S1–S4 and cif files. CCDC 1901201–1901203 For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra05276c |
| This journal is © The Royal Society of Chemistry 2019 |